

Abstract
Knowledge concerning mechanisms that control proliferation and differentiation of preadipocytes is essential to our understanding of adipocyte hyperplasia and the development of obesity. Evidence has shown that temporal regulation of mitogen-activated protein kinase (MAPK) phosphorylation and dephosphorylation is critical for coupling extracellular stimuli to cellular growth and differentiation. Using differentiating 3T3-L1 preadipocytes as a model of adipocyte hyperplasia, we examined a role for dual-specificity phosphatase 1 (DUSP1) on the timely modulation of MAPK signaling during states of growth arrest, proliferation, and differentiation. Using real-time reverse transcription PCR (qRT-PCR), we report that DUSP1 is induced during early preadipocyte proliferation concomitant with ERK and p38 dephosphorylation. As deactivation of ERK and p38 is essential for the progression of adipocyte differentiation, we further showed that de novo mRNA synthesis was required for ERK and p38 dephosphorylation, suggesting a role for “inducible” phosphatases in regulating MAPK signaling. Pharmacological and genetic inhibition of DUSP1 markedly increased ERK and p38 phosphorylation during early adipocyte differentiation. Based on these findings, we postulated that loss of DUSP1 would block adipocyte hyperplasia. However, genetic loss of DUSP1 was not sufficient to prevent preadipocyte proliferation or differentiation, suggesting a role for other phosphatases in the regulation of adipogenesis. In support of this, qRT-PCR identified several MAPK-specific DUSPs induced during early (DUSP2, −4, −5,&−6), mid (DUSP4&−16) and late (DUSP9) stages of adipocyte differentiation. Collectively, these data suggest an important role for DUSPs in regulating MAPK dephosphorylation, with an emphasis on DUSP1, during early adipogenesis.
Obesity is a major risk factor for the development of stroke, heart disease, and type 2 diabetes (Haslam and James, 2005). Obesity develops from an increase in adipose tissue (AT) mass due to enlargement of adipocyte size (hypertrophy) and cell number (hyperplasia) (Hirsch and Batchelor, 1976; Knittle et al., 1979). Mature adipocytes are post-mitotic; therefore hyperplasia refers to the recruitment of new adipocytes through proliferation and subsequent differentiation of adipocyte precursor cells referred to as “preadipocytes.” Preadipocyte replication slows during adulthood, yet adipocyte hyperplasia has been shown to play a critical role in the onset of obesity throughout all stages of life, particularly during childhood obesity and in morbidly obese adults (Faust et al., 1978; Bjorntorp et al., 1982; Hirsch et al., 1989; Hausman et al., 2001). Unlike hypertrophic obesity, exercise, and caloric restriction are less effective in treating obesity derived from adipocyte hyperplasia (Faust et al., 1978; Yang et al., 1980; Mandenoff et al., 1982). Despite evidence suggesting that hyperplastic obesity is associated with the poorest prognosis of treatment, mechanisms regulating adipocyte hyperplasia remain poorly understood.
Intracellular signaling molecules play a critical role in linking environmental cues to increased adipocyte hyperplasia. Of the major signal transduction cascades, mitogen-activated protein kinases (MAPKs) have been shown to be essential for both adipocyte proliferation and differentiation (Bost et al., 2005a,b; Aouadi et al., 2007). In the adipocyte, extracellular signal-regulated kinase (ERK) and p38 are the best characterized MAPKs known to play a pivotal role in adipogenesis (Bost et al., 2005a). Timely phosphorylation as well as dephosphorylation of ERK and p38 is critical for adipocyte hyperplasia as loss of ERK phosphorylation as well as prolonged ERK phosphorylation has been shown to inhibit adipogenesis (Engelman et al., 1998, 1999; Prusty et al., 2002; Bost et al., 2005a,b; Aouadi et al., 2007). In addition to temporal regulation, MAPK nuclear localization has been shown to regulate adipocyte hyperplasia through transcriptional mechanisms involving adipocyte gene expression (Prusty et al., 2002; Park et al., 2004), demonstrating that compartmentalized signaling events can impact different properties of adipocyte differentiation.
It is well established that MAPK activity is regulated by a cascade of phosphorylation events mediated by sequential activation of upstream kinases, where MAPK is phosphorylated by upstream MAPK kinases (MAP2Ks), which are phosphorylated by MAPK kinase kinases (MAP3Ks) in response to diverse external stimuli. Phosphorylation of both threonine and tyrosine residues of the (T-X-Y) motif within the activation loop is essential and sufficient for MAPK activity (Pearson et al., 2001; Tanoue and Nishida, 2003; Zhang and Dong, 2007). While MAPK activity has traditionally been attributed to upstream kinases, recent evidence suggests that MAPK dephosphorylation via phosphatases plays a central role in controlling MAPK-dependent biological processes (Ebisuya et al., 2005; Dickinson and Keyse, 2006; Kondoh and Nishida, 2007). Dual-specificity phosphatases (DUSPs) are a subclass of protein tyrosine phosphatases that specifically inhibit MAPK activity through dephosphorylation of both threonine and tyrosine residues. Within this subclass, there are ten DUSPs that contain a MAP Kinase Binding (MKB) domain that selectively confers catalytic activity toward ERK, c-Jun N-terminal kinases (JNK) or p38 (Dickinson and Keyse, 2006; Kondoh and Nishida, 2007; Zhang and Dong, 2007). These MAPK-specific DUSPs are further classified based on gene structure, substrate specificity, and subcellular localization into three groups composed of nuclear DUSPs, cytosolic ERK-specific DUSPs, and DUSPs that selectively inactivate JNK and p38 MAPKs (Table 1). Lastly, DUSPs are highly inducible at the level of gene expression in a cell-type and stimulus-dependent manner (Dickinson and Keyse, 2006; Jeffrey et al., 2007; Keyse, 2008). For adipocytes, these stimuli include insulin and the synthetic glucocorticoid dexamethasone (Kusari et al., 1997; Kassel et al., 2001).
TABLE 1.
DUSP and inflammatory genes analyzed in this study
| Symbol | Name/alias | Accession | ABI number | CT | |||||
|---|---|---|---|---|---|---|---|---|---|
| Dual specificity phosphatases | GP | MKB | NLS | NES | EJP | ||||
| Dusp1 | MKP-1,hVH1 | I | ● | ● | NM_013642 | Mm00457274_g1 | 24 | ||
| Dusp2 | PAC-1 | I | ● | ● | NM_010090 | Mm00839675_g1 | 30 | ||
| Dusp4 | MKP-2, hVH2 | I | ● | ● | NM_176933 | Mm00723761_m1 | 25 | ||
| Dusp5 | hVH3 | I | ● | ● | NM_001085390 | Mm01266104_m1 | 27 | ||
| Dusp6 | MKP-3, rVH6 | II | ● | ● | E | NM_026268 | Mm00650255_g1 | 24 | |
| Dusp7 | MKP-X | II | ● | ● | E | NM_153459 | Mm00463228_m1 | 27 | |
| Dusp9 | MKP-4 | II | ● | ● | E | NM_029352 | Mm00512646_m1 | 27 | |
| Dusp8 | M3/6, hVH5 | III | ● | ● | ● | J/P | NM_008748 | Mm00456230_m1 | 27 |
| Dusp10 | MKP-5 | III | ● | J/P | NM_022019 | Mm00517678_m1 | 24 | ||
| Dusp16 | MKP-7, MKP-M | III | ● | ● | ● | J/P | NM_130447 | Mm00459935_m1 | 25 |
| Inflammatory genes | |||||||||
| IL-6 | Interleukin-6 | NM_031168 | Mm99999064_m1 | 32 | |||||
| TNFα | Tumor necrosis factor-alpha | NM_013693 | Mm99999068_m1 | 28 | |||||
| Reference gene | |||||||||
| 18S | 18 Ribosomal RNA | X03205 | 4342930E | 9 | |||||
DUSP group (GP), MAPK binding domain (MKB), nuclear localization sequence (NLS), nuclear export sequence (NES), ERK (E), JNK (J), p38 (P), threshold cycle (CT) measured in lean adipose tissue.
Increasing attention has been given to MAPK-specific DUSPs regarding adipocyte differentiation and insulin resistance (IR) (Xu et al., 2003b; Bazuine et al., 2004; Sakaue et al., 2004; Wu et al., 2006; Emanuelli et al., 2008; Feng et al., 2014). Of the ten DUSPs examined, the archetypal member, DUSP1 has emerged as a key player in diet-induced obesity and IR (Wu et al., 2006). Considering the role for DUSPs on MAPK signaling, we explored the functional role for DUSP1 on MAPK activity and adipocyte differentiation. Initial characterization of DUSP1 demonstrated increased expression of the phosphatase in animal models of genetic and diet-induced obesity in the presence of increasing inflammation. Using the murine 3T3-L1 cell line as a model for adipocyte hyperplasia, we further show that dephosphorylation of ERK and p38 during early preadipocyte proliferation was strongly correlated to DUSP1 expression. Consistent with this observation, pharmacological and genetic inhibition of DUSP1 resulted in sustained phosphorylation of ERK and p38. Paradoxically, the sustained MAPK phosphorylation observed with DUSP1 knockdown was not sufficient to block adipogenesis. Based on these findings, we further showed that additional DUSPs were induced during early (DUSP2, −4, −5, & −6), mid (DUSP4 & −16) and late (DUSP9) stages of adipocyte differentiation. Collectively, these data support a role for DUSPs, in particular DUSP1, as negative regulators of MAPK activity and highlight redundancies among these phosphatases in the regulation of MAPK signaling and adipogenesis.
Materials and Methods
Mice and experimental diets
Animals used for this study include genetically obese male B6.VLepob/J (B6-ob/ob) mice and their lean littermates as well as C57BL/6J mice rendered obese by diet and their lean controls. All mice were housed and treated by the supplier (Jackson Laboratories, Bar Harbor, Maine) until shipment 1 week prior to tissue harvest. B6-ob/ob mice and lean littermates were purchased for experimentation at 6 and 10 weeks of age and given free access to a standard laboratory chow diet. According to Jackson Labs, mice homozygous for the obese mutation exhibit hyperplasia with ensuing obesity notable at 1 month of age and transient glucose intolerance that begins at ~6 weeks of age and subsides between 12 to 16 weeks of age. C57BL/6J mice subjected to diet-induced obesity (DIO) were fed a high fat diet (HFD) consisting of 60% kcal from fat (Research Diets Inc. D12492) from 6 weeks of age. Lean C57BL/6J control mice were fed a control low fat diet (LFD) consisting of 10% kcal from fat (Research Diet Inc. D12450B) from 6 weeks of age. Both diets contained 10% kcal from protein with the balance in caloric value provided by differences in carbohydrate content. Mice receiving both diets were given free access to food and shipped for experimentation at 18 and 24 weeks of age. All animals were euthanized by CO2 gas asphyxiation and epididymal white AT (WAT) tissue collected and processed for preparation of total RNA. Animal care and use was in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Use and Care Committee.
Materials.
Dulbecco’s Modified Eagle’s Medium (DMEM), calf bovine serum (CS), and trypsin-EDTA were purchased from Invitrogen. Fetal bovine serum (FBS) was obtained from HyClone. Antibodies used for immunoblotting include phospho-ERK, phosho-p38, total p38, aP2, GAPDH, and α-tubulin purchased from Cell Signaling and DUSP1, PPARγ, C/EBPβ and C/EBPα purchased from Santa Cruz Biotechnology. Enhanced chemiluminescence (ECL) reagents were obtained from Perkin-Elmer Life Sciences. Actinomycin D was purchased from Sigma Aldrich. Triptolide was obtained from Calbiochem. All Taqman primer probes (Table 1) used in this study were purchased from Applied Biosystems. The murine 3T3-L1 cell line was purchased from Howard Green, Harvard Medical School (Djian et al., 1985).
Cell culture.
