

Abstract
Objective
To assess whether IV immunoglobulins (IVIgs) as a first-line treatment for chronic inflammatory demyelinating polyneuropathy (CIDP) have a regulative effect on natural killer (NK) cells that is related to clinical responsiveness to IVIg.
Methods
In a prospective longitudinal study, we collected blood samples of 29 patients with CIDP before and after initiation of IVIg treatment for up to 6 months. We used semiquantitative PCR and flow cytometry in the peripheral blood to analyze the effects of IVIg on the NK cells. The results were correlated with clinical aspects encompassing responsiveness.
Results
We found a reduction in the expression of several typical NK cell genes 1 day after IVIg administration. Flow cytometry furthermore revealed a reduced cytotoxic CD56dim NK cell population, whereas regulatory CD56bright NK cells remained mostly unaffected or were even increased after IVIg treatment. Surprisingly, the observed effects on NK cells almost exclusively occurred in IVIg-responsive patients with CIDP.
Conclusions
The correlation between the altered NK cell population and treatment efficiency suggests a crucial role for NK cells in the still speculative mode of action of IVIg treatment. Analyzing NK cell subsets after 24 hours of treatment initiation appeared as a predictive marker for IVIg responsiveness. Further studies are warranted investigating the potential of NK cell status as a routine parameter in patients with CIDP before IVIg therapy.
Classification of evidence
This study provides Class I evidence that NK cell markers predict clinical response to IVIg in patients with CIDP.
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired chronic autoimmune disorder of the peripheral nervous system.1,2 A broad spectrum of subtypes has been described, and its heterogeneity poses challenges to diagnostics, treatment, and pathogenic concepts.3,4 Although etiopathogenesis and autoimmune targets have not been fully elucidated, there is strong evidence for the involvement of cellular and humoral immune responses.5–7
Immunomodulatory therapies can improve clinical signs, and up to 80% of patients respond to 1 of the 3 first-line treatments: corticosteroids, plasma exchange, and IV immunoglobulins (IVIgs).8 Although IVIg is therapeutically efficacious in close to 70% of patients, it takes 2–6 months before nonresponders can be identified.9 Although key opinion leaders in the field tend to switch to the next treatment option even within the first 3 months of ineffective treatment, there is an underestimated time window in IVIg nonresponding patients without effective therapy. Given the heterogeneity of therapeutic response, patients would greatly benefit from the availability of prognostic markers and surrogate markers, which predict treatment response.10,11
Reduced numbers of natural killer (NK) cells or a diminution in cytotoxic NK cell activity has been reported in various autoimmune conditions such as MS, systemic lupus erythematosus, rheumatoid arthritis, or type I diabetes.12,13 However, the pathophysiologic contribution of NK cells in the context of CIDP has not been addressed in detail.
To further understand the mode of action of IVIg in patients with CIDP and to decipher the alterations, which are responsible for its therapeutic effect, we took a prospective longitudinal approach to collect blood samples of patients with CIDP before and after treatment initiation with IVIg and investigated immune cell populations in detail.
Methods
Standard protocol approvals, registrations, and patient consents
The study was performed in accordance with the principles of the Declaration of Helsinki, and the local ethics committees approved the study plan (Ethics Committee University of Essen and Ethics Committee University of Düsseldorf). Participants who provided written informed consent were included. All participants were older than 18 years. A total of 29 patients with CIDP were investigated.
Diagnosis and classification of patients with CIDP
A total of 29 patients with CIDP (age range 34–78 years, mean age 55 years) consented to be enrolled, and peripheral blood samples were obtained before treatment initiation and 1, 2, 3, and 6 months after the first infusion of IVIg. CIDP was diagnosed according to the European Federation of Neurological Societies/Peripheral Nerve Society criteria.14 All patients had not received treatments other than methylprednisolone or plasma exchange before sampling and had been without immunomodulatory or immunosuppressive treatment for at least 4 weeks before sampling. Patients were observed in monthly intervals for up to 2 years. Samples of patients were excluded when they did not meet quality standards (cell viability, messenger RNA [mRNA] quality, and false-negative or false-positive controls during measurements of the samples).
Summed Inflammatory Neuropathy Cause and Treatment (INCAT) disability scores were assessed as previously described at each visit.15 Briefly, the INCAT disability score assesses functional disability on an ordinal scale ranging from 0 (no disability) to 5 (no purposeful movement possible) for upper and lower extremities. The summed INCAT score is added from both values and can thus range from 0 to 10. Patients were classified as responders if the INCAT sum score declined at least 1 point during 6 months of treatment. Furthermore, they were classified as responders if they stabilized under IVIg treatment with a progression of at least 2 points of the INCAT sum score over a period of 6 months before IVIg initiation.
