

Abstract
Rationale:
Cardiac myosin binding protein-C (cMyBP-C) is a critical regulator of heart contraction, but the mechanisms by which cMyBP-C affects actin and myosin are only partly understood. A primary obstacle is that cMyBP-C localization on thick filaments may be a key factor defining its interactions, but most in vitro studies cannot duplicate the unique spatial arrangement of cMyBP-C within the sarcomere.
Objective:
The goal of this study was to validate a novel hybrid genetic/protein engineering approach for rapid manipulation of cMyBP-C in sarcomeres in situ.
Methods and Results:
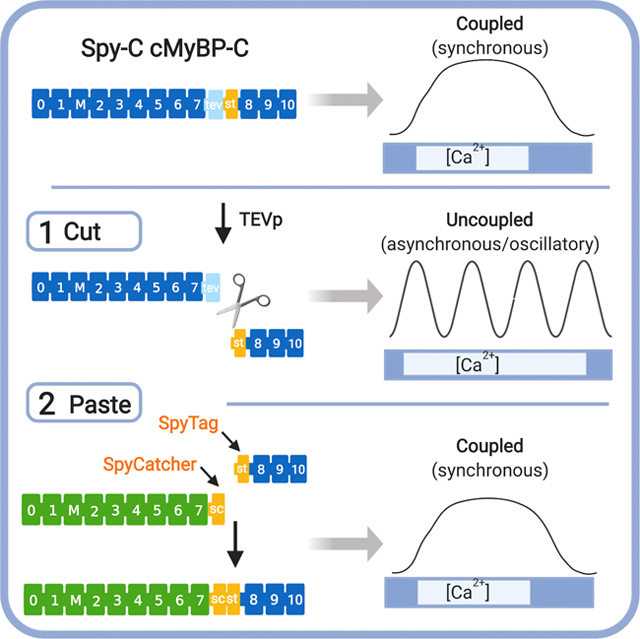
We designed a novel “cut and paste” approach for removal and replacement of cMyBP-C N’-terminal domains (C0-C7) in detergent-permeabilized cardiomyocytes from gene-edited “Spy-C” mice. Spy-C mice express a tobacco etch virus protease (TEVp) cleavage site and a “SpyTag” between cMyBP-C domains C7 and C8. A “cut” is achieved using TEVp which cleaves cMyBP-C to create a soluble N’-terminal gC0C7 fragment and an insoluble C’-terminal SpyTag (st)-C8-C10 fragment that remains associated with thick filaments. “Paste” of new recombinant (r)C0C7 domains is achieved by a covalent bond formed between SpyCatcher (-sc) (encoded at the C’-termini of recombinant proteins) and SpyTag. Results show that loss of gC0C7 reduced myofilament Ca2+ sensitivity and increased cross bridge cycling (ktr) at submaximal [Ca2+]. Acute loss of gC0C7 also induced auto-oscillatory contractions at submaximal [Ca2+]. Ligation of rC0C7-sc returned pCa50 and ktr to control values and abolished oscillations, but phosphorylated (p)-rC0C7-sc did not completely rescue these effects.
Conclusions:
We describe a robust new approach for acute removal and replacement of cMyBP-C in situ. The method revealed a novel role for cMyBP-C N’-terminal domains to damp sarcomere-driven contractile waves (so called “SPOC”). Because phosphorylated (p)-rC0C7-sc was less effective at damping contractile oscillations, results suggest that SPOC may contribute to enhanced contractility in response to inotropic stimuli.
Keywords: Cardiac myosin binding protein-C, SPOC, actin, myosin, SpyCatcher, SpyTag, mouse model, mechanics, myosin binding protein, sarcomere, myocyte, phosphorylation, Cardiomyopathy, Contractile Function, Hypertrophy, Myocardial Biology
Graphical Abstract
INTRODUCTION
Cardiac myosin binding protein-C (cMyBP-C) is a tunable regulator of heart function that influences the rate at which myosin force-generating cross-bridges interact with actin. cMyBP-C is necessary for normal cardiac function and for enhanced contractility in response to “fight-or-flight” stimuli that speed cross-bridge kinetics through phosphorylation of cMyBP-C1–3. Conversely, reduced cMyBP-C phosphorylation occurs in heart failure4–6 and mutations in MYBPC3, the gene encoding cMyBP-C, are the most common cause of hypertrophic cardiomyopathy (HCM)7,8. Although the significance of cMyBP-C to heart health is now well-established, it remains less certain how cMyBP-C regulates myosin cross-bridge interactions with actin or how these interactions are altered by phosphorylation or mutations that cause HCM.
cMyBP-C is a modular protein composed of 11 folded domains (named C0-C10 starting from the N’-terminus as shown in Fig. 1). An additional “M”-domain, unique to myosin binding protein-C isoforms, is located between C1 and C2 and encodes serine residues that are targets of common kinases in multiple signaling pathways9,10. Whereas the C8-C10 domains anchor cMyBP-C tightly to thick filaments and determine its characteristic localization in sarcomere A-bands11,12, N’-terminal domains of cMyBP-C (e.g., C0-C1-M-C2) contain multiple binding sites for both myosin and actin that are dynamically regulated by phosphorylation13,14. Whereas cMyBP-C interactions with myosin are thought to be primarily inhibitory15,16, cMyBP-C binding to actin can potently activate actomyosin interactions by directly shifting the position of tropomyosin to its open state on the thin filament thereby allowing cross-bridge formation17–19.
Figure 1. A “cut and paste” method for replacing N’-terminal domains of cMyBP-C in situ.

Top, cMyBP-C consists of 11 folded domains numbered C0 to C10 starting at the N’-terminus of the molecule plus an additional “M”-domain unique to MyBP-C proteins between C1 and C2. Gene-edited Spy-C mice express a modified cMyBP-C with a TEV protease (TEVp) recognition site (light blue rectangle) and a SpyTag (orange rectangle) inserted between domains C7 and C8. (1) CUT: TEVp treatment of detergent-permeabilized homozygous (HO) Spy-C myocytes releases genetically encoded (γ)C0-C7. (2) PASTE: New recombinant (r)C0-C7-sc domains (green) (encoding any desired modification such as point mutations, deletions, fluorescent probes) are added to the bath where they are covalently attached to st-C8C10 on the thick filament via a spontaneous bond formed between SpyCatcher and SpyTag. Figure created with BioRender.com.
A wealth of information on the complex regulatory effects of cMyBP-C on actomyosin interactions has been obtained through in vitro studies. However, translating in vitro results into a complete picture of how cMyBP-C regulates cardiac muscle contraction in working sarcomeres has proven much more challenging. A primary obstacle is the complexity of the sarcomere itself, a semi-crystalline array of interdigitating thick and thin filaments wherein cMyBP-C is an integral component of the thick filaments, albeit with a limited distribution to 9 discrete stripes spaced 43 nm apart in each ½ A-band20–22. The significance of the limited occurrence of cMyBP-C remains unclear, but is thought to be a key factor in determining how cMyBP-C interacts with its binding partners. However, until now most studies have used truncated proteins such as “C0C2” (C0-C1-M-C2) or “C1C2” (C1-M-C2) that lack the C’-terminal domains (C8-C10) needed for cMyBP-C localization into sarcomeres. While these studies are invaluable for defining residues that control cMyBP-C binding to actin or myosin17, determining effects of cMyBP-C when it is confined to its precise position in sarcomeres has remained an ongoing challenge.