3T3-L1 preadipocytes were propagated in DMEM supplemented with 10% CS until density-induced growth arrest, as previously described (Morrison and Farmer, 1999b). At 2 days post-confluence, growth medium was replaced with DMEM supplemented with 10% FBS, 0.5 mM 1-methyl-3-isobutylxanthine, 1 μM dexamethasone, and 1.7 μM insulin (MDI). Throughout the study, “time 0” refers to density arrested cells immediately before the addition of MDI to the culture medium. Experiments described herein were conducted within the period of differentiation spanning from density arrest (d0) through 8 days (d8) post-MDI. All experiments were repeated 2–3 times to validate results and ensure reliability.
Immunoblotting.
Cell monolayers were washed with phosphate-buffered saline (PBS) and scraped into ice-cold lysis buffer containing 1.0 M Tris, pH 7.4, 150 mM NaCl, 1% Triton X, 0.5% Nonidet P-40 (NP40), 1 mM EDTA, 1 mM EGTA, and 10 mM N-ethylmaleimide. Phosphatase (20 mM ß-glycerophosphate, 10 mM NaFl, and 2 μM sodium vanadate), as well as protease (0.3 μM aprotonin, 21 μM leupeptin, 1 μM pepstatin, 50 μM phenanthroline, and 0.5 μM phenylmethylsulfonyl fluoride) inhibitors were added to lysis buffer prior to cell harvest. Cell lysates were sonicated, centrifuged (13,000g, 10 min, 4°C), and the supernatant transferred to a fresh tube. Bicinchoninic acid assay (Pierce, Rockford, IL) was used to determine protein concentration. Cell lysates were resuspended in loading buffer containing 0.25 M Tris, pH 6.8, 4% sodium dodecyl sulfate (SDS), 10% glycerol, 10% dithiothreitol, and 0.01% bromophenol blue and heated for 5 min at 80°C. Proteins were resolved on SDS-polyacrylamide gel electrophoresis gels (PAGE) and transferred to polyvinylidene fluoride membranes (Millipore Corp., Billerica, MA). After transfer, membranes were blocked with 4% milk and probed with indicated primary antibodies overnight at 4°C. Membranes were subsequently probed with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Immunoblots were immersed in ECL and visualized by autoradiography using CL-XPosure film (Pierce).
Real-time qRT-PCR.
Total RNA was extracted and genomic DNA contamination was removed using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s protocol. Total RNA was quantified with a Nanodrop ND-1000 spectrophotometer and reverse-transcribed to cDNA in a 10 ml reaction volume using a high capacity cDNA reverse transcription kit (Applied Biosystems). The reverse transcription (RT) master mix containing RT buffer, deoxyribonucleotide triphosphate (dNTP) mix, RT random primers, RNase inhibitor (1.0 U/μl), and MultiScribe RT was added to 1 μg RNA and RNase-free water. Reverse transcription reaction conditions followed the protocol (25°C for 10 min, 37°C for 120 min, 85°C for 5 sec, followed by 4°C indefinitely/RT complete) and utilized the Gene Amp PCR System 9700 thermal cycler (Applied Biosystems) for cDNA synthesis.
PCR amplification was run utilizing the 7500 fast system (Applied Biosystems) that consisted of enzyme activation at 95°C for 20 sec, followed by 40 cycles of denaturation at 95°C for 3 sec combined with annealing/extension at 60°C for 30 sec All data were analyzed with the ABI 7500 real time PCR system (Applied Biosystems). Data were recorded and analyzed with Sequence Detector Software (Applied Biosystems) and graphs visualized with SigmaPlot software. All data were presented as mean ± standard error of the mean (SEM) and representative of at least two experiments performed in duplicate. Data were normalized to 18S previously validated by this lab as a suitable reference gene under these experimental conditions (Ferguson et al., 2010). Relative differences between treated and untreated control samples were analyzed by the 2−ΔΔCT method as previously described (Livak and Schmittgen, 2001; Ferguson et al., 2010).
Statistical analyses were conducted using SPSS v18. Phenotypic differences were determined via student’s t-test where a P-value of <0.05 was considered significant. Knockdown and Inhibitor data were analyzed using analysis of variance, with Tukey’s post-hoc analysis used when the p value for the respective parameter was statistically significant (P < 0.05). MDI component data were analyzed using analysis of variance, with Dunnet’s post-hoc analysis used when the P value for the respective parameter was statistically significant (P < 0.05).
Flow cytometry.
Cell monolayers were washed with PBS and trypsinized. Detached cells were diluted in ice-cold PBS, and gently pelleted by centrifugation (300g, 5 min, 4°C). PBS was decanted, and cells were fixed and permeabilized by drop-wise addition of 70% ethanol at −20°C while vortexing. Cells were washed in PBS and incubated in the dark for 30 min with a propidium iodide staining solution (50 μg/ml propidium iodide and 100 μg/ml RNase A in PBS). DNA fluorescence was measured with using the Guava easyCyte HT flow cytometer (Millipore) equipped with a 488-nm argon laser. Width (FL2W) and area (FL2A) of propidium iodide fluorescence were recorded for at least 5,000 counts, and DNA histograms were extracted from FL2W-FL2A dot plots. Histograms were analyzed with Modfit software.
RNA interference
SMARTpools containing four different short interfering RNAs (siRNAs) for dusp1 specific sequences as well as non-targeting sequences were transfected using DharmaFect 3 transfection reagent according to manufacturer’s (Dharmacon) protocol. Briefly, 3T3-L1 preadipocytes were propagated in 6-well culture dishes until reaching density-induced growth arrest. Growth medium was then replaced with DMEM supplemented with 10% CS, 3 μl DharmaFect 3 reagent and either 100 nM DUSP1 specific siRNA or non-targeting siRNA for 72 h Growth medium was subsequently switched to differentiation medium containing MDI as described above.
Results
Regulation of DUSP1 in adipose tissue of genetic and diet-induced obesity
Others have reported that DUSP1 null mice were resistant to diet-induced obesity (Wu et al., 2006). In addition, it has recently been reported that DUSP1 is elevated in adipose tissue of obese, non-diabetic patients, concomitant to p38 dephosphorylation (Khadir et al., 2015). Thus, we initially sought to examine the developmental role of obesity on DUSP1 gene expression. For genetic obesity development, B6-ob/ob mice and wildtype littermates were purchased (Jackson Laboratory) for studies conducted at 6 and 10 weeks of age. The age of mice chosen for this study represented developmental stages of obesity which were arbitrarily designated as stage I (6 weeks) and stage II (10 weeks) for comparison (see Materials and Methods). Consistent with progressive development of obesity, 6 and 10 weeks old B6-ob/ob mice presented with 67% and 83% increases in body weight, respectively, relative to lean (control) littermates (Table 2). For development of diet-induced obesity (DIO), C57BL/6J males were given ad libitum access to a high fat diet (HFD; 60% kcal) starting at six weeks of age. Control males were fed a control low fat diet (LFD) containing 10% kcal from fat and the same protein content and total caloric value as the HFD. Studies were conducted at 18 weeks (stage I) and 24 weeks (stage II) of age, representing 12 weeks and 18 weeks of specialized diet, respectively. As shown in Table 2, 18 and 24 week old mice fed a HFD in this study presented with 13% and 26% increases in body weight, respectively, relative to lean mice fed the LFD.
TABLE 2.
Anthropometrics of mice in this study
| Stage | Age (week) | Final BW (g) | % Δ | |
|---|---|---|---|---|
| Genetic obesity | lean | ob/obb | ||
| I | 6 | 21.8 | 36.4 | 67 |
| II | 10 | 28.6 | 52.4 | 83 |
| Diet-induced obesity | LFD | HFD | ||
| I | 18 | 30.8 | 34.7 | 13 |
| II | 24 | 32.2 | 40.6 | 26 |
Percent difference in body weight (BW) between lean and obese mice (%Δ).
RNA was isolated from white adipose tissue (WAT) of epididymal fat pads in male B6-ob/ob and DIO mice and assessed by qRT-PCR, normalized to 18S rRNA, and presented relative to lean controls. Obesity is characterized by a progressive increase in chronic, low-grade inflammation (Xu et al., 2003a; Strissel et al., 2007; Gregor and Hotamisligil, 2011). Therefore, we first assessed the relative mRNA abundance of two major cytokines, IL-6 and TNFα. Both cytokines were elevated only in stage II of B6-ob/ob mice fed standard chow consistent with the premise of progressive obesity-induced inflammation (Fig. 1A and B). IL-6 and TNFα were elevated in DIO mice at both stages of obesity, but with a progressive increase in TNFα correlating with subsequent stages of obesity (Fig. 1C and D). Finally, relative DUSP1 gene expression was examined in adipose tissue during the development of obesity and inflammation as assessed above. DUSP1 was elevated in obese adipose tissue only during conditions that also displayed an increase in TNFα (Fig. 1A–D).
Fig. 1.
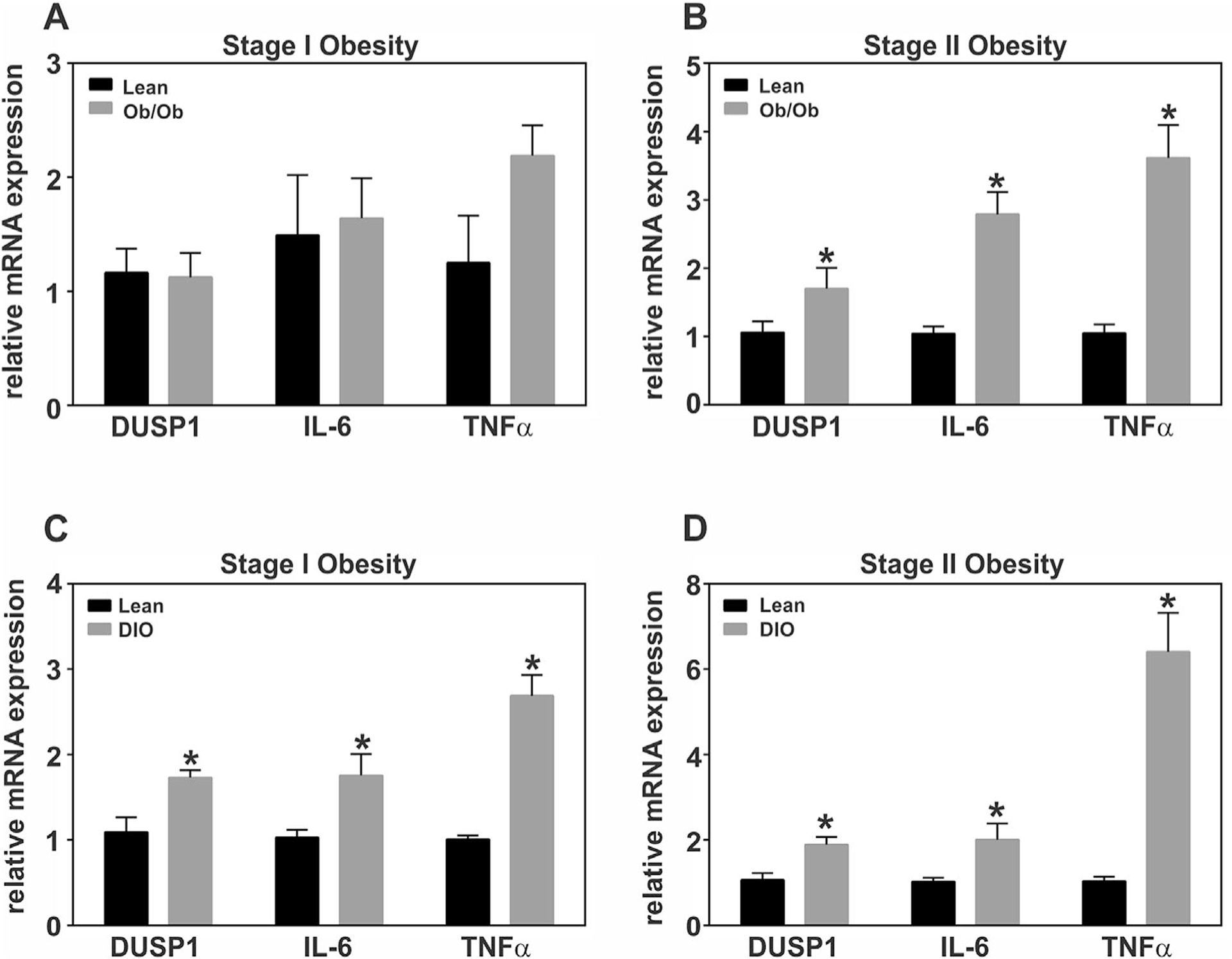
Adipose tissue specific regulation of DUSP1 and inflammatory mediators during the development of obesity under conditions of genetic and diet-induced obesity. Total RNA was prepared from white adipose tissue (WAT) harvested during development of genetic obesity at (A) 6 and (B) 10 weeks of age from male B6-ob/ob mice and their lean litter mates. For development of diet-induced obesity (DIO), C57BL/6J males were given ad libitum access to a high fat diet (HFD; 60% kcal) starting at 6 weeks of age. Control males were fed a control low fat diet (LFD) containing 10% kcal from fat and the same protein content as the HFD. RNA was isolated from WAT at (C) 18 and (D) 24 weeks of age. Relative DUSP mRNA abundance was measured via qRT-PCR and statistical significance determined by student’s t-test (P<0.05). All data were normalized to 18S rRNA and expressed relative to lean littermates.