Peripheral blood mononuclear cell preparation
Ethylenediaminetetraacetic acid (EDTA) tubes were used to collect peripheral blood mononuclear cells (PBMCs) and PAXgene tubes to collect RNA. Fresh blood samples in EDTA tubes were purified using Ficoll (Invitrogen, Mannheim, Germany) gradient centrifugation according to the manufacturer's protocol on the day of collection. Cells were divided into aliquots; one was freshly used for flow cytometry analyses, and the others were stored in 10% dimethyl sulfoxide/fetal calf serum in liquid nitrogen until further use.
Real-time PCR
Total RNA was isolated using the PAXgene preparation kit according to the manufacturer's instructions and quantified (Nanodrop 2000; Thermo Scientific, Waltham, MA). Synthesis of complementary DNA (cDNA) was performed using 1 μg of total RNA with Moloney murine leukemia virus reverse transcriptase, RNAse OUT, deoxynucleoside triphosphates, and oligo dT primer (all Invitrogen).
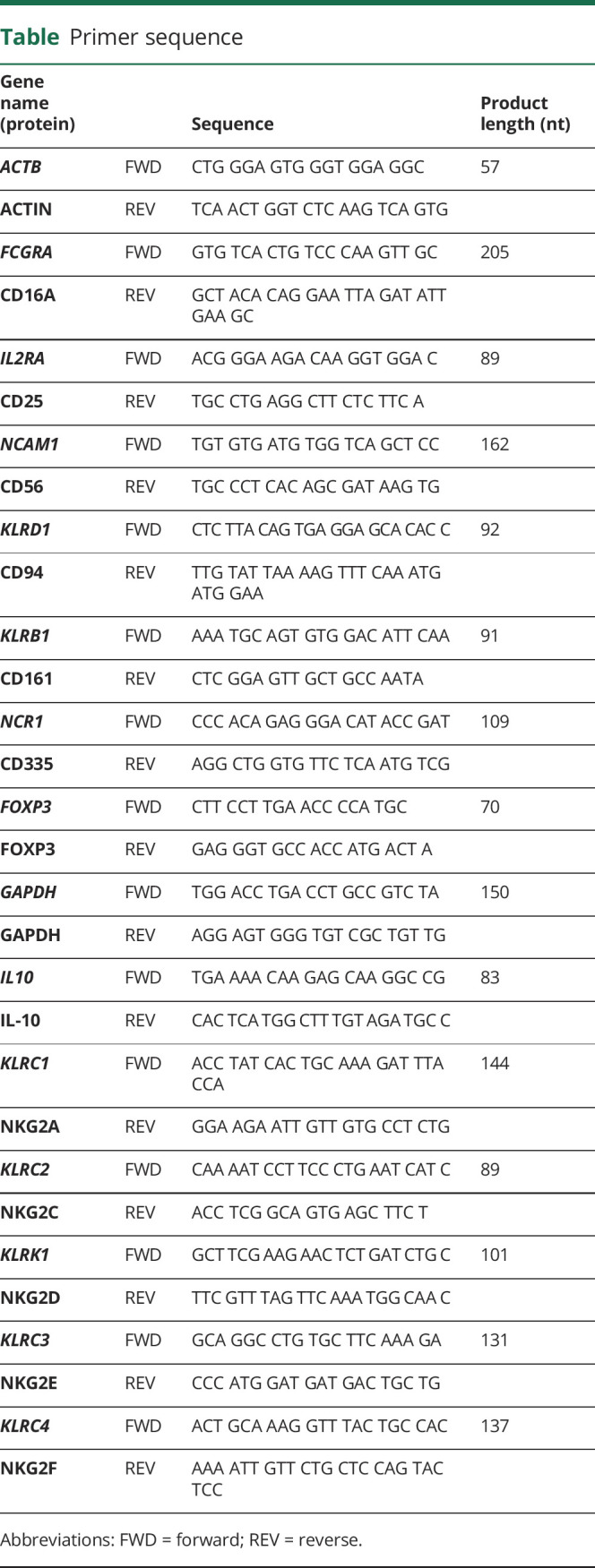
For real-time PCR, the thermal cycler (AbiPrism7000; Applied Biosystems, Foster City, CA) was set to run for 2 minutes at 50°C and 10 minutes at 95°C; 40 cycles for 15 seconds at 95°C; and 1 minute at 60°C using Power SYBR Green PCR Master Mix (Applied Biosystems). cDNA was amplified in the presence of a final concentration of 0.625 μM for each primer (sequences in the table). To affirm the specificity of the product, PCR was followed by a melting curve analysis. The housekeeping gene GAPDH was used as the control gene to relate mRNA content.
Table.
Primer sequence
Flow cytometry
Cells were surface stained using antibodies according to standard procedures.16 For extracellular staining, 1 × 106 cells were stained, and fluorescence of cells was analyzed using the FACSCanto II flow cytometer with BD FACSDiva 8.0 software (BD Biosciences, Heidelberg, Germany). Distinct cell populations were analyzed using FlowJo 10.0 (LCC, Eugene, OR). For intracellular stainings of cytokines, the BD Cytofix/Cytoperm Plus kit (BD Biosciences) was used after extracellular staining according to the manufacturer's protocols. Intracellular staining was performed using FoxP3 Fixation/Permeabilization (Thermo Fisher).