To overcome limitations intrinsic to many in vitro approaches, here we designed a novel method that fills a methodological gap that merges the speed and convenience of in vitro methods with ex vivo approaches that preserve the spatial and stoichiometric relationship of cMyBP-C in sarcomeres in situ. As shown in Fig. 1, our novel “cut and paste” method uses a single gene-edited mouse model (“Spy-C” mice) as a platform for rapid replacement of genetically encoded cMyBP-C domains (g)C0-C7 with new recombinant (r) N’-terminal domains at their precise position in sarcomeres in situ. Briefly, gene-edited Spy-C mice express a 20 amino acid insertion between domains C7 and C8 that encodes i) a TEV protease recognition site (tobacco etch virus protease, TEVp) followed by ii) a SpyTag (st)23. Importantly, SpyTag is ½ of a split protein pair that when combined with SpyCatcher, the other half of the pair, the two catalyze an instantaneous covalent isopeptide bond. To achieve cMyBP-C replacement, skinned (Triton X-100 detergent-permeabilized) myocytes from Spy-C mice are treated with TEVp to cleave and release genetically encoded N’-terminal (g)C0-C7 domains of cMyBP-C while the C’-terminal SpyTag-C8-C10 fragment remains anchored to the thick filament12. Next, new recombinant (r)C0C7-sc proteins encoding SpyCatcher (-sc) at their C’-termini are covalently ligated to the st-C8C10 domains via an instantaneous isopeptide bond formed between SpyCatcher and SpyTag23.
Here we report validation of the “cut and paste” approach by demonstrating rapid and efficient replacement of cMyBP-C N’-terminal domains (rC0-C7) at the position of genetically encoded cMyBP-C in sarcomeres and show that the method recapitulates effects obtained using traditional knockout and transgenic mouse models of cMyBP-C. Furthermore, we show that the “cut and paste” approach revealed a new regulatory role for cMyBP-C to damp contractile oscillations. Because phosphorylated p-rC0C7-sc was less effective at damping contractile oscillations, results suggest that regulation of contractile waves by cMyBP-C contributes to cardiac contraction during inotropic challenges. Based on the robustness and ease of use of the “cut and paste” approach, the method should be broadly applicable to other proteins that have proven difficult or impossible to manipulate in protein complexes.
METHODS
An expanded Materials and Methods section can be found online in Supplemental Materials.
Data that support the findings of this study are available from the corresponding author upon reasonable request.
A list of major reagents and their sources can be found in the Major Resources Table in the Online Supplemental Materials.
Gene-editing and generation of Spy-C mice.
Spy-C mice were created by the GEMM core at the University of Arizona using CRISPR/Cas9 based gene-editing and Homology Directed Repair. Briefly, Spy-C mice were created by inserting a 60-nucleotide cassette into the MYBPC3 gene locus (between domains C7 and C8 of the cMyBP-C protein, Fig. 1) using a synthetic single guide RNA injected into fertilized mouse zygotes (strain C57BL6/NJ). The 60 nucleotide insert encodes i) a TEVp consensus recognition sequence (ENLYFQG) followed by ii) a SpyTag sequence (AHIVMVDAYKPTK)23. 3 male Spy-C founder mice were obtained following gene-editing of zygotes; 2 males (7E and 2G) were homozygous for the insertion and 1 male (1F) was heterozygous. All 3 founders survived into adulthood, were fertile, and generated progeny that were maintained as separate lines. All lines produced offspring in expected Mendelian inheritance ratios (Online Fig. I). cMyBP-C protein expression and echocardiography was analyzed independently for each line.
Cut and paste replacement of cMyBP-C N’-terminal domains in permeabilized myocytes.
For batch removal and replacement of cMyBP-C N’-terminal domains in permeabilized myocytes from HO Spy-C mice, myocytes were prepared as described for force measurements (see Online Supplemental Materials) and rinsed 3x in fresh relax buffer without detergents. To remove genetically encoded (g) domains C0C7 of endogenous cMyBP-C, myocytes were incubated with purified recombinant TEVp (20 mM, 30 min at room temperature) followed by 3 rinses in fresh relax buffer to remove TEVp and gC0C7. Covalent ligation of new recombinant (r) cMyBP-C-sc N’-terminal domains (with SpyCatcher [-sc] encoded at their C’-terminal ends) was achieved by incubating with 20 mM of the desired recombinant protein for 20 min. In some cases, TEVp treatment was omitted and recombinant proteins were directly added to permeabilized myocytes. Excess recombinant proteins not covalently ligated to SpyTag [st] were removed by 3x washes in fresh relax buffer without added protein. Myocyte samples were then dissolved in a urea sample buffer and analyzed by western blotting as described in the expanded Online Supplemental Materials.
RESULTS
Normal cMyBP-C expression, localization, and cardiac function in Spy-C mice.
Online Fig. I shows that wild type (WT), heterozygous (HT), and homozygous (HO) Spy-C mice were born in expected Mendelian ratios and there were no significant differences in cMyBP-C expression across the three different founder lines or genotypes as determined by western blots of left ventricle (LV) homogenates. cMyBP-C localization visualized by immunofluorescence staining of cMyBP-C was also similar in WT, HT, and HO myocytes (Online Fig. I). Cardiac size and morphology appeared normal and there were no significant differences in heart to body-weight ratios indicating a lack of overt cardiac hypertrophy (Online Fig. I). Longitudinal echocardiography performed on mice 8–52 weeks of age showed no consistent differences in either cardiac wall dimensions or functional indices (Online Figs. II–III). Active and passive steady state force and Ca2+ sensitivity of tension were also similar in permeabilized myocytes (Online Fig. IV). Collectively, these data establish that the TEV/SpyTag insertion in cMyBP-C of Spy-C mice had minimal impact on cardiac function under resting conditions.
Validation of the cut and paste method.
To determine whether cMyBP-C N’-terminal domains can be selectively removed and replaced in cardiomyocytes from Spy-C mice as hypothesized, we treated permeabilized myocytes from HO Spy-C mice with TEV protease (TEVp) to remove the genetically encoded N’-terminal domains of cMyBP-C (gC0C7). Fig. 2A shows that TEVp cleaved cMyBP-C in HO myocytes from Spy-C mice resulting in loss of the full-length cMyBP-C band and the appearance of a lower molecular weight band corresponding to the size expected for gC0C7 (~106 kDa). The gC0C7 band disappeared after rinsing myocytes with fresh bath solutions demonstrating that the gC0C7 fragment is soluble and can be eliminated from sarcomeres following cleavage with TEVp (yellow box in Fig. 2A). The remaining st-C8-C10 band (~45 kDa) was detected using an antibody raised against SpyTag (Fig. 2D) because the antibody against cMyBP-C preferentially recognizes N’-terminal domains of cMyBP-C24. Unlike the gC0C7 band, however, the st-C8-C10 band persisted on western blots even after washing with fresh buffers, as expected if the st-C8-C10 domains remain adhered to the thick filament following TEVp proteolysis11,12,25. Importantly, TEVp was specific for cMyBP-C only in engineered Spy-C myocytes and did not affect cMyBP-C function in WT myocytes (Fig. 2B, Fig 3) and did not affect other sarcomeric proteins as visualized by SDS-PAGE (Online Fig. VII). Quantitative proteomics further confirmed that cMyBP-C was the most significantly affected protein following TEVp treatment in skinned LV homogenates from HO Spy-C mice and that cMyBP-C was the only sarcomeric protein with significantly reduced abundance after TEVp treatment (Online Fig. VIII, Online Tables IV, V). These results confirm the high specificity of TEVp for Spy-C cMyBP-C in our system and are consistent with high specificity reported for TEVp in a variety of in vitro and in vivo systems26,27.
