

Abstract
Chronic, low-grade inflammation is linked to aging and has been termed “inflammaging”. Inflammaging is considered a key contributor to the development of metabolic dysfunction and a broad spectrum of diseases or disorders including declines in brain and heart function. Genome-wide association studies (GWAS) coupled with epigenome-wide association studies (EWAS) have shown the importance of diet in the development of chronic and age-related diseases. Moreover, dietary interventions e.g. caloric restriction can attenuate inflammation to delay and/or prevent these diseases. Common themes in these studies entail the use of phytochemicals (plant-derived compounds) or the production of short chain fatty acids (SCFAs) as epigenetic modifiers of DNA and histone proteins. Epigenetic modifications are dynamically regulated and as such, serve as potential therapeutic targets for the treatment or prevention of age-related disease. In this review, we will focus on the role for natural products that include phytochemicals and short chain fatty acids (SCFAs) as regulators of these epigenetic adaptations. Specifically, we discuss regulators of methylation, acetylation and acylation, in the protection from chronic inflammation driven metabolic dysfunction and deterioration of neurocognitive and cardiac function.
1. Introduction: aging-associated inflammation
“Inflammaging” is a phenomena of chronic low grade inflammation experienced with advanced age that is closely linked with the severity and progression of a variety of diseases.1,2 Inflammation is a tightly controlled and broadly conserved defensive response to injury and infection. The inflammatory response can be superficially characterized as swelling, redness, pain, heat and dysfunction around the affected area.3 On a cellular level, several mediators act to isolate the affected area, attack foreign pathogens and clear damaged cells or irritants. These mediators include immune cells (e.g., macrophages and leukocytes), recognition receptors (e.g., toll-like receptors (TLR)) and signaling molecules (e.g., pro-inflammatory cytokines and chemokines). These signals activate intracellular cascades, such as necrosis factor kappa B (NF-kB), mitogen-activated protein kinases (MAPKs) and Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathways, to upregulate the inflammatory response.4 Ultimately, in healthy individuals, this response leads to tissue repair, which triggers resolution of inflammation and restoration of homeostasis. Over time, however, as observed in aging, the inflammatory response loses its ability to properly reach resolution, which results in a progressive state of chronic inflammation, i.e., chronic, low-grade inflammation (CLGI). CLGI can cause cellular/molecular dysregulation as observed with cell senescence, disrupted proteolysis and apoptosis and subsequent tissue dysfunction and degeneration.5 Conversely, a homeostatic inflammatory environment promotes health and longevity.6,7
Recent reports link inflammatory dysregulation with epigenetic modifications.5 Traditionally, epigenetic modifications regulate differential gene expression independently from altering DNA sequences. Well-known epigenetic modifications occur on DNA and histone proteins and include DNA methylation as well as histone methylation, acetylation and acylation. Moreover, there are well-described catalytic enzymes that add/remove these epigenetic modifications, which include: DNA methyltransferases (DNMTs) for DNA methylation; histone methyltransferases for histone methylation; histone demethylases (HDMs) for histone demethylation; histone acetyltransferases (HATs) for histone acetylation; and histone deacetylases (HDACs) for histone deacetylation. DNMTs, such as DNMT1, promote gene repression by forming hypermethylation-induced heterochromatin (chromatin condensation),8 inhibiting transcription factors from binding at promoter sites on genomic DNA9 as well as coupling with other gene repressing proteins such as HDACs.10 Histone deacetylation is regulated by HDACs, which mediate histone hypoacetylation towards heterochromatin and gene suppression, and HATs, which mediate histone hyperacetylation towards euchromatin and gene upregulation. In contrast to histone acetylation, which primarily occurs on lysine residues, histone methylation can occur on lysine, arginine and several other amino acid residues on histone protein tails, subsequently resulting in either gene expression or suppression.
Understanding epigenetic modifications in human-related diseases is becoming more relevant, and advancements in “-omics” technologies have allowed scientists to gather data regarding epigenetic modification in age-associated diseases. For example, epigenome-wide association studies (EWAS) correlated DNA hypomethylation with increased inflammation and frailty.11,12 Conversely, delayed hypomethylation was noted in children of centenarians as was better health status when compared to their counterparts.13,14 Moreover, several inflammatory signaling cascades are regulated in part via epigenetic modifications.15–18 Thus, epigenetic modifications play an integral part in healthy aging and inflammation. Below we will discuss in detail how diet, lifestyle and natural products can alter metabolites that contribute to changes in the epigenome during ‘inflammaging’ (Fig. 1).
Fig. 1.
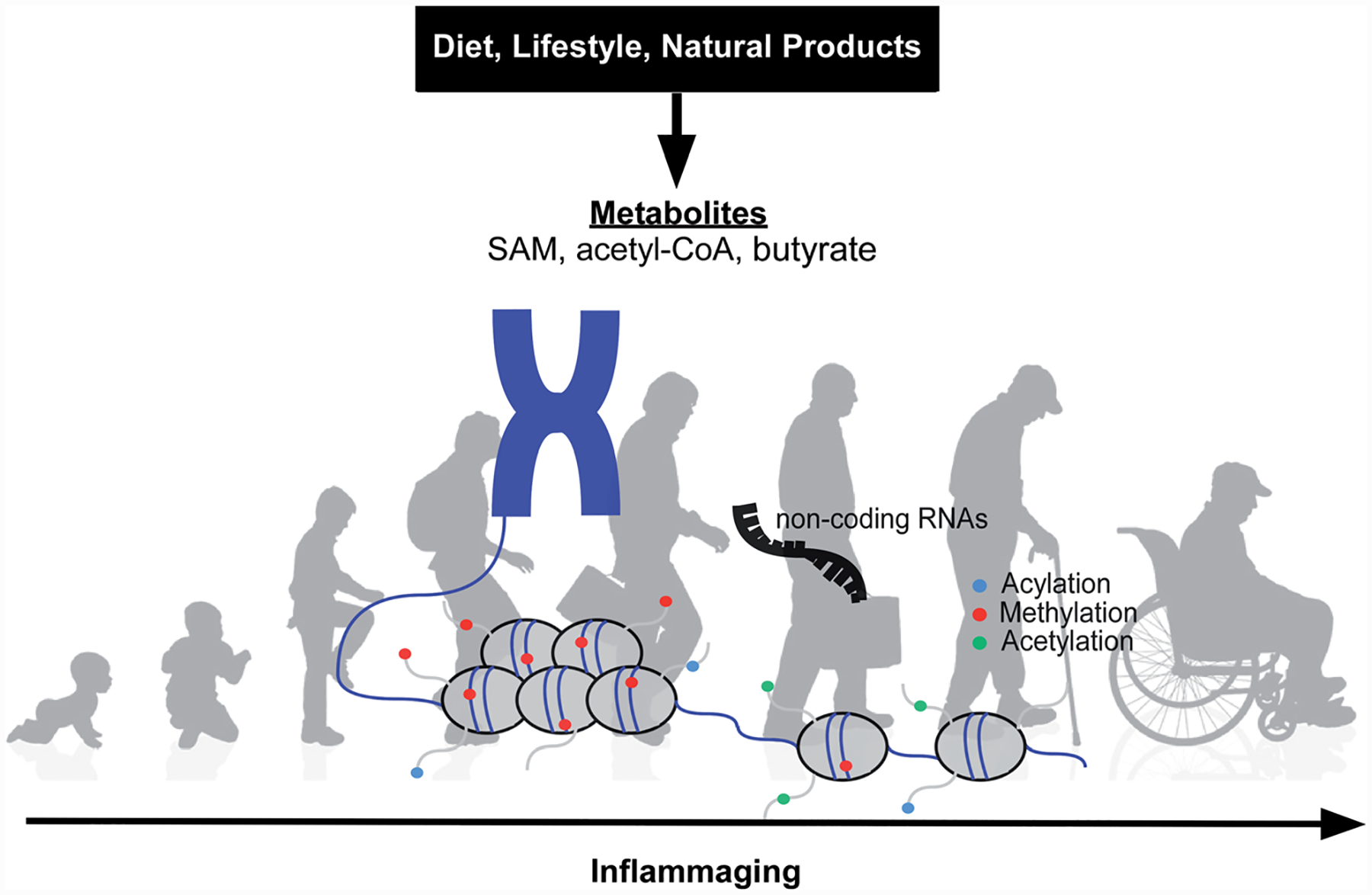
Diet, lifestyle and natural product supplementation can alter key metabolites necessary for histone and DNA modifications; these epigenetic adaptations contribute towards the prevention (delay) or promotion of ‘inflammaging’ that ultimately drives age-related chronic diseases (e.g. CVD, diabetes, Alzheimer’s Disease).
1.1. Aging-related metabolic dysfunction
As with inflammation, systemic control over metabolic processes decline with age and affect adipose, liver, pancreas and skeletal muscle function. A primary outcome of metabolic dysfunction is insulin resistance, resulting in an impaired ability to regulate blood glucose through critical signaling pathways (i.e., type 2 diabetes (T2D)). Additional outcomes include dysregulated gluconeogenesis in liver; mitochondrial dysfunction in many tissues; lipogenesis that occurs in adipose, liver and skeletal muscle; as well as dyslipidemia and glycogenolysis in liver.19 Outside of T2D, metabolic dysregulation can result in obesity, non-alcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD) risk and other age-associated diseases including neurodegeneration. NAFLD, often comorbid with obesity and T2D,20 is associated with increased production of damaging cytokines and chemokines.21 Several cellular mediators link age-associated metabolic dysfunction to inflammation. For example, the peroxisome proliferator-activated receptors (PPAR; PPARγ, PPARα, PPARd) are a family of nuclear receptors that drive transcriptional events involved in fatty acid and glucose metabolism but, importantly, decline with age.22 Moreover, PPARs play a critical role in combating liver- and adipocyte-derived pro-inflammatory cytokines and chemokines.23 This is interesting as pro-inflammatory pathways (e.g., NF-kB,24 JNK25 and the NOD-like receptor protein 3 (NLRP3) inflammasome26) hinder insulin signaling. Therefore, metabolic dysfunction is observed with age and inflammation exacerbates substrate metabolism defects.
Several pathologies outside of obesity and T2D are associated with substrate metabolism dysfunction. For example, while the brain primarily uses glucose as its source for ATP production, metabolic dysregulation of glucose occurs in the brain of patients with age-associated neurodegenerative diseases.27,28 Conversely, the heart predominantly utilizes fatty acid oxidation (60–90%) for its energy source but shifts to glucose in pathological settings.29 Metabolic dysregulation of all major substrates (i.e., fatty acids, glucose and protein) is also observed in cancer.30 Not surprisingly, it has been suggested that influencing differential genomic expression of factors, such as enzymes, involved in metabolic regulation may offer some benefit.31 Therefore, dietary interventions that modulate such factors via epigenetic actions are worth further exploration.
Acetyl Coenzyme A (Acetyl CoA), nicotinamide adenine dinucleotide (NAD+) and S-adenosyl methionine (SAM) are metabolites regulated by energy metabolism, yet also function in epigenetic modifications. Availability of these metabolites regulate differential acetylation and methylation status, which impacts gene expression.32–34 This is interesting because substrate overconsumption (i.e. carbohydrates, protein and fat) can initiate metabolic dysfunction, implying that diet-derived DNA and/or histone modification influences obesity, T2D or NAFLD. Indeed, studies have uncovered alterations in epigenetic enzymes and modifications that delineate metabolic abnormalities. For example, the class III, NAD+-dependent HDAC, sirtuin1 was reduced in obese individuals.35 The sirtuin family has been well-described in age-associated metabolic dysfunction, partially due to its actions in mitochondria.36 Another report associated DNA hypomethylation with T2D.37 From these data, it is clear that ‘inflammaging’ contributes to epigenetic modifications resulting in metabolic disease; this ultimately can contribute to neuro-impairment and cardiovascular disease.
1.2. Aging, inflammation and neurodegeneration
Incidences of neurodegenerative diseases have increased significantly in recent years due to increased life span; these include Parkinson’s Disease (PD) and Alzheimer’s Disease (AD), the latter of which is a leading cause of mortality.38 Accumulating evidence now demonstrates that inflammation is a pivotal driver of neurodegeneration.39
Barriers separate the brain from the circulatory system, namely the blood–brain barrier (BBB) and the blood-cerebrospinal fluid barrier (BCSFB), which ultimately prevent outside offenders, such as mediators of systemic inflammation, from brain intrusion. A tightly controlled system at these brain barriers exists, which includes transport proteins and receptor-mediated signaling to allow an influx and efflux of nutrients, molecules and waste between the brain and the circulatory system. The brain contains a specialized inflammatory response system with similar characteristics and mediators to the immune system found in the periphery. In the brain, astrocytes, microglia and perivascular macrophages and leukocytes engage molecular receptors such as toll-like receptors (TLRs) in response to a pathological assault,40 which upregulates NF-kB, MAPK and JAK/STAT pathways.4 T cells further regulate the inflammatory response by mediating cytokine and chemokine production in the brain, and can force macrophage hyperpolarization to a pro-inflammatory state.41 As with the periphery, microglia macrophages and other mechanisms clear unwanted debris resulting from the insult. These defense systems in the brain can also become dysfunctional over time. Barriers separating the brain from the body become compromised thus allow leakage and pathogenic infiltration.42 Chronic, low grade inflammation stemming from the circulatory system can stimulate inflammation in the brain,43 and thus contribute to a pro-inflammatory environment. Brain inflammation contributes to receptor-mediated degradation of myelinated axons and neurons.44 Unwanted, and oftentimes damaging, proteins aggregate in the brain due to dysfunctional phagosomes, as observed with β (Aβ) and tau amyloid protein accumulation in neurodegenerative diseases. Such dysfunctions affect cognitive performance and downstream systemic performance such as muscle control; this may even result in death.45,46
Several epigenetic modifications are affected in neurodegenerative environments. For example, DNA methylation status of genes involved in neurogenesis were altered in patients with PD.47,48 In agreement, differential DNA methylation of CpG islands in the genes, reelin and protein phosphatase 1 (PP1), was shown to be important for cognitive performance.49 In relation to histone acetylation, the repressor element-1 silencing transcription factor (REST) is a corepressor complex that binds and recruits other components, namely HDACs 1 and 2, to transcriptional regions towards gene downregulation.50 While important during embryogenesis,51 REST has been implicated in mature neuron cell death within ischemia,52 epileptic seizure53 and Huntington’s Disease models.54 REST suppressed genes associated with neuronal synaptic function and drove neuron cell death in an in vivo clinical model of global ischemia, while HDAC inhibitor treatment abated these insults.50 This is interesting because HDAC inhibitor treatment typically dampens inflammation and the HDAC inhibitor, valproic acid, was the first of its kind to be offered as a prescription for neurological dysfunction.55 Highlighting the cooperative nature of acetylation and methylation in gene expression regulation, blocking histone hypermethylation via methyltransferase-inhibition has also been shown to reduce neuroinflammation.56,57 Class III HDACs, the sirtuin family, have a more complex role in neurodegeneration. Sirt1 overexpression inhibited NF-kB-mediated inflammation and Aβ-mediated neurodegenerative actions in microglia.58 Moreover, Sirt6 was depleted in brains of AD patients, while Sirt6 overexpression prevented agonist-induced DNA damage in mouse neurons.59 In contrast, nicotinamide, a pan sirtuin inhibitor, blocked neurodegeneration and cognitive defects concomitant with increased acetylation of common sirtuin targets in AD mice.60 Similarly confounding, Sirt6 overexpression in the hippocampal CA1 region was shown to impair cognitive performance.61 It would seem whether sirtuin-mediated actions are neuroprotective or neurodegenerative is contextual. The cumulative evidence, still, suggests epigenetic regulation of neuro-inflammation and neurodegeneration is promising and worth further exploration.
1.3. Aging, inflammation and cardiovascular disease
Cardiovascular diseases (CVD) remain the leading cause of death world-wide and include heart attack, arrhythmias, atherosclerosis and heart failure. Many well-characterized pathogeneses of the heart manifest from inflammation, and pro-inflammatory markers in circulation are used to predict CVD risk.62 One of the most common of these inflammation-induced heart diseases is atherosclerosis. Chronic, low grade inflammation results in endothelium insult that upregulates the inflammatory response around the affected site.63 There, macrophages take up oxidized apolipoprotein B (ApoB)-containing lipoproteins that accumulate in the subendothelial space. The macrophages then transform into foam cells, which are primary constituents of atherosclerotic plaques. Intriguingly, rupture or erosion of atherosclerotic plaques initiate myocardial infarction (MI), both of which mediate an ongoing inflammatory response. Following MI, cardiomyocytes suffer ischemic injury or death, which further exacerbates inflammation and cardiac remodeling,64,65 a common hallmark of heart failure. Cytokines produced from aforementioned cardiac insults activate signaling cascades involved in heart failure pathology, including cardiac hypertrophy, fibrosis and dysfunction.66,67 These data suggest inflammation drives cardiac pathologies and is a prime target for CVD therapy.