The following fluorescently labeled antibodies were used: CD4 (RPA-T4), CD8 (RPA-T8), CD16 (3G8), CD19 (SJ25C1), and CD56 (NCAM16.2; all BD Biosciences); CD3 (Okt 03), CD25 (M-A251), CD107a (H4-A3), CD161 (HP 3G10), CD335(9E2), FOXP3 (150D), interferon gamma (IFNγ) (4S.B3), and tumor necrosis factor alpha (TNFα) (MAb11; all BioLegend, San Diego, CA); and NKG2A (REA110), NKG2C (REA205), and NKG2D (BAT221; all Miltenyi, Bergisch Gladbach, Germany).
Cytotoxicity assay
2 × 105 PBMCs were preincubated for 16 hours before the K562 cell line was cocultivated for an hour to stimulate NK cell activity in the presence of CD107a antibody (H4-A3; BioLegend). Cultivation was prolonged for 4 hours in the presence of GolgiStop and GolgiPlug (BD Biosciences). After cultivation, cells were harvested and flow cytometry analyzed as described before. Shortly, cell populations were determined as well as CD107 degranulation and expression of the intracellular cytokines IFNγ and TNFα.
Statistics
The primary research questions of this prospective study were whether IVIg as a first-line treatment for CIDP have a regulatory effect on NK cells and their markers and whether these changes are related to clinical responsiveness. The classification of evidence assigned to these questions is Class I. The Shapiro-Wilk test was applied to assess normal distribution. The Mann-Whitney U test was performed for unrelated (unpaired) samples, the Wilcoxon rank test was performed for related (paired) samples, the Kruskal-Wallis analysis of variance (ANOVA) was performed for multiple testing, and the Dunn-Bonferroni post hoc test was used to test for statistically significant differences. Correlation analysis was performed by calculation of Spearman ρ. Differences were considered significant at p values <0.05 with a CI of 95%. GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA) and SPSS version 25 (IBM Corp. 2017, Armonk, NY) were used for statistical analysis.
Data availability
All data generated or analyzed during this study are included in this published article.
Results
Treatment of patients with CIDP with IVIg reduces NK cell frequency, but not the frequency of T cells and B cells
Administration of IVIg is known to have strong effects on various subpopulations of leukocytes in human blood. To investigate distinct changes, flow cytometry of peripheral blood cells was performed before and 24 hours after IVIg administration.
Lymphocytes were identified based on size and granularity (figure 1A). Gated on this population staining with CD3 vs CD56 allowed the discrimination of NKT cells (CD3+/CD56+) vs NK cells (CD3−/CD56+). NK cells were further subclassified using CD16 and CD56. NK cells with regulatory functions express CD16−/CD56bright, whereas predominantly cytotoxic NK cells are CD16+, but expression of CD56 is only intermediate (CD16+/CD56dim). The frequency of total NK cells (CD3−/CD56+) was reduced after IVIg infusions in almost all patients (figure 1B; p < 0.001). Subdivision of NK cells into CD16−/CD56bright and CD16+/CD56dim NK cells demonstrated that significant reduction is mainly due to reduction within the CD16+/CD56dim populations (figure 1C), whereas the smaller CD16−/CD56bright population in most patients increased 24 hours after IVIg treatment (figure 1D; p < 0.001).
Figure 1. Treatment with IVIg reduces NK cell frequency, but not the frequency of T cells and B cells.

Flow cytometry analysis of PBMCs from patients with CIDP (n = 14) before treatment initiation and 24 hours after IVIg treatment. (A) Representative gating strategy to define NK cells. Lymphocytes were identified based on the size and granularity of cells. To exclude NKT cells, CD56 and CD3 costaining was performed. Gated on CD56+/CD3− cells, subpopulations of CD16+/CD56dim and CD16−/CD56bright NK cells were determined. (B) Changes in NK cells gated on lymphocytes in individual IVIg-naive patients and 24 hours after IVIg infusion. (C) Proportion of CD56bright and CD56dim NK cell subpopulations within lymphocytes. Depicted is mean ± SEM of the analyzed patients. (D) Changes in CD56bright subpopulation gated on NK cells in individual IVIg-naive patients and 24 hours after IVIg infusion. (E) Changes in frequencies of T cells and B cells gated on lymphocytes in individual IVIg-naive patients and 24 hours after IVIg infusion. (F) Depicted are changes in the ratio of lymphocytes and monocytes within the leukocyte population in individual IVIg-naive patients and 24 hours after IVIg infusion (asterisks indicate significance: ***p < 0.001; nonparametric distribution; Wilcoxon rank test for paired samples). CIDP = chronic inflammatory demyelinating neuropathy; IVIg = IV immunoglobulin; NK = natural killer; PBMC = peripheral blood mononuclear cell.