Figure 2. Validation of the cut and paste method for rapid removal and replacement of cMyBP-C N’-terminal domains.

A) Western blot of LV homogenates probed with an antibody to cMyBP-C before and after TEVp treatment and after covalent bond formation with rC0C7-sc. Uncut cMyBP-C is visible as a band (green) in the untreated lane (left). After TEVp treatment the gC0C7 proteolytic fragment (yellow box) can be removed by washing with fresh solutions. Newly ligated rC0C7-sc-st-C8C10 protein is visible as a band (green) in lanes with added rC0C7-sc. Excess (un-ligated) rC0C7-sc is visible as a lower molecular weight band (green) below cMyBP-C. α-actinin (red) served as a loading control. B) Control experiment showing a western blot of cMyBP-C in WT myocytes before and after TEVp treatment and after addition of rC0C7-sc. WT cMyBP-C was not cleaved by TEVp and addition of rC0C7-sc did not affect the native (WT) cMyBP-C band. C) Immunofluorescence staining showing the normal doublet (green) pattern of cMyBP-C staining in HO myocytes (left panel), loss of green doublets after TEVp treatment (middle panel), and reappearance of doublets after ligation with rC0C7-sc (right panel). Control experiments showed TEVp had no effect on the doublet pattern of cMyBP-C localization in WT myocytes (Inset, middle panel) and addition of rC0C7 (without SpyCatcher) did not restore cMyBP-C doublets (Inset, right panel). D) Representative western blots showing ligation efficiency of rC0C7-sc in TEVp treated LV homogenates from HO Spy-C mice. Top, Homogenates were probed with an antibody against cMyBP-C. Middle and bottom panels, western blots of LV homogenates probed with a custom antibody against SpyTag. The SpyTag antibody recognized SpyTag in cMyBP-C prior to TEVp treatment (middle panel, green band in left untreated lane) and also recognized SpyTag in the smaller st-C8C10 (~35 kDa) fragment after TEVp treatment (bottom panel). However, the SpyTag antibody did not recognize SpyTag in the ligated rC0C7-sc-st-C8C10 protein after covalent bond formation with SpyCatcher (note the absence of green cMyBP-C bands in the middle panel in all TEVp treated + rC0C7-sc lanes). E) Summary data from western blots as in D to quantify efficiency of rC0C7-sc ligation. The cMyBP-C/α-actinin ratio in each lane was normalized to the cMyBP-C/α-actinin ratio in the untreated lane. > 90% ligation was achieved relative to uncut cMyBP-C when [rC0C7-sc] was > 5 μM. F) Addition of rC0C7-sc to HO myocytes without first treating with TEVp resulted in the appearance of a high MW band (> 250 kDa) consistent with formation of a branched (“Y”-shaped) cMyBP-C (right lane).
Figure 3. Tension-pCa relationships in WT and HO myocytes before and after treatment with TEVp, and after ligation with rC0C7-sc or phosphorylated rC0C7-sc (p-rC0C7-sc).

A) Normalized tension-pCa curves measured in WT myocytes before (circles) and after TEVp (squares) treatment and after incubation with rC0C7-sc (diamonds). Inset, bars show average pCa50 values under each condition (N=5, n=11). B) Normalized tension-pCa curves measured in HO myocytes before and after TEVp treatment and after incubation with rC0C7-sc. Inset, bars show average pCa50 values under each condition (N=5, n=7). C) Normalized tension-pCa curves measured in WT myocytes before and after TEVp treatment and after incubation with p-rC0C7-sc (triangles). Inset, bars show average pCa50 values under each condition (N=4, n=4). D) Normalized tension-pCa curves measured in HO myocytes before and after TEVp treatment and after incubation with p-rC0C7-sc. Inset, bars show average pCa50 values under each condition (N=7, n=8). *** p< 0.0005.
Replacement of gC0C7 with recombinant rC0C7-sc was achieved by adding rC0C7-sc directly to myocyte bath solutions after TEVp treatment (Figs 2A, C, D). Spontaneous covalent ligation of the added recombinant domains was confirmed by the appearance of a band at the molecular weight predicted for the newly formed rC0C7-sc-st-C8C10 complex (~152 kDa) slightly higher than native cMyBP-C. This new band persisted after washing, consistent with covalent ligation of rC0C7-sc to st-C8-C10 anchored to thick filaments (Figs. 2A, D).
Fig. 2C shows immunofluorescent localization of cMyBP-C in Spy-C myocytes before TEVp treatment, after TEVp cleavage and washout of gC0C7, and again after covalent ligation of rC0C7-sc. Results show that the normal doublet pattern of cMyBP-C in each ½ sarcomere was lost after TEVp treatment but was restored following ligation of rC0C7-sc as expected if rC0C7-sc ligates to st-C8C10 at the position of native cMyBP-C on the thick filament. Negative control experiments (Fig. 2C, inset) showed that addition of rC0C7 alone (without encoded SpyCatcher) did not recapitulate the doublet pattern of cMyBP-C localization, confirming that the SpyCatcher-SpyTag bond was specifically required for proper localization of rC0C7-sc. To quantify efficiency of rC0C7-sc replacement, we performed ligations with increasing [rC0C7-sc] or [p-rC0C7-sc]. Figs. 2D, E shows that the ~152 kDa ligation product increased with [rC0C7-sc] until a plateau was reached between 10–30 mM where efficiency was >90% relative to genetic cMyBP-C expression measured prior to TEVp treatment. Similar ligation efficiencies were obtained for phosphorylated (p)-rC0C7-sc (Online Fig. V). All ligations therefore used 20 mM rC0C7-sc or (p)-rC0C7-sc unless otherwise noted.
In a separate set of experiments, we explored whether the SpyTag sequence in cMyBP-C of Spy-C mice was recognizable by rC0C7-sc without first cleaving with TEVp. Fig. 2F shows results from western blots where rC0C7-sc was added directly to permeabilized myocytes without prior treatment with TEVp. Under these conditions rC0C7-sc caused loss of the native full-length cMyBP-C and appearance of a high molecular weight band migrating >250 kDa. The latter presumably represents a branched “Y”-shaped protein formed by ligating rC0C7-sc directly to the SpyTag sequence between C7 and C8 of Spy-C cMyBP-C. The appearance of this high molecular weight band indicates that the SpyTag sequence is accessible to SpyCatcher even in uncut cMyBP-C and is consistent with the ability of the SpyTag antibody to recognize the SpyTag sequence in uncut cMyBP-C from Spy-C myocytes (Fig. 2D). In principle, accessibility of SpyTag in uncut Spy-C cMyBP-C offers an opportunity to attach probes in parallel with genetically encoded N’-terminal C0-C7 domains.