Early induction of DUSP1 tightly correlates with MAPK dephosphorylation during early adipocyte differentiation
It is well established that ERK and p38 play functional roles during adipocyte differentiation; however the extent (i.e., magnitude and duration) of MAPK activity during distinct stages of differentiation remains uncertain (Bost et al., 2005a,b; Aouadi et al., 2007). It has been reported that MAPK activity is required for early stages of differentiation but inhibits late stages of differentiation, suggesting that “timing” of activity is critical for proper adipocyte differentiation. To elucidate a role for phosphorylation and dephosphorylation mechanisms regarding MAPK activity during adipocyte hyperplasia, we utilized the well-established 3T3-L1 murine cell line for this study. This cell line synchronously progresses through distinct stages of the cell cycle before entering a state of terminal growth arrest. Concomitant with growth arrest is the upregulation of a cascade of transcription factors that mediate late stage adipocyte gene expression and acquisition of the functional adipocyte phenotype.
To determine the extent of MAPK activity during these distinct stages of adipocyte differentiation, density-arrested preadipocytes were stimulated with a hormonal cocktail (i.e., MDI) and cell lysates harvested over early (0–24 hr) and late (d0–d8) stages of differentiation and immunoblotted with phosphospecific antibodies that recognize MAPKs only when bisphosphorylated on the T-x-Y motif of the activation loop. As shown in Figure 2, ERK and p38 were rapidly phosphorylated (15 min) and then completely dephosphorylated (<4 hr) during early adipocyte differentiation (panel A). Also shown is the phosphorylation state of MEK, the upstream MAP2K responsible for ERK phosphorylation, demonstrating that ERK was completely dephosphorylated 6–10 hrs before MEK phosphorylation returned to baseline (Fig. 2A). It should be noted that ERK dephosphorylation occurred with induction of PPARγ gene expression, which is a known target of ERK whose transcriptional activity is suppressed when phosphorylated by ERK (Hu et al., 1996). Lastly, while p38 phosphorylation was nearly identical to ERK during the first 4 hrs of differentiation (Fig. 2A), the phosphorylation state of p38 displayed a biphasic pattern that was not similar to ERK during later stages of differentiation (Fig. 2C). Immunoblot analysis of total p38 remained unchanged during all stages of adipocyte differentiation and served as a loading control (Fig. 2A–C). These data suggest a role for phosphatases in the regulation of MAPK dephosphorylation. Consistent with this hypothesis, we further showed that DUSP1 mRNA (Fig. 2B) and protein (Fig. 2A) expression increased during early adipocyte differentiation in a manner consistent with MAPK dephosphorylation. However, DUSP1 expression was markedly reduced during late stages of adipocyte differentiation (note difference in scale of Y-axis between panels B and D), highlighting a potential role for this phosphatase during early, but not late stages of adipogenesis (Fig. 2C and D).
Fig. 2.
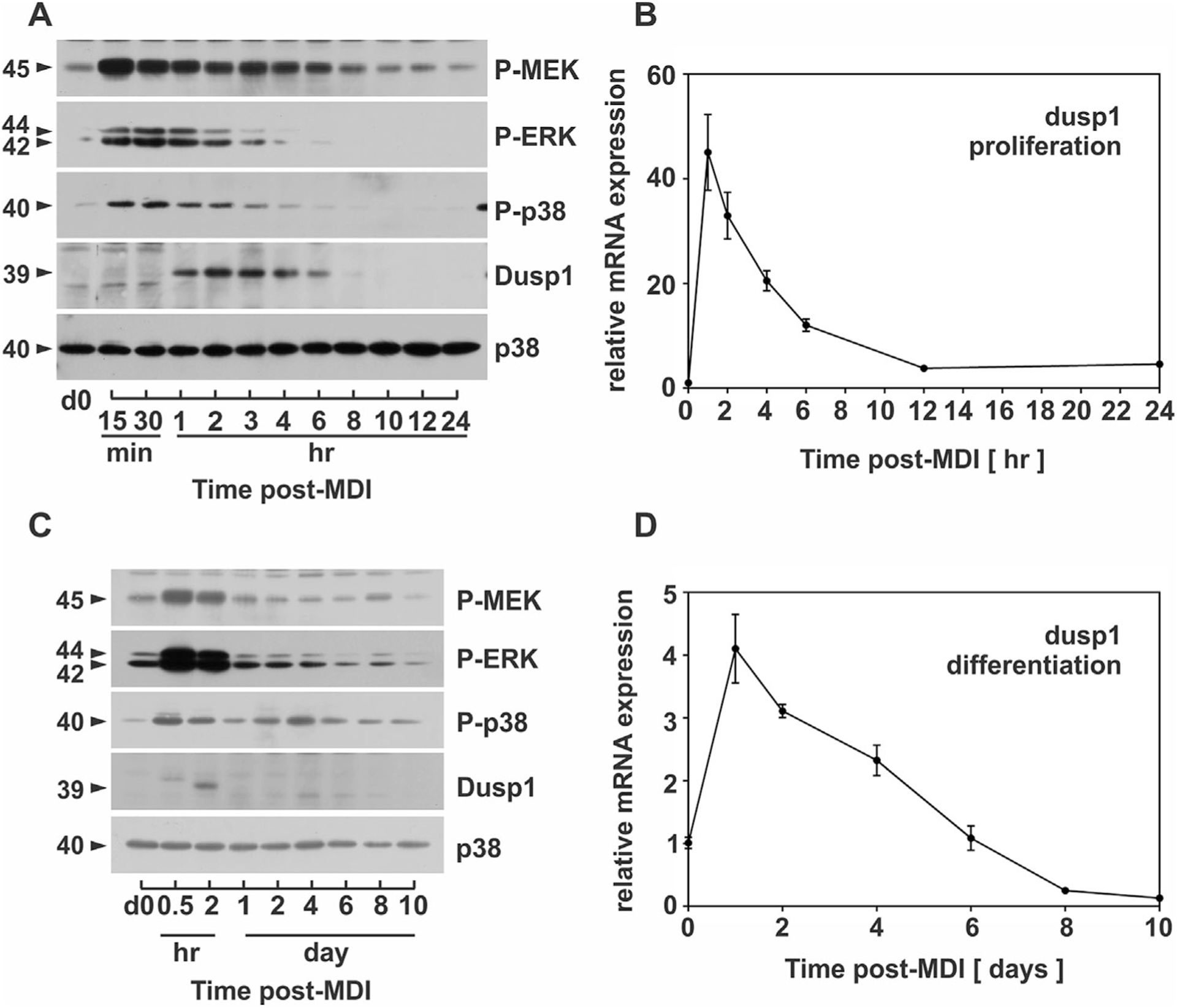
MAPK dephosphorylation is strongly correlated with DUSP1 expression. PAs were stimulated with MDI and cell lysates harvested during (A) early (0–24 h) and (C) late differentiation (d0–d8) prior to immunoblot analysis of phosphorylated MEK, ERK, and p38 as well as DUSP1, total p38 and PPARγ. In addition, PAs were stimulated with MDI and RNA isolated during (B) early (0–24h) and (D) late differentiation (d0–d8) prior to qRT-PCR analysis of DUSP1. DUSP1 was normalized to 18S rRNA and expressed relative to untreated cells (0 h or 0 d). DUSP1 was considered inducible when upregulated above a twofold criterion.
Pharmacological inhibition of DUSP1 amplified MAPK phosphorylation during early adipocyte differentiation
Data presented above demonstrated that DUSP1 expression strongly correlates with the deactivation of MAPK signaling. As others have shown that DUSP1 acts as an immediate early gene to regulate MAPK signaling (Dickinson and Keyse, 2006), we initially examined if ERK and p38 signaling was involved in de novo mRNA synthesis. For this, preadipocytes were pretreated with and without 1 ng/ml actinomycin D (Actino D) for 30 mins to inhibit mRNA synthesis prior to stimulation with MDI. Cell lysates were harvested over time during early adipocyte differentiation (<24 h) and immunoblotted for phosphorylated ERK and p38 as well as DUSP1 as discussed above. Blocking RNA synthesis resulted in a marked decrease in DUSP1 protein expression concomitant with an increase in ERK phosphorylation extending from less than 4 h in the absence of Actino D to more than 24 h when RNA synthesis was inhibited (Fig. 3A). Inhibition of RNA synthesis also had a marked effect of increased p38 phosphorylation (Fig. 3A). While MAPK dephosphorylation was dependent on de novo mRNA synthesis, initial MAPK phosphorylation (0 hr) was unaffected by Actino D (Fig. 3A).
Fig. 3.

Inhibition of DUSP1 enhances ERK and p38 phosphorylation during early adipocyte differentiation. (A) PAs were pretreated (30 min) in the absence or presence of 1 ng/ml Actinomycin D (AD) prior to MDI stimulation. Cell lysates were collected over time and protein expression of phosphorylated ERK and p38 along with DUSP1 and GAPDH analyzed via immunoblotting. (B) In addition, PAs were pretreated (30 min) in the presence or absence of 1 μM triptolide (TRP) prior to MDI stimulation. Cell lysates were collected over time post-MDI and protein expression of phosphorylated ERK and p38 along with DUSP1 and α-Tubulin examined via immunoblotting.
To further determine a role for DUSP1 on the extent of MAPK phosphorylation during early adipocyte differentiation, preadipocytes were pretreated with or without 1 μM triptolide (TRP) for 30 min prior to MDI stimulation. TRP is a bioactive compound purified from the Chinese herb Tripterygium wilfordii and shown to be effective as an immunosuppressant and anti-inflammatory agent as well as a potent inhibitor of DUSP1 gene expression (Liu, 2011). To determine if TRP influenced MAPK phosphorylation, cell lysates were harvested over time and immunoblotted for phosphorylated ERK and p38 as well as DUSP1. MDI stimulation markedly increased ERK and p38 phosphorylation. In the absence of TRP, MDI stimulation also resulted in the accumulation of DUSP1 protein in a manner consistent with the dephosphorylation of both MAPKs (Fig. 3B). In contrast, TRP pretreatment abolished the accumulation of DUSP1 and prolonged phosphorylated ERK and p38 through 24 hrs post-MDI stimulation. Pretreatment with TRP did not block the phosphorylation of either MAPK by upstream MAPK kinases following MDI stimulation. Together, these data support a role for DUSP1 in modulating the extent of MAPK phosphorylation during early adipocyte differentiation.