In contrast, IVIg treatment had no significant effect on the proportion of CD3+ T cells and CD19+ B cells (figure 1E), and lymphocyte/monocyte ratios remained stable as well (figure 1F), indicating no specific expansion/reduction of the investigated subpopulations of cells. In single patients, no significant changes were observed, but a slight trend toward increase or reduction was detectable.
INCAT correlates with NK cells and their markers
We next addressed the question as to whether disease severity correlates with the NK cell population size (figure 2). Patients were subgrouped according to the INCAT score (0–3) and the percentage of NK cells within the lymphocyte population (figure 2A), and the relative expression of NK cell–specific genes in peripheral blood (figure 2B) was graphed. Patients with low motor disabilities (INCAT = 0) had higher NK cell number in tendency compared with patients with a higher disability (INCAT = 3). Significant differences were obtained for mRNA expression. Relative mRNA expression of neural cell adhesion molecule 1 (NCAM1) (CD56) and KLRD1 (CD94) were significantly lower in patients with higher INCAT score (figure 2B; Spearman ρ −0.465, p = 0.017 for KLRD1 and −0.467, p = 0.014 for NCAM1). In line with the reduced NK cell numbers, patients with a higher disease severity also had lower NK cell–specific transcription levels detectable in the peripheral blood.
Figure 2. Correlation of INCAT with NK cells and their markers.
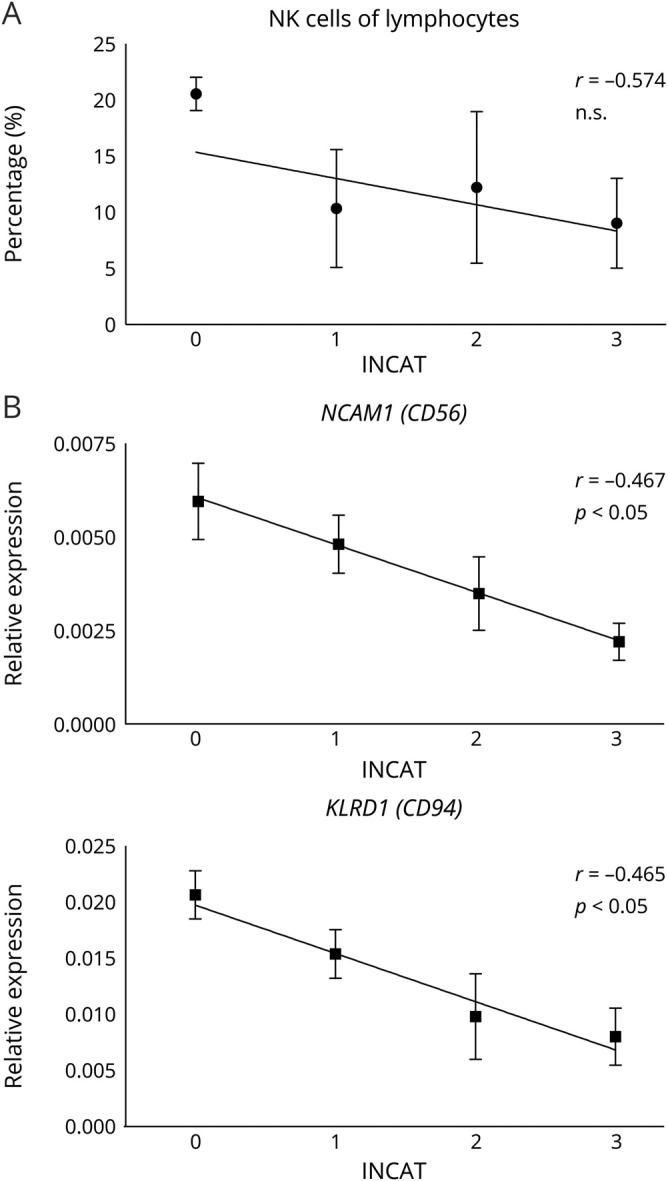
Correlation of the INCAT score with the percentage of NK cells within lymphocytes (A, n = 16) and the expression level of NK-specific genes CD56 and CD94 (B, n = 27). Patients were classified in terms of the INCAT disability score. Depicted is mean ± SD and correlation with Spearman correlation coefficient r, significance level p is as stated. INCAT = Inflammatory Neuropathy Cause and Treatment; NK = natural killer.
Treatment with IVIg reduces NK cells and NK-specific markers, but values recover between IVIg administrations
To validate the flow cytometry data and additionally investigate functional markers of NK cells, the relative mRNA expression of different NK cell genes was measured in patients with CIDP before and 24 hours after IVIg infusion (figure 3A). NK cell–specific markers NCAM1 (CD56), KLRD1 (CD94), KLRB1 (CD161), and NCR1 (CD335) were analyzed as well as functional NKG family receptors either with activating (KLRC2 [NKG2C] and KLRK1 [NKG2D]) or inhibiting (KLRC1 [NKG2A]) function or with unknown function (KLRC4 [NKG2F]).