Epigenetic modifications have been characterized in CVD. A recent epigenome-wide association study (EWAS) in myocardial infarct patients associated DNA methylation at several gene promoter regions with CVD risk including associated blood lipid levels that contribute to atherosclerotic events.68 Further studies have also linked hypertension and inflammation with methylation status.69 While these studies focused on methylation, others have reported changes in acetylation. For example, sirtuin levels were reduced in CVD patients, linking changes in acetylation to CVD.70 Much like in age-associated diseases described in above sections, sirtuin deacetylase activity has been shown to have cardioprotective effects.71 Consistent with these findings, histone acetyltransferase activity of p300 was shown to be necessary for agonist-induced cardiac hyper-trophy.72 Conversely, HDAC inhibition and histone hyperacetylation have been shown to attenuate cardiac/cardiomyocyte hypertrophy and associated fetal gene re-expression,73,74 cardiac fibrosis75 and cardiac dysfunction.76 Class I HDACs seem to be necessary for the pathological cardiac hypertrophy as well as inflammatory signaling.18,77 Likewise, recent reports demonstrate an important role for HDACs in age-related diastolic heart failure, where HDAC inhibition improved diastolic function.78 These data combined, demonstrate an epigenetic role for DNA methylation and histone modification in CVD risk and development, yet further studies are needed to determine the various molecular targets of the HAT and HDAC enzymes that relay different outcomes.
2. Phytochemicals in aging-associated inflammation
Phytochemicals are compounds synthesized by plants for protection against pests, pathogens and UV light.79 Upon human consumption, phytochemicals interact in biological processes such as redox reactions,80–82 cell signaling83 and, of interest, inflammation.82 Indeed, diets rich in phytochemicals have been associated with reduced inflammation and, in doing so, may mitigate inflammaging-associated diseases.84 Indeed, plant based diets have been shown to reduce the risk of developing metabolic dysfunction,85 neurodegeneration,86 and cardiovascular disease.87 An emerging body of evidence suggests that bioactive compounds impart advantageous environments, such as homeostatic inflammation, through differential gene expression.88,89 This suggests phytochemicals can mitigate deleterious gene expression (e.g., pro-inflammatory genes) and promote beneficial gene expression (e.g., anti-inflammatory genes). Moreover, phytochemicals have been implicated in regulating gene expression through epigenetic modifications. In this section, we will discuss current studies linking phytochemical-mediated epigenetic modifications to age-associated diseases. These and additional compounds are described in Table 1 and basic structures for these compounds discussed are depicted in Fig. 2.
Table 1.
Natural product regulators of the epigenome, their gene targets and their structures
| Epigenetic modification | Natural product | Polyphenolic group | Epigenetic mode of action | Target and effect | References |
|---|---|---|---|---|---|
| DNA methylation | Curcumin | Curcuminoid | ↓PPARα promoter region DNA methylation | ↑PPARα expression; ↓DNMT activity, liver steatosis, abdomical fat, LDL cholesterol and triglycerides | 92 and 93 |
| ↓DNMT activity | Reversed pathological DNMT hyperactivity in retina cells | 94 | |||
| Sulforaphane | Isothiocyanate | ↓DNMT activity and DNA methylation | ↑Nrf2 expression; ↓neurological inflammation (IL-1β and IL-6) and Aβ accumulation | 109 | |
| EGCG | Catechin | ↓DNA hypermethylation; ↓Grhl3, Pax3, and Tulp3 CpG island hypermethylation; ↓DNMT3a and 3b expression | ↓Diabetes-induced neural tube defects; ↑Grhl3, Pax3, and Tulp3 expression | 136 and 137 | |
| Quercetin | Flavonol | ↓DNA hypermethylation at Pgc-lα promoter | ↓HFD-induced Pgc-lα expression in rodents | 138 | |
| Resveratrol | Stilbene | ↑DNMT activity; ↑DNMT1, 3a and 3b expression; ↑LINE-1 methylation | Reversed oxidative stress- and inflammation-induced changes in ARPE-19 cells | 139 | |
| Ascorbic acid | NA; micronutrient | ↑TET (DNA demethylase) activity; ↓methylation of Foxp3 CpG enhancer region | ↑Foxp3 expression and Treg cell differentiation; ↓Treg senescence | 121, 122, 127 and 128 | |
| Histone methylation | Emodin | Anthraquinone | ↑H3 K27 trimethylation | ↓TNFα, IL6, iNOS, IRF4, Arg1 and YM1; ↓IRF5, NF-κBp65 and STAT1 nuclear activity in M1 macrophages; ↓STAT6 and IRF4 nuclear activity in M2 macrophages | 146 |
| Ellagic acid | Hydroxybenzoic acid | ↓CARM1 activity and H3 R17 dimethylation | ↓PPARγ activity, adipose inflammation and adipose differentiation | 158 and 161 | |
| Ascorbic acid | NA; micronutrient | ↑JMJD3 histone demethylase activity; ↓methylation of H3K9me2, H3K27me2, H3K36me2/3 | ↓Cell senescence via Ink4/Arf; ↑cell cycle progression | 129, 133 and 134 | |
| Histone acetylation | Resveratrol | Stilbene | ↑Sirtuin activity | ↑Longevity in numberous cell types | 167 and 169–176 |
| ↑Sirtuin activity | ↑PPARγ, PGC-1α, GLUT4 and AMPK/IRS/Akt expression; ↓ adipocyte inflammation, triglyceride accumulation and insulin resistance in adipocytes | 177–179 | |||
| ↑Sirtuin activity; ↓MMP9 promoter region acetylation | ↓MAPK, NF-κB and MMP9 signaling; potentially neuro- and metabolically protective | 181 and 186–188 | |||
| ↑Sirtuin activity | ↓Oxidative stress in T2D patients | 193 and 194 | |||
| Sulforaphane | Isothiocyanate | ↑H3 and H4 acetylation at promoter regions I, II and IV of BDNF | ↑BDNF, TrkB and TrkB target expression in cortical neurons and AD-induced mice | 196 | |
| Emodin | Anthraquinone | ↓Class I & II HDAC activity; ↑ histone acetylation | Potentially cardioprotective, particularly through NF-κB | 205 | |
| Curcumin | Curcuminoid | ↓p300 HAT activity | ↓GATA4 signaling, cardio hypertrophic gene expression and cardiac hypertrophy | 209–211 | |
| ↓HDAC1 activity; ↑TIMPl promoter region acetylation | ↑TIMP1 expression; ↓MMP2, cardiac fibrosis and inflammation | 218 | |||
| EGCG | Catechin | ↓Histone acetylation, p300 and HDACs 1 & 2 binding at NF-κBp65 promoter regions | ↓Inflammation in endothelial cells | 225 | |
| ↓Histone acetylation at FOXO1 promoter region | ↓FOXO1 nuclear activity and autophagy in hypoxic cardiomyoblasts | 229 | |||
| ↑Acetylation at cTnI promoter region | ↑cTnI expression and cardiac diastolic function | 233 | |||
| Anthocyanins | Flavanoid | ↑H3 acetylation at K9, K14 and K18; ↓HDAC activity; ↓HAT activity | ↓Fibrosis, AST, ALT, IL1β, MCP1, MIP-2, TIMP1, MMP-9 (damage) in rodent liver disease model; ↓TNFα; ↑PPARα and fatty acid catabolism | 239, 240 and 244–246 |
Fig. 2.
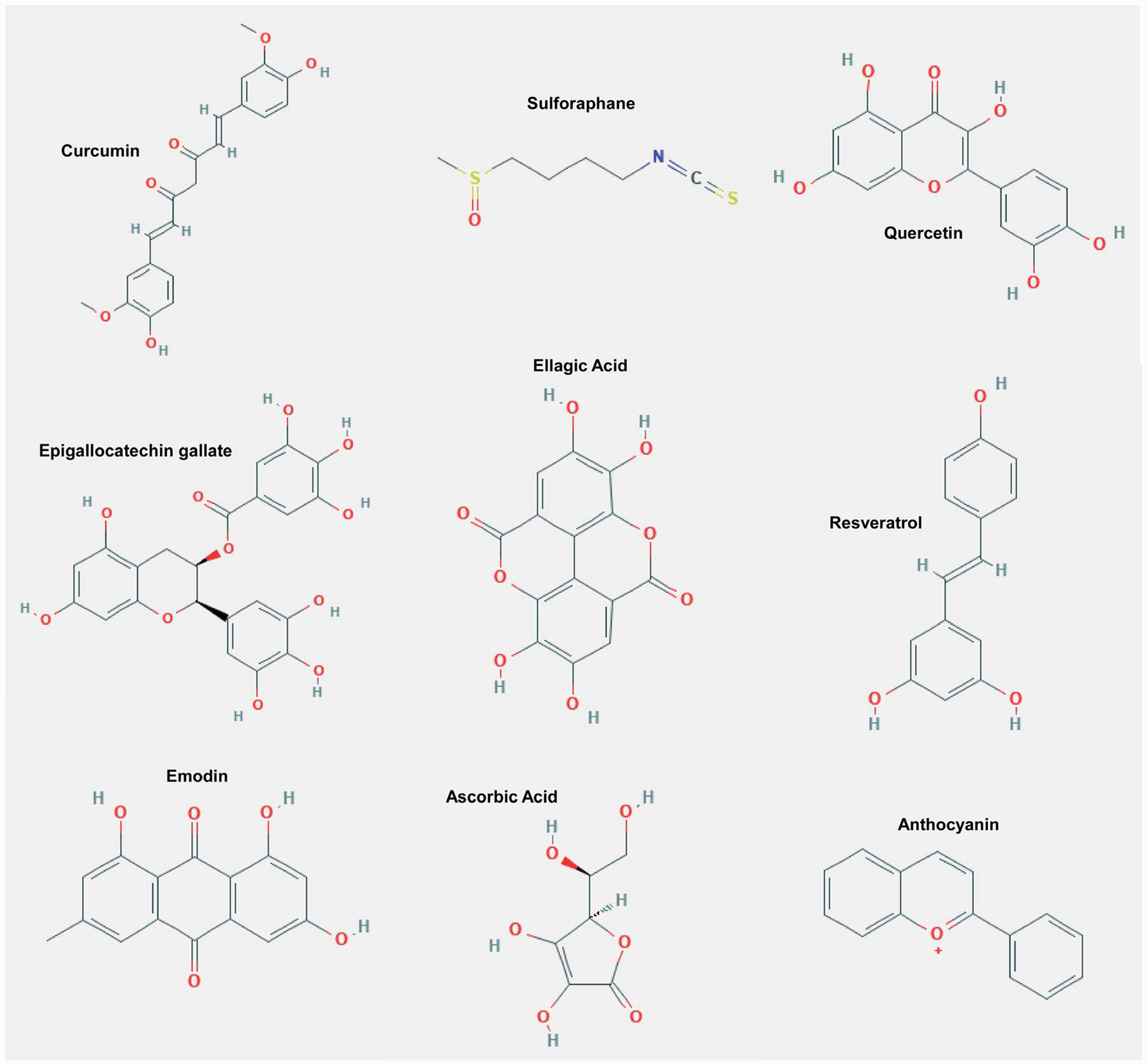
The basic structure of various phytochemicals and micronutrients important for histone and DNA modifications (epigenetic adaptations) that have been shown to prevent or delay age-related chronic diseases.
2.1. Phytochemicals – DNA methylation and gene expression
2.1.1. Curcumin and DNA methylation.
Curcumin is a polyphenolic curcuminoid found in the yellow pigmented plant, turmeric, and has been used for centuries as a medicinal herb. Curcumin is one of the more well-studied phytochemicals and has been shown to attenuate metabolic dysfunction.90,91 Of interest, several studies suggest curcumin is metabolically efficacious by regulating DNA methylation.92 For example, curcumin was shown to reduce DNA hypermethylation at CpG sites [C0] 360, [C0]341, [C0]329, [C0]316 and [C0]307 at the promoter region of peroxisome proliferator-activated receptor-alpha (PPARα) in a non-alcoholic fatty liver disease (NAFLD) rat model.93 These data correlated with increased PPARα expression and reduced signs of liver cell death.93 The PPARα-dependent protective effects of curcumin in rat liver congruently occurred in steatosis-induced liver cells.93 As reversal of DNA methyl transferase (DNMT)-mediated DNA hypermethylation is associated with gene repression, curcumin likely upregulated PPARα by inhibiting DNMTs.94 PPARα is a transcription factor that predominates in the liver where it regulates expression of genes involved in inflammation and several metabolic processes including gluconeogenesis, cholesterol metabolism and xeno-biotic bioavailability.95–99 Reduced PPARα results in increased adiposity and pro-inflammatory adipokines and is correlated with aging.23,100,101 Moreover, liver steatosis is prevented by PPARα-dependent fatty acid catabolism.97,99 Consistent with this, curcumin-induced PPARα expression resulted in differential expression of several PPARα targeted genes involved in cholesterol metabolism and insulin sensitivity in NAFLD-induced liver of broiler chickens.102 These results were linked to reduced abdominal fat, LDL cholesterol and triglycerides, suggesting that the PPARα-dependent effects of curcumin were observed on a physiological level.102 Others have also reported that curcumin mediates metabolic protection by regulating DNA methylation.94 In this report, curcumin attenuated high-glucose-induced DNMT mRNA overexpression, which subsequently reduced DNMT activity in a human cell model of diabetic retinopathy.94 This is important because diabetic retinopathy is a common complication for inflammaging-associated metabolic dysfunction and is implicated in CVD.103 These combined data support the postulate that curcumin is metabolically protective by mitigating DNMT activity. Furthermore, as PPARα is reduced with aging, curcumin may attenuate the deleterious actions associated with age-related depletion of PPARα, including liver dysfunction and adiposity.
2.1.2. Sulforaphane and DNA methylation.
Sulforaphane is an isothiocyanate predominantly found in cruciferous vegetables such as broccoli. Sulforaphane has repeatedly been shown to inhibit cancer cell growth as well as attenuate other age-associated diseases; these actions were regulated in part by inhibiting inflammation by changing DNA methylation.104–108 It is of no surprise, then, that sulforaphane was shown to attenuate DNMT-mediated DNA hypermethylation at the promoter region of NF-E2-related factor 2 (Nrf2), which subsequently reactivated nuclear Nrf2 in a cell model of Alzheimer’s Disease (AD).109 Nrf2 is a transcription factor that regulates reduction– oxidation homeostasis upon nuclear translocation and has been implicated in inflammation and neurodegeneration.110–114 From the report above, researchers further showed that sulforaphane inhibited pro-inflammatory signaling (IL-1β and IL-6) and β amyloid (Aβ) protein accumulation as well as oxidative damage.109 Moreover, the anti-inflammatory actions of sulforaphane diminished upon Nrf2 knockdown, demonstrating that DNA hypomethylation of the Nrf2 promoter with sulforaphane treatment was sufficient to inhibit Aβ accumulation.109 Others have also shown that sulforaphane regulates inflammation via Nrf2-dependent changes in gene expression.115,116 These data suggest that sulforaphane protects from AD-like neurodegeneration by blocking pro-inflammatory signaling through Nrf2-dependent actions and that sulforaphane regulates Nrf2 activity via DNA hypomethylation.
2.1.3. Ascorbic acid and DNA methylation.
Ascorbic acid (i.e., vitamin C) is a micronutrient, which cannot be synthesized by humans due to L-gulono-g-lactone oxidase dysfunction. Thus, the ascorbic acid deficiency disease, scurvy occurs when ascorbic acid-rich foods (e.g., fruits, vegetables and some animal organs) are underconsumed and ascorbic acid plasma concentrations reach less than 11 mM.117 Outside of preventing its deficiency disease, ascorbic acid is a potent reducing agent with four hydroxyl groups in its chemical structure. It comes as no surprise, then, that ascorbic acid is a suggested remedy for inflammaging-associated diseases,118,119 as oxidation is a key driver of inflammation.120 However, emerging evidence suggests that ascorbic acid attenuates ‘inflamaging’ independent of anti-oxidation;121 instead ascorbic acid has been shown to alter the epigenome. For example, ascorbic acid was shown to demethylate CpG-rich conserved noncoding sequence 2 (CNS2) of the transcription factor forkhead box protein 3 (Foxp3) in T regulatory cells (Treg).122 These actions were dependent on the ten-eleven translocation (TET) oxidative demethylation protein, Tet2.122 CNS2 methylation inversely correlates with Foxp3 expression,123 which was shown to regulate the physiological function of Treg cells.124 These data are interesting as functional non-senescent Treg cells play an important anti-inflammatory role.125 Moreover, immunesenescence and senescence of other cell types, indicative of inflammaging, increases in the aging populace.126 Thus, ascorbic acid may combat an aging pro-inflammatory state by regulating immune cell function that is dependent on demethylase activation.
Others have also linked inhibited cell senescence and enhanced cell pluripotency and differentiation with DNA or histone demethylase-dependent ascorbic acid-induced demethylation.121,127–129 Indeed, hyper di- and/or tri-methylation on histone H3 lysine residues K9, K27 and K36, which increased senescence and inhibited cell pluripotency and differentiation,130–132 is reversed by ascorbic acid.129,133,134 Ascorbic acid-induced hypomethylation and differentiation has further been linked to reduced DNA methyltransferase (DNMT) expression in cardiac stem cells.135 These data suggest that ascorbic acid may not only attenuate inflammaging-associated diseases through DNA and histone demethylation, but may also be a (co-)therapeutic for stem cell rejuvenation of tissues susceptible to age-associated dysfunction.
While this section only focused on two well-studied phytochemicals and one micronutrient, it should be noted that many natural products have been reported to regulate DNA methylation including epigallocatechin gallate (EGCG),136,137 quer cetin138 and resveratrol.139 As DNA methylation methodology is commonly used outside of experimental animal models, it would be interesting to examine the effects of individual natural products on DNA methylation and inflammaging-associated cascades in humans.