DUSP1 knockdown enhanced MAPK signaling during 3T3-L1 preadipocyte differentiation
To further establish a mechanistic role for DUSP1 on MAPK signaling, 3T3-L1 PAs were transfected with siRNA targeted to DUSP1 or non-targeting control sequences for 72 h prior to stimulation with MDI. Total RNA and whole cell lysates were harvested over time and analyzed for DUSP1 mRNA abundance and MAPK phosphorylation. DUSP1 gene expression was markedly induced during early differentiation in cells transfected with control siRNA (Fig. 4A). In contrast, both basal (>70%) and MDI-stimulated (>60%) expression of DUSP1 mRNA was significantly suppressed in cells treated with DUSP1 specific siRNA. Specific knockdown of DUSP1 mRNA nearly ablated accumulation of DUSP1 protein resulting from MDI stimulation (Fig. 4B). Moreover, specific knockdown of DUSP1 markedly increased ERK as well as p38 phosphorylation following MDI stimulation. Total p38 and α-Tubulin served as loading controls and remained unchanged (Fig. 4B).
Fig. 4.
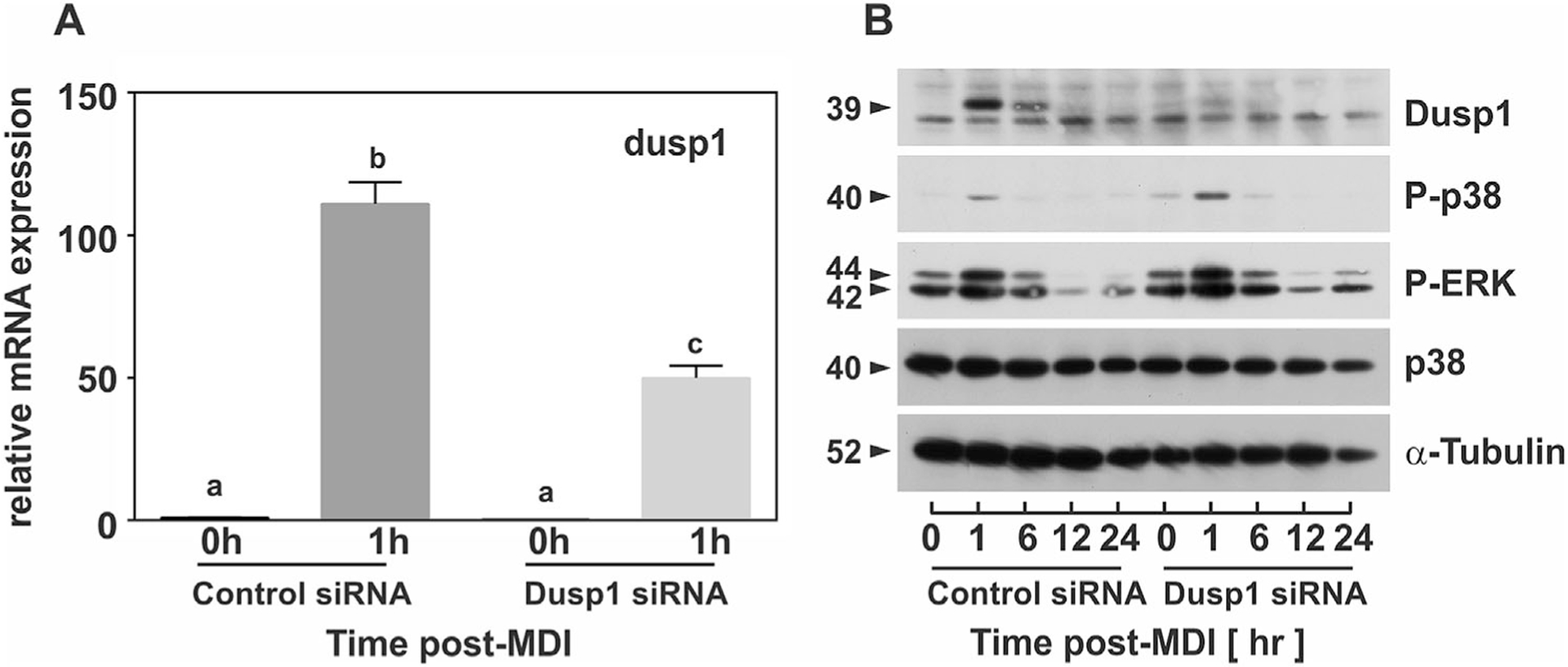
DUSP1 knockdown enhances ERK and p38 signaling magnitude and duration during early adipocyte differentiation. PAs were transfected with non-targeting control siRNA or DUSP1-specific siRNA for 72 h prior to stimulation with MDI. (A) Cell lysates were harvested for RNA at 0 and 1 h post-MDI and mRNA expression of DUSP1 analyzed via qRT-PCR. Data were normalized to 18S rRNA and changes in gene expression measured as fold differences relative to untreated control siRNA (0h). Statistical differences were determined by ANOVA, with Tukey’s post-hoc analysis performed when the p value for the respective parameter was statistically significant (P<0.05). (B) Cell lysates were collected over time post-MDI for control siRNA and DUSP1 siRNA and protein expression of phosphorylated ERK and p38 along with DUSP1, total p38, and α-Tubulin examined via immunoblotting.
PKA and ERK signaling regulate DUSP1 expression
Data presented above demonstrated a role for DUSP1 as an immediate early gene that regulates MAPK dephosphorylation during early adipogenesis. However, how DUSP1 is regulated under conditions of adipocyte differentiation remain unknown. Reports suggest a canonical feedback loop whereby MAPK-mediated induction of DUSP gene expression results in the expression of targeted phosphatases that specifically inhibit MAPK activity. To initially address this question, 3T3-L1 preadipocytes were treated with components of the adipogenic cocktail, 1-methyl-3-isobutylxanthine, dexamethasone, and insulin (MDI), individually or in combination and DUSP1 mRNA expression was analyzed at 1 h post-stimulation via qRT-PCR. As shown in Figure 5A, methyl-isobutylxanthine (Mix), a known activator of cAMP/PKA signaling, was the prominent regulator of DUSP1 mRNA expression. DUSP1 mRNA expression was also significantly elevated by treatment with dexamethasone (Fig. 5A). However, it should be noted that DUSP1 message was significantly upregulated in all conditions in which Mix was present (Fig. 5A). Intriguingly, induction of DUSP1 mRNA expression mirrored that of C/EBPβ, a key transcriptional regulator of early adipogenesis (Fig. 5B) (Morrison and Farmer, 1999a). Consistent with a role for cAMP signaling, MDI-mediated induction of DUSP1 mRNA expression was significantly attenuated in the presence of the cAMP/PKA inhibitor H-89 (Fig. 5C). DUSP1 mRNA expression was also reduced in the presence of the specific MEK inhibitor, UO126, albeit to a significantly lesser extent, suggesting cross-talk between cAMP/PKA and ERK pathways (Fig. 5C). Consistent with the MDI experiment above, complete loss of DUSP1 and C/EBPβ protein expression occurred in the presence of PKA inhibition (Fig. 5D).
Fig. 5.
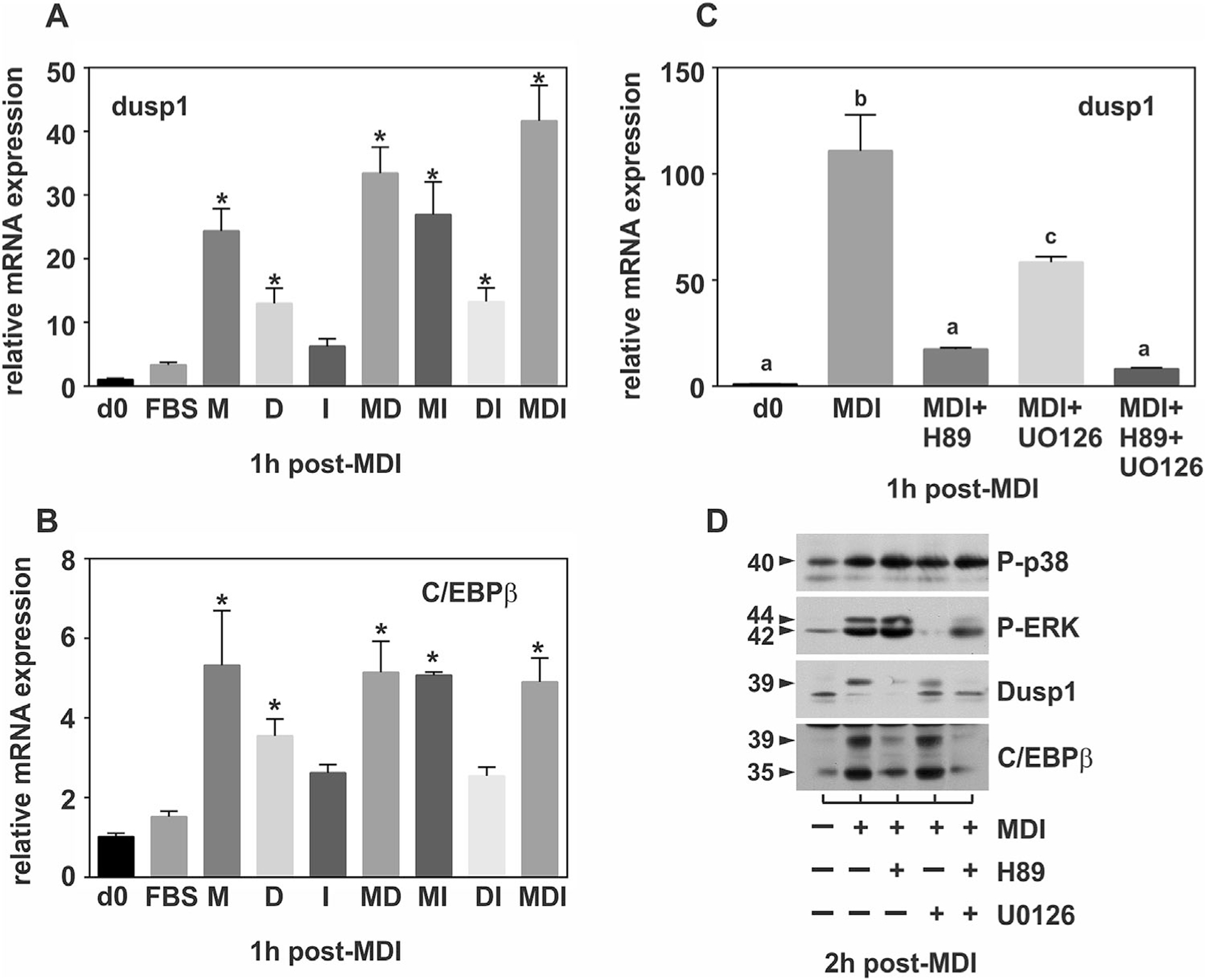
Regulation of DUSP1 by PKA and ERK signaling. PAs were stimulated in the absence or presence of the adipogenic cocktail for 1 h either individually or in combination prior to qRT-PCR analysis of (A) DUSP1 or (B) C/EBPβ. In addition, PAs were pretreated with the PKA inhibitor H-89 (30 min) or MEK inhibitor UO126 (30 min) prior to (C) qRT-PCR analysis of DUSP1 or (D) immunoblot analysis of phosphorylated ERK, p38, DUSP1 and C/EBPβ. All RT-PCR data were normalized to 18S rRNA and changes in gene expression measured as fold differences relative to untreated cells (0 h). For panels A&B, statistical differences between treated and control (d0) were determined by ANOVA with Dunnet’s post-hoc analysis performed when the P value for the respective parameter was statistically significant (P<0.05). For panel C, statistical differences treated and control (d0) were determined by ANOVA with Tukey’s post-hoc analysis performed when the P value for the respective parameter was statistically significant (P<0.05).