Figure 3. Treatment with IVIg reduces NK cells and NK-specific markers in a reversible manner.

Analysis of changes in relative expression levels of the indicated typical NK cell–specific genes in PBMCs from patients with CIDP before treatment initiation and 24 hours after IVIg treatment. (A) Depicted are changes of relative expression of indicated genes in individual patients (n = 17) related to the house keeping gene GAPDH (2−ΔCt, asterisks indicate significance: *p < 0.05, **p < 0.01, ***p < 0.001; nonparametric distribution, Wilcoxon rank test for paired samples). Changes of the indicated genes. (B) Representative expression levels of the indicated genes of a representative patient over time before IVIg treatment initiation and 24 hours after monthly infusion of IVIg. CIDP = chronic inflammatory demyelinating neuropathy; IVIg = IV immunoglobulin; NK = natural killer; PBMC = peripheral blood mononuclear cell.
The relative expression of NCAM1 was significantly reduced 24 hours after the first IVIg infusion (p = 0.0038, reduction observed in 12 of 16 patients). The same reduction of relative expression of NK cell–specific markers was detected regarding KLRD1 (p = 0.0012) and KLRB1 (p = 0.0087) transcripts. Diminished relative expression of NCR1 was most prominent among NK cell–specific markers and was observed in 15 of 16 patients (p < 0.001). The relative reduction was also significant for most of the NKG family members (KLRC2, p = 0.0239 and KLRC4, p = 0.0348). The KLRC1 marker was decreased in tendency after 24 hours of IVIg infusion. In summary, transcripts of NK cell markers and receptors were diminished 24 hours after IVIg infusion.
In contrast to that, we did not find relevant changes in longitudinal analysis of samples prior treatment initiation compared with 1, 2, 3, and 6 months of treatment (data not shown), neither with flow cytometry nor transcriptional analysis. To further understand the observations of the 24-hour samples, we confirmed that changes are reversible within the monthly treatment intervals. Therefore, changes of different NK cell genes were analyzed over 3 months in 3 responding patients, and representative data of 1 patient are depicted in figure 3B. As expected, the relative expression of the analyzed genes was markedly reduced 24 hours after infusion. However, the levels returned to the initial value between the treatments and were again lowered after subsequent infusions. Over a period of 3 months, patients revealed a stable recurring pattern.
In treatment-naive patients, no difference in NK cell marker expression is detectable between responders and nonresponders
Patients with CIDP differ in their responsiveness to IVIg. Retrospectively, enrolled patients were subdivided into responders and nonresponders after 3–6 months of IVIg treatment. NK cell lineage and functional markers were compared between the 2 groups before IVIg treatment initiation (responders: dark gray bars; nonresponders: light gray bars, figure 4). Flow cytometry analysis of NK cells and relevant NK cell markers (figure 4A) revealed no significant differences between the 2 groups. Frequencies of NK cell lineage–specific markers were in a comparable range, and also, functional receptors did not differ in IVIg responders vs nonresponders. These results were confirmed on the mRNA level (figure 4B) and revealed no significant difference in NK cell markers. In summary, treatment-naive responders vs nonresponders had no distinct NK cell profile. NK cell function was tested in an ex vivo assay and in line with the above described results. Neither the cytotoxicity measured by a CD107 flow cytometry assay nor the cytokine production of TNFα and IFNγ after stimulation with K562 target cells was different between responders and nonresponders (figure e-1).
Figure 4. In treatment-naive patients, no difference in NK cell marker expression is detectable between responders and nonresponders.

Comparison of NK cell markers of PBMCs from patients with CIDP before IVIg treatment that were subsequently classified as responders (n = 11) or nonresponders (n = 6) depending on the treatment efficacy of IVIg. Depicted is mean ± SEM. (A) Changes of frequencies of NK cells, CD56bright and CD56dim NK cell subpopulations, and CD161+ and NKG2A+ populations in patients with CIDP. In addition, mean fluorescence intensity was analyzed of surface protein expression levels of CD161 and CD335. (B) Longitudinal changes in transcriptions levels of the indicated genes after real-time qPCR analysis was performed on mRNA preparations of PBMCs. CIDP = chronic inflammatory demyelinating neuropathy; IVIg = IV immunoglobulin; NK = natural killer; mRNA = messenger RNA; PBMC = peripheral blood mononuclear cell.