2.2. Phytochemicals – histone methylation and gene expression
2.2.1. Emodin and histone methylation.
Emodin is an anthraquinone found in plants used in traditional Chinese medicines, such as buckthorn, knotweed, rhubarb and Da Huang, which is a primary ingredient for the decoction, Dahuang Fuzi,140 as well as other plant-based foods like beans and cabbage.141,142 As herbal remedies, emodin-rich plants have been used for viral and bacterial insults, kidney disorders and gastrointestinal distress in traditional Chinese medicine. In relation to epigenetics, early reports showed emodin was efficacious in cancer models by regulating DNA methylation.143–145 However, recent evidence showed that emodin was able to inhibit the removal of histone H3K27 trimethylation in macrophages exposed to inflammatory stress.146 This was particularly apparent at promoter regions of inflammatory signaling molecules, inducible nitric oxide synthase (iNOS), tumor necrosis factor alpha (TNFa), interleukin 6 (IL6), interferon regulatory factor 4 (IRF4), arginase 1 (Arg1) and chitinase 3-like protein 3 (YM1) in M1 or M2 macrophage phenotypes.146 Further, emodin inhibited nuclear translocation of interferon regulatory factor 5 (IRF5), nuclear factor kappa B p65 (NF-kBp65) and signal transducer and activator of transcription 1 (STAT1) in M1 macrophages as well as STAT6 and IRF4 in M2 macrophages.146 Activation of macrophages by pro-inflammatory signaling molecules is important to the anti-inflammatory response.147 However, macrophage hyperpolarization into either M1 or M2 phenotypes is deleterious to health.147,148 The M1 hyperpolarized macrophage is of particular interest in neurodegenerative disease and has been suggested as a target for phytochemicals.149 Indeed, emodin has been shown to be protective in multiple neurodegenerative disease models, including cerebral ischemic stroke,150 traumatic brain injury151 and AD.152 In the above report, emodin restored the balance away from M1 and M2 hyperpolarization phenotypes146 and thus suggests that emodin may provide protection against brain pathologies through epigenetic balancing of macrophage activation versus hyperpolarization.
2.2.2. Ellagic acid and histone methylation.
Ellagic acid is a polyphenolic hydroxybenzoic acid derivative found ubiquitously in many fruits, such as raspberries and strawberries, nuts, such as walnuts and pecans and mushrooms, namely the ox tongue mushroom. Ellagic acid has profound anti-inflammatory effects.153–156 Of interest, Western diet-induced inflammation and adiposity have been shown to be reversed with ellagic acid treatment.153,156 Excess accumulation of adipose tissue, or obesity, commonly initiates downstream inflammation and metabolic dysfunction157 and has become a primary target to resolve metabolic syndrome, T2D and CVD. In relation to epigenetic regulation of metabolic dysfunction, ellagic acid was reported to inhibit coactivator-associated arginine methyltransferase 1 (CARM1) activity,158 which is an intricate methyltransferase enzyme implicated in both NF-κB-mediated inflammation and inflammation-associated metabolic dysfunction.159,160 Indeed, ellagic acid has been shown to attenuate NF-κB activity.155 Consistent with this, ellagic acid was reported to attenuate differentiation-induced hyper-dimethylation of histone 3 arginine 17 independent from CARM1 expression in human adipose-derived stem cells.161 However, PPARγ, a CARM1 target, was also downregulated upon ellagic acid treatment in differentiated adipocytes.161 PPARγ is an important regulator of adipogenesis and adipocyte function, agonistically targeted for T2D and atherosclerosis treatment and is coactivated in part by CARM1-mediated histone methylation.162–164 However, PPARγ activation results in increased adiposity,165 and thus some question the overall efficacy of PPARγ agonists for metabolic dysfunction.163 Thus, ellagic acid-mediated downregulation of PPARγ may, in fact, be chronically advantageous. On the other hand, others have shown the anti-inflammatory actions of ellagic acid were partially PPARγ-dependent.166 Further exploration is nevertheless required to fully elucidate the molecular targets of ellagic acid, if its efficacy outweigh the detriments in chronic inflammation/age-associated diseases and how it regulates these actions through epigenetic-mediated mechanisms.
2.3. Phytochemicals – histone acetylation and gene expression
2.3.1. Resveratrol and histone acetylation.
Arguably one of the most well-studied natural dietary products that regulates histone acetylation and health is the stilbene, resveratrol. Resveratrol is found mainly in grapes and wine but also in berries, nuts and cocoa. Resveratrol potently activates sirtuins, which are the nicotinamide adenine dinucleotide (NAD+)-dependent class III HDACs.167 Sirtuins are ubiquitously expressed and can be found localized to the nucleus, cytoplasm and mitochondria; thus, sirtuins are involved in chromatin remodeling, DNA stability, differential gene expression and metabolic regulation.168 Studies across numerous models have reported the anti-aging effects of resveratrol-mediated sirtuin activation.167,169–176 These studies suggest resveratrol promotes longevity and health in a conserved manner, and does so mostly through Sirt1-dependent mechanisms.177,178
Indeed, resveratrol has been shown to reverse age-associated repression of genes that regulate free fatty acid metabolism and β-oxidation (PPARγ and its coactivator, PGC-1α) in differentiated adipocytes.177 These data were linked with reduced triglyceride accumulation upon resveratrol-mediated sirtuin activation.177 Consistently, resveratrol upregulated genes responsible for glucose sensitivity (AMPK/IRS/Akt signaling, PPARγ and GLUT4), while it inhibited adipokine inflammation within insulin-resistant adipocytes in vitro and in vivo.178 Others have also reported that resveratrol-induced sirtuin activation improved glucose sensitivity by upregulating GLUT4 in liver and muscle of metabolically compromised mice.179 These data suggest resveratrol attenuated age-associated metabolic dysregulation.
In addition to its role in regulating insulin signaling, resveratrol has been shown to inhibit inflammation in part through regulation of the signaling cascades NF-κB and mitogen-activated protein kinases (MAPKs)180 as well as through regulation of histone deacetylation-dependent gene expression.181 Specifically, resveratrol was shown to deacetylate the promoter region of matrix metalloproteinase 9 (MMP9) and subsequently downregulate MMP9 expression.181 Upon Sirt1 knockout, resveratrol no longer attenuated MMP9 expression.181 MMP9 is an endoproteinase involved in inflammation-induced tissue remodeling that is overly activated in CVD, neurodegeneration and diabetes.182–184 While no study has linked the cardioprotective benefits of resveratrol and MMP9,185 several reports suggest that resveratrol-mediated suppression of MMP9 is protective in models of diabetes and neurodegeneration.186–188 The MAPK, c-Jun N-terminal kinases (JNK), and NF-κB bind the promoter region of, and thus activate, MMP9.189 As resveratrol has been shown to inhibit JNK and NF-kB,190,191 the stilbene may further attenuate MMP9 activity independent of its histone deacetylase activity. However, it should be noted that JNK18 and NF-kB16,17 have been shown to bind to HDAC complexes and their activity regulated by HDACs or methyltransferases, suggesting that resveratrol may also regulate the activity of these molecules by acting on writer or eraser enzymes (e.g. HDACs). Consistent with this, resveratrol has been reported to regulate other epigenetic modifying enzymes, including zinc-dependent HDACs and DNMTs, in age-associated disease models.139,192
Despite the positive findings in animal models however, clinical studies using resveratrol have been less exciting. For example, resveratrol supplementation over a six-month period increased sirtuin activation and reduced oxidative stress, yet did not improve markers of metabolic dysfunction in T2D patients.193,194 One problem in animal models of T2D involves the age of the animal; most are young animals. NAD+ has been reported to decrease with aging.195 As sirtuins require NAD+ for catalytic deacetylase activity, resveratrol treatment may do little in the way of promoting sirtuin activation with declining NAD+ levels in human subjects.
2.3.2. Sulforaphane and histone acetylation.
Aside from DNA methylation, sulforaphane has also been reported to promote brain health by regulating brain-derived natriuretic factor (BDNF) via histone acetylation.196 In primary cortical neurons, sulforaphane increased histone H3 and H4 acetylation at promoter regions I, II and IV of BDNF and subsequently increased BDNF expression.196 These actions were likely due to reduced HDAC2 expression and HDAC activity upon sulforaphane exposure; HDAC inhibition would be expected to increase histone acetylation and gene expression.196 Additionally, sulforaphane was shown to upregulate the BDNF receptor, tyrosine kinase receptor B (TrkB), and increase activity of TrkB downstream targets (i.e., cAMP-responsive element-binding protein (CREB), Ca2+/calmodulin-dependent protein kinase II (CaMKII), extracellular signal-regulated kinase (ERK) and protein kinase B (Akt)).196 Consistent with these cell data, sulforaphane also increased BDNF and its downstream targets in a triple-transgenic mouse model of AD.196 Increased TrkB expression and its activated targets are associated with synaptic plasticity and strength, neurite outgrowth and differentiation, improved cognitive function and neuronal survival in models of neurodegeneration.197,198 Moreover, HDAC2 has been shown to impair cognitive function by binding, and thus inhibiting, several genes important for memory and synaptic plasticity.199 Indeed, several studies have shown sulforaphane improves cognitive impairment and attenuates neuronal degradation,200–202 consistent with its inhibition of HDAC2.196 In conjunction with all data provided in this review, sulforaphane is a multifaceted regulator of DNA and histone modifications, DNA methylation and histone acetylation in particular, and may protect the brain from age-associated diseases through these epigenetic-mediated actions.
2.3.3. Emodin and histone acetylation.
Emodin was recently shown to dose-dependently attenuate the NOD-, LRR- and pyrin-domain containing protein 3 (NLRP3) inflammasome pathway in hypoxic-exposed hearts and heart cells.203 The NLRP3 inflammasome synthesizes pro-inflammatory byproducts and mediates inflammation-induced cell death, or pyroptosis.204 Indeed, this report showed emodin blocked pro-inflammatory byproduct signaling, namely NF-kB, and pyroptosis concomitantly with reduced scar tissue formation in the heart.203 As NF-κB activity is regulated by HDACs, emodin may have blocked NF-κB activity through HDAC inhibition. In this regard, our lab recently reported that emodin dose-dependently inhibited cardiac-based HDAC activity and increased histone acetylation in cardiac myoblasts.205 HDAC inhibition is well-known to prevent and treat cardiac dysfunction in pre-clinical animal models of heart failure, as described above.73–76 Emodin likely inhibits HDAC activity by chelating zinc ions within HDAC catalytic domains due to its chelating properties.206 Furthermore, other well-characterized HDAC inhibitors have been shown to block NF-κB signaling,207 thus, further suggesting emodin is an HDAC inhibitor that regulates pro-inflammatory signaling cascades. Finally, histone acetylation mediates differential gene expression.73,205,208 This suggests that emodin may reverse stress-induced changes in the transcriptome. Experiments aimed at elucidating the epigenetic-dependent actions of emodin in CVD models are currently underway. Nevertheless, these data collectively suggest emodin inhibits pro-inflammatory signaling and downstream insults; these actions potentially depend on emodin’s role as an HDAC inhibitor.
2.3.4. Curcumin and histone acetylation.
Curcumin is a pleiotropic compound that has been shown to regulate multiple molecular targets; one such target involves the regulation of histone acetylation. Specifically, curcumin has been shown to inhibit p300 histone acetyltransferase (HAT) activity in experimental models of CVD.209–211 Curcumin-dependent p300 inhibition was shown to reduce histone acetylation on promoter regions of the GATA binding protein (GATA4) as well as other pro-hypertrophic genes subsequently resulting in decreased expression of cardio-hypertrophic genes.211 This is in agreement with the canonical role of HATs in catalyzing histone hyperacetylation and gene upregulation. In addition, the GATA4 transcription factor can be acetylated by p300, increasing GATA4 DNA binding to the promoter regions of other cardio hypertrophic genes; this leads to pathological cardiac enlargement.212 GATA4 can further induce cardiac hypertrophy by promoting the pro-hypertrophic transcription factor, nuclear factor of activated T cells (NFAT).213 Just as important, GATA4 mediates inflammation and age-associated cell dysregulation, or cell senescence, through NF-κB-dependent mechanisms.214,215 As such, both NFAT and NF-κB signaling are blocked upon curcumin exposure.216,217 More so, NF-κB activity is regulated in part via p300-dependent actions.16 It would be interesting to see if curcumin mitigates NFAT or NF-κB signaling pathways through p300/GATA4-dependent regulation.
In addition to its HAT inhibitory actions, curcumin has also been shown to inhibit inflammation-driven cardiac remodeling by inhibiting HDACs.218 In these studies, curcumin inhibited HDAC1-dependent hypoacetylation at the promoter region of tissue inhibitor of metalloproteinase 1 (TIMP1); this led to increased histone acetylation and subsequent TIMP1 gene expression, as well as attenuation of cardiac fibrosis and inflammation.218 Consistent with these data, curcumin reduced expression of the TIMP1 inhibitory target, metalloproteinase 2 (MMP2).218 MMPs contribute to inflammatory signaling,219 heart remodeling220 and other diseases detailed within this review.221,222 Therefore, MMP inhibition may be efficacious in numerous diseased states. In agreement with the data by Hu et al.,218 other reports have shown that curcumin inhibits HDAC activity223 and protects against inflammaging-associated CVD by regulating TIMP/MMPs.224 Collectively, these data suggest that curcumin may mitigate pathological molecular imbalances in the heart through several epigenetic modifications, including histone acetylation.
2.3.5. EGCG and histone acetylation.
Epigallocatechin gallate (EGCG) is a polyphenol found in tea varieties that has been shown to have anti-inflammatory properties. Thus, EGCG is an intriguing therapeutic agent against inflammaging-associated diseases including heart disease. Recent evidence suggests that EGCG can attenuate inflammation by regulating histone acetylation.225 Specifically, Liu et al.225 showed that EGCG modulated differential binding of p300 and HDACs 1 and 2 at promoter regions of pro-inflammatory genes, including NF-κB subunit p65, which was linked with reversed hyperacetylation and suppressed pro-inflammatory gene expression in stress-induced endothelial cells.225 Indeed, the pro inflammatory activity of NF-κB is, in part, dependent upon p300-, HDAC1- and HDAC2-mediated actions.15,16 Moreover, endothelial cells are a first line of defense in response to inflammation but will become dysfunctional with over-exposure to inflammation, which can result in fibrogenesis and atherosclerosis.226,227 This is interesting as EGCG was shown to be efficacious for endothelial dysfunction in patients with coronary artery disease.228 Therefore, EGCG may prevent inflammation-induced cardiac dysfunction through regulation of histone acetylation at pro-inflammatory gene promoters. In line with this cardioprotective postulate, EGCG reduced acetylation at the promoter region of forkhead box protein O1 (FOXO1) in hyperglycemia-induced cardiomyoblasts.229 Of particular interest, nuclear FOXO1 acetylation was reduced upon EGCG treatment.229 This is important because nuclear acetylated FOXO1 induces autophagy in cardiac cells230,231 and overly active autophagy is linked to cardiac dysmorphology and atherosclerosis.232 Indeed, EGCG was further shown to block hyperglycemia-induced autophagy in H9C2 heart cells.229 Finally, EGCG was shown to attenuate age-induced hypoacetylation at the proximal promoter region of cardiac troponin I (cTnI) in aged mice.233 This is interesting as cTnI is a critical regulator of diastolic function, and EGCG treatment increased cTnI expression and cardiac diastolic function.233 These data combined suggest EGCG is a natural product that regulates both inflammation homeostasis and cardiac function via histone acetylation-dependent mechanisms.
2.3.6. Anthocyanins and histone acetylation.
Anthocyanins are flavonoids with three phenolic rings and credited for the pigmentation of foods such as berries (e.g., blueberry, raspberry and blackberry), grapes, beans and potatoes. As of late, anthocyanin-rich foods have received attention for their metabolic efficacy in humans234–236 and have even shown favorable outcomes against fatty liver and inflammation.237,238 Here, anthocyanins were shown to protect the liver in part via changes in histone acetylation. For example, Zhan and colleagues showed that an anthocyanin-rich blueberry extract increased histone H3 acetylation at lysine residues K9, K14 and K18 and reduced liver fibrosis and damage to rats exposed to carbon tetrachloride.239,240 Acetylation of K9 and K14 was shown to be important for proper liver function; liver dysfunction and fibrosis is increased with aging.241 Furthermore, histone H3 hyperacetylation results in euchromatin formation and subsequent differential gene expression.242 These data suggest that anthocyanins improve liver function and block liver fibrosis by regulating gene expression through histone acetylation. In addition to these studies, others have shown that anthocyanins reduce expression of pro-inflammatory transcripts.243 Unfortunately, Zhan et al. did not link anthocyanin-mediated histone acetylation with HDAC or HAT activity.239,240 However, it should be noted that anthocyanins have been shown to modulate HDAC and HAT activity.244–246 Intriguingly, H3K9/14 hyperacetylation at transcriptional start sites of TNFa was associated with liver inflammation, a common aggressor of fibrosis, in obesity-induced mice.247 This was likely due to an increase in HAT activity.248 Indeed, the hepatoprotective actions of other phytochemicals have been associated with HAT inhibition.249 As anthocyanins have been shown to concomitantly reduce HAT activity and pro-inflammatory TNFa signaling outside of liver244 as well as mitigate pro-inflammatory gene expression and TNFa signaling in liver of aging mice,243 future research should focus on if anthocyanins block inflammaging-associated liver fibrosis via HAT inhibition.