DUSP1 knockdown does not inhibit adipocyte hyperplasia
Data presented above demonstrated that DUSP1 regulates MAPK dephosphorylation during early adipocyte differentiation. As recent reports differ on the role of DUSP1 during adipogenesis (Sakaue et al., 2004; Wu et al., 2006), we sought to examine the biological role of DUSP1 on preadipocyte proliferation and differentiation as essential components of adipocyte hyperplasia. A continuum of sequential events culminates in the activation of PPARγ and C/EBPα in order to regulate adipocyte gene expression and acquisition of the mature adipocyte phenotype during adipogenesis (Morrison and Farmer, 1999a; Rosen and Spiegelman, 2000). Among the early events leading to PPARγ and C/EBPα, is an ‘obligatory’ prerequisite period of mitotic clonal expansion, in which density-arrested preadipocytes synchronously re-enter the cell cycle prior to activation of adipocyte-specific genes and terminal growth arrest (Tang et al., 2003). To determine if DUSP1 plays a regulatory role during clonal expansion, density-arrested preadipocytes were transfected with DUSP1 specific or non-targeting siRNA for 72 hrs prior to stimulation with MDI. Cells were harvested at 0 and 20 h post-MDI, fixed, and stained with propidium iodide for flow cytometric analysis of cell cycle progression. Two DNA peaks in the 2n and 4n range, representing G0/G1 and G2/M cell populations, were present in all histograms (Fig. 6A). Stimulation with MDI resulted in similar shifts from G0/G1 to S and G2/M phase populations demonstrating synchronous cell cycle progression in cells transfected with DUSP1 or control siRNA. To confirm these results, cell lysates were harvested under identical conditions and analyzed for cyclin A protein expression, which we have previously shown to accumulate during S phase in this cell model (Auld et al., 2007). Cyclin A protein accumulated with both DUSP1-specific and control siRNA, with modestly elevated cyclin A protein accumulation with DUSP1 knockdown (Fig. 6B). Collectively, these data demonstrate that DUSP1 does not block cell cycle progression, which is a known prerequisite for adipocyte differentiation. To the contrary, the modest increase in cyclin A following DUSP1 knockdown would be indicative of more efficient S-phase entry, possibly resulting from elevated MAPK phosphorylation.
Fig. 6.
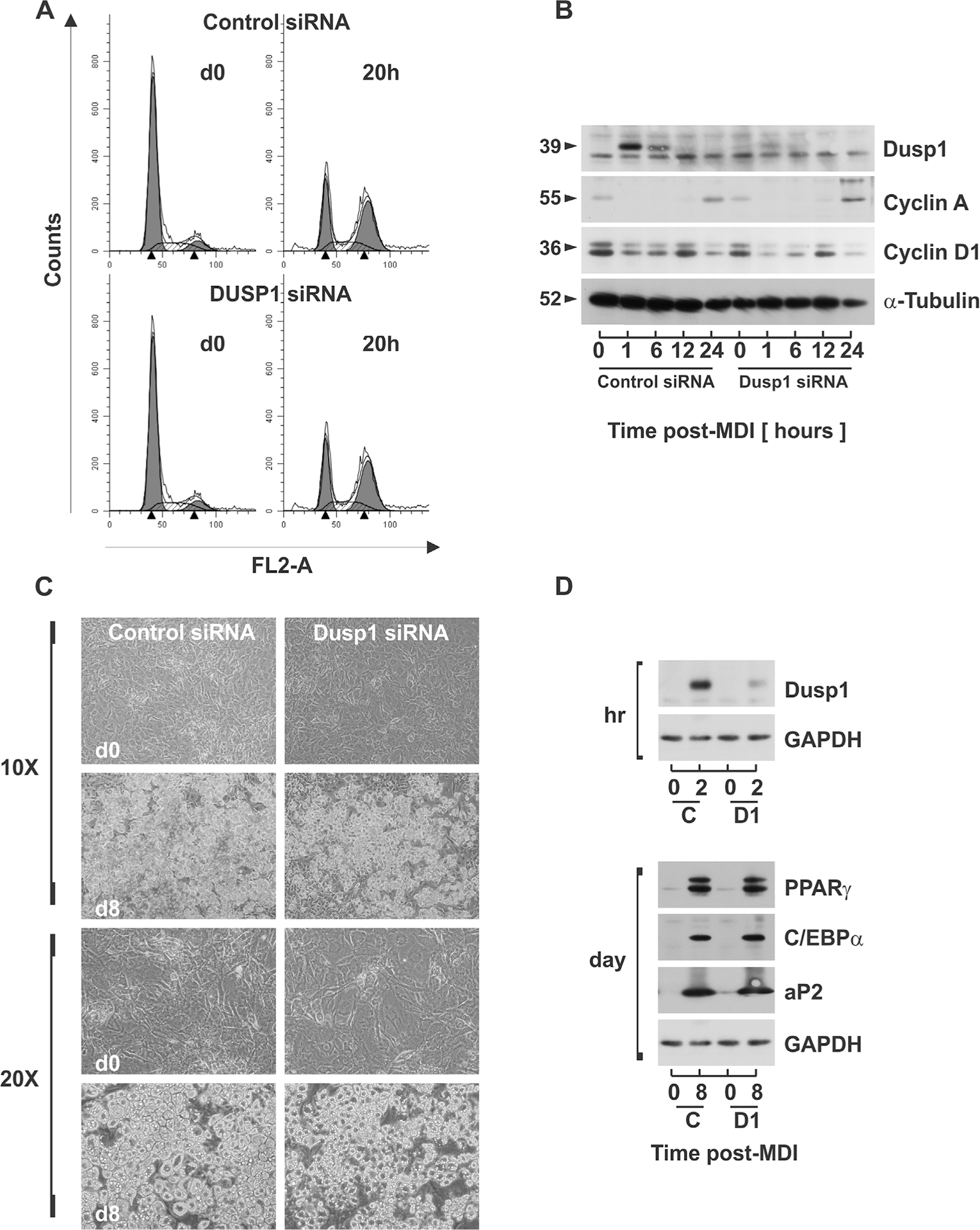
DUSP1 knockdown does not inhibit 3T3-L1 adipogenesis. PAs were transfected with non-targeting control siRNA or dusp1-specific siRNA for 72 hrs prior to stimulation with MDI. (A) Cells were fixed at d0 and 20 h post-MDI and DNA stained with propidium iodide. DNA histograms were assessed via flow cytometry. (B) Cell lysates were collected over time and protein expression of DUSP1, cyclin A, and α-Tubulin examined via immunoblotting. (C) Examination of cell morphology of untreated preadipocytes and mature adipocytes treated with control siRNA or dusp1 siRNA was assessed for lipid accumulation using phase contrast microscopy at 10× and 20× magnitudes. (D) Cell lysates were collected from PAs (d0) and mature ADs (d8) treated with control siRNA (C) or DUSP1 siRNA (D1) and protein expression of adipocyte genes PPARγ, C/EBPα, and aP2 examined via immunoblot analysis.
To elucidate a role for DUSP1 on adipocyte differentiation, density-arrested preadipocytes were transfected with control siRNA or DUSP1 siRNA 72 hrs prior to stimulation with MDI. Phase contrast microscopy was used to assess changes in cell morphology and lipid accumulation between undifferentiated preadipocytes (d0) and fully mature adipocytes (d8). Preadipocytes (d0) transfected with siRNA for DUSP1 or non-targeting sequences displayed nearly identical fibroblast-like morphology. Similarly, transfection with DUSP1-specific or non-targeting sequences prior to differentiation resulted in no discernable differences in the lipid-filled morphology of fully mature adipocytes (d8) (Fig. 6C). Consistent with the above findings, PPARγ, C/EBPα, and aP2 increased similarly with differentiation in cells transfected with DUSP1-specific or non-targeting control siRNA (Fig. 6D). PPARγ and C/EBPα represent major transcription factors that mediate the expression of functional proteins such as the fatty acid binding protein aP2 during adipogenesis. As DUSP1 protein accumulation was markedly attenuated at 2 h post-MDI with DUSP1 siRNA, these data demonstrate that knockdown of DUSP1 alone was not sufficient to block adipogenesis.
Inducible DUSP gene expression during adipocyte differentiation
Data presented above suggest the possibility of redundant or collective roles between multiple phosphatases in limiting the extent of MAPK phosphorylation that is essential for adipocyte differentiation. Therefore, we examined relative mRNA expression of all known MAPK-specific DUSPs during adipocyte differentiation using qRT-PCR. All ten DUSPs were measurable in preadipocytes within the detectable limits of 36 threshold cycles (CT), with dusp2, dusp8, and dusp9 least abundantly expressed at 32 cycles and dusp1, dusp6, and dusp7 most abundantly expressed at 24 cycles. This 8 cycle difference was equivalent to a 256-fold difference (i.e., 28) in base-line mRNA abundance in preadipocytes (Table 1). To determine which DUSPs were induced at the level of gene expression during differentiation, preadipocytes were stimulated with MDI and relative mRNA abundance assessed by qRT-PCR over time representing early (Panel A and B) and late stage (Panel C and D) differentiation. Prior to these studies, we set an arbitrary threshold of twofold change as a conservative measure of biological differences versus technical variation as measured by qRT-PCR (Gorzelniak et al., 2001). Thus, DUSP gene expression was only considered “inducible” when relative mRNA levels exceeded this threshold.
As illustrated in Figure 7A, DUSP2, −4, −5, and −6 were rapidly and transiently induced following induction of differentiation with DUSP2 and −5 reaching greater than 10-fold changes in mRNA by 2 hrs post-MDI. DUSP2 and −5 mRNA rapidly declined with values remaining at or below baseline from 12 hrs through d10 of differentiation (Fig. 7A–D). DUSP4 was unique among these phosphatases with biphasic expression with a transient, smaller peak (threefold) occurring at 4 hrs and a more sustained, larger peak (sevenfold) occurring at 4 d and remaining above baseline through d10 (Fig. 7A–C). While the kinetic profile of these DUSPs was consistent with the timing of MAPK dephosphorylation during early stages of differentiation, the mid-stage biphasic induction of DUSP4 occurred concomitantly with the time period when ERK phosphorylation falls below baseline levels and PPARγ is expressed. DUSP6 was the only cytosolic phosphatase determined to be rapidly and transiently induced during early adipocyte differentiation where a 3.5-fold induction peaked at 1 h and returned to baseline within 12 hrs post-MDI (Fig. 7A). In contrast, DUSP9 displayed a reciprocal pattern of expression where mRNA fell below baseline during early stages of differentiation (Fig. 7B) followed by a gradual induction during late stages remaining markedly elevated (eightfold) through d10 of differentiation (Fig. 7C). Finally, DUSP16 was transiently induced during late stages with peak mRNA abundance observed at d4 of differentiation (Fig. 7C). Intriguingly, DUSP2, 5, 6 were also induced in response to obesity as observed with DUSP1 (Fig. 8). Together, these data support redundant or collective roles for multiple DUSPs in the regulation of MAPK signaling and adipogenesis.
Fig. 7.