Reduction of NK cell marker expression 24 hours after IVIg treatment initiation is more pronounced in responding patients
We further analyzed whether the changes within the NK cell population differed in responders and nonresponders. Therefore, frequency changes of NK cell were compared in responders and nonresponders 24 hours after IVIg infusion (figure 5A). Compared with naive samples, we found a significant increase of the regulatory CD56bright population within the NK cells after IVIg infusion, whereas the frequency did not increase in nonresponders. Most interestingly, this difference is significant between responders and nonresponders (p = 0.0047). Within the lymphocytes, the regulatory CD56bright population even decreased in nonresponders. In general, the frequencies of NK cells were reduced in responders and nonresponders with higher reduction in responders. This decrease of NK cells within the lymphocyte population was also confirmed on the mRNA expression level (figure 5B). The majority of NK cell genes were significantly downregulated in responders, whereas in nonresponders, only a moderate downregulation could be detected or even an upregulation of some mRNA transcripts (KLRB1, KLRK1, and KLRC4) 24 hours after IVIg infusion. This resulted in a significant difference in responders and nonresponders (KLRB1: p = 0.027, KLRK1: p = 0.031, KLRC4: p = 0.024).
Figure 5. Reduction of NK cell marker expression after 24-hour IVIg treatment is more pronounced in responding patients.

(A) Changes of frequencies of relevant NK cell markers in PBMCs of patients with CIDP 24 hours after IVIg infusion related to baseline in responders (n = 11) and nonresponders (n = 6). Frequency of CD56bright NK cells was determined within the total NK cell population and within lymphocytes. Asterisks above bars indicate significant changes after IVIg treatment compared with control (*p < 0.05, **p < 0.01, ***p < 0.001; nonparametric distribution, Wilcoxon rank test for paired samples). Asterisks between bars indicate significant changes between the responder and nonresponder cohort. Depicted is mean ± SEM (*p < 0.05, **p < 0.01; nonparametric distribution, unpaired; Mann-Whitney U test). (B) Changes of the transcription level of relevant NK cell genes in PBMCs of patients with CIDP 24 hours after IVIg infusion related to baseline in responders (n = 11) and nonresponders (n = 6). Asterisks above bars indicate significant changes after IVIg treatment compared with control (*p < 0.05, **p < 0.01; nonparametric distribution, Wilcoxon rank test for paired samples). Asterisks between bars indicate significant changes between the responder and nonresponder cohort. Depicted is mean ± SEM (*p < 0.05, **p < 0.01; nonparametric distribution, unpaired; Mann-Whitney U test). (C) Changes in PBMCs of patients with CIDP after 24 hours of IVIg infusion in representative markers of NK cells on gene expression and surface expression in the individual patients related to baseline (responders n = 8, nonresponders n = 6). CIDP = chronic inflammatory demyelinating neuropathy; IVIg = IV immunoglobulin; NK = natural killer; PBMC = peripheral blood mononuclear cell.
To better discriminate responders from nonresponders, 2 candidate markers were depicted for representative single responding and nonresponding patients (figure 5C). Although most of the responding patients showed decreased KLRC2 mRNA expression after IVIg treatment, in nonresponders, the shift was not as pronounced, or the expression was even slightly increased. In most of the responding patients, CD56bright NK cells were strongly increased, whereas this phenomenon could not be observed in the nonresponder cohort.
To predict the responsiveness of IVIg, a combinational approach of more than 1 candidate marker could increase the significance. To test this hypothesis, CD56bright flow cytometry data where combined with KLRB1 and KLRK1 transcriptional data. For the adjustment of multiple testing, we performed 1-way Kruskal-Wallis ANOVA. In this model, responders were significantly discriminated from nonresponders (F = 6.974, p = 0.013). In pairwise comparisons with the Dunn-Bonferroni post hoc test, CD56bright showed the highest significance level (p = 0.007), followed by KLRK1 (p = 0.010) and KLRB1 (p = 0.027). Thus, the combination of these 3 candidate markers increases the level of significance for CD56bright and KLRK1, improving prediction for the responsiveness of IVIg.
Discussion
Despite generally high efficacy of IVIg, a significant proportion of patients with CIDP fail to respond to this treatment. In these instances, treatment failure is confirmed after 3–6 months of unaltered disease progression. Patients refractory to IVIg treatment would highly benefit from surrogates capable of predicting treatment efficacy earlier. Although a majority of patients respond well to treatment with IVIg (responders), around 30% of patients do not benefit at all (nonresponders).9 Here, we demonstrate changes within the lymphocyte populations in patients with CIDP, induced by IVIg treatment and—furthermore—a distinct change in the NK cell profile 24 hours after treatment depending on clinical responsiveness to IVIg treatment. The effect was predominantly observed within NK populations and for NK cell receptors. IVIg responders had a relative reduction of CD16+/CD56dim cells in the peripheral blood and a relative increase in CD16−/CD56bright cells. In nonresponders, these effects were less pronounced or absent. We did not check for IgG4 subclasses or antibodies to paranodal antigens. Because up to 10% of the nonresponders have antibodies against paranodal antigens,17 some of our patients may not relate to NK transcripts but rather to a different autoantibody status. To increase the prediction of IVIg responsiveness, we tested a combinatorial approach of 3 strong candidate markers. The combination of CD56bright flow cytometry data with the transcriptional data of KLRB1 and KLRK1 increased the level of significance, improving prediction for the responsiveness of IVIg. Nevertheless, establishing a test in the clinical routine could be an ambitious goal.