Loss of gC0C7 reduces myofilament Ca2+ sensitivity of tension and exaggerates transient force responses in permeabilized Spy-C myocytes.
We next assessed effects of gC0C7 removal and replacement on force in permeabilized myocytes by measuring tension-pCa relationships in individual myocytes before and after TEVp treatment, and again following ligation with rC0C7-sc. Removal of gC0C7 caused a significant rightward shift in Ca2+ sensitivity of tension, but had no significant effect on maximal force at saturating Ca2+ (pCa 4.5) or on passive force in the absence of Ca2+ (pCa 9.0) (Fig. 3, Online Table II). Ligation of new rC0C7-sc fully reversed the effect on Ca2+ sensitivity of tension by inducing a significant leftward shift in Ca2+ sensitivity of tension. However, ligation of phosphorylated rC0C7-sc (p-rC0C7-sc) did not reverse the rightward shift caused by loss of gC0C7. Ca2+ sensitivity of tension was not significantly different in WT myocytes after treatment with TEVp or after incubation with rC0C7-sc, indicating effects of TEVp in HO myocytes were due specifically to removal and replacement of cMyBP-C and cannot be attributed to non-specific effects on other proteins (Fig. 3, Online Figs. VII–VIII, Online Tables II, IV-V).
To assess the impact of gC0C7 removal and ligation of rC0C7-sc on cross-bridge kinetics we measured the rate of tension re-development following a release and re-stretch maneuver (ktr) before and after digestion with TEVp. Loss of gC0C7 after TEVp treatment increased the apparent rate of tension recovery at submaximal pCa values near the pCa50 for tension development, whereas ligation of rC0C7-sc reversed these effects and returned ktr values to pre-TEVp values (Fig. 4). However, comparison of ktr values before and after TEVp treatment was complicated by the presence of force “overshoots” after TEVp treatment in HO myocytes where force transiently exceeded the steady state force prior to the slack and stretch maneuver at submaximal [Ca2+] near the pCa50 for tension development. Overshoots disappeared after ligation with rC0C7-sc, whereas (p)-rC0C7-sc reduced but did not completely eliminate overshoots (Fig. 4). Force overshoots were not observed in WT myocytes before or after TEVp treatment or after incubation with rC0C7-sc (not shown).
Figure 4. Rates of tension redevelopment (ktr) and force overshoots in WT and HO myocytes before and after treatment with TEVp and after ligation with rC0C7-sc or phosphorylated rC0C7-sc (p-rC0C7-sc).

A) Representative trace of motor position showing ktr protocol. After steady state force was reached in a given pCa solution, myocytes were slackened by 20% for 20 ms followed by a brief (1 ms) 5% overstretch before returning to their starting length. B) Representative force traces from the same HO myocyte before and after TEVp treatment and following ligation with rC0C7-sc. Note the appearance of a force “overshoot” following TEVp treatment (dashed trace). C) Schematic diagram describing calculation of force overshoots where PT0 is the maximum overshoot and Px is the isometric force in a given pCa solution. D) Summary ktr data for WT myocytes showing no significant difference in ktr before or after TEVp treatment or after incubation with rC0C7-sc (N=5, n=11). E) Summary ktr data for HO myocytes before and after treatment with TEVp and after ligation with rC0C7-sc (N=5, n=7). F) Summary ktr data for HO myocytes before and after treatment with TEVp and after ligation with p-rC0C7-sc (N=7, n=8). G) Force overshoots (PT0) before TEVp, after TEVp, and after treatment with rC0C7-sc in WT myocytes expressed as a percentage over steady state force (Px) (N=5, n=11). H) and I) Force overshoots (PT0) in HO myocytes expressed as a percentage of steady state force (Px) before TEVp, after TEVp, and after ligation with rC0C7-sc (N=5, n=7) or p-rC0C7-sc (N=7, n=8), respectively. Before TEVp vs. After TEVp: *p< 0.05; **p< 0.005, After TEVp vs. After ligation # p< 0.05; ## p< 0.005.
Loss of cMyBP-C N’-terminal domains induces sustained spontaneous auto-oscillatory contractions (SPOC).
Following TEVp digestion and washout of gC0C7 there was a significant increase in the appearance of spontaneous oscillatory contractions evident as contractile waves propagated across multiple sarcomeres during constant Ca2+ activation (Fig. 5, Online Video I). Spontaneous Ca2+ induced oscillations (referred to as “SPOC”) have been described previously where SPOC is attributed to intrinsic oscillatory properties of contractile systems that occur in the absence of other cyclic changes such as Ca2+ release from the sarcoplasmic reticulum (SR)28–31. Consistent with this, force oscillations observed here were not attributable to cyclic changes in [Ca2+] because the SR was removed by detergent permeabilization with Triton X-100 and oscillations occurred despite constant bath [Ca2+] (e.g., during activation in a single pCa solution). SPOC was most frequent at intermediate [Ca2+] near the pCa50 for force development, although oscillations were sometimes evident even in maximal activating solutions at pCa 4.5 (not shown). SPOC was sometimes observed in WT and HO myocytes before TEVp treatment, but under these conditions SPOC was transient, lasting only briefly (seconds) during the pre-steady state period when force was rapidly rising up to a plateau value. By contrast, SPOC persisted for long periods (>45 min) after TEVp treatment in HO myocytes once force had reached a steady state value at a given [Ca2+] (Online Video I). Ligation of new rC0C7-sc abolished SPOC at all [Ca2+] during steady state force development (Online Video I). By contrast, PKA phosphorylated p-rC0C7-sc was less effective and did not completely eliminate SPOC (Online Video II). However, SPOC following p-rC0C7-sc ligation was also long lasting (i.e., sustained for periods up to 60 minutes, not shown). TEVp treatment and exposure to rC0C7-sc had no effect in WT myocytes (Online Video III).
Figure 5. Contractile oscillations in HO myocytes after TEVp treatment recorded during steady state force activations.

A) Representative force traces from a single HO myocyte before TEVp treatment, after TEVp treatment, and after ligation of rC0C7-sc. Traces were arbitrarily shifted along the Y-axis for clarity. Force traces after TEVp treatment showed variations in steady state force due to underlying oscillatory contractions visible in contracting myocytes (see Online Video I). B) PIVLab image analysis of a contracting HO myocyte to represent force oscillations as changes in vector direction (black arrows) over time (inset, 1, 2, 3, 4) (see also Online Video I). White boxed region indicates area chosen for analysis in C. C) Results of speckle tracking analysis using Tracker 5.0.7 of 4 speckles in the white boxed region shown in B was performed and average displacement was plotted over time. Individual sarcomeres and contiguous groups of sarcomeres showed organized wave-like oscillations when activated at a constant pCa.
The first 4 N’-terminal domains of cMyBP-C rescue functional effects due to loss of gC0C7.