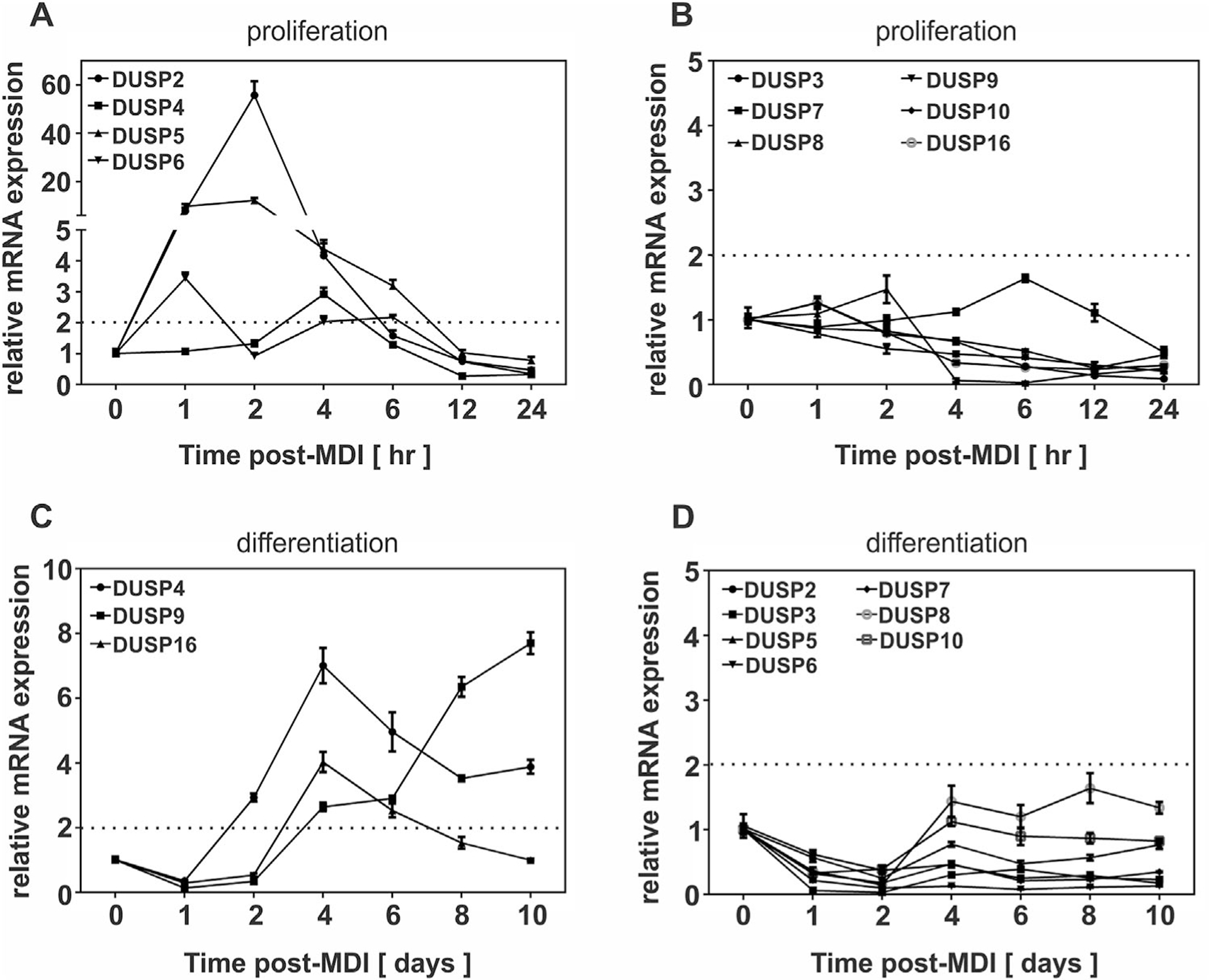
Mitogen-mediated induction of DUSPs during adipocyte differentiation. PAs (d0) were stimulated with MDI for adipocyte differentiation. Total RNA was harvested from PAs (d0) stimulated with MDI to examine (A) early inducible or (B) non-inducible DUSPs and (C) late inducible and (D) non-inducible DUSPs during differentiation. DUSP mRNA was assessed by qRT-PCR. All data were normalized to 18S rRNA and expressed relative to untreated cells (0 h). Genes selected as inducible were upregulated above a twofold criterion indicated by a dashed line on the graph.
Fig. 8.
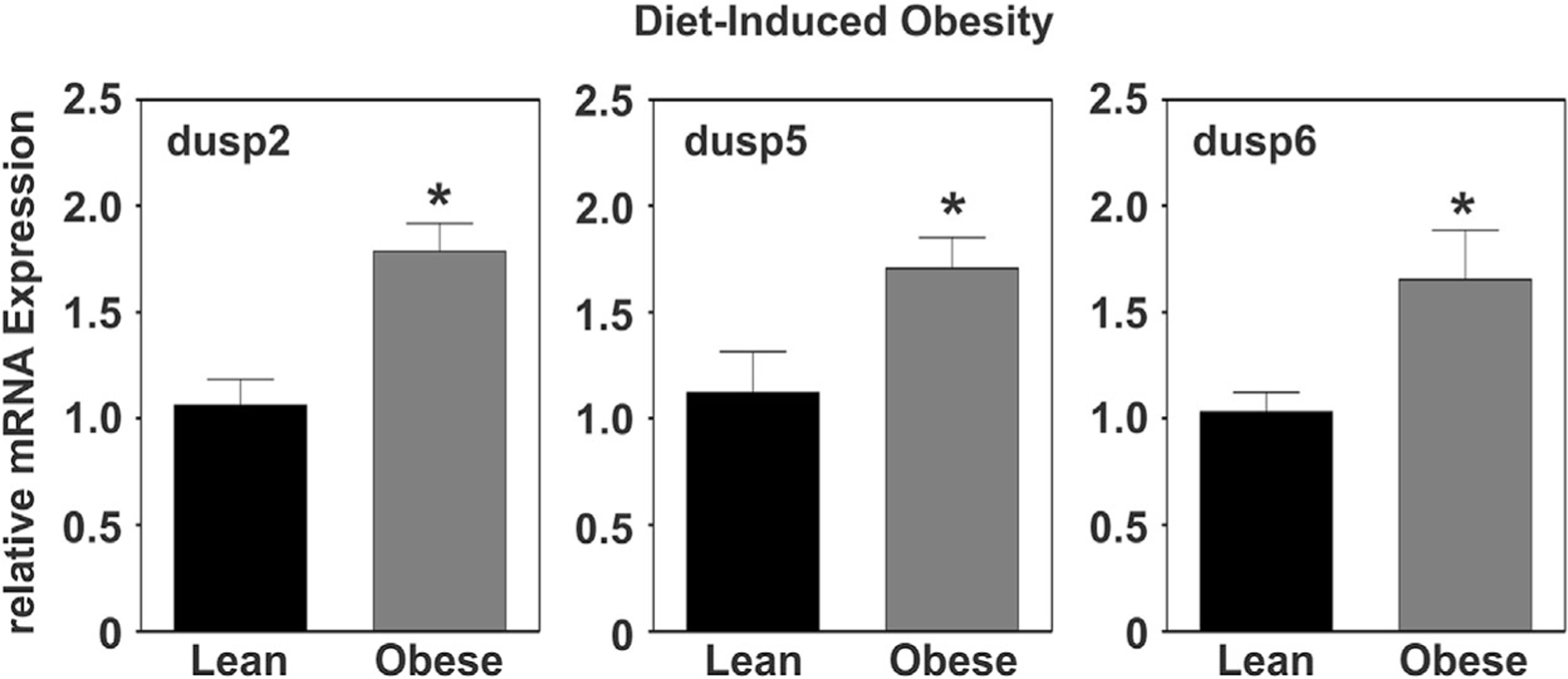
Adipose tissue specific regulation of DUSPs under conditions of diet-induced obesity. Total RNA was prepared from white adipose tissue (WAT) harvested at 18 weeks of age from diet-induced obesity (DIO), C57BL/6J males given ad libitum access to a high fat diet (HFD; 60% kcal) starting at 6 weeks of age. Control males were fed a control low fat diet (LFD) containing 10% kcal from fat and the same protein content as the HFD. Relative DUSP mRNA abundance was measured via qRT-PCR and statistical significance determined by student’s t-test (P<0.05). All data were normalized to 18S rRNA and expressed relative to lean littermates.
Discussion
This report highlights the mechanistic actions of DUSP1 as a negative regulator of ERK and p38 signaling during early adipocyte differentiation. Moreover, we present evidence that suggest a redundant and/or co-operative role for “inducible” DUSPs as negative regulators of MAPK activity during adipogenesis. We propose a model in which mitogenic signaling promotes DUSP expression in order to regulate MAPK activity throughout adipocyte differentiation (Fig. 9). We further showed that agonist-mediated induction of DUSP1 was dependent on cAMP/PKA and ERK signaling demonstrating feedback regulation of mitogenic signaling cascades by targeted phosphatases. These findings define a previously unexplored role for DUSP1 on the regulation of MAPK signaling during early adipogenesis.
Fig. 9.
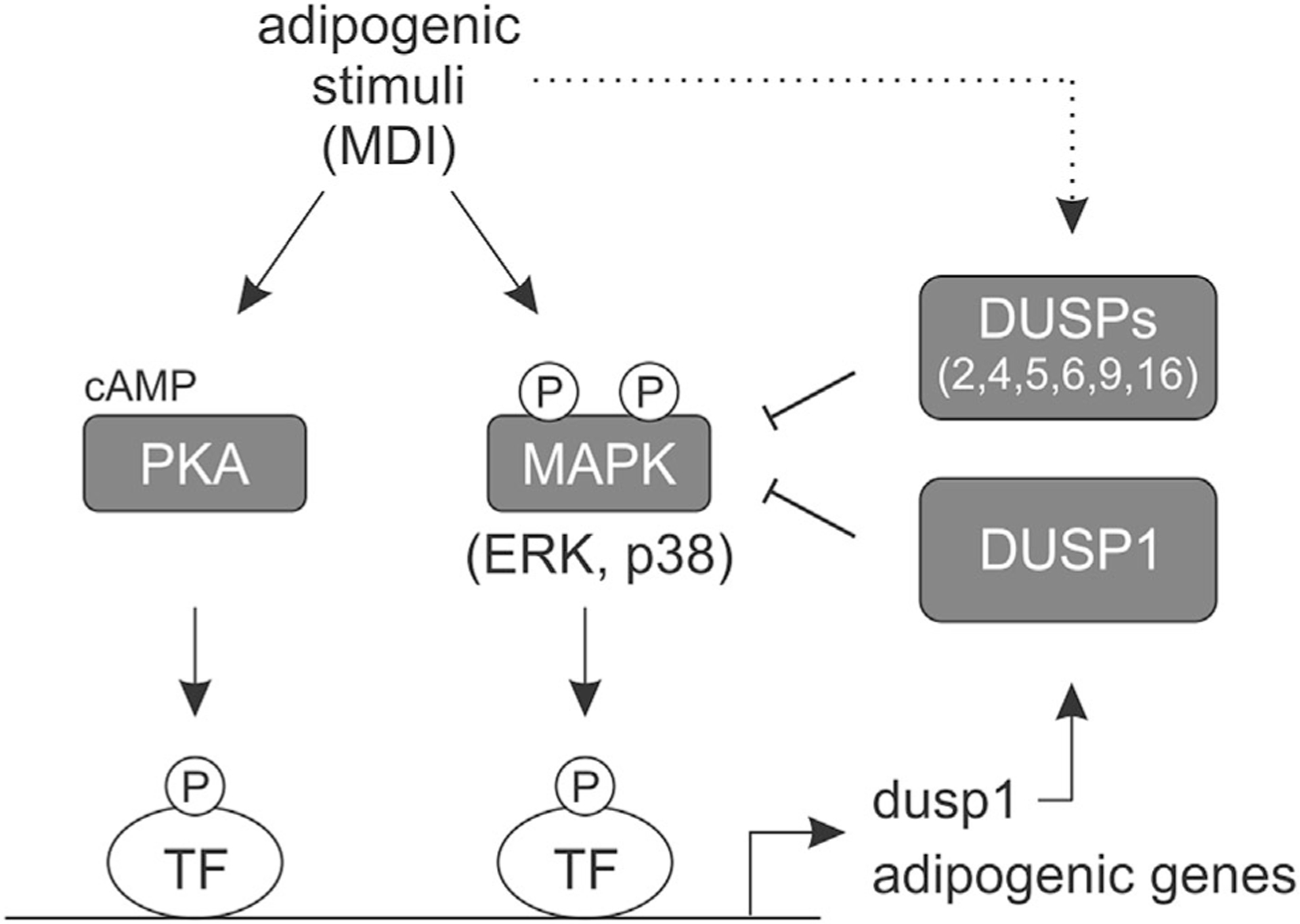
Model for the regulation of MAPK signaling and adipocyte differentiation by DUSPs. Adipogenic stimuli (MDI) increase PKA/MAPK-dependent dusp1 expression leading to dephosphorylation and inhibition of ERK and p38 activity. MDI-dependent stimulation of additional MAPK-specific DUSPs also inhibit MAPK activation (dotted line). Together, DUSP-mediated regulation of MAPK signaling can alter adipogenic gene expression and adipocyte differentiation.