The role of NK cells was not investigated so far in context of autoimmune neuropathies. NK cells are innate immune cells and recognize virus-infected or pathologically altered cells. Target cells are either eliminated by release of cytolytic granules or secrete cytokines to regulate the immune response. NK cells are also involved in limiting immune response after inflammatory reactions.18 Several NK cell receptors activate or inhibit their activity (such as the NKG2 receptor family), and subpopulations can be identified via the expression level of the surface molecule CD56 (encoded by NCAM1). The CD56dim cell subpopulation has cytotoxic activity, whereas CD56bright cells fulfill regulatory functions partly via cytokine secretion.19
IVIgs achieve their anti-inflammatory effect through a broad range of target mechanisms.20 The therapeutic effect of IVIgs is in part mediated via the Fc gamma receptor, which is also expressed on NK cells. Another mode of action that has been proposed is the upregulation of target cell killing by antibody-dependent cytotoxicity (ADCC).20 There is also evidence for a reduction of cytotoxic NK cells immediately after IVIg treatment in patients with CIDP most likely due to lowering of the number of circulating NK cells.21 The possible link between IVIg efficacy and NK cells appears obvious but has not been further investigated in CIDP.
One possible mechanism of the IVIg effect on NK cells is the activation/induction of their cytotoxic phenotype. In our study, particularly CD56dim NK cells were reduced in peripheral blood, predominantly in responders. It is known that recruitment of CD56dim cells into inflamed tissue is mandatory for limiting immune reaction in experimental autoimmune encephalomyelitis (EAE).12,22 An increased fraction of CD56dim cytotoxic cells migrating into inflamed tissue could therefore lead to a local reduction of autoreactive T cells, eliminated via cytotoxic lysis by the NK cells.23 This mechanism is well known; thus, recruitment of CD56dim NK cells is a potent defense mechanism against inflammation in various tissues. It influences dendritic cell (DC) activation, regulates the adaptive immune system, and reduces local tissue destruction.24,25 Although mature DCs are protected by an upregulation of major histocompatibility complex class I molecules,26 NK cells are known to lyse immature DCs.27 IVIg enhances lysis of mature DCs via CD16+/CD56dim NK cells and ADCC.28 This may terminate autoimmune reactions due to limited T-cell activation via restricted DC antigen presentation.
We and others have shown6,29 that patients with CIDP display changes in their T-cell receptor repertoire, but these alterations do hardly explain the treatment effect of IVIg and do not answer the question why a distinct group of patients with CIDP do not respond to therapy. It is known from an animal model of MS that NK cells are crucial for the therapeutic effect of IVIg. NK cell depletion in EAE leads to a loss of IVIg efficacy.30 Furthermore, a recent study on the CSF of patients with inflammatory neuropathies revealed a distinctive increase in NK cell numbers—indicating an underestimated role of NK cells in inflammatory neuropathies.31
One limitation of the study is the comparable low INCAT score of 1.42. This can be explained by the fact that the study mainly included patients with a newly diagnosed CIDP. Still, an INCAT of 2 is a relevant functional limitation for patients, and INCAT mainly reflects motor symptoms. Because the INCAT score is a well-established scale, we choose this motor functional score to classify response and clinical course.
Our observations add evidence of an IVIg-induced migration of cytotoxic CD16+/CD56dim NK cells into the inflamed peripheral nerve where they might induce suppression of autoreactive immune cells and therefore limit autoimmunity. However, in nonresponders, concurrent homing of CD56bright NK cells may augment autoimmune reactions, counteracting this mechanism. In addition, NK cell–mediated lysis of activated macrophages32 or autoreactive T cells33 may further curtail autoimmune reactions following IVIg treatment. The observation of the monthly recovery of NK cell markers is of high interest, and further studies will correlate the NK cell course with the clinical end-of-dose effect in IVIg-treated patients.34 In addition, monitoring the NK transcripts during the treatment interval of 3–4 weeks will add knowledge to the role of NK cells in pathophysiology of CIDP.
Taken together, this comprehensive study provides evidence for a so far unknown relevant contribution of NK cells to the pathogenesis of CIDP. Furthermore, the differential response of subpopulations of NK cells in responders and nonresponders to IVIg treatment may serve as a surrogate marker in predicting the outcome of IVIg treatment in patients with CIDP after reproduction of the results in an independent cohort.
Acknowledgment
The authors thank Zippora Kohne for excellent technical assistance.
Glossary
- ADCC
antibody-dependent cytotoxicity
- ANOVA
analysis of variance
- BNB
blood-nerve barrier
- cDNA
complementary DNA
- CIDP
chronic inflammatory demyelinating neuropathy
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- EDTA
ethylenediaminetetraacetic acid
- IFNγ
interferon gamma
- INCAT
Inflammatory Neuropathy Cause and Treatment
- IVIg
IV immunoglobulin
- mRNA
messenger RNA
- NCAM1
neural cell adhesion molecule 1
- NK
natural killer
- PBMC
peripheral blood mononuclear cell
- TNFα
tumor necrosis factor alpha
Appendix. Authors
Study funding
G. Meyer Zu Horste received grant support from the Deutsche Forschungsgemeinschaft (DFG, ME4050/4-1, ME4050/8-1), from the Grant for Multiple Sclerosis Innovation (GMSI, Merck), from the Innovative Medical Research (IMF) program of the University Münster, and from the Ministerium für Innovation, Wissenschaft und Forschung (MIWF) of the state Nordrhein-Westfalen.