Because most regulatory effects of cMyBP-C are attributed to its first 4 N’-terminal domains of cMyBP-C (e.g., C0C2 = C0-pal-C1-m-C2), we next determined whether addition of rC0C2-sc alone was sufficient to rescue changes in force after TEVp treatment. As shown in Online Fig. VI and Online Table III, ligation of rC0C2-sc increased Ca2+ sensitivity of tension, eliminated force overshoots and eliminated SPOC (Online Video IV). However, unlike full-length rC0C7-sc, rC0C2-sc did not return ktr to control values prior to TEVp treatment so that ktr values remained elevated even after ligation of rC0C2-sc (Online Fig. VI). By contrast, phosphorylated (p)-rC0C2-sc reduced ktr values at submaximal [Ca2+] near the pCa50 for force development and caused a small but significant leftward shift in Ca2+ sensitivity of tension. p-rC0C2-sc also eliminated force overshoots (Online Fig. VI, Online Table III), although p-rC0C2-sc did not completely eliminate SPOC (Online Video V). Taken together, these results show that the first 4 N’-terminal domains are sufficient to confer most of the phosphorylation dependent effects of cMyBP-C on force, but domains C3-C7 may confer additional regulatory effects on cross-bridge kinetics assessed by ktr.
Finally, because cMyBP-C is subject to proteolytic cleavage during cardiac stress causing loss of a ~29 kD N’-terminal fragment (i.e., “C0C1f”, containing the first 2 N’-terminal domains of cMyBP-C and the first 17 amino acids of the M-domain32,33, we next investigated whether loss of the ~29 kDa fragment affected force or SPOC activity. As shown in Online Fig. VI, ligation of a recombinant protein that lacked the C0C1f fragment (i.e. “r-mDfC2C7-sc”) did not rescue the rightward shift in Ca2+ sensitivity after TEVp treatment and did not eliminate force overshoots or damp SPOC (Online Table III, Online Video VI). Similar results were obtained following addition of p-r-mDfC2C7-sc (Online Fig VI, Online Table III). Addition of the ~29 kD fragment alone (rC0C1f-sc) also had no effect on Ca2+ sensitivity or on force overshoots (Online Fig. VI) and SPOC activity (Online Table III, Online Video VII). These results show that proteolytic cleavage of cMyBP-C N’-terminal domains reduces the contractile effects of cMyBP-C N’-terminal domains and diminishes the damping effects of cMyBP-C on SPOC.
DISCUSSION
Results from this study establish a novel “cut and paste” approach for rapidly manipulating cMyBP-C in permeabilized myocytes from genetically engineered Spy-C mice. Results show that the method is robust, thus filling a methodological gap between in vitro biochemical assays that are expedient but lack spatial and mechanical constraints and ex vivo approaches that preserve cMyBP-C localization in sarcomeres but that are time consuming and costly. Validation of the new method included recapitulation of effects on cross-bridge kinetics reported for traditional cMyBP-C knockout models15. However, the ability to remove and replace cMyBP-C rapidly in the absence of secondary remodeling effects revealed a previously unrecognized role for cMyBP-C to damp oscillatory contractions in a phosphorylation dependent manner. Implications of these findings are that regulated oscillatory contractions may contribute to enhanced relaxation kinetics in response to inotropic stimuli, whereas dysregulation of oscillatory contractions may contribute to contractile dysfunction under conditions of cardiac stress or in diseases related to cMyBP-C. Application of the cut and paste approach described here further suggests that the method may be broadly applicable to other proteins that are difficult or impossible to manipulate using traditional methods.
Success of the cut and paste approach was facilitated by 3 conditions. First, the 20 amino acid insertion encoding the TEVp consensus site and SpyTag sequence had little or no obvious impact on cardiac function in Spy-C mice under resting conditions (Online Figs. I–III). The absence of an overt phenotype simplified data analysis because results could be interpreted without confounding factors arising from cardiac remodeling. However, even if Spy-C mice had shown a phenotype different than WT mice, the cut and paste method could still be useful because each myocyte served as its own control, allowing for repeated measure study designs. Second, selective cleavage of cMyBP-C in Spy-C myocytes by TEVp was rapid and complete, requiring only a brief incubation for complete digestion of Spy-C cMyBP-C without significant loss of other sarcomeric proteins (Fig. 2, Online Figs. VII–VIII, Online Tables IV-V). Third, the SpyTag sequence (st) between domains C7 and C8 of cMyBP-C of Spy-C myocytes was easily accessible to recombinant proteins encoding the cognate SpyCatcher (sc) allowing rapid and specific ligation of added proteins at the position of native cMyBP-C.
The ability to manipulate cMyBP-C in sarcomeres in situ using the cut and paste approach thus overcomes a major limitation of most previous studies that typically used partial cMyBP-C N’-terminal proteins such as C0C2 in force assays, namely the inability to restrict exogenous proteins exclusively to positions normally occupied by native cMyBP-C. Thus, despite the profound effects of recombinant cMyBP-C N’-terminal domains to increase myofilament Ca2+ sensitivity in a variety of assays34–36, it has remained an open question whether cMyBP-C N’-terminal domains exert activating effects when properly localized in sarcomeres in situ. Results from the current study resolve this question by showing that removal of gC0C7 after TEVp treatment caused a significant rightward shift in tension-pCa relationships (Fig. 3) as expected if gC0C7 indeed sensitizes myofilaments to Ca2+ when properly localized in sarcomeres. The rightward shift was most apparent in mice that had been given propranolol to blunt adrenergic responses prior to sacrifice4,37,38. The latter suggests that cMyBP-C phosphorylation reduces Ca2+ sensitizing effects of the N’-terminal domains in concert with TnI phosphorylation following b-adrenergic stimuli2,39. We directly confirmed Ca2+ sensitizing effects of N’-terminal domains by showing that ligation of rC0C7-sc induced a leftward shift in Ca2+ sensitivity, whereas phosphorylation blunted this effect (Fig. 3). Furthermore, we showed the first 4 N’-terminal domains of cMyBP-C (i.e., C0C2 = C0-pal-C1-M-C2) are sufficient to increase Ca2+ sensitivity of tension (Online Fig. VI), in agreement with previous in vitro studies using truncated C0C2 or C1C234,36.
Effects of N’-terminal domains to increase Ca2+ sensitivity of tension are consistent with results from traditional cMyBP-C knockout models which also showed trends towards decreased Ca2+ sensitivity in the absence of cMyBP-C40. However, others reported either no change or increased Ca2+ sensitivity in myocytes from knockout mice41,42. Because changes in Ca2+ sensitivity may be modest (as reported here following acute TEVp treatment), differences may reflect differences in mouse models or other experimental conditions. Another possibility is that activating effects of cMyBP-C N’-terminal domains may be less apparent under isometric conditions than during isotonic or auxotonic shortening. For example, activating effects of N’-terminal domains may be most relevant during the shortening phase of systole to offset shortening induced deactivation of the thin filament4 as seen in cMyBP-C in knockout mice43.