2.4. Phytochemicals – histone acylation and gene expression
To our knowledge, no reports have currently been published examining the role of phytochemicals on histone acylation. A recent report showed that histone crotonylation is increased at transcriptional start sites of neuroglial cell-mediated endocytosis-related genes in a progressive AD cell model.250 As neuroglial cells clear Aβ, these data suggest that histone hypercrotonylation may serve as a compensatory mechanism to stimulate the expression endocytosis-related genes in order to clear Aβ in AD patients; of course further investigation is warranted to address this question. Of interest, the HAT, p300/CBP regulates crotonyl-CoA-dependent histone crotonylation.251 Within this report, we described curcumin as a HAT and HDAC inhibitor.209–211,218 As curcumin has been shown to block inflammation and Aβ-dependent neurodegeneration,252 curcumin may mediate its neuroprotective effects by regulating histone crotonylation. However, further study is imperative to elucidate how phytochemicals, in particular curcumin, regulate histone acylation and if these actions are efficacious for inflammaging, neurodegeneration and other age-associated diseases.
3. Short chain fatty acids (SCFAs) and aging-associated inflammation
Short chain fatty acids (SCFAs) are carboxyl group (COOH) containing chains of 2–6 carbon atoms. SCFAs can be saturated or unsaturated and have a variety of side chain structures. Nomenclature for the SCFA and its corresponding ester/salt are frequently used interchangeably, and will be here, as at physiological pH, the SCFA carboxyl hydrogen is typically lost to form the associated ester. For instance, butyric acid rapidly becomes butyrate in most biological conditions. A number of molecular mechanisms engaged by SCFAs have been described in the literature. These include regulating signaling cascades via interaction with G-protein coupled receptors, modulating the activity of epigenetic modifying enzymes, serving as molecular substrates for metabolism, and serving as molecules used in protein post translational modification.
SCFAs are produced endogenously in mammalian systems when cellular glucose levels are low. This can occur in fasting, diabetes, and with some low carbohydrate diets. As oxaloacetate is diverted for gluconeogenesis, the TCA cycle slows and excess acetyl-CoA is processed into 4-carbon SCFA ketones. SCFAs are also produced by a variety of microbes, including in the digestive tracks of animals, where they have been shown to influence host physiology.
Interestingly, in humans, specific circulating SCFAs under fasting conditions are negatively correlated with BMI and positively correlated with insulin sensitivity.253 Experiments in rodents indicate that with age, the ability to produce ketones in fasting conditions is diminished or slowed254 and importantly, a 220 subject human study showed that serum concentrations of total free fatty acid and hydroxybutyrate (unfasted) were decreased with age.255 These point toward a negative correlation between SCFA concentrations and conditions associated with inflammation, specifically advanced age and obesity/diabetes.
3.1. SCFAs – histone acetylation and gene expression
It has long been known that valproate (C:8 medium chain fatty acid sometimes classified as a branched SCFA) and butyrate function as HDAC inhibitors. In 2012, it was shown that beta-hydroxy butyrate (β-OHB), a SCFA generated by ketogenic diet or caloric restriction/fasting, could inhibit HDAC activity and decrease inflammation, specifically in the kidney.256 Given that acetylation at specific lysine residues on histone and non-histone proteins is diminished with age,257–259 acetylation regulating effects of pro-longevity strategies, including SCFA generation from diet manipulation, have become a growing area of research interest. However, a recent report demonstrates that butyrate has a much more dramatic effect on histone acetylation than does β-OHB. In fact β-OHB showed no effect on histone acetylation and increased inflammatory gene expression in cultured endothelial cells.260 One potential explanation for this discrepancy might be found in regulation of SCFA induced histone modifications by pre-existing modifications,261 which are likely cell type and culture condition specific.
Ketogenic diet, caloric restriction and intermittent fasting have all been shown to improve recovery in rodents following acute spinal cord injury.262–265 In each model, the concentration of β-OHB was elevated in the plasma and spinal fluid. These diet manipulations corresponded with increased histone acetylation and oxidative stress resistance factor expression (e.g. Foxo3a, Mt2, catalase, mnSOD), while reducing HDAC activity and lipid peroxidation enzyme expression. β-OHB has been repeatedly shown to increase production of another neuroprotective molecule, brain derived neurotrophic factor (BDNF). It was recently demonstrated that b-OHB induced cAMP/PKA dependent phosphorylation of CREB and activation of the BDNF promoter to increase BDNF expression in hippocampal neurons.266
In addition to the common pattern of anti-ROS gene expression seen with SCFA generating diets or direct treatment with SCFAs, these strategies also drive calbindin expression in the brain. This is potentially beneficial via buffering of calcium influx, which occurs following exposure to inflammatory cytokines, and excitatory neurotransmitters.267 There is also evidence that DNA damage repair is governed by histone acetylation and stimulated with HDAC inhibition.268 b-OHB also demonstrates neuroprotective properties via inhibition of the NLRP3 inflammasome.269 These data sum to indicate that SCFAs are potently anti-inflammatory, including in the brain, through a variety of mechanisms including HDAC/histone acetylation mediated regulation of gene expression.
3.2. SCFAs – histone acylation and gene expression
SCFAs are also used in the production of acyl-CoA molecules important for protein post-translational modifications. Many short, medium, and even long chain fatty acids have been identified via mass spectrometry as modifications on histone and non-histone proteins. Crotonylation of histone lysine residues appears to impact gene expression in a similar manner as does histone lysine acetylation.270 However, the magnitude of gene expression changes seen in response to histone crotonylation appears to be larger than that for histone acetylation at the select gene loci that have been studied.251 It is unclear if this pattern will be conserved when chromatin wide studies are conducted. Crotonate is a larger molecule than acetate, with crotonate having two additional carbon atoms, and crotonate also possesses a double bond between the second and third carbon atoms, that is predicted to provide a more rigid structure. This limiting of three dimensional movement of crotonyllysine may stabilize protein–protein interactions relative to acetyl-lysine. Surprisingly, class I HDACs can catalyze removal of both acetyl and crotonyl modifications and similarly, P300 can catalyze addition of both to protein lysine residues.251,271 Several metabolic pathways have been pointed to as potential sources of crotonyl-CoA, including ketogenesis.272
Recently, it was shown that b-OHB, once bound to coA, serves as a P300 catalyzed post translational modification. Remarkably, P300 selectively adds hydroxybutyrate to target proteins that do not overlap with other p300 acetyl target proteins.273 The addition of hydroxyl butyrate to key lysine residues increased the activity of glycolysis pathway enzymes. Clearly, the field is just beginning to uncover the relevance of these and other less traditionally studied acyl modifications, and their impact on cellular functions.
3.3. Caloric restriction, SCFAs and aging-associated inflammation
Caloric restriction (CR) has been shown in a variety of species to prolong lifespan and healthspan.274,275 Anti-oxidative and anti-inflammatory effects of CR have been well documented by many independent groups in many tissues, including the brain and cardiovascular systems.276–278 CR is also effective at reducing the risks of diabetes, cardiovascular disease, cancer, and neurodegenerative diseases.279 These effects are often attributed to CR mediated changes to activity of mTOR, HDACs, and other metabolic enzymes. SCFA generation and SCFA molecular targets may help explain shared anti-aging, anti-inflammatory and disease protective effects of caloric restriction, ketogenic diets, exogenous SCFA supplementation, and mTOR and HDAC inhibition.
SCFAs also bind G-protein coupled receptors (GPCRs), specifically Free Fatty Acid Receptors (FFARs) FFAR2 and FFAR3, and the niacin receptor (HCA2). These receptors were previously orphan receptors and are also known as GPCR43, GPCR41, and GPR109A respectively. HCA2 receptors are found in monocytes, macrophages, T cells, and microglia, among others. Binding of HCA2 by butyrate, β-OHB, and niacin, is potently anti-inflammatory and protective effects have been described in a number of neurological and neurodegenerative disease models.280 Given a plethora of potential mechanisms engaged by these ligands it was important to show that these benefits were dependent on the HCA2 receptor as was accomplished in stroke and Parkinson’s models.281–283 β-OHB is an inhibitor of FFAR2 and FFAR3, while other SCFAs (e.g. propio-nate) can act as activators of the receptors, leading to opposite downstream inflammation regulated effects.284
Another potential mechanism for SCFA mediated anti-inflammatory effects is via use as metabolic substrates. SCFAs relative to lipids are able to bypass multiple β-oxidation cycles before entry to the TCA cycle, which minimizes oxygen consumption and reactive oxidation generation. This is due to avoidance of increased production of FADH2 relative to NADH + H, which occurs during β-oxidation driven metabolism. FADH2 provides electron motive force to complex II of the ETC, which can lead to reverse electron flow through complex I and generation of reactive oxygen species (ROS). ROS accumulation is positively associated with inflammation.120 Interestingly, propionate and butyrate, which both increased histone acetylation in cultured eosinophils, blocked eosinophil migration and activation and caused mitochondrial depolarization and apoptosis in eosinophils from donors with allergies but not from control donors.285 This may suggest altered ETC composition or spatial arrangement in the eosinophils from allergic donors.
It is becoming increasingly apparent that cue dependent expansion or differentiation of T regulatory cells (Treg) is critical for limiting post inflammation tissue damage.286,287 Butyrate has been shown to induce expansion of Treg cells through inhibition of HDAC8 and was beneficial in a model of rheumatoid arthritis.288 In support of this notion, work from colon cancer cells suggests potential synergy between SCFAs and aryl hydrocarbon receptor (Ahr) activation.289 Ahr activation is known in other contexts to be anti-inflammatory via triggered expansion of Treg populations.290,291
Several other neurologic conditions are also associated with neuro-inflammation. Here also SCFAs or SCFA generating dietary approaches appear to provide benefit. Butyrate, B-OHB, and other SCFA combinations been shown to have anti-depressant and anti-anxiety effects,292–294 while valproate has proven effective in treating bipolar depression and PTSD.295,296 HDAC inhibition and exogenous ketone supplementation have also been shown to prevent protein accumulation and memory impairment in mouse models of dementia/AD.199,297–299 Similarly, a ketogenic diet was shown to increase lifespan and improve memory in aged mice. Again, this corresponded with decreased mTOR activity, increased lysine acetylation and increased antioxidant gene expression.300
3.4. Pro- and prebiotics, SCFAs and gut–brain axis
SCFAs are synthesized by gut-specific microbiota and have been shown to play a critical role in the gut–brain axis as described above.301 The gut–brain axis is an intricate communication network between the central nervous system (CNS) in the brain, the autonomic nervous system (ANS), the enteric nervous system (ENS) in the gut and the hypothalamic pituitary adrenal (HPA) axis, within which gut microbiota modulate inflamma-tory, metabolic and neurological signaling.301 Indeed, dysbiosis, an imbalance in microbiota that synthesize SCFAs and promotes pathogenesis, is observed with aging and has been implicated in age-associated diseases including metabolic dysfunction,302,303 neurological disorders304 and CVD.305 Moreover, aging-linked dysbiosis has been shown to increase the pro-inflammatory systemic environment,306 which may further drive neuro-inflammation and neurodegeneration. While the anti-pathogenic effects of the microbiota are still being elucidated, it has been postulated that the protective effects of gut bacteria are imparted through epigenetic modifications.307 Consistent with this, dysbiosis is associated with reduced bioavailability of epigenetic modifying SCFAs (see above for SCFAs and histone modification).308 These data, thus, suggest that creating a balanced microbiota, eubiosis, would deter chronic, low-grade inflammation and its co-morbidities, in part, through epigenetic changes in gene expression. To this end, prebiotics and probiotics have been used to re-establish eubiosis in the gut.
3.4.1. Prebiotics, SCFAs and aging-linked disease.
Prebiotics are natural products that provide substrates for gut microbiota fermentation; for example, Bifidobacterium and Lactobacillus produce SCFAs through prebiotic fermentation. Thus, prebiotic bacterial fermentation to SCFAs can promote histone acetylation via HDAC inhibition contributing to differential gene expression that imparts neuro-protection.309–311 Indeed, a recent report linked increased colonic butyric acid, a SCFA HDAC inhibitor, with transcriptome-wide changes in gene expression in aged rats fed the prebiotic fructo-oligosaccharide.312 Of significance, fructo-oligosaccharide supplementation regulated genes associated with immune function in aged rats, improved cecum pH as well as promoted healthy bacterial growth by increasing the numbers of Bifidobacterium and Lactobacillus.312 Colonic pH is critical for the growth of Bifidobacterium and Lactobacillus, which mainly ferment carbohydrate or prebiotics and thus increase SCFA synthesis.313 As this study only looked at 12 week prebiotic supplementation,312 it is unclear if constant prebiotic consumption is required for preventing age-associated dysbiosis long-term. In addition, the researchers in this study did not examine changes in histone modifications linked to SCFAs such as HDAC inhibition or histone hyperacetylation; this makes it difficult to associate the observed differential gene expression with any one epigenetic mark.312
In contrast to above, prebiotic supplementation with galacto-oligosaccharide was shown to inhibit HDAC activity in the brains of obese rats;309 this was linked to increased Bifidobacterium microbiota and increased SCFAs that led to improved cognitive function and reduced neuro-inflammation.314–316 Further studies have reported that prebiotic supplementation contributed to gut eubiosis and subsequently improved cognitive impairment,317 metabolic function318 and gut inflammation.318 Combined, these data suggest that prebiotics confer gut eubiosis that contributes to SCFA synthesis; this subsequently results in HDAC inhibition, changes in histone modifications (e.g., acetylation/acylation) and anti-inflammaging actions in the gut–brain axis. Interestingly, prebiotics have also been suggested to regulate the epigenome independent of SCFA synthesis.309 For example, while galacto-oligosaccharide treatment increased the SCFA, acetate, in plasma and inhibited HDAC activity in cortex/hippocampus brain regions, lone acetate treatment was shown to have no such effect in the brain.309 It may have been other SCFAs that galacto-oligosaccharide synthesized to attenuate neurological HDAC activity, however, no other plasma SCFAs were reported. Given these recent findings, further investigation is needed to elucidate the chromatin remodeling and transcriptomic effects of prebiotics that do not involve butyrate, acetate or propionate.
3.4.2. Probiotics, SCFAs and inflammaging.
While prebiotics “feed” the gut microbiota, probiotics are living microorganisms that promote microbial growth and deter competing bacterial invasion,319 thus allowing improved production of SCFAs. Indeed, Bifidobacterium supplementation improved microbiota composition in geriatric individuals.320 Probiotics further promote intestinal barrier function and anti-inflammatory actions.321 These actions appear, in part, to be epigenetically controlled. For example, Bifidobacterium or Lactobacillus reduced histone acetylation and increased DNA methylation in LPS-induced epithelial cells.322 These epigenetic modifications were linked to reduced pro-inflammatory cytokine expression as well as nuclear NF-κB and MAPK signaling.322 As described throughout this manuscript, NF-κB and MAPK signaling are partially regulated by epigenetic enzymes.18,323,324 In agreement with these data, histone hypoacetylation and DNA hypermethylation have been associated with gene suppression, suggesting that Bifidobacterium or Lactobacillus suppress pro-inflammatory genes.322 -acetylation -, co- and post-treated with Lactobacilli probiotics.325 These data suggest probiotic treatment can both prevent and reverse pathological microbiota-mediated epigenetic alterations that are associated with gut and systemic inflammation.
Finally, probiotic treatment has also been shown to attenuate CVD. Here, probiotic treatment reduced myocardial infarct size and pro-inflammatory cytokines TNFa and IL-6 as well as the chemokine monocyte chemoattractant protein-1 (MCP-1) in a mouse model of CVD.326 In this model, probiotic treatment contributed to T regulatory cell histone hyperacetylation and thus anti-inflammatory function, whereas T regulatory cell depletion attenuated the protective effects of probiotic treatment.326 T regulatory cells are regulated by the forkhead box P3 (FOXP3), and their acetylation status is critical for immuno-suppression during the inflammatory response.327 Combined, these data suggest that probiotic supplementation increased T regulatory cell hyperacetylation and thus anti-inflammatory function critical for cardioprotection in this model of CVD. Others have reported that probiotic supplementation blocked matrix metalloproteinase-2 (MMP-2) activity and cardiac fibrosis in a mouse model of dysbiosis-induced CVD.328 While no epigenetic modifications such as histone hyperacetylation were assessed, fecal levels of the SCFA acetate were increased in dysbiotic mice supplemented with probiotics.328 Acetate is a predominant SCFA in both the gut and systemic circulation that can inhibit HDAC activity329,330 as well as promote HAT activity.331 As MMP-2 activity seems to be dependent upon its acetylation status,332 this suggests that probiotic treatment regulated acetate-mediated MMP-2 inhibition via histoneacetylation-dependent mechanisms.328 Further investigation is still required to fully elucidate the mechanisms by which probiotics regulate MMP-2 activity and if these mechanisms are, in fact, epigenetically related. However, these and the above data suggest that probiotics can regulate histone modifications and thus play a key role in the regulation of aging, inflammation, neurodegeneration and CVD.