Data presented here demonstrate that DUSP1 was divergently regulated at the level of gene expression based on the inflammatory environment resulting from obesity. Chronic-low grade inflammation is a cardinal hallmark of obesity, where increased inflammation (e.g., TNFα and IL-6) has been shown to drive adipocyte dysfunction and block adipogenesis (Torti et al., 1989; Stephens et al., 1992; Stephens and Pekala, 1992). We show that DUSP1 was induced in adipose tissue under conditions of genetic or diet-induced obesity where inflammatory genes (i.e., IL-6 and TNFα) were elevated. Conversely, DUSP1 was not elevated during early stage (i.e., stage I) of B6-ob/ob obesity even though these mice presented with a 67% increase in body weight in obese versus lean adipose tissue, suggesting that inflammation-mediated induction of DUSP1 is necessary for MAPK dephosphorylation in order to maintain adipocyte function and adipogenesis. While our data are the first to demonstrate elevated DUSP1 gene expression in adipose tissue of obese mice, others have shown that mice fed a HFD present with increased DUSP1 accumulation in the liver (Reddy et al., 2004) and that whole body DUSP1 gene ablation protects against diet-induced obesity in mice (Wu et al., 2006). Interestingly, DUSP1 has recently been identified as being differentially expressed in visceral adipose tissue of severely obese men with versus without metabolic syndrome, while others have shown that single nucleotide polymorphisms (SNPs) in the DUSP1 gene of obese individuals were associated with modulation of plasma glucose and HDL-cholesterol levels (Bouchard et al., 2007; Guenard et al., 2013). More recent data demonstrated that increased adipose tissue and circulating DUSP1 in obese, nondiabetic, patients positively correlated to increased inflammatory markers IP-10 and RANTES (Khadir et al., 2015). Taken together, obesity-associated inflammation and not obesity per se, appear responsible for elevated DUSP1 gene expression in adipose tissue. Moreover, this increase in DUSP1 may be critical for the regulation of metabolic function associated with obesity but not adipogenesis per se.
It is well established that DUSP1 acts as an immediate early gene whose expression is induced rapidly and transiently by various extracellular stimuli including agents used to differentiate 3T3-L1 preadipocytes such as insulin, glucocorticoids (e.g., dexamethasone), and cAMP analogs (Kusari et al., 1997; Schliess et al., 2000; Kassel et al., 2001). Notably, we show significant induction of DUSP1 in all cases where Mix is present. Historical data has defined Mix in the regulation of PKA and ERK signaling in order to drive C/EBPβ expression and adipogenesis (Morrison and Farmer, 1999a, 2000). In keeping we found that inhibition of PKA and ERK phosphorylation blocked DUSP1 expression (Fig. 5C and D). Recent data however has implicated protein kinase G (PKG) signaling in the regulation of adipogenesis (Zhang et al., 2010). Mix is a well-known phosphodiesterase inhibitor that stimulates PKA and PKG signaling pathways (Huai et al., 2004). As such, we cannot completely rule out a role for cAMP/PKA/PKG pathways in the regulation of DUSP1 expression. Consistent with a role for PKA and ERK signaling pathways in the regulation of DUSP1, we further showed that expression of C/EBPβ mirrored that of the DUSP1 gene. Together, these data suggest a role for the C/EBPβ transcription factor as a regulator of DUSP1 gene expression. Consistent with this hypothesis, reports have identified putative binding sites for C/EBPβ within the DUSP1 promoter and further show that binding of C/EBPβ to these sites promotes DUSP1 mRNA expression (Cho et al., 2008; Johansson-Haque et al., 2008). In addition, others have shown consensus phosphorylation sites within C/EBPβ downstream of PKA (S299) and ERK (T188) that facilitate nuclear translocation and transcriptional activation of C/EBPα and adiponectin, respectively (Chinery et al., 1997; Park et al., 2004). Importantly, C/EBPβ is an essential transcription factor for adipocyte differentiation that acts to induce PPARg and C/EBPα gene expression (Wu et al., 1996; Park et al., 2004). Of note, loss of C/EBPβ prevents adipocyte differentiation even in the presence of PPARg and C/EBPα, demonstrating that adipocyte differentiation requires C/EBPβ (Tanaka et al., 1997). Together, these data would suggest that C/EBPβ can further serve as a regulator of adipogenesis by upregulating the negative inhibitor of MAPK signaling, DUSP1, to modulate MAPK dephosphorylation.
Timely phosphorylation as well as dephosphorylation of ERK and p38 are necessary for adipogenesis (Engelman et al., 1998, 1999; Prusty et al., 2002; Bost et al., 2005a,b; Aouadi et al., 2006, 2007), where rapid and transient activation (i.e., phosphorylation) of ERK and p38 is essential for preadipocyte proliferation as well as the induction and activation of C/EBPβ, both of which are required for adipocyte hyperplasia (Sale et al., 1995; Belmonte et al., 2001; Prusty et al., 2002; Tang et al., 2003). Conversely, prolonged ERK signaling is known to phosphorylate and subsequently inhibit PPARγ activity, thus blocking adipocyte differentiation (Hu et al., 1996). Unlike prolonged ERK activation, chronic p38 activity results in cell death (Engelman et al., 1998, 1999; Aouadi et al., 2007). However, the extent of MAPK phosphorylation during distinct stages of adipocyte differentiation remains unclear. Data presented here demonstrate a rapid and transient activation of ERK signaling during early adipocyte differentiation. Consistent with previous reports (Bost et al., 2005b), data presented here further demonstrate that ERK phosphorylation is suppressed during mid and late stages of adipocyte differentiation falling below basal expression, concomitant with the induction of PPARγ. As prolonged ERK phosphorylation inhibits PPARγ activity and subsequently adipogenesis (Hu et al., 1996), these data suggest that transient activation during early differentiation and suppression of ERK phosphorylation during late differentiation are necessary for the mature adipocyte phenotype. While phosphorylation of p38 during early adipocyte differentiation was similar to ERK, p38 displayed a biphasic pattern of signaling during late stage differentiation. While some have reported suppression of p38 phosphorylation during late adipocyte differentiation (Engelman et al., 1998, 1999), others have noted similar biphasic activity (Aouadi et al., 2006). Antithetical to observations that sustained p38 results in cell death; ectopic expression of p38 has been shown sufficient to promote adipocyte differentiation in 3T3-L1 preadipocytes and NIH 3T3 fibroblasts (Engelman et al., 1998, 1999; Aouadi et al., 2007). However, it remains uncertain if biphasic p38 activity or loss of activity during late differentiation is required for adipocyte differentiation and maintenance of the mature adipocyte phenotype.
Phosphatases are now being recognized as key modulators and even controllers of cellular signaling and biological processes including proliferation and differentiation. This is clearly evident in reports that demonstrate that DUSPs control MAPK dephosphorylation of myocyte, osteocyte, and neuronal cells (Bennett and Tonks, 1997; Reffas and Schlegel, 2000; Matsuguchi et al., 2009), where timely MAPK phosphorylation and dephosphorylation dictates cellular growth and differentiation of these cell types (Bennett and Tonks, 1997; Matsuguchi et al., 2009; Datta et al., 2010). For instance, DUSP1-mediated ERK dephosphorylation has been shown necessary for muscle-specific gene expression and myogenesis. Conversely, prolonged ERK phosphorylation due to loss of DUSP1 blocked myogenesis (Bennett and Tonks, 1997). Likewise, loss of DUSP1 markedly increased ERK phosphorylation during late osteoblast differentiation, in which ERK dephosphorylation is required for terminal growth arrest and bone formation (Datta et al., 2010).
Recent reports have begun highlighting the potential role for DUSPs as negative regulators of the mature adipocyte phenotype (Xu et al., 2003b; Sakaue et al., 2004; Wu et al., 2006; Feng et al., 2014). Already, reports have shown that mice globally lacking DUSP1 or DUSP6 are protected from DIO. However, decreases in adiposity in these knockout mice were attributed to increases in energy expenditure and not inhibition of adipocyte differentiation per se (Wu et al., 2006; Feng et al., 2014). While a few reports have linked ectopic expression of DUSPs, in particular DUSP1 and DUSP9, to inhibition of IR in adipocytes (Xu et al., 2003b; Bazuine et al., 2004; Emanuelli et al., 2008), only three papers have examined a role for DUSPs on adipocyte differentiation (Xu et al., 2003b; Sakaue et al., 2004; Wu et al., 2006). In these reports, DUSP1 and DUSP9 have again been the only DUSPs examined as negative regulators of MAPK signaling and adipogenesis (Xu et al., 2003b; Sakaue et al., 2004; Wu et al., 2006). Converse to our findings that DUSP1 acts as an immediate early gene to regulate MAPK dephosphorylation, Sakaue and Colleagues (Sakaue et al., 2004) reported that DUSP1 expression increased during late stages of adipocyte differentiation, with peak expression observed at day 6 of differentiation. In addition, they report that loss of DUSP1 with anti-sense RNA blocked adipocyte differentiation (Sakaue et al., 2004). In contrast to Sakaue et al. (2004), DUSP1 knockout mouse embryonic fibroblasts (MEFs) were reported to differentiate equally as well when compared to wild-type counterparts, suggesting that DUSP1 is not essential for adipocyte differentiation (Wu et al., 2006). Consistent with this report, we found that genetic loss of DUSP1 did not block adipocyte differentiation. In fact, examination of cyclin A in our study suggested that S-phase transition was more efficient in the presence of increased MAPK signaling. This observation is consistent with Schroder et al. (Schroder et al., 2009) where DUSP1 knockdown enhanced preadipocyte proliferation. Thus, while DUSP1 appears essential for MAPK dephosphorylation, its role with adipocyte differentiation remains unresolved.
In summary, our data suggest that chronic, low-grade inflammation associated with obesity drives DUSP1 expression in adipose tissue. Based on our findings, we would postulate that induction of DUSP1 gene expression acts in a compensatory manner to mitigate MAPK signaling and inflammation in adipose tissue. In the preadipocyte (stromal vascular fraction of adipose tissue), increased DUSP1 potentially acts to maintain MAPK signaling to favor adipocyte differentiation and prevent preadipocyte dysfunction. Indeed PKA and ERK signaling induced DUSP1 expression to feedback inhibit ERK and p38 phosphorylation. While knockdown of DUSP1 alone was not sufficient to block adipogenesis, induction other MAPK-specific DUSPs (Fig. 7) suggest redundancy for MAPK signaling regulation. In keeping, several of these ‘inducible’ DUSPs were also significantly upregulated in obese animals (Fig. 8), suggesting a co-operative and compensatory role for these ‘inducible’ phosphatases on the maintenance of adipocyte function in response to obesity and chronic, low-grade inflammation. Future research into the role of other early ‘inducible’ DUSPs, identified in this study, in the regulation of adipocyte differentiation will be of great interest. In addition, while some evidence suggests that ectopic expression of DUSP9 blocks adipocyte differentiation (Xu et al., 2003b), a role for late stage ‘inducible’ DUSPs regarding MAPK signaling, adipogenesis, and insulin resistance will also be of keen interest.
Acknowledgments
The authors are grateful for superb technical assistance from Dr. Robin G. Hopkins (UNC Greensboro). Study was supported by NIH funding to BSF (F31-DK847702), JMS (R01-DK52968) and RFM (R15-DK082799).
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest.