Effects of cMyBP-C N’-terminal domains to accelerate cross bridge cycling (ktr) at submaximal [Ca2+] following TEVp treatment (Fig. 4) are also in good agreement with studies in knockout mice41,44,45. However, ktr force traces observed here after removal of gC0C7 showed force “overshoots” where force transiently exceeded steady state force measured prior to the maneuver especially at intermediate pCa values closest to the pCa50 for force development (Fig. 4). Ligation of rC0C7-sc returned ktr to control values and eliminated the overshoots, but phosphorylated p-rC0C7-sc was less effective and reduced, but did not abolish overshoots. Phosphorylation dependence of overshoots may thus be similar to PKA-induced force overshoots seen in slow skeletal muscle46 where overshoots were attributed to transient increases in filament compliance leading to compliant re-alignment of actin binding sites to allow increased numbers of myosin heads to temporarily bind to the thin filament47,48. Because binding of N’-terminal cMyBP-C domains to actin can reduce thin filament torsional flexibility49,50 similar mechanisms may apply here.
Loss of gC0C7 using the cut and paste approach further revealed a novel role for cMyBP-C to damp oscillatory contractile waves. Spontaneous oscillatory contractions (so-called “SPOC”) have been described in skinned skeletal and cardiac muscles where SPOC is characterized by alternating cycles of slow sarcomere shortening followed by rapid relaxation and sarcomere re-lengthening30,51. SPOC thus differs fundamentally from sustained isometric contraction in the presence of constant activating [Ca2+] by the occurrence of repetitive cycles of sarcomere relaxation (e.g., sarcomere “give” compare Online Video I, left panel prior to TEVp treatment to middle panel after TEVp treatment). Our finding that loss of cMyBP-C induces SPOC is thus in good qualitative agreement with studies showing cMyBP-C extends the duration of systolic ejection and slows relaxation4,43,52,53. If so, loss of cMyBP-C may directly speed the inter-sarcomeric fast phase of relaxation54 by promoting sarcomere “give” causing multiple cycles of relaxation even during conditions of constant [Ca2+] (Fig. 6). cMyBP-C thus appears to play a critical role in coupling contraction and relaxation to the rise and fall of Ca2+ as previously suggested55, potentially by preventing premature relaxation during Ca2+ activation or the shortening phase of systole when [Ca2+] is falling43. Conversely, loss of cMyBP-C appears to uncouple contraction/relaxation by inducing premature sarcomere relaxation and giving rise to oscillatory SPOC behavior even during steady Ca2+ activation (Fig. 6). The overall effect may be analogous to stretch activation effects in asynchronous insect flight muscles which undergo multiple cycles of contraction and relaxation once a threshold level of activating [Ca2+] is achieved56,57. If so, then increased thin filament torsional flexibility (as suggested above to account for force overshoots) provides a testable mechanism by which cross-bridge energy could be elastically stored and released across multiple sarcomeres58,59.
Figure 6. cMyBP-C normally inhibits sarcomere relaxation leading to coupled contraction, whereas loss of cMyBP-C uncouples contraction and leads to SPOC.

Left, cMyBP-C normally prevents premature sarcomere relaxation, causing contraction and relaxation to be coupled to an increase or decrease in [Ca2+], respectively. Right, loss of cMyBP-C and/or phosphorylation of cMyBP-C accelerates sarcomere relaxation, leading to oscillatory cycles of contraction and relaxation even when [Ca2+] is constant. Figure created with BioRender.com.
Because SPOC was completely damped by ligation of unphosphorylated rC0C7-sc, but not by phosphorylated p-rC0C7-sc (compare Online Videos I and II), results imply that SPOC (and/or the underlying processes that give rise to it) is regulated by cMyBP-C phosphorylation. Until now SPOC was not considered to be under any specific regulatory control. However, the current results suggest that SPOC may contribute to increased cardiac contractility during b-adrenergic signaling. Theoretically, advantages of increased SPOC under conditions of increased inotropic drive could include mechanical acceleration of intra- and inter- sarcomere relaxation kinetics resulting in increased energetic efficiency54,60. Inter-sarcomere propagation of SPOC across Z-disks of adjacent sarcomeres59 could also more broadly impact mechano-chemical signaling61,62 or provide mechanical-electrical feedback to influence SR Ca2+ release or action potential properties63,64. Conversely, dysregulation of SPOC could contribute to cardiac dysfunction and arrhythmogenesis in HCM patients with cMyBP-C haploinsufficiency65, under cardiac stress that results in proteolysis and loss of cMyBP-C N’-terminal domains32, or in heart failure where cMyBP-C phosphorylation is reduced5. The novel cut and paste approach described here should be useful in testing these new hypotheses of cMyBP-C function as well as in defining the role of SPOC in cardiac contractility during health and disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
-
Cardiac myosin binding protein-C (cMyBP-C) is a critical regulator of heart contraction, but the mechanisms by which cMyBP-C affects processes which could alter cardiomyopathies only partly understood.
Cardiac myosin binding protein-C (cMyBP-C) is critical for “fight-or-flight” responses, while mutations in cMyBP-C (MYBPC3) cause hypertrophic cardiomyopathy (HCM).
Transgenic mouse models established that “fight-or-flight” stimuli phosphorylate cMyBP-C, increase myosin cross-bridge kinetics, and speed diastolic relaxation.
Approaches that complement transgenic models are needed to understand the molecular mechanisms by which cMyBP-C regulates contraction.
What New Information Does This Article Contribute?
We designed a novel “cut and paste” method to remove and replace cMyBP-C in muscle sarcomeres in situ.
Loss of cMyBP-C caused spontaneous oscillatory contractions (“SPOC”) that were damped by replacement with unphosphorylated cMyBP-C, but phosphorylated cMyBP-C was less effective.
Results suggest a new role for cMyBP-C phosphorylation in regulating mechanical oscillations with implications for cross-bridge kinetics, inter-sarcomere dynamics, and mechano-electrical signaling.
A major challenge in understanding how cMyBP-C regulates contraction is that the spatial distribution of cMyBP-C in sarcomeres is limited and cannot be duplicated in most cell-free systems while transgenic models that preserve cMyBP-C localization are costly and time consuming. Here we designed a novel system to remove and replace (“cut and paste”) cMyBP-C at its native position in permeabilized myocytes from “Spy-C” mice. The method requires only minutes to test functional effects of cMyBP-C carrying any desired mutation or modification. Loss of cMyBP-C using the new method induced spontaneous oscillatory contractions (SPOC) that were damped by cMyBP-C replacement. Results reveal a new role for cMyBP-C in regulating mechanical force oscillations that have the potential to impact cross-bridge cycling kinetics, inter-sarcomere kdynamics, and mechano-electrical feedbac
ACKNOWLEDGMENTS
The authors thank Drs. Thomas Doetschman and Teodora Georgieva for creating the Spy-C mice founder lines at the UArizona GEMM core and Dr. Mark Howarth (Oxford University) for generous assistance with SpyCatcher/SpyTag system. We thank Dr. Sabine J. van Dijk for assistance in establishing the Spy-C mouse colony.
SOURCES OF FUNDING
This work was supported by American Heart Association 17IRG33411051 and National Institutes of Health HL080367 and HL140925. A research award from the Precision Mouse Modeling Core at UArizona College of Medicine provided support for creation of the Spy-C mice.