4. Conclusion
While the effects of individual products are often examined in a lone experimental environment, phytochemicals and fiber-produced SCFAs are frequently consumed together. Notably, a majority of fiber-rich foods are abundant in several phytochemicals. This brings to question, in what ways do phytochemicals interact with other phytochemicals or SCFAs? Few studies have examined the phytochemical–phytochemical or phytochemical–SCFA interactions, and whether these interactions are competitive, synergistic or compounding in their epigenetic actions. In one report that used colon cancer cells, EGCG and butyrate in combination reduced HDAC activity as well as CpG methylation and were more anti-oncogenic than their individual treatments.333 While others have also reported the potential for using phytochemical–SCFA combinations in cancer models,334 no mechanistic study, to our knowledge, has examined it in other inflammaging-associated diseases that were discussed in this review. However, several epigenome-wide association studies (EWAS) have reported that differential methylation of inflammatory and metabolic genes were associated with diets rich in phytochemicals and fiber in peripheral white blood cells isolated from geriatric individuals.335–337 One EWAS inversely correlated dietary fiber-intake with methylation of lysophosphatidylcholine acyltransferase 1 (LPCAT1) and Ras GTPase-activating protein 3 (RASA3) genes in young African Americans.338 Furthermore, LPCAT1 methylation was positively associated with visceral adiposity and increased inflammation in the blood.338 This is important because LPCAT1 mitigates pro-inflammatory signaling cascades associated with metabolic dysfunction339 and may be regulated by diet-driven epigenetic modifications like DNA methylation. Conversely, another report associated fiber and fruit intake with TNFα hypomethylation in isolated human white blood cells.340 EWAS studies are often limited to DNA methylation due to many variables including costs of sequencing technologies such as chromatin immuno-precipitation sequencing (ChIP-seq) vs. bisulfite sequencing. It should be further noted that epidemiology-based studies like ones described above infer association but not causation. Therefore, future study is required to fully elucidate the epigenomic roles of diet-derived products in regulating inflammation and inflammaging-associated events.
Another consideration is that absorption, distribution, metabolism and excretion of natural products vary.341,342 This suggests the interaction between compounds that are distributed to the same location and at the same time may only be of interest. Even still, compounds can affect tissues to which they are not distributed; for example, emodin is not distributed to the heart in considerable amounts341 yet has been shown to be cardioprotective in vivo.203 Therefore, the metabolites of parent compounds, which enter circulation may be responsible for efficacious actions observed in tissues that have unmeasurable concentrations of their parent compounds. However, a majority of phytochemical metabolites are rendered bio-unavailable upon synthesis.342,343 While “more is better” comes to mind towards addressing bio-unavailability issues, phytochemicals are also toxic at higher concentrations.141,344,345 Technologies aimed at increasing bioavailability of natural products at lower concentrations may resolve these dilemmas.346 Alternatively, cells of the immune system that travel through regions of high compound concentration (e.g. mucosa/submucosa of the intestine), may broadcast anti-inflammatory signals as they return to lymph or general circulation, or by secreted signals. Adding further complexity to this diet-epigenome discussion, animal-based foods further contain epigenetic modifying compounds, namely micro RNAs (miRNAs), that may influence health and disease upon consumption.347 Animal-based compounds may also interact with phytochemicals and/or SCFAs that could alter cellular outcomes, for better or for worse. These are purely speculative but reflect how little is understood about whole food-gene and natural product-gene regulation of health and disease. Nevertheless, researchers and clinicians alike are hopeful that future studies will begin to provide evidence for which foods or natural products are best for optimizing health and deterring inflammaging-associated metabolic dysregulation, neurodegenerative diseases and CVD.
Finally, evidence suggests that dietary interventions may compliment pharmaceutical treatment in ameliorating pathologies, and that the therapeutic actions may be epigenetically-related. For example, vorinostat (SAHA), which is an FDA-approved HDAC inhibitor used for cancer, in combination with curcumin proved more efficacious than their lone treatments for Aβ-dependent neurodegeneration.252 Moreover, co-treatment of these compounds resulted in differential gene expression,252 thus hinting that epigenetic-mediated actions were at play. In other studies, using high-fat diet-induced diabetic mice, the anti-diabetic drug, metformin in combination with resveratrol ameliorated inflammation, glucose intolerance and other metabolic insults.348,349 However, in neither study were DNA methylation or histone modifications reported. Further investigation for common medicine-natural product co-therapies are, therefore, warranted.
In conclusion, there is strong evidence supporting the postulate that foods and natural products influence health and disease through epigenetic modification. While caloric and macronutrient composition govern cofactors involved in non-sequence alterations on DNA and histone proteins, more and more studies suggest (by)product composition in food is also important. This report described how phytochemicals and SCFAs regulate DNA methylation and histone modifications (methylation, acetylation and acylation) in experimental models of age-associated diseases. Moreover, observational data has consistently associated foods rich in phytochemicals and SCFA-producing fiber with health and longevity. Nevertheless, few studies have translated the epigenetic-mediated actions of these food derivatives to human models.
6. Acknowledgements
This work is supported by the USDA NIFA (Hatch-NEV00727, Hatch-NEV00767), the Dennis Meiss & Janet Ralston Fund for Nutri-epigenetic Research, the National Institute for General Medical Sciences (NIGMS) of the NIH (P20 GM130459) and the National Heart, Lung, and Blood Institute of the NIH (R15 HL143496) and NSF EPSCOR Track II (OIA-1826801) to B. S.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
7. References
- 1.Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP, Listi F, Nuzzo D,Lio D and Caruso C, Mech. Ageing Dev, 2007, 128, 83–91. [DOI] [PubMed] [Google Scholar]
- 2.Fagiolo U, Cossarizza A, Santacaterina S, Ortolani C, Monti D, Paganelli R and Franceschi C, Ann. N. Y. Acad. Sci, 1992, 663, 490–493. [DOI] [PubMed] [Google Scholar]
- 3.Freund A, V Orjalo A, Desprez PY and Campisi J, Trends Mol. Med, 2010, 16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X and Zhao L, Oncotarget, 2018, 9, 7204–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T and Sierra F, Cell, 2014, 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH, Heimovitz H, Cohen HJ and Wallace R, Am. J. Med, 1999, 106, 506–512. [DOI] [PubMed] [Google Scholar]
- 7.Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J, Tipping RW, Ford CE, Pressel SL, Walldius G, Jungner I, Folsom AR, Chambless L, Ballantyne CM, Panagiotakos D, Pitsavos C, Chrysohoou C, Stefanadis C, Knuiman MW, Goldbourt U, Benderly M, Tanne D, Whincup P, Wannamethee SG, Morris RW, Kiechl S, Willeit J, Mayr A, Schett G, Wald N, Ebrahim S, Lawlor D, Yarnell J, Gallacher J, Casiglia E, Tikhonoff V, Nietert PJ, Sutherland SE, Bachman DL, Keil JE, Cushman M, Psaty BM, Tracy R, Tybjærg-Hansen A, Nordestgaard BG, Zacho J, Frikke-Schmidt R, Giampaoli S, Palmieri L, Panico S, Vanuzzo D, Pilotto L, De La Cámara AG, Gómez Gerique JA, Simons L, McCallum J, Friedlander Y, Fowkes FGR, Lee A, Taylor J, Guralnik JM, Phillips CL, Wallace RB, Blazer DG, Khaw KT, Brenner H, Raum E, Müller H, Rothenbacher D, Jansson JH, Wennberg P, Nissinen A, Donfrancesco C, Harald K, Jousilahti P, Vartiainen E, Woodward M, D’Agostino RB, Wolf PA, Vasan RS, Benjamin EJ, Bladbjerg EM, Jørgensen T, Salomaa V, Jespersen J, Dankner R, Chetrit A, Lubin F, Rosengren A, Wilhelmsen L, Lappas G, Eriksson H, Björkelund C, Lissner L, Bengtsson C, Cremer P, Nagel D, Tilvis RS, Strandberg TE, Kiyohara Y, Arima H, Doi Y, Ninomiya T, Rodriguez B, Dekker J, Nijpels G, Stehouwer CDA, Rimm E, Pai JK, Sato S, Iso H, Kitamura A, Noda H, Salonen JT, Nyyssönen K, Tuomainen TP, Laukkanen JA, Deeg DJH, Bremmer MA, Meade TW, Cooper JA, Hedblad B, Berglund G, Engström G, Verschuren WMM, Blokstra A, Shea S, Döring A, Koenig W, Meisinger C, Bueno-De-Mesquita HB, Kuller LH, Grandits G, Selmer R, Tverdal A, Nystad W, Gillum RF, Mussolino M, Hankinson S, Manson JE, Knottenbelt C, Bauer KA, Davidson K, Kirkland S, Shaffer J, Korin MR, Naito Y, Holme I, Nakagawa H, Miura K, Ducimetiere P, Jouven X, Luc G, Crespo CJ, Garcia-Palmieri MR, Amouyel P, Arveiler D, Evans A, Ferrieres J, Schulte H, Assmann G, Packard CJ, Sattar N, Westendorp RG, Buckley BM, Cantin B, Lamarche B, Després JP, Dagenais GR, Barrett-Connor E, Wingard DL, Bettencourt RR, Gudnason V, Aspelund T, Sigurdsson G, Thorsson B, Trevisan M, Witteman J, Kardys I, Breteler MMB, Hofman A, Tunstall-Pedoe H, Tavendale R, Howard BV, Zhang Y, Best L, Umans J, Ben-Shlomo Y, Davey-Smith G, Onat A, Njølstad I, Mathiesen EB, Løchen ML, Wilsgaard T, Ingelsson E, Basu S, Cederholm T, Byberg L, Gaziano JM, Stampfer M, Ridker PM, Ulmer H, Diem G, Concin H, Tosetto A, Rodeghiero F, Wassertheil-Smoller S, Marmot M, Clarke R, Fletcher A, Brunner E, Shipley M, Buring J, Shepherd J, Cobbe S, Ford I, Robertson M, He Y, Marin Ibanez A, Feskens EJM, Walker M, Watson S, Erqou S, Lewington S, Pennells L, Perry PL, Ray KK, Sarwar N, Alexander M, Thompson A, White IR and Wood AM, Lancet, 2010, 375, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyes J and Bird A, Cell, 1991, 64, 1123–1134. [DOI] [PubMed] [Google Scholar]
- 9.Serra RW, Fang M, Park SM, Hutchinson L and Green MR, Elife, 2014, 2014, e02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rountree MR, Bachman KE and Baylin SB, Nat. Genet, 2000, 25, 269–277. [DOI] [PubMed] [Google Scholar]
- 11.Jylhävä J, Nevalainen T, Marttila S, Jylhä M, Hervonen A and Hurme M, Aging Cell, 2013, 12, 388–397. [DOI] [PubMed] [Google Scholar]
- 12.Bellizzi D, D’Aquila P, Montesanto A, Corsonello A, Mari V, Mazzei B, Lattanzio F and Passarino G, Age, 2012, 34, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucci L, Ostan R, Cevenini E, Pini E, Scurti M, Vitale G, Mari D, Caruso C, Sansoni P, Fanelli F, Pasquali R, Gueresi P, Franceschi C and Monti D, Aging, 2016, 8, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gentilini D, Mari D, Castaldi D, Remondini D, Ogliari G, Ostan R, Bucci L, Sirchia SM, Tabano S, Cavagnini F, Monti D, Franceschi C, Di Blasio AM and Vitale G, Age, 2013, 35, 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashburner BP, Westerheide SD and Baldwin AS, Mol. Cell. Biol, 2001, 21, 7065–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong H, May MJ, Jimi E and Ghosh S, Mol. Cell, 2002, 9, 625–636. [DOI] [PubMed] [Google Scholar]
- 17.Wei H, Wang B, Miyagi M, She Y, Gopalan B, Bin Huang D, Ghosh G, Stark GR and Lu T, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 13516–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, Holson EB and McKinsey TA, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 9806–9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barzilai N, Huffman DM, Muzumdar RH and Bartke A, Diabetes, 2012, 61, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig J, Viggiano TR, McGill DB and Oh BJ, Mayo Clin. Proc, 1980, 55, 434–438. [PubMed] [Google Scholar]
- 21.Syn WK, Choi SS and Diehl AM, Clin. Liver Dis, 2009, 13, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung B, Park S, Yu BP and Chung HY, J. Gerontol., Ser. A, 2004, 59, B997–B1006. [DOI] [PubMed] [Google Scholar]
- 23.Youssef J and Badr M, J. Biomed. Biotechnol, 2004, 2004, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J and Karin M, Nat. Med, 2005, 11, 191–198. [DOI] [PubMed] [Google Scholar]
- 25.Lanuza-Masdeu J, Isabel Arévalo M, Vila C, Barberà A, Gomis R and Caelles C, Diabetes, 2013, 62, 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao H, Ou J, Ma Y, Guo F, Yang Z, Wiggins M, Liu C, Song W, Han X, Wang M, Cao Q, Chung BHF, Yang D, Liang H, Xue B, Shi H, Gan L and Yu L, Cell Rep, 2014, 7, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedland RP, Budinger TF, Ganz E, Yano Y, Mathis CA, Koss B, Ober BA, Huesman RH and Derenzo SE, J. Comput. Assist. Tomogr, 1983, 7, 590–598. [DOI] [PubMed] [Google Scholar]
- 28.Dastur DK, Cereb J. Blood Flow Metab, 1985, 5, 1–9. [DOI] [PubMed] [Google Scholar]
- 29.Stanley WC and Chandler MP, Heart Failure Rev, 2002, 7, 115–130. [DOI] [PubMed] [Google Scholar]
- 30.Pavlova NN and Thompson CB, Cell Metab, 2016, 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taegtmeyer H, Circulation, 2002, 106, 2043–2045. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi H, McCaffery JM, Irizarry RA and Boeke JD, Mol. Cell, 2006, 23, 207–217. [DOI] [PubMed] [Google Scholar]
- 33.Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME, Cogger EA and James SJ, J. Nutr, 2001, 131, 2811–2818. [DOI] [PubMed] [Google Scholar]
- 34.Borra MT, Langer MR, Slama JT and Denu JM, Biochemistry, 2004, 43, 9877–9887. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen SB, Ølholm J, Paulsen SK, Bennetzen MF and Richelsen B, Int. J. Obes, 2008, 32, 1250–1255. [DOI] [PubMed] [Google Scholar]
- 36.Hall JA, Dominy JE, Lee Y and Puigserver P, J. Clin. Invest, 2013, 123, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, Wainstein J, Friedlander Y, Levy-Lahad E, Glaser B and Hellman A, Hum. Mol. Genet, 2012, 21, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K and Bennett DA, Neurology, 2014, 82, 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao HM and Hong JS, Trends Immunol, 2008, 29, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chitnis T and Weiner HL, American Society for Clinical Investigation, J. Clin. Invest, 2017, 127, 3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez FO and Gordon S, F1000Prime Rep, 2014, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farrall AJ and Wardlaw JM, Neurobiol. Aging, 2009, 30, 337–352. [DOI] [PubMed] [Google Scholar]
- 43.Noh H, Jeon J and Seo H, Neurochem. Int, 2014, 69, 35–40. [DOI] [PubMed] [Google Scholar]
- 44.Fakhoury M, Neurodegener. Dis, 2015, 15, 63–69. [DOI] [PubMed] [Google Scholar]
- 45.Xie H, Hou S, Jiang J, Sekutowicz M, Kelly J and Bacskai BJ, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 7904–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glenner GG and Wong CW, Biochem. Biophys. Res. Commun, 1984, 120, 885–890. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Deng J, Pan Q, Zhan Y, Fan JB, Zhang K and Zhang Z, J. Genet. Genomics, 2016, 43, 587–592. [DOI] [PubMed] [Google Scholar]