Literature Cited
- Aouadi M, Jager J, Laurent K, Gonzalez T, Cormont M, Binetruy B, Le Marchand-Brustel Y, Tanti JF, Bost F. 2007. P38MAP Kinase activity is required for human primary adipocyte differentiation. FEBS Lett 581:5591–5596. [DOI] [PubMed] [Google Scholar]
- Aouadi M, Laurent K, Prot M, Le Marchand-Brustel Y, Binetruy B, Bost F. 2006. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes 55:281–289. [DOI] [PubMed] [Google Scholar]
- Auld CA, Fernandes KM, Morrison RF. 2007. Skp2-mediated p27(Kip1) degradation during S/G2 phase progression of adipocyte hyperplasia. J Cell Physiol 211:101–111. [DOI] [PubMed] [Google Scholar]
- Bazuine M, Carlotti F, Tafrechi RS, Hoeben RC, Maassen JA. 2004. Mitogen-activated protein kinase (MAPK) phosphatase-1 and −4 attenuate p38 MAPK during dexamethasone-induced insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol 18:1697–1707. [DOI] [PubMed] [Google Scholar]
- Belmonte N, Phillips BW, Massiera F, Villageois P, Wdziekonski B, Saint-Marc P, Nichols J, Aubert J, Saeki K, Yuo A, Narumiya S, Ailhaud G, Dani C. 2001. Activation of extracellular signal-regulated kinases and CREB/ATF-1 mediate the expression of CCAAT/enhancer binding proteins beta and -delta in preadipocytes. Mol Endocrinol 15:2037–2049. [DOI] [PubMed] [Google Scholar]
- Bennett AM, Tonks NK. 1997. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278:1288–1291. [DOI] [PubMed] [Google Scholar]
- Bjorntorp P, Karlsson M, Pettersson P. 1982. Expansion of adipose tissue storage capacity at different ages in rats. Metabolism 31:366–373. [DOI] [PubMed] [Google Scholar]
- Bost F, Aouadi M, Caron L, Binetruy B. 2005a. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 87:51–56. [DOI] [PubMed] [Google Scholar]
- Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, Dani C, Hofman P, Pages G, Pouyssegur J, Le Marchand-Brustel Y, Binetruy B. 2005b. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 54:402–411. [DOI] [PubMed] [Google Scholar]
- Bouchard L, Tchernof A, Deshaies Y, Marceau S, Lescelleur O, Biron S, Vohl MC. 2007. ZFP36: A promising candidate gene for obesity–related metabolic complications identified by converging genomics. Obes Surg 17:372–382. [DOI] [PubMed] [Google Scholar]
- Chinery R, Brockman JA, Dransfield DT, Coffey RJ. 1997. Antioxidant-induced nuclear translocation of CCAAT/enhancer-binding protein beta. A critical role for protein kinase A-mediated phosphorylation of Ser299. J Biol Chem 272:30356–30361. [DOI] [PubMed] [Google Scholar]
- Cho IJ, Woo NR, Kim SG. 2008. The identification of C/EBPbeta as a transcription factor necessary for the induction of MAPK phosphatase-1 by toll-like receptor-4 ligand. Arch Biochem Biophys 479:88–96. [DOI] [PubMed] [Google Scholar]
- Datta NS, Kolailat R, Fite A, Pettway G. Abou-Samra AB (2010). Distinct roles for mitogen-activated protein kinase phosphatase-1 (MKP-1) and ERK-MAPK in PTH1R signaling during osteoblast proliferation and differentiation. Cell Signal 22:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RJ, Keyse SM. 2006. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci 119:4607–4615. [DOI] [PubMed] [Google Scholar]
- Djian P, Phillips M, Green H. 1985. The activation of specific gene transcription in the adipose conversion of 3T3 cells. J Cell Physiol 124:554–556. [DOI] [PubMed] [Google Scholar]
- Ebisuya M, Kondoh K, Nishida E. 2005. The duration, magnitude and compartmentalization of ERK MAP kinase activity: Mechanisms for providing signaling specificity. J Cell Sci 118:2997–3002. [DOI] [PubMed] [Google Scholar]
- Emanuelli B, Eberle D, Suzuki R, Kahn CR. 2008. Overexpression of the dual-specificity phosphatase MKP-4/DUSP-9 protects against stress-induced insulin resistance. Proc Natl Acad Sci USA 105:3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Berg AH, Lewis RY, Lin A, Lisanti MP, Scherer PE. 1999. Constitutively active mitogen-activated protein kinase kinase 6 (MKK6) or salicylate induces spontaneous 3T3-L1 adipogenesis. J Biol Chem 274:35630–35638. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Lisanti MP, Scherer PE. 1998. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J Biol Chem 273:32111–32120. [DOI] [PubMed] [Google Scholar]
- Faust IM, Johnson PR, Stern JS, Hirsch J. 1978. Diet-induced adipocyte number increase in adult rats: A new model of obesity. Am J Physiol 235:E279–E286. [DOI] [PubMed] [Google Scholar]
- Feng B, Jiao P, Helou Y, Li Y, He Q, Walters MS, Salomon A, Xu H. 2014. MAP kinase phosphatase 3 (MKP-3) deficient mice are resistant to diet induced-obesity. Diabetes 63:2924–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BS, Nam H, Hopkins RG, Morrison RF. 2010. Impact of reference gene selection for target gene normalization on experimental outcome using real-time qRT-PCR in adipocytes. PLoS ONE 5:e15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorzelniak K, Janke J, Engeli S, Sharma AM. 2001. Validation of endogenous controls for gene expression studies in human adipocytes and preadipocytes. Horm Metab Res 33:625–627. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. 2011. Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445. [DOI] [PubMed] [Google Scholar]
- Guenard F, Bouchard L, Tchernof A, Deshaies Y, Hould FS, Lebel S, Marceau P, Perusse L, Vohl MC. 2013. DUSP1 Gene polymorphisms are associated with obesity-related metabolic complications among severely obese patients and impact on gene methylation and expression. Int J Genomics 2013:609748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam DW, James WP. 2005. Obesity. Lancet 366:1197–1209. [DOI] [PubMed] [Google Scholar]
- Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. 2001. The biology of white adipocyte proliferation. Obes Rev 2:239–254. [DOI] [PubMed] [Google Scholar]
- Hirsch J, Batchelor B. 1976. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab 5:299–311. [DOI] [PubMed] [Google Scholar]
- Hirsch J, Fried SK, Edens NK, Leibel RL. 1989. The fat cell. Med Clin North Am 73:83–96. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, Spiegelman BM. 1996. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274:2100–2103. [DOI] [PubMed] [Google Scholar]
- Huai Q, Wang H, Zhang W, Colman RW, Robinson H, Ke H. 2004. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc Natl Acad Sci USA 101:9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey KL, Camps M, Rommel C, Mackay CR. 2007. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 6:391–403. [DOI] [PubMed] [Google Scholar]
- Johansson-Haque K, Palanichamy E, Okret S. 2008. Stimulation of MAPK-phosphatase 1 gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J Mol Endocrinol 41:239–249. [DOI] [PubMed] [Google Scholar]
- Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. 2001. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J 20:7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM. 2008. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev 27:253–261. [DOI] [PubMed] [Google Scholar]
- Khadir A, Tiss A, Abubaker J, Abu-Farha M, Al-Khairi I, Cherian P, John J, Kavalakatt S, Warsame S, Al-Madhoun A, Al-Ghimlas F, Elkum N, Behbehani K, Dermime S, Dehbi M. 2015. MAP kinase phosphatase DUSP1 is overexpressed in obese humans and modulated by physical exercise. Am J Physiol Endocrinol Metab 308:E71–E83. [DOI] [PubMed] [Google Scholar]
- Knittle JL, Timmers K, Ginsberg-Fellner F, Brown RE, Katz DP. 1979. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J Clin Invest 63:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh K, Nishida E. 2007. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta 1773:1227–1237. [DOI] [PubMed] [Google Scholar]
- Kusari AB, Byon J, Bandyopadhyay D, Kenner KA, Kusari J. 1997. Insulin-induced mitogen-activated protein (MAP) kinase phosphatase-1 (MKP-1) attenuates insulin-stimulated MAP kinase activity: A mechanism for the feedback inhibition of insulin signaling. Mol Endocrinol 11:1532–1543. [DOI] [PubMed] [Google Scholar]
- Liu Q 2011. Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol 11:377–383. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Mandenoff A, Lenoir T, Apfelbaum M. 1982. Tardy occurrence of adipocyte hyperplasia in cafeteria-fed rat. Am J Physiol 242:R349–R351. [DOI] [PubMed] [Google Scholar]
- Matsuguchi T, Chiba N, Bandow K, Kakimoto K, Masuda A, Ohnishi T. 2009. JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J Bone Miner Res 24:398–410. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. 1999a. Insights into the transcriptional control of adipocyte differentiation. J Cell Biochem S32–33:59–67. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. 1999b. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J Biol Chem 274:17088–17097. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. 2000. Hormonal signaling and transcriptional control of adipocyte differentiation. J Nutr 130:3116S–3121S. [DOI] [PubMed] [Google Scholar]
- Park BH, Qiang L, Farmer SR. 2004. Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol Cell Biol 24:8671–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH. 2001. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 22:153–183. [DOI] [PubMed] [Google Scholar]
- Prusty D, Park BH, Davis KE, Farmer SR. 2002. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem 277:46226–46232. [DOI] [PubMed] [Google Scholar]
- Reddy ST, Nguyen JT, Grijalva V, Hough G, Hama S, Navab M, Fogelman AM. 2004. Potential role for mitogen-activated protein kinase phosphatase-1 in the development of atherosclerotic lesions in mouse models. Arterioscler Thromb Vasc Biol 24:1676–1681. [DOI] [PubMed] [Google Scholar]
- Reffas S, Schlegel W. 2000. Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem J 352:701–708. [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. 2000. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 16:145–171. [DOI] [PubMed] [Google Scholar]
- Sakaue H, Ogawa W, Nakamura T, Mori T, Nakamura K, Kasuga M. 2004. Role of MAPK phosphatase-1 (MKP-1) in adipocyte differentiation. J Biol Chem 279:39951–39957. [DOI] [PubMed] [Google Scholar]
- Sale EM, Atkinson PG, Sale GJ. 1995. Requirement of MAP kinase for differentiation of fibroblasts to adipocytes, for insulin activation of p90 S6 kinase and for insulin or serum stimulation of DNA synthesis. EMBO J 14:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliess F, Kurz AK, Haussinger D. 2000. Glucagon-induced expression of the MAP kinase phosphatase MKP-1 in rat hepatocytes. Gastroenterology 118:929–936. [DOI] [PubMed] [Google Scholar]
- Schroder K, Wandzioch K, Helmcke I, Brandes RP. 2009. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29:239–245. [DOI] [PubMed] [Google Scholar]
- Stephens JM, Butts MD, Pekala PH. 1992. Regulation of transcription factor mRNA accumulation during 3T3-L1 preadipocyte differentiation by tumour necrosis factor-alpha. J Mol Endocrinol 9:61–72. [DOI] [PubMed] [Google Scholar]
- Stephens JM, Pekala PH. 1992. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J Biol Chem 267:13580–13584. [PubMed] [Google Scholar]
- Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, DeFuria J, Jick Z, Greenberg AS, Obin MS. 2007. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56:2910–2918. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S. 1997. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J 16:7432–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. 2003. Mitotic clonal expansion: A synchronous process required for adipogenesis. Proc Natl Acad Sci USA 100:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Nishida E. 2003. Molecular recognitions in the MAP kinase cascades. Cell Signal 15:455–462. [DOI] [PubMed] [Google Scholar]
- Torti FM, Torti SV, Larrick JW, Ringold GM. 1989. Modulation of adipocyte differentiation by tumor necrosis factor and transforming growth factor beta. J Cell Biol 108:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, Neufer PD, Shulman GI, Kim JK, Bennett AM. 2006. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metab 4:61–73. [DOI] [PubMed] [Google Scholar]
- Wu Z, Bucher NL, Farmer SR. 1996. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol Cell Biol 16:4128–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. 2003a. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Dembski M, Yang Q, Yang D, Moriarty A, Tayber O, Chen H, Kapeller R, Tartaglia LA. 2003b. Dual specificity mitogen-activated protein (MAP) kinase phosphatase-4 plays a potential role in insulin resistance. J Biol Chem 278:30187–30192. [DOI] [PubMed] [Google Scholar]
- Yang SP, Martin LJ, Schneider G. 1980. Weight reduction utilizing a protein-sparing modified fast. J Am Diet Assoc 76:343–346. [PubMed] [Google Scholar]
- Zhang X, Ji J, Yan G, Wu J, Sun X, Shen J, Jiang H, Wang H. 2010. Sildenafil promotes adipogenesis through a PKG pathway. Biochem Biophys Res Commun 396:1054–1059. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dong C. 2007. Regulatory mechanisms of mitogen-activated kinase signaling. Cell Mol Life Sci 64:2771–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]