Nonstandard Abbreviations and Acronyms:
- gC0C7
endogenous (genetically encoded) N’-terminal domains C0 to C7 of cardiac myosin binding protein-C
- rC0C7
exogenous (recombinant) N’-terminal domains C0 to C7 of cardiac myosin binding protein-C
- (p)-rC0C7
phosphorylated recombinant N’-terminal domains C0 to C7 of cardiac myosin binding protein-C
- sc
SpyCatcher
- st
SpyTag
- TEVp
tobacco etch virus protease
- SPOC
spontaneous oscillatory contractions
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Moss RL, Fitzsimons DP, Ralphe CJ. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ Res. 2015;116:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gresham KS, Stelzer JE. The contributions of cardiac myosin binding protein C and troponin I phosphorylation to β-adrenergic enhancement of in vivo cardiac function. J Physiology. 2016;594:669–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanft LM, Cornell TD, McDonald CA, Rovetto MJ, Emter CA, McDonald KS. Molecule specific effects of PKA-mediated phosphorylation on rat isolated heart and cardiac myofibrillar function. Arch Biochem Biophys. 2016;601:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dijk SJ, Kooiker KB, Napierski NC, Touma KD, Mazzalupo S, Harris SP. Point mutations in the tri-helix bundle of the M-domain of cardiac myosin binding protein-C influence systolic duration and delay cardiac relaxation. J Mol Cell Cardiol. 2018;119:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuster DW, Bawazeer A, Zaremba R, Goebel M, Boontje NM, van der Velden J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J Muscle Res Cell M. 2012;33:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J Mol Cell Cardiol. 2013;60:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glazier AA, Thompson A, Day SM. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflügers Archiv - European Journal of Physiology. 2019;471:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bardswell SC, Cuello F, Kentish JC, Avkiran M. cMyBP-C as a promiscuous substrate: phosphorylation by non-PKA kinases and its potential significance. J Muscle Res Cell M. 2012;33:53–60. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert R, Cohen J, Pardo S, Basu A, Fischman D. Identification of the A-band localization domain of myosin binding proteins C and H (MyBP-C, MyBP-H) in skeletal muscle. Journal of cell science. 1999;112 ( Pt 1):69–79. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert R, Kelly M, Mikawa T, Fischman D. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. Journal of cell science. 1996;109 ( Pt 1):101–111. [DOI] [PubMed] [Google Scholar]
- 13.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? The EMBO journal. 2004;14:1952–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. 2009;284:12318–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korte SF, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circulation Res. 2003;93:752–758. [DOI] [PubMed] [Google Scholar]
- 16.Kensler RW, Craig R, Moss RL. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc National Acad Sci. 2017;114:E1355–E1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risi C, Belknap B, Forgacs-Lonart E, Harris SP, Schröder GF, White HD, Galkin VE. N-Terminal Domains of Cardiac Myosin Binding Protein C Cooperatively Activate the Thin Filament. Structure (London, England : 1993). 2018;26:1604–1611.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mun J, Previs MJ, Yu HY, Gulick J, Tobacman LS, Previs S, Robbins J, Warshaw DM, Craig R. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc National Acad Sci. 2014;111:2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris SP, Belknap B, Sciver RE, White HD, Galkin VE. C0 and C1 N-terminal Ig domains of myosin binding protein C exert different effects on thin filament activation. P Natl Acad Sci Usa. 2016;113:1558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luther PK, Bennett PM, Knupp C, Craig R, Padron R, Harris SP, Patel J, Moss RL. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J Mol Biol. 2008;384:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett P, Craig R, Starr R, Offer G. The ultrastructural location of C-protein, X-protein and H-protein in rabbit muscle. Journal of muscle research and cell motility. 1986;7:550–567. [DOI] [PubMed] [Google Scholar]
- 22.Tonino P, Kiss B, Gohlke J, Smith JE, Granzier H. Fine mapping titin’s C-zone: Matching cardiac myosin-binding protein C stripes with titin’s super-repeats. J Mol Cell Cardiol. 2019;133:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc National Acad Sci. 2012;109:E690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee K, Harris SP, Sadayappan S, Craig R. Orientation of myosin binding protein C in the cardiac muscle sarcomere determined by domain-specific immuno-EM. J Mol Biol. 2015;427:274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flashman E, Watkins H, Redwood C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem J. 2007;401:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauli A, Althoff F, Oliveira RA, Heidmann S, Schuldiner O, Lehner CF, Dickson BJ, Nasmyth K. Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Developmental cell. 2008;14:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cesaratto F, Burrone OR, Petris G. Tobacco Etch Virus protease: A shortcut across biotechnologies. Journal of biotechnology. 2016;231:239–249. [DOI] [PubMed] [Google Scholar]
- 28.Fabiato A, Fabiato F. Myofilament-generated tension oscillations during partial calcium activation and activation dependence of the sarcomere length-tension relation of skinned cardiac cells. The Journal of general physiology. 1978;72:667–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagemoto T, Li A, Remedios C, Ishiwata S. Spontaneous oscillatory contraction (SPOC) in cardiomyocytes. Biophysical Rev. 2015;7:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishiwata S, Shimamoto Y, Fukuda N. Contractile system of muscle as an auto-oscillator. Prog Biophysics Mol Biology. 2011;105:187–198. [DOI] [PubMed] [Google Scholar]
- 31.Wolfe J, Ishiwata S, Braet F, Whan R, Su Y, Lal S, Remedios CG. SPontaneous Oscillatory Contraction (SPOC): auto-oscillations observed in striated muscle at partial activation. Biophysical Rev. 2011;3:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witayavanitkul N, Mou Y, Kuster DW, Khairallah RJ, Sarkey J, Govindan S, Chen X, Ge Y, Rajan S, Wieczorek DF, Irving T, Westfall MV, de Tombe PP, Sadayappan S. Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J Biol Chem. 2014;289:8818–8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Govindan S, McElligott A, Muthusamy S, Nair N, Barefield D, Martin JL, Gongora E, Greis KD, Luther PK, Winegrad S, Henderson KK, Sadayappan S. Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J Mol Cell Cardiol. 2012;52:154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razumova MV, Bezold KL, Tu A-Y, Regnier M, Harris SP. Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J Gen Physiology. 2008;132:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razumova MV, Shaffer JF, Tu A-Y, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846–35854. [DOI] [PubMed] [Google Scholar]
- 36.Belknap B, Harris SP, White HD. Modulation of Thin Filament Activation of Myosin ATP Hydrolysis by N-Terminal Domains of Cardiac Myosin Binding Protein-C. Biochemistry-us. 2014;53:6717–6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herron TJ, Korte FS, McDonald KS. Power Output Is Increased After Phosphorylation of Myofibrillar Proteins in Rat Skinned Cardiac Myocytes. Circ Res. 2001;89:1184–1190. [DOI] [PubMed] [Google Scholar]