- 48.Young JI, Sivasankaran SK, Wang L, Ali A, Mehta A, Davis DA, Dykxhoorn DM, Petito CK, Beecham GW, Martin ER, Mash DC, Pericak-Vance M, Scott WK, Montine TJ and Vance JM, Neurol.: Genet, 2019, 5, e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller CA and Sweatt JD, Neuron, 2007, 53, 857–869. [DOI] [PubMed] [Google Scholar]
- 50.Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MVL and Zukin RS, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, E962–E971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen ZF, Paquette AJ and Anderson DJ, Nat. Genet, 1998, 20, 136–142. [DOI] [PubMed] [Google Scholar]
- 52.Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MVL and Zukin RS, J. Neurosci, 2003, 23, 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palm K, Belluardo N, Metsis M and Timmusk T, J. Neurosci, 1998, 18, 1280–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D and Cattaneo E, Nat. Genet, 2003, 35, 76–83. [DOI] [PubMed] [Google Scholar]
- 55.Terbach N and Williams RSB, Biochem. Soc. Trans, 2009, 37, 1126–1132. [DOI] [PubMed] [Google Scholar]
- 56.Arifuzzaman S, Das A, Kim SH, Yoon T, Lee YS, Jung KH and Chai YG, Biochem. Pharmacol, 2017, 137, 61–80. [DOI] [PubMed] [Google Scholar]
- 57.Yadav R and Weng HR, Neuroscience, 2017, 349, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L and Gan L, J. Biol. Chem, 2005, 280, 40364–40374. [DOI] [PubMed] [Google Scholar]
- 59.Jung ES, Choi H, Song H, Hwang YJ, Kim A, Ryu H and Mook-Jung I, Sci. Rep, 2016, 6, 25628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM and LaFerla FM, J. Neurosci, 2008, 28, 11500–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin X, Gao Y, Shi HS, Song L, Wang JC, Shao J, Geng XH, Xue G, Li JL and Hou YN, Sci. Rep, 2016, 6, 18982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, Dinarello CA and Maseri A, Circulation, 1999, 99, 2079–2084. [DOI] [PubMed] [Google Scholar]
- 63.Ferrucci L and Fabbri E, Nat. Rev. Cardiol, 2018, 15, 505–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riad A, Jäger S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, Pauschinger M, Schultheiss HP and Tschöpe C, J. Immunol, 2008, 180, 6954–6961. [DOI] [PubMed] [Google Scholar]
- 65.Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y, Hiramori K and Nakamura M, Int. J. Cardiol, 2006, 109, 226–234. [DOI] [PubMed] [Google Scholar]
- 66.Kuusisto J, Kärjä V, Sipola P, Kholová I, Peuhkurinen K, Jääskeläinen P, Naukkarinen A, Ylä-Herttuala S, Punnonen K and Laakso M, Heart, 2012, 98, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fang L, Ellims AH, Beale AL, Taylor AJ, Murphy A and Dart AM, Am. J. Transl. Res, 2017, 9, 5063–5073. [PMC free article] [PubMed] [Google Scholar]
- 68.Nakatochi M, Ichihara S, Yamamoto K, Naruse K, Yokota S, Asano H, Matsubara T and Yokota M, Clin. Epigenet, 2017, 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Westerman K, Sebastiani P, Jacques P, Liu S, DeMeo D and Ordovás JM, bioRxiv, 2018, 471722. [Google Scholar]
- 70.Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J and Bennett M, Circulation, 2013, 127, 386–396. [DOI] [PubMed] [Google Scholar]
- 71.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J, Circulation, 2010, 122, 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gusterson RJ, Jazrawi E, Adcock IM and Latchman DS, J. Biol. Chem, 2003, 278, 6838–6847. [DOI] [PubMed] [Google Scholar]
- 73.Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ and Olson EN, J. Biol. Chem, 2003, 278, 28930–28937. [DOI] [PubMed] [Google Scholar]
- 74.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN and Hill JA, Circulation, 2006, 113, 2579–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR and McKinsey TA, J. Mol. Cell. Cardiol, 2014, 67, 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, Scellini B, Piroddi N, Tesi C, Poggesi C, Jeong MY and McKinsey TA, Am. J. Physiol.: Heart Circ. Physiol, 2014, 307, H252–H258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, Cho GW, Kim SY, Wang ZV, Rotter D, Rothermel BA, Schneider JW, Lavandero S, Gillette TG and Hill JA, Sci. Signaling, 2016, 9, ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA and McKinsey TA, Sci. Transl. Med, 2018, 10, eaao0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Isah T, Biol. Res, 2019, 52, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Egert S, Bosy-Westphal A, Seiberl J, Kürbitz C, Settler U, Plachta-Danielzik S, Wagner AE, Frank J, Schrezenmeir J, Rimbach G, Wolffram S and Müller MJ, Br. J. Nutr, 2009, 102, 1065–1074. [DOI] [PubMed] [Google Scholar]
- 81.Seifried HE, Anderson DE, Fisher EI and Milner JA, J. Nutr. Biochem, 2007, 18, 567–579. [DOI] [PubMed] [Google Scholar]
- 82.Islam MA, Alam F, Solayman M, Khalil MI, Kamal MA and Gan SH, Oxid. Med. Cell. Longevity, 2016, 2016, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perez-Gregorio R and Simal-Gandara J, Curr. Pharm. Des, 2017, 23, 2731–2741. [DOI] [PubMed] [Google Scholar]
- 84.Eichelmann F, Schwingshackl L, Fedirko V and Aleksandrova K, Obes. Rev, 2016, 17, 1067–1079. [DOI] [PubMed] [Google Scholar]
- 85.Chen Z, Schoufour JD, Rivadeneira F, Lamballais S, Ikram MA, Franco OH and Voortman T, Epidemiology, 2019, 30, 303–310. [DOI] [PubMed] [Google Scholar]
- 86.Gao X, Chen H, Fung TT, Logroscino G, Schwarzschild MA, Hu FB and Ascherio A, Am. J. Clin. Nutr, 2007, 86, 1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim H, Caulfield LE, Garcia-Larsen V, Steffen LM, Coresh J and Rebholz CM, J. Am. Heart Assoc, 2019, 8, e012865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dong B, Liu C, Xue R, Wang Y, Sun Y, Liang Z, Fan W, Jiang J, Zhao J, Su Q, Dai G, Dong Y and Huang H, J. Nutr. Biochem, 2018, 62, 221–229. [DOI] [PubMed] [Google Scholar]
- 89.Strunz CMC, Roggerio A, Cruz PL, Pacanaro AP, Salemi VMC, Benvenuti LA, Mansur A. d. P. and Irigoyen MC, J. Nutr. Biochem, 2017, 40, 219–227. [DOI] [PubMed] [Google Scholar]
- 90.Shao W, Yu Z, Chiang Y, Yang Y, Chai T, Foltz W, Lu H, Fantus IG and Jin T, PLoS One, 2012, 7, e28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rashid K, Chowdhury S, Ghosh S and Sil PC, Biochem. Pharmacol, 2017, 143, 140–155. [DOI] [PubMed] [Google Scholar]
- 92.Teiten MH, Dicato M and Diederich M, Mol. Nutr. Food Res, 2013, 57, 1619–1629. [DOI] [PubMed] [Google Scholar]
- 93.Li YY, Tang D, Du YL, Cao CY, Nie YQ, Cao J and Zhou YJ, J. Dig. Dis, 2018, 19, 421–430. [DOI] [PubMed] [Google Scholar]
- 94.Maugeri A, Mazzone MG, Giuliano F, Vinciguerra M, Basile G, Barchitta M and Agodi A, Oxid. Med. Cell. Longevity, 2018, 2018, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B and Wahli W, J. Clin. Invest, 1999, 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang W, Lin Q, Lin R, Zhang J, Ren F, Zhang J, Ji M and Li Y, Exp. Cell Res, 2013, 319, 1523–1533. [DOI] [PubMed] [Google Scholar]
- 97.Stienstra R, Duval C, Müller M and Kersten S, PPAR Res, 2007, 2007, 95974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mandard S, Müller M and Kersten S, Cell. Mol. Life Sci, 2004, 61, 393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patsouris D, Reddy JK, Müller M and Kersten S, Endocrinology, 2006, 147, 1508–1516. [DOI] [PubMed] [Google Scholar]
- 100.Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T and Kadowaki T, Diabetes, 2005, 54, 3358–3370. [DOI] [PubMed] [Google Scholar]
- 101.Costet P, Legendre C, Moré J, Edgar A, Galtier P and Pineau T, J. Biol. Chem, 1998, 273, 29577–29585. [DOI] [PubMed] [Google Scholar]
- 102.Xie Z, Shen G, Wang Y and Wu C, Poult. Sci, 2019, 98, 422–429, DOI: 10.3382/ps/pey315. [DOI] [PubMed] [Google Scholar]
- 103.Yau JWY, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund J, Haffner S, Hamman RF, Ikram MK, Kayama T, Klein BEK, Klein R, Krishnaiah S, Mayurasakorn K, O’Hare JP, Orchard TJ, Porta M, Rema M, Roy MS, Sharma T, Shaw J, Taylor H, Tielsch JM, Varma R, Wang JJ, Wang N, West S, Zu L, Yasuda M, Zhang X, Mitchell P and Wong TY, Diabetes Care, 2012, 35, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaufman-Szymczyk A, Majewski G, Lubecka-Pietruszewska K and Fabianowska-Majewska K, Int. J. Mol. Sci, 2015, 16, 29732–29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lubecka-Pietruszewska K, Kaufman-Szymczyk A, Stefanska B, Cebula-Obrzut B, Smolewski P and Fabianowska-Majewska K, J. Nutrigenet. Nutrigenomics, 2015, 8, 91–101. [DOI] [PubMed] [Google Scholar]
- 106.Myzak MC, Hardin K, Wang R, Dashwood RH and Ho E, Carcinogenesis, 2006, 27, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH and Ho E, Clin. Epigenet, 2011, 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Senanayake GVK, Banigesh A, Wu L, Lee P and Juurlink BHJ, Am. J. Hypertens, 2012, 25, 229–235. [DOI] [PubMed] [Google Scholar]
- 109.Zhao F, Zhang J and Chang N, Eur. J. Pharmacol, 2018, 824, 1–10. [DOI] [PubMed] [Google Scholar]
- 110.Kumar H, Kim IS, More SV, Kim BW and Choi DK, Nat. Prod. Rep, 2014, 31, 109–139. [DOI] [PubMed] [Google Scholar]
- 111.Paladino S, Conte A, Caggiano R, Pierantoni GM and Faraonio R, Cell. Physiol. Biochem, 2018, 47, 1951–1976. [DOI] [PubMed] [Google Scholar]
- 112.Schmidlin CJ, Dodson MB, Madhavan L and Zhang DD, Free Radicals Biol. Med, 2019, 134, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K and Yamamoto M, Mol. Cell. Biol, 2004, 24, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT and Jordan-Sciutto KL, J. Neuropathol. Exp. Neurol, 2007, 66, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang C, Su ZY, Khor TO, Shu L and Kong ANT, Biochem. Pharmacol, 2013, 85, 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.An YW, Jhang KA, Woo SY, Kang JL and Chong YH, Neurobiol. Aging, 2016, 38, 1–10. [DOI] [PubMed] [Google Scholar]
- 117.Hampl JS, Taylor CA and Johnston CS, Am. J. Public Health, 2004, 94, 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Monacelli F, Acquarone E, Giannotti C, Borghi R and Nencioni A, Nutrients, 2017, 9, E670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ellulu MS, Rahmat A, Patimah I, Khaza’Ai H and Abed Y, Drug Des., Dev. Ther, 2015, 9, 3405–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mittal M, Siddiqui MR, Tran K, Reddy SP and Malik AB, Antioxid. Redox Signaling, 2014, 20, 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Esteban MA, Wang T, Qin B, Yang J, Qin D, Cai J, Li W, Weng Z, Chen J, Ni S, Chen K, Li Y, Liu X, Xu J, Zhang S, Li F, He W, Labuda K, Song Y, Peterbauer A, Wolbank S, Redl H, Zhong M, Cai D, Zeng L and Pei D, Cell Stem Cell, 2010, 6, 71–79. [DOI] [PubMed] [Google Scholar]
- 122.Sasidharan Nair V, Song MH and Oh KI, J. Immunol, 2016, 196, 2119–2131. [DOI] [PubMed] [Google Scholar]
- 123.Kim HP and Leonard WJ, J. Exp. Med, 2007, 204, 1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, Nakayama M, Rosenthal W and Bluestone JA, Nat. Immunol, 2009, 10, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pangrazzi L, Meryk A, Naismith E, Koziel R, Lair J, Krismer M, Trieb K and Grubeck-Loebenstein B, Eur. J. Immunol, 2017, 47, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Olsson J, Wikby A, Johansson B, Löfgren S, Nilsson BO and Ferguson FG, Mech. Ageing Dev, 2001, 121, 187–201. [DOI] [PubMed] [Google Scholar]
- 127.Chung TL, Brena RM, Kolle G, Grimmond SM, Berman BP, Laird PW, Pera MF and Wolvetang EJ, Stem Cells, 2010, 28, 1848–1855. [DOI] [PubMed] [Google Scholar]
- 128.Minor EA, Court BL, Young JI and Wang G, J. Biol. Chem, 2013, 288, 13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, Qin B, Zeng L, Esteban MA, Pan G and Pei D, Cell Stem Cell, 2011, 9, 575–587. [DOI] [PubMed] [Google Scholar]
- 130.Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Chen J, Wu Y, Guo L, Zhu J, Zhao X, Peng T, Zhang Y, Chen S, Li X, Li D, Wang T and Pei D, Nat. Genet, 2013, 45, 34–42. [DOI] [PubMed] [Google Scholar]
- 131.Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann-Zalcenstein D, Maza I, Geula S, Viukov S, Holtzman L, Pribluda A, Canaani E, Horn-Saban S, Amit I, Novershtern N and Hanna JH, Nature, 2012, 488, 409–413. [DOI] [PubMed] [Google Scholar]
- 132.He J, Kallin EM, Tsukada YI and Zhang Y, Nat. Struct. Mol. Biol, 2008, 15, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ebata KT, Mesh K, Liu S, Bilenky M, Fekete A, Acker MG, Hirst M, Garcia BA and Ramalho-Santos M, Epigenet. Chromatin, 2017, 10, DOI: 10.1186/s13072-017-0143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang T, Huang K, Zhu Y, Wang T, Shan Y, Long B, Li Y, Chen Q, Wang P, Zhao S, Li D, Wu C, Kang B, Gu J, Mai Y, Wang Q, Li J, Zhang Y, Liang Z, Guo L, Wu F, Su S, Wang J, Gao M, Zhong X, Liao B, Chen J, Zhang X, Shu X, Pei D, Nie J and Pan G, J. Biol. Chem, 2019, 294, 13657–13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abbey D and Seshagiri PB, Differentiation, 2017, 96, 1–14. [DOI] [PubMed] [Google Scholar]
- 136.Zhong J, Xu C, Reece EA and Yang P, Am. J. Obstet. Gynecol, 2016, 215, 368.e1–368.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nandakumar V, Vaid M and Katiyar SK, Carcinogenesis, 2011, 32, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Devarshi PP, Jones AD, Taylor EM, Stefanska B and Henagan TM, PPAR Res, 2017, 2017, 3235693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maugeri A, Barchitta M, Mazzone MG, Giuliano F, Basile G and Agodi A, Int. J. Mol. Sci, 2018, 19, 2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tu Y, Sun W, Wan YG, Gao K, Liu H, Yu BY, Hu H and Huang YR, J. Ethnopharmacol, 2014, 156, 115–124. [DOI] [PubMed] [Google Scholar]
- 141.Dong X, Fu J, Yin X, Cao S, Li X, Lin L and Ni J, Phyther. Res, 2016, 30, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mueller SO, Schmitt M, Dekant W, Stopper H, Schlatter J, Schreier P and Lutz WK, Food Chem. Toxicol, 1999, 37, 481–491. [DOI] [PubMed] [Google Scholar]
- 143.Cha TL, Chuang MJ, Tang SH, Wu ST, Sun KH, Chen TT, Sun GH, Chang SY, Yu CP, Ho JY, Liu SY, Huang SM and Yu DS, Mol. Carcinog, 2015, 54, 167–177. [DOI] [PubMed] [Google Scholar]
- 144.Zhang H, Chen L, Bu HQ, Yu QJ, Jiang DD, Pan FP, Wang Y, Liu DL and Lin SZ, Oncol. Rep, 2015, 33, 3015–3023. [DOI] [PubMed] [Google Scholar]
- 145.Pan FP, Zhou HK, Bu HQ, Chen ZQ, Zhang H, Xu LP, Tang J, Yu QJ, Chu YQ, Pan J, Fei Y, Lin SZ, Liu DL and Chen L, Oncol. Rep, 2016, 35, 1941–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iwanowycz S, Wang J, Altomare D, Hui Y and Fan D, J. Biol. Chem, 2016, 291, 11491–11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C and Li J, Cell. Signalling, 2014, 26, 192–197. [DOI] [PubMed] [Google Scholar]
- 148.Wynn TA, Chawla A and Pollard JW, Nature, 2013, 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jin X, Liu MY, Zhang DF, Zhong X, Du K, Qian P, Gao H and Wei MJ, Pharmacol. Res, 2019, 145, 104253. [DOI] [PubMed] [Google Scholar]
- 150.Li Y, Xu QQ, Shan CS, Shi YH, Wang Y and Zheng GQ, Front. Pharmacol, 2018, 9, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ma Y, Xia X, Cheng J. m., Kuang Y. q., Yang T, Yang L. b., Fan K and Gu J. w., Neurochem. Res, 2014, 39, 1809–1816. [DOI] [PubMed] [Google Scholar]
- 152.Liu T, Jin H, Sun QR, Xu JH and Hu HT, Brain Res, 2010, 1347, 149–160. [DOI] [PubMed] [Google Scholar]
- 153.Panchal SK, Ward L and Brown L, Eur. J. Nutr, 2013, 52, 559–568. [DOI] [PubMed] [Google Scholar]
- 154.Marín M, María Giner R, Ríos JL and Carmen Recio M, J. Ethnopharmacol, 2013, 150, 925–934. [DOI] [PubMed] [Google Scholar]
- 155.Rosillo MA, Sánchez-Hidalgo M, Cárdeno A, Aparicio-Soto M, Sánchez-Fidalgo S, Villegas I and De La Lastra CA, Pharmacol. Res, 2012, 66, 235–242. [DOI] [PubMed] [Google Scholar]
- 156.Gourineni V, Shay NF, Chung S, Sandhu AK and Gu L, J. Agric. Food Chem, 2012, 60, 7674–7681. [DOI] [PubMed] [Google Scholar]
- 157.Shulman GI, J. Clin. Invest, 2000, 106, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Selvi BR, Batta K, Kishore AH, Mantelingu K, Varier RA, Balasubramanyam K, Pradhan SK, Dasgupta D, Sriram S, Agrawal S and Kundu TK, J. Biol. Chem, 2010, 285, 7143–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Porta M, Amione C, Barutta F, Fornengo P, Merlo S, Gruden G, Albano L, Ciccarelli M, Ungaro P, Durazzo M, Beguinot F, Berchialla P, Cavallo F and Trento M, Endocrine, 2019, 63, 284–292. [DOI] [PubMed] [Google Scholar]
- 160.Miao F, Li S, Chavez V, Lanting L and Natarajan R, Mol. Endocrinol, 2006, 20, 1562–1573. [DOI] [PubMed] [Google Scholar]
- 161.Kang I, Okla M and Chung S, J. Nutr. Biochem, 2014, 25, 946–953. [DOI] [PubMed] [Google Scholar]
- 162.Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM and Bedford MT, EMBO Rep, 2008, 9, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Larsen TM, Toubro S and Astrup A, Int. J. Obes, 2003, 27, 147–161. [DOI] [PubMed] [Google Scholar]
- 164.Wang M and Tafuri S, J. Cell. Biochem, 2003, 89, 38–47. [DOI] [PubMed] [Google Scholar]
- 165.Harrington WW, Britt CS, Wilson JG, Milliken NO, Binz JG, Lobe DC, Oliver WR, Lewis MC and Ignar DM, PPAR Res, 2007, 2007, 97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kuo MY, Ou HC, Lee WJ, Kuo WW, Hwang LL, Song TY, Huang CY, Chiu TH, Tsai KL, Tsai CS and Sheu WHH, J. Agric. Food Chem, 2011, 59, 5100–5108. [DOI] [PubMed] [Google Scholar]
- 167.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B and Sinclair DA, Nature, 2003, 425, 191–196. [DOI] [PubMed] [Google Scholar]
- 168.Houtkooper RH, Pirinen E and Auwerx J, Nat. Rev. Mol. Cell Biol, 2012, 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee Y, Drake AC, Thomas NO, Ferguson LG, Chappell PE and Shay KP, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol, 2018, 208, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fischer N, Büchter C, Koch K, Albert S, Csuk R and Wätjen W, J. Pharm. Pharmacol, 2017, 69, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Thompson AM, Martin KA and Rzucidlo EM, PLoS One, 2014, 9, e85495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yu W, Wan Z, Qiu XF, Chen Y and Dai YT, Asian J. Androl, 2013, 15, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K and Zhou Z, Cell Metab, 2012, 16, 738–750. [DOI] [PubMed] [Google Scholar]
- 174.Porquet D, Casadesús G, Bayod S, Vicente A, Canudas AM, Vilaplana J, Pelegŕı C, Sanfeliu C, Camins A, Pallàs M and Del Valle J, Age, 2013, 35, 1851–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kao CL, Chen LK, Chang YL, Yung MC, Hsu CC, Chen YC, Lo WL, Chen SJ, Ku HH and Hwang SJ, J. Atheroscler. Thromb, 2010, 17, 970–979. [DOI] [PubMed] [Google Scholar]
- 176.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P and Auwerx J, Cell, 2006, 127, 1109–1122. [DOI] [PubMed] [Google Scholar]
- 177.Imamura H, Nagayama D, Ishihara N, Tanaka S, Watanabe R, Watanabe Y, Sato Y, Yamaguchi T, Ban N, Kawana H, Ohira M, Endo K, Saiki A, Shirai K and Tatsuno I, Mol. Genet. Metab. Rep, 2017, 12, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen S, Zhao Z, Ke L, Li Z, Li W, Zhang Z, Zhou Y, Feng X and Zhu W, J. Nutr. Biochem, 2018, 55, 209–218. [DOI] [PubMed] [Google Scholar]
- 179.Yonamine CY, Pinheiro-Machado E, Michalani ML, Alves-Wagner AB, V Esteves J, Freitas HS and Machado UF, Molecules, 2017, 22, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.de Sá Coutinho D, Pacheco MT, Frozza RL and Bernardi A, Int. J. Mol. Sci, 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gao Z and Ye J, Biochem. Biophys. Res. Commun, 2008, 376, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xie Z, Singh M and Singh K, J. Biol. Chem, 2004, 279, 39513–39519. [DOI] [PubMed] [Google Scholar]
- 183.Mohammad G, Mairaj Siddiquei M, Imtiaz Nawaz M and Abu El-Asrar AM, J. Diabetes Res, 2013, 2013, 658548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P and Henderson CE, Neuron, 2014, 81, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yar AS, Menevse S and Alp E, Genet. Mol. Res, 2011, 10, 2962–2975. [DOI] [PubMed] [Google Scholar]
- 186.Kowluru RA, Santos JM and Zhong Q, Invest. Ophthalmol. Visual Sci, 2014, 55, 5653–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gao D, Zhang X, Jiang X, Peng Y, Huang W, Cheng G and Song L, Life Sci, 2006, 78, 2564–2570. [DOI] [PubMed] [Google Scholar]
- 188.Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS and Turner RS, J. Neuroinflammation, 2017, 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cheng CY, Hsieh HL, Der Hsiao L and Yang CM, Stem Cell Res, 2012, 9, 9–23. [DOI] [PubMed] [Google Scholar]
- 190.Ren Z, Wang L, Cui J, Huoc Z, Xue J, Cui H, Mao Q and Yang R, Pharmazie, 2013, 68, 689–694. [PubMed] [Google Scholar]
- 191.Kawabata T, Tokuda H, Fujita K, Kainuma S, Sakai G, Matsushima-Nishiwaki R, Kozawa O and Otsuka T, Cell. Physiol. Biochem, 2017, 43, 1025–1036. [DOI] [PubMed] [Google Scholar]
- 192.Venturelli S, Berger A, Böcker A, Busch C, Weiland T, Noor S, Leischner C, Schleicher S, Mayer M, Weiss TS, Bischoff SC, Lauer UM and Bitzer M, PLoS One, 2013, 8, e73097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bo S, Togliatto G, Gambino R, Ponzo V, Lombardo G, Rosato R, Cassader M and Brizzi MF, Acta Diabetol, 2018, 55, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bo S, Ponzo V, Ciccone G, Evangelista A, Saba F, Goitre I, Procopio M, Pagano GF, Cassader M and Gambino R, Pharmacol. Res, 2016, 111, 896–905. [DOI] [PubMed] [Google Scholar]
- 195.Clement J, Wong M, Poljak A, Sachdev P and Braidy N, Rejuvenation Res, 2019, 22, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kim J, Lee S, Choi BR, Yang H, Hwang Y, Park JHY, LaFerla FM, Han JS, Lee KW and Kim J, Mol. Nutr. Food Res, 2017, 61, 1600194. [DOI] [PubMed] [Google Scholar]
- 197.Minichiello L, Nat. Rev. Neurosci, 2009, 10, 850–860. [DOI] [PubMed] [Google Scholar]
- 198.Kemppainen S, Rantamäki T, Jerónimo-Santos A, Lavasseur G, Autio H, Karpova N, Kärkkäinen E, Stavén S, Vicente Miranda H, Outeiro TF, Diógenes MJ, Laroche S, Davis S, Sebastîao AM, Castrén E and Tanila H, Neurobiol. Aging, 2012, 33, 1122.e23–1122.e39. [DOI] [PubMed] [Google Scholar]
- 199.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJF, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R and Tsai LH, Nature, 2009, 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dash PK, Zhao J, Orsi SA, Zhang M and Moore AN, Neurosci. Lett, 2009, 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kim HV, Kim HY, Ehrlich HY, Choi SY, Kim DJ and Kim YS, Amyloid, 2013, 20, 7–12. [DOI] [PubMed] [Google Scholar]
- 202.Zhang R, Miao QW, Zhu CX, Zhao Y, Liu L, Yang J and An L, American Journal of Alzheimer’s Disease & Other Dementiasr, 2015, 30, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ye B, Chen X, Dai S, Han J, Liang X, Lin S, Cai X, Huang Z and Huang W, Drug Des., Dev. Ther, 2019, 13, 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Swanson KV, Deng M and Ting JP-Y, Nat. Rev. Immunol, 2019, 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Godoy LD, Lucas JE, Bender AJ, Romanick SS and Ferguson BS, Mol. Nutr. Food Res, 2016, 1600744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pal T, Jana NR and Das PK, Analyst, 1992, 117, 791–793. [Google Scholar]
- 207.Ooi JYY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ and El-Osta A, Epigenetics, 2015, 10, 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kaneda R, Ueno S, Yamashita Y, Choi YL, Koinuma K, Takada S, Wada T, Shimada K and Mano H, Circ. Res, 2005, 97, 210–218. [DOI] [PubMed] [Google Scholar]
- 209.Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T and Hasegawa K, J. Clin. Invest, 2008, 118, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Feng B, Chen S, Chiu J, George B and Chakrabarti S, Am. J. Physiol.: Endocrinol. Metab, 2008, 294, E1119–E1126. [DOI] [PubMed] [Google Scholar]
- 211.Sun H, Yang X, Zhu J, Lv T, Chen Y, Chen G, Zhong L, Li Y, Huang X, Huang G and Tian J, Life Sci, 2010, 87, 707–714. [DOI] [PubMed] [Google Scholar]
- 212.Yanazume T, Hasegawa K, Morimoto T, Kawamura T, Wada H, Matsumori A, Kawase Y, Hirai M and Kita T, Mol. Cell. Biol, 2003, 23, 3593–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bueno OF, Van Rooij E, Molkentin JD, Doevendans PA and De Windt LJ, Cardiovasc. Res, 2002, 53, 806–821. [DOI] [PubMed] [Google Scholar]
- 214.Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, O’Brien CA, Jilka RL, Zhou D and Almeida M, Aging Cell, 2017, 16, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J and Elledge SJ, Science, 2015, 349, aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hernández M, Wicz S and Corral RS, Phytomedicine, 2016, 23, 1392–1400. [DOI] [PubMed] [Google Scholar]
- 217.Singh S and Aggarwal BB, J. Biol. Chem, 1995, 270, 24995–25000. [DOI] [PubMed] [Google Scholar]
- 218.Hu J, Shen T, Xie J, Wang S, He Y and Zhu F, Exp. Ther. Med, 2017, 14, 5896–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Manicone AM and McGuire JK, Semin. Cell Dev. Biol, 2008, 19, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Papazafiropoulou A and Tentolouris N, Hippokratia, 2009, 13, 76–82. [PMC free article] [PubMed] [Google Scholar]
- 221.Hopps E and Caimi G, Eur. J. Intern. Med, 2012, 23, 99–104. [DOI] [PubMed] [Google Scholar]
- 222.Mroczko B, Groblewska M and Barcikowska M, J. Alzheimer’s Dis, 2013, 37, 273–283. [DOI] [PubMed] [Google Scholar]
- 223.Liu HL, Chen Y, Cui GH and Zhou JF, Acta Pharmacol. Sin, 2005, 26, 603–609. [DOI] [PubMed] [Google Scholar]
- 224.Ma J, Ma S. y. and Ding C. h., Chin. J. Integr. Med, 2017, 23, 362–369. [DOI] [PubMed] [Google Scholar]
- 225.Liu D, Perkins JT and Hennig B, J. Nutr. Biochem, 2016, 28, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pober JS and Sessa WC, Nat. Rev. Immunol, 2007, 7, 803–815. [DOI] [PubMed] [Google Scholar]
- 227.Ross R, Engl N. J. Med, 1999, 340, 115–126. [DOI] [PubMed] [Google Scholar]
- 228.Widlansky ME, Hamburg NM, Anter E, Holbrook M, Kahn DF, Elliott JG, Keaney JF and Vita JA, J. Am. Coll. Nutr, 2007, 26, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu J, Tang Y, Feng Z, Hou C, Wang H, Yan J, Liu J, Shen W, Zang W, Liu J and Long J, Metabolism, 2014, 63, 1314–1323. [DOI] [PubMed] [Google Scholar]
- 230.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA and Sadoshima J, Circ. Res, 2010, 107, 1470–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sengupta A, Molkentin JD and Yutzey KE, J. Biol. Chem, 2009, 284, 28319–28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sun Y and Guan X, Frontiers in Laboratory Medicine, 2018, 2, 68–71. [Google Scholar]
- 233.Pan B, Quan J, Liu L, Xu Z, Zhu J, Huang X and Tian J, J. Cell. Mol. Med, 2017, 21, 2481–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Stote K, Corkum A, Sweeney M, Shakerley N, Kean T and Gottschall-Pass K, Nutrients, 2019, 11, E202, DOI: 10.3390/nu11010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yang L, Ling W, Yang Y, Chen Y, Tian Z, Du Z, Chen J, Xie Y, Liu Z and Yang L, Nutrients, 2017, 9, E1104, DOI: 10.3390/nu9101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Castro-Acosta ML, Stone SG, Mok JE, Mhajan RK, Fu CI, Lenihan-Geels GN, Corpe CP and Hall WL, J. Nutr. Biochem, 2017, 49, 53–62. [DOI] [PubMed] [Google Scholar]
- 237.Zhang PW, Chen FX, Li D, Ling WH and Guo HH, Medicine, 2017, 94, E758, DOI: 10.1097/md.0000000000000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Habanova M, Saraiva JA, Haban M, Schwarzova M, Chlebo P, Predna L, Gažo J and Wyka J, Nutr. Res, 2016, 36, 1415–1422. [DOI] [PubMed] [Google Scholar]
- 239.Zhan W, Liao X, Xie RJ, Tian T, Yu L, Liu X, Liu J, Li P, Han B, Yang T, Zhang B, Cai LJ, Li R and Yang Q, Oncotarget, 2017, 8, 96761–96773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zhan W, Liao X, Tian T, Yu L, Liu X, Li B, Liu J, Han B, Xie RJ, Ji QH and Yang Q, Genet. Mol. Res, 2017, 16, DOI: 10.4238/gmr16019188. [DOI] [PubMed] [Google Scholar]
- 241.Shi Y, Sun H, Bao J, Zhou P, Zhang J, Li L and Bu H, Am. J. Pathol, 2011, 179, 1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kouzarides T, Cell, 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 243.Wei J, Zhang G, Zhang X, Xu D, Gao J and Fan J, Plant Foods Hum. Nutr, 2017, 72, 425–431. [DOI] [PubMed] [Google Scholar]
- 244.Seong AR, Yoo JY, Choi KC, Lee MH, Lee YH, Lee J, Jun W, Kim S and Yoon HG, Biochem. Biophys. Res. Commun, 2011, 410, 581–586. [DOI] [PubMed] [Google Scholar]
- 245.Rahnasto-Rilla M, Tyni J, Huovinen M, Jarho E, Kulikowicz T, Ravichandran S, Bohr VA, Ferrucci L, Lahtela-Kakkonen M and Moaddel R, Sci. Rep, 2018, 8, 4163, DOI: 10.1038/s41598-018-22388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Downing LE, Heidker RM, Caiozzi GC, Wong BS, Rodriguez K, Del Rey F and Ricketts ML, PLoS One, 2015, 10, e0140267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mikula M, Majewska A, Ledwon JK, Dzwonek A and Ostrowski J, Int. J. Mol. Med, 2014, 34, 1647–1654. [DOI] [PubMed] [Google Scholar]
- 248.Yao W, Wang T and Huang F, BioMed Res. Int, 2018, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chung MY, Song JH, Lee J, Shin EJ, Park JH, Lee SH, Hwang JT and Choi HK, Molecular Metabolism, 2019, 19, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Wang Z, Zhao Y, Xu N, Zhang S, Wang S, Mao Y, Zhu Y, Li B, Jiang Y, Tan Y, Xie W, Yang BB and Zhang Y, Cell. Mol. Life Sci, 2019, 76, 3005–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, Roeder RG and Allis CD, Mol. Cell, 2018, 69, 533, DOI: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Meng J, Li Y, Camarillo C, Yao Y, Zhang Y, Xu C and Jiang L, PLoS One, 2014, 9, e85570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Müller M, Hernández MAG, Goossens GH, Reijnders D, Holst JJ, Jocken JWE, van Eijk H, Canfora EE and Blaak EE, Sci. Rep, 2019, 9, 12515, DOI: 10.1038/s41598-019-48775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Okuda Y, Kawai K and Yamashita K, Endocrinology, 1987, 120, 2152–2157, DOI: 10.1210/endo-120-5-2152. [DOI] [PubMed] [Google Scholar]
- 255.Nakayama H, Tokubuchi I, Wada N, Tsuruta M, Ohki T, Oshige T, Sasaki Y, Iwata S, Kato N, Ohtsuka Y, Matsuo Y, Tajiri Y and Yamada K, Endocr. J, 2015, 62, 235–241, DOI: 10.1507/endocrj.ej14-0431. [DOI] [PubMed] [Google Scholar]
- 256.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, De Cabo R, Ulrich S, Akassoglou K and Verdin E, Science, 2013, 339, 211–214, DOI: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ryan JM and Cristofalo VJ, Biochem. Biophys. Res. Commun, 1972, 48, 735–742, DOI: 10.1016/0006-291x(72)90668-7. [DOI] [PubMed] [Google Scholar]