- 38.Hanft LM, McDonald KS. Sarcomere length dependence of power output is increased after PKA treatment in rat cardiac myocytes. American journal of physiology Heart and circulatory physiology. 2009;296:H1524–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kögler H, Rüegg J. Cardiac contractility: modulation of myofibrillar calcium sensitivity by beta-adrenergic stimulation. Israel journal of medical sciences. 1997;33:1–7. [PubMed] [Google Scholar]
- 40.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. [DOI] [PubMed] [Google Scholar]
- 41.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of Myosin-Binding Protein-C Accelerates Force Development in Mouse Myocardium. Biophys J. 2006;90:4119–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraysse B, Weinberger F, Bardswell SC, Cuello F, Vignier N, Geertz B, Starbatty J, Krämer E, Coirault C, Eschenhagen T, Kentish JC, Avkiran M, Carrier L. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol. 2012;52:1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer BM, Georgakopoulos D, Janssen PM, Wang Y, Alpert NR, Belardi DF, Harris SP, Moss RL, Burgon PG, Seidman CE, Seidman J, Maughan DW, Kass DA. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circulation research. 2004;94:1249–1255. [DOI] [PubMed] [Google Scholar]
- 44.Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol-heart C. 2005;289:H801–H812. [DOI] [PubMed] [Google Scholar]
- 45.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006;99:884–890. [DOI] [PubMed] [Google Scholar]
- 46.Robinett JC, Hanft LM, Geist J, Kontrogianni-Konstantopoulos A, McDonald KS. Regulation of myofilament force and loaded shortening by skeletal myosin binding protein C. The Journal of general physiology. 2019;151:645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell KS. Tension Recovery in Permeabilized Rat Soleus Muscle Fibers after Rapid Shortening and Restretch. Biophys J. 2006;90:1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell KS. Filament Compliance Effects Can Explain Tension Overshoots during Force Development. Biophys J. 2006;91:4102–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunch TA, Kanassatega R-S, Lepak VC, Colson BA. Human cardiac myosin-binding protein C restricts actin structural dynamics in a cooperative and phosphorylation-sensitive manner. Journal of Biological Chemistry. 2019;294:16228–16240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colson BA, Rybakova IN, Prochniewicz E, Moss RL, Thomas DD. Cardiac myosin binding protein-C restricts intrafilament torsional dynamics of actin in a phosphorylation-dependent manner. Proc National Acad Sci. 2012;109:20437–20442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato K, Ohtaki M, Shimamoto Y, Ishiwata S. A theory on auto-oscillation and contraction in striated muscle. Prog Biophysics Mol Biology. 2011;105:199–207. [DOI] [PubMed] [Google Scholar]
- 52.Tong CW, Nair NA, Doersch K, Liu Y, Rosas PC. Cardiac myosin-binding protein-C is a critical mediator of diastolic function. Pflügers Archiv - European J Physiology. 2014;466:451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, Mukhopadhyay D, Kumar R, Baker KM, Mitchell BM, Powers PA, Fitzsimons DP, Patel BG, Warren CM, Solaro JR, Moss RL, Tong CW. Phosphorylation of cardiac Myosin-binding protein-C is a critical mediator of diastolic function. Circulation Hear Fail. 2015;8:582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stehle R, Krüger M, Pfitzer G. Force kinetics and individual sarcomere dynamics in cardiac myofibrils after rapid ca(2+) changes. Biophys J. 2002;83:2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janssen PM. Kinetics of cardiac muscle contraction and relaxation are linked and determined by properties of the cardiac sarcomere. Am J Physiol-heart C. 2010;299:H1092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bullard B, Pastore A. Through thick and thin: dual regulation of insect flight muscle and cardiac muscle compared. Journal of muscle research and cell motility. 2019;40:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwamoto H. Structure, function and evolution of insect flight muscle. Biophysics (Nagoya-shi, Japan). 2011;7:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yasuda K, Shindo Y, Ishiwata S. Synchronous behavior of spontaneous oscillations of sarcomeres in skeletal myofibrils under isotonic conditions. Biophys J. 1996;70:1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linke W, Bartoo M, Pollack G. Spontaneous sarcomeric oscillations at intermediate activation levels in single isolated cardiac myofibrils. Circulation research. 1993;73:724–734. [DOI] [PubMed] [Google Scholar]
- 60.Vitale G, Ferrantini C, Piroddi N, Scellini B, Pioner J, Colombini B, Tesi C, Poggesi C. The relation between sarcomere energetics and the rate of isometric tension relaxation in healthy and diseased cardiac muscle. Journal of muscle research and cell motility. 2019; 10.1007/s10974-019-09566-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pyle GW, Solaro JR. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circulation research. 2004;94:296–305. [DOI] [PubMed] [Google Scholar]
- 62.Samarel A, Koshman Y, Swanson ER, Russell B. Biophysical Forces Modulate the Costamere and Z-Disc for Sarcomere Remodeling in Heart Failure. Biophysics of the Failing Heart Physics and Biology of Heart Muscle. 2013;141–174. [Google Scholar]
- 63.ter Keurs HE, Wakayama Y, Miura M, Shinozaki T, Stuyvers BD, Boyden PA, Landesberg A. Arrhythmogenic Ca(2+) release from cardiac myofilaments. Prog Biophysics Mol Biology. 2006;90:151–171. [DOI] [PubMed] [Google Scholar]
- 64.ter Keurs HE, Wakayama Y, Sugai Y, Price G, Kagaya Y, Boyden PA, Miura M, uyvers BD. Role of sarcomere mechanics and Ca2+ overload in Ca2+ waves and arrhythmias in rat cardiac muscle. Ann Ny Acad Sci. 2006;1080:248–267. [DOI] [PubMed] [Google Scholar]
- 65.Kraft T, Montag J, Radocaj A, Brenner B. Hypertrophic Cardiomyopathy: Cell-to-Cell Imbalance in Gene Expression and Contraction Force as Trigger for Disease Phenotype Development. Circ Res. 2016;119:992–995. [DOI] [PubMed] [Google Scholar]
- 66.Kapust R, Tözsér J, Fox J, Anderson D, Cherry S, Copeland T, Waugh D. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein engineering. 2001;14:993–1000. [DOI] [PubMed] [Google Scholar]
- 67.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bezold KL, Shaffer JF, Khosa JK, Hoye ER, Harris SP. A Gain of Function Mutation in the M-domain of Cardiac Myosin Binding Protein-C Increases Binding to Actin. J Biol Chem. 2013;288: 21496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown D. Tracker Video Analysis and Modeling Tool. https://physlets.org/tracker/.
- 70.Thielicke W, Stamhuis E. PIVlab - Time-Resolved Digital Particle Image Velocimetry Tool for MATLAB (version: 2.02). Journal of Open Research Software. 2014; 10.6084/M9.FIGSHARE.1092508.V5. [DOI] [Google Scholar]
- 71.Thielicke W, Stamhuis E. Towards User-friendly, Affordable and Accurate Digital Particle Image Velocimetry in MATLAB. Journal of Open Research Software, 2(1), p.e30 DOI: 10.5334/jors.bl [DOI] [Google Scholar]
- 72.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak M-Y, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, us D, MacCoss M, Tabb DL, Mallick P. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 2012;30:918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parker SS, Krantz J, Kwak E-A, Barker NK, Deer CG, Lee NY, Mouneimne G, Langlais PR. Insulin Induces Microtubule Stabilization and Regulates the Microtubule Plus-end Tracking Protein Network in Adipocytes. Molecular & cellular proteomics : MCP. 2019;18:1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.