- 258.Lu JY, Lin YY, Sheu JC, Wu JT, Lee FJ, Chen Y, Lin MI, Chiang FT, Tai TY, Berger SL, Zhao Y, Tsai KS, Zhu H, Chuang LM and Boeke JD, Cell, 2011, 146, 969–979, DOI: 10.1016/j.cell.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lin Y. y., Lu J. y., Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL and Zhu H, Cell, 2009, 136, 1073–1084, DOI: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chriett S, Dabek A, Wojtala M, Vidal H, Balcerczyk A and Pirola L, Sci. Rep, 2019, 9, 742, DOI: 10.1038/s41598-018-36941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wang T, Holt MV and Young NL, Epigenetics, 2018, 13, 519–535, DOI: 10.1080/15592294.2018.1475979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Plunet WT, Streijger F, Lam CK, Lee JHT, Liu J and Tetzlaff W, Exp. Neurol, 2008, 213, 28–35, DOI: 10.1016/j.expneurol.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 263.Plunet WT, Lam CK, Lee JHT, Liu J and Tetzlaff W, Ann. N. Y. Acad. Sci, 2010, 1198, E1–E11. [DOI] [PubMed] [Google Scholar]
- 264.Streijger F, Plunet WT, Lee JHT, Liu J, Lam CK, Park S, Hilton BJ, Fransen BL, Matheson KAJ, Assinck P, Kwon BK and Tetzlaff W, PLoS One, 2013, 8, E78765, DOI: 10.1371/journal.pone.0078765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang X, Wu X, Liu Q, Kong G, Zhou J, Jiang J, Wu X, Huang Z, Su W and Zhu Q, Neuroscience, 2017, 366, 36–43, DOI: 10.1016/j.neuroscience.2017.09.056. [DOI] [PubMed] [Google Scholar]
- 266.Hu E, Du H, Zhu X, Wang L, Shang S, Wu X, Lu H and Lu X, Neuroscience, 2018, 386, 315–325, DOI: 10.1016/j.neuroscience.2018.06.036. [DOI] [PubMed] [Google Scholar]
- 267.Noh HS, Sang SK, Dong WK, Young HK, Chang HP, Jae YH, Gyeong JC and Choi WS, Epilepsy Res, 2005, 65, 153–159, DOI: 10.1016/j.eplepsyres.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 268.Haldar S, Dru C, Mishra R, Tripathi M, Duong F, Angara B, Fernandez A, Arditi M and Bhowmick NA, Sci. Rep, 2016, 6, 39257, DOI: 10.1038/srep39257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C, Morozova-Roche LA, Herzog RI, Iwasaki A and Dixit VD, Cell Rep, 2017, 18, 2077–2087, DOI: 10.1016/j.celrep.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B and Zhao Y, Cell, 2011, 146, 1016–1028, DOI: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kelly RDW, Chandru A, Watson PJ, Song Y, Blades M, Robertson NS, Jamieson AG, Schwabe JWR and Cowley SM, Sci. Rep, 2018, 8, 14690, DOI: 10.1038/s41598-018-32927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Mihaylova MM and Stratton MS, in Nutritional Epigenomics, 2019. [Google Scholar]
- 273.Huang H, Tang S, Ji M, Tang Z, Shimada M, Liu X, Qi S, Locasale JW, Roeder RG, Zhao Y and Li X, Mol. Cell, 2018, 70, 663–678, DOI: 10.1016/j.molcel.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Wood JG, Regina B, Lavu S, Hewitz K, Helfand SL, Tatar M and Sinclair D, Nature, 2004, 430, 686–689, DOI: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 275.Mattison JA, Roth GS, Mark Beasley T, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK and De Cabo R, Nature, 2012, 489, 318–321, DOI: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mattson MP and Wan R, J. Nutr. Biochem, 2005, 16, 129–137. [DOI] [PubMed] [Google Scholar]
- 277.Meydani SN, Das SK, Pieper CF, Lewis MR, Klein S, Dixit VD, Gupta AK, Villareal DT, Bhapkar M, Huang M, Fuss PJ, Roberts SB, Holloszy JO and Fontana L, Aging, 2016, 8, 1416–1431, DOI: 10.18632/aging.100994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Fontana L, Meyer TE, Klein S and Holloszy JO, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 6659–6663, DOI: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Li Y, Daniel M and Tollefsbol TO, BMC Med, 2011, 9, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Graff EC, Fang H, Wanders D and Judd RL, Metabolism, 2016, 65, 102–113. [DOI] [PubMed] [Google Scholar]
- 281.Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Müller-Fielitz H, Pokorná B, Vollbrandt T, Stölting I, Nadrowitz R, Okun JG, Offermanns S and Schwaninger M, Nat. Commun, 2014, 5, 3944, DOI: 10.1038/ncomms4944. [DOI] [PubMed] [Google Scholar]
- 282.Zhou E, Li Y, Yao M, Wei Z, Fu Y and Yang Z, Int. Immunopharmacol, 2014, 23, 121–126, DOI: 10.1016/j.intimp.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 283.Fu SP, Wang JF, Xue WJ, Liu HM, Liu B. r., Zeng YL, Li SN, Huang BX, Lv QK, Wang W and Liu JX, J. Neuroinflammation, 2015, 12, 9, DOI: 10.1186/s12974-014-0230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A and Tsujimoto G, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 8030–8035, DOI: 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Theiler A, Bärnthaler T, Platzer W, Richtig G, Peinhaupt M, Rittchen S, Kargl J, Ulven T, Marsh LM, Marsche G, Schuligoi R, Sturm EM and Heinemann A, J. Allergy Clin. Immunol, 2019, 144, 764–776, DOI: 10.1016/j.jaci.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 286.Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A and Fallon PG, J. Allergy Clin. Immunol, 2010, 125, 1114–1124, DOI: 10.1016/j.jaci.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 287.Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, De Zoete MR, Licona-Limón P, Paiva RS, Ching T, Weaver C, Zi X, Pan X, Fan R, Garmire LX, Cotton MJ, Drier Y, Bernstein B, Geginat J, Stockinger B, Esplugues E, Huber S and Flavell RA, Nature, 2015, 523, 221–225, DOI: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kim DS, Kwon JE, Lee SH, Kim EK, Ryu JG, Jung KA, Choi JW, Park MJ, Moon YM, Park SH, La Cho M and Kwok SK, Front. Immunol, 2018, 9, 1525, DOI: 10.3389/fimmu.2018.01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Jin UH, Cheng Y, Park H, Davidson LA, Callaway ES, Chapkin RS, Jayaraman A, Asante A, Allred C, Weaver EA and Safe S, Sci. Rep, 2017, 7, 10163, DOI: 10.1038/s41598-017-10824-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, Griffith AE, Patel B, Shouval DS, Weiner HL, Snapper SB and Quintana FJ, Cell Rep, 2016, 17, 1318–1329, DOI: 10.1016/j.celrep.2016.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lv Q, Shi C, Qiao S, Cao N, Guan C, Dai Y and Wei Z, Cell Death Dis, 2018, 9, 890, DOI: 10.1038/s41419-018-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.van de Wouw M, Boehme M, Lyte JM, Wiley N, Strain C, O’Sullivan O, Clarke G, Stanton C, Dinan TG and Cryan JF, J. Physiol, 2018, 596, 4923–4944, DOI: 10.1113/JP276431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Schroeder FA, Lin CL, Crusio WE and Akbarian S, Biol. Psychiatry, 2007, 62, 55–64, DOI: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 294.Kovács Z, D’Agostino DP and Ari C, Front. Behav. Neurosci, 2018, 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Calabrese JR and Delucchi GA, J. Clin. Psychiatry, 1989, 50, 30–34. [PubMed] [Google Scholar]
- 296.Smith LA, Cornelius VR, Azorin JM, Perugi G, Vieta E, Young AH and Bowden CL, J. Affective Disord, 2010, 122, 1–9. [DOI] [PubMed] [Google Scholar]
- 297.Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD and Rumbaugh G, Neuropsychopharmacology, 2010, 35, 870–880, DOI: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Sartor GC, Malvezzi AM, Kumar A, Andrade NS, Wiedner HJ, Vilca SJ, Janczura KJ, Bagheri A, Al-Ali H, Powell SK, Brown PT, Volmar CH, Foster TC, Zeier Z and Wahlestedt C, J. Neurosci, 2019, 39, 612–626, DOI: 10.1523/jneurosci.1604-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Yin JX, Maalouf M, Han P, Zhao M, Gao M, Dharshaun T, Ryan C, Whitelegge J, Wu J, Eisenberg D, Reiman EM, Schweizer FE and Shi J, Neurobiol. Aging, 2016, 39, 25–37, DOI: 10.1016/j.neurobiolaging.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 300.Roberts MN, Wallace MA, Tomilov AA, Zhou Z, Marcotte GR, Tran D, Perez G, Gutierrez-Casado E, Koike S, Knotts TA, Imai DM, Griffey SM, Kim K, Hagopian K, Haj FG, Baar K, Cortopassi GA, Ramsey JJ and Lopez-Dominguez JA, Cell Metab, 2018, 27, 1156, DOI: 10.1016/j.cmet.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Carabotti M, Scirocco A, Maselli MA and Severi C, Ann. Gastroenterol, 2015, 28, 203–209. [PMC free article] [PubMed] [Google Scholar]
- 302.Chang C-J, Lin C-S, Lu C-C, Martel J, Ko Y-F, Ojcius DM, Tseng S-F, Wu T-R, Chen Y-YM, Young JD and Lai H-C, Nat. Commun, 2015, 6, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Wang J, Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, Lechatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K and Wang J, Nature, 2012, 490, 55–60. [DOI] [PubMed] [Google Scholar]
- 304.Kadowaki A, Miyake S, Saga R, Chiba A, Mochizuki H and Yamamura T, Nat. Commun, 2016, 7, 11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mahler A, Balogh A, Marko L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Fratzer C, Krannich A, Gollasch M, Grohme DA, Corte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ and Muller DN, Nature, 2017, 551, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larché MJ, Davidson DJ, Verdú EF, Surette MG and Bowdish DME, Cell Host Microbe, 2017, 21, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sook Lee E, Ji Song E and Do Nam Y, Journal of Clinical Epigenetics, 2019, 139, 191–198, DOI: 10.21767/2472-1158.100048. [DOI] [Google Scholar]
- 308.de la Cuesta-Zuluaga J, Mueller NT, Alvarez-Quintero R, Velásquez-Mejía EP, Sierra JA, Corrales-Agudelo V, Carmona JA, Abad JM and Escobar JS, Nutrients, 2018, 11, E51, DOI: 10.3390/nu11010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kao ACC, Chan KW, Anthony DC, Lennox BR and Burnet PW, Neuropharmacology, 2019, 150, 184–191. [DOI] [PubMed] [Google Scholar]
- 310.Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang YL, Hennig KM, Gale J, Zhao WN, Reis S, Barker DD, Berry-Scott E, Kim SW, Clore EL, Hooker JM, Holson EB, Haggarty SJ and Petryshen TL, PLoS One, 2013, 8, e71323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB and Hong JS, Int. J. Neuropsychopharmacol, 2008, 11, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lima GC, Vieira VCC, Cazarin CBB, Ribeiro RR, Junior SB, de Albuquerque CL, Vidal RO, Netto CC, Yamada AT, Augusto F and Maróstica MR Junior, Eur. J. Nutr, 2018, 57, 1499–1510. [DOI] [PubMed] [Google Scholar]
- 313.Kerckhoffs APM, Samsom M, van Berge Henegouwen GP, Akkermans LMA, Nieuwenhuijs VB and Visser MR, in Gastrointestinal Microbiology, CRC Press, 2006, pp. 25–50. [Google Scholar]
- 314.Gronier B, Savignac HM, Di Miceli M, Idriss SM, Tzortzis G, Anthony D and Burnet PWJ, Eur. Neuropsychopharmacol, 2018, 28, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Yang XD, Wang LK, Wu HY and Jiao L, BMC Anesthesiol, 2018, 18, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kao ACC, Spitzer S, Anthony DC, Lennox B and Burnet PWJ, Transl. Psychiatry, 2018, 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wang S, Huang XF, Zhang P, Wang H, Zhang Q, Yu S and Yu Y, J. Nutr. Biochem, 2016, 36, 42–50. [DOI] [PubMed] [Google Scholar]
- 318.Hira T, Suto R, Kishimoto Y, Kanahori S and Hara H, Eur. J. Nutr, 2018, 57, 965–979. [DOI] [PubMed] [Google Scholar]
- 319.Cha BK, Jung SM, Choi CH, Do Song I, Lee HW, Kim HJ, Hyuk J, Chang SK, Kim K, Chung WS and Seo JG, J. Clin. Gastroenterol, 2012, 46, 220–227. [DOI] [PubMed] [Google Scholar]
- 320.Ahmed M, Prasad J, Gill H, Stevenson L and Gopal P, J. Nutr., Health Aging, 2007, 11, 26–31. [PubMed] [Google Scholar]
- 321.Ng SC, Hart AL, Kamm MA, Stagg AJ and Knight SC, Inflammatory Bowel Dis, 2009, 15, 300–310. [DOI] [PubMed] [Google Scholar]
- 322.Ghadimi D, Helwig U, Schrezenmeir J, Heller KJ and de Vrese M, J. Leukocyte Biol, 2012, 92, 895–911. [DOI] [PubMed] [Google Scholar]
- 323.Leus NG, Zwinderman MR and Dekker FJ, Curr. Opin. Chem. Biol, 2016, 33, 160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lu T and Stark GR, Cancer Res, 2015, 75, 3692–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Bhat MI, Kumari A, Kapila S and Kapila R, Ann. Microbiol, 2019, 69, 603–612. [Google Scholar]
- 326.Danilo CA, Constantopoulos E, McKee LA, Chen H, Regan JA, Lipovka Y, Lahtinen S, Stenman LK, Nguyen T-VVV, Doyle KP, Slepian MJ, Khalpey ZI and Konhilas JP, Benefic. Microbes, 2017, 8, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wang L, De Zoeten EF, Greene MI and Hancock WW, Nat. Rev. Drug Discovery, 2009, 8, 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.George AK, Singh M, Pushpakumar S, Homme RP, Hardin SJ and Tyagi SC, J. Cell. Physiol, 2020, 235, 2590–2598, DOI: 10.1002/jcp.29163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Bolduc J-F, Hany L, Barat C, Ouellet M and Tremblay MJ, J. Virol, 2017, 91, DOI: 10.1128/jvi.01943-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Soliman ML and Rosenberger TA, Mol. Cell. Biochem, 2011, 352, 173–180. [DOI] [PubMed] [Google Scholar]
- 331.Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Bin Yang H, Jiang SX, Yan GQ, Wang D, Wang Y, Liu Y, Cai Z, Xu YY, Chen J, Yu W, Yang PY and Lei QY, Nat. Commun, 2016, 7, 11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Debret R, Brassart-Pasco S, Lorin J, Martoriati A, Deshorgue A, Maquart FX, Hornebeck W, Rahman I and Antonicelli F, Biochim. Biophys. Acta, Mol. Cell Res, 2008, 1783, 1718–1727. [DOI] [PubMed] [Google Scholar]
- 333.Saldanha SN, Kala R and Tollefsbol TO, Exp. Cell Res, 2014, 324, 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.McBride J, Zhu Y, Wang P and Sang S, FASEB J, 2015, 29, 394.4.25376834 [Google Scholar]
- 335.Arpón A, Milagro FI, Razquin C, Corella D, Estruch R, Fitó M, Marti A, Martínez-González MA, Ros E, Salas-Salvadó J, Riezu-Boj JI and Martínez JA, Nutrients, 2018, 10, 15. [Google Scholar]
- 336.Corella D, Ordovas J, Sorli J, Asensio E, Ortega C, Carrasco P, Portoles O and Coltell O, FASEB Journal.Conference Exp. Biol, 2015, 29, LB242. [Google Scholar]
- 337.Arpón A, Riezu-Boj JI, Milagro FI, Marti A, Razquin C, Martínez-González MA, Corella D, Estruch R, Casas R, Fitó M, Ros E, Salas-Salvadó J and Martínez JA, J. Physiol. Biochem, 2016, 73, 445–455. [DOI] [PubMed] [Google Scholar]
- 338.Chen L, Dong Y, Wang X, Hao G, Huang Y, Gutin B and Zhu H, Mol. Nutr. Food Res, 2018, 62, 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Cheng L, Han X and Shi Y, Am. J. Physiol.: Endocrinol. Metab, 2009, 297, E1276–E1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Carraro JCC, Hermsdorff HHM, Mansego ML, Zulet MA, Milagro FI, Bressan J and Martínez JA, J. Nutrigenet. Nutrigenomics, 2016, 9, 95–105. [DOI] [PubMed] [Google Scholar]
- 341.Lin SP, Chu PM, Tsai SY, Wu MH and Hou YC, J. Ethnopharmacol, 2012, 144, 671–676. [DOI] [PubMed] [Google Scholar]
- 342.Lampe JW and Chang JL, Semin. Cancer Biol, 2007, 17, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Rein MJ, Renouf M, Cruz-Hernandez C, Actis-Goretta L, Thakkar SK and da Silva Pinto M, Br. J. Clin. Pharmacol, 2013, 75, 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wilk-Zasadna I, Bernasconi C, Pelkonen O and Coecke S, Toxicology, 2015, 332, 8–19. [DOI] [PubMed] [Google Scholar]
- 345.Morgan S, Stair EL, Martin T, Edwards WC and Morgan GL, Am. J. Vet. Res, 1988, 49, 493–499. [PubMed] [Google Scholar]
- 346.Liu W, Zhai Y, Heng X, Che FY, Chen W, Sun D and Zhai G, J. Drug Targeting, 2016, 24, 694–702. [DOI] [PubMed] [Google Scholar]
- 347.Fatima A and Morris DG, Physiol. Genomics, 2013, 45, 685–696. [DOI] [PubMed] [Google Scholar]
- 348.Frendo-Cumbo S, MacPherson REK and Wright DC, Physiol. Rep, 2016, 4, e12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Zhao W, Li A, Feng X, Hou T, Liu K, Liu B and Zhang N, Cell. Signalling, 2016, 28, 1401–1411. [DOI] [PubMed] [Google Scholar]