Disclosure
A.K. Mausberg and M.K. Heininger report no disclosures relevant to the manuscript. G. Meyer Zu Horste has received speaker honoraria and served in advisory boards for LFB Pharma. S. Cordes, M. Fleischer, F. Szepanowski, and C. Kleinschnitz report no disclosures relevant to the manuscript. H.-P. Hartung received honoraria for serving on steering committees from CSL Behring, LFB, and Octapharma, outside this work from Bayer HealthCare, Biogen, Celgene Receptos, GeNeuro, Merck, Novartis, Roche, and TG Therapeutics with approval by the Rector of Heinrich-Heine-University. B.C. Kieseier reports no disclosures relevant to the manuscript. M. Stettner served on the scientific advisory and/or received speaker honoraria or travel funding from UCB, Biogen Idec, Grifols, Genzyme, Roche, Merck, Novartis, Octapharma, Sanofi-Aventis, Teva, and Bayer HealthCare. Go to Neurology.org/NN for full disclosures.
References
- 1.Köller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med 2005;352:1343–1356. [DOI] [PubMed] [Google Scholar]
- 2.Vallat J-M, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol 2010;9:402–412. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat RevNeurol 2011;7:507–517. [DOI] [PubMed] [Google Scholar]
- 4.Bunschoten C, Jacobs BC, Van den Bergh PYK, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol 2019;18:784–794. [DOI] [PubMed] [Google Scholar]
- 5.Querol L, Siles AM, Alba-Rovira R, et al. Antibodies against peripheral nerve antigens in chronic inflammatory demyelinating polyradiculoneuropathy. Sci Rep 2017;7:14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mausberg AK, Dorok M, Stettner M, et al. Recovery of the T-cell repertoire in CIDP by IV immunoglobulins. Neurology 2013;80:296–303. [DOI] [PubMed] [Google Scholar]
- 7.Mathey EK, Park SB, Hughes RAC, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatr 2015;86:973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Léger J-M, Guimarães-Costa R, Muntean C. Immunotherapy in peripheral neuropathies. Neurotherapeutics 2015;13:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes RAC, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008;7:136–144. [DOI] [PubMed] [Google Scholar]
- 10.Brannagan TH. Current diagnosis of CIDP: the need for biomarkers. J Peripher Nerv Syst 2011;16:3–13. [DOI] [PubMed] [Google Scholar]
- 11.Kieseier BC, Mathey EK, Sommer C, Hartung HP. Immune-mediated neuropathies. Nat Rev Dis Primers 2018;4:31. [DOI] [PubMed] [Google Scholar]
- 12.Yoshii F, Shinohara Y. Natural killer cells in patients with Guillain-Barré syndrome. J Neurol Sci 1998;157:175–178. [DOI] [PubMed] [Google Scholar]
- 13.Fogel LA, Yokoyama WM, French AR. Natural killer cells in human autoimmune disorders. Arthritis Res Ther 2013;15:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van den Bergh PYK, Hadden RDM, Bouche P, et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. Eur J Neurol 2010;17:356–363. [DOI] [PubMed] [Google Scholar]
- 15.Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol 2001;50:195–201. [DOI] [PubMed] [Google Scholar]
- 16.Warnke C, Stettner M, Lehmensiek V, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler 2015;21:1036–1044. [DOI] [PubMed] [Google Scholar]
- 17.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014;82:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol 2019;19:282–290. [DOI] [PubMed] [Google Scholar]
- 19.Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 2009;126:458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol 2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 21.Bohn AB, Nederby L, Harbo T, et al. The effect of IgG levels on the number of natural killer cells and their Fc receptors in chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol 2011;18:919–924. [DOI] [PubMed] [Google Scholar]
- 22.Hertwig L, Hamann I, Suarez SR, et al. CX3CR1‐dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur J Immunol 2016;46:1984–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leavenworth JW, Wang X, Wenander CS, Spee P, Cantor H. Mobilization of natural killer cells inhibits development of collagen-induced arthritis. Proc Natl Acad Sci USA 2011;108:14584–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell JJ, Qin S, Unutmaz D, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol 2001;166:6477–6482. [DOI] [PubMed] [Google Scholar]
- 25.Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol 2002;2:957–965. [DOI] [PubMed] [Google Scholar]
- 26.Ferlazzo G, Semino C, Melioli G. HLA Class I molecule expression is up-regulated during maturation of dendritic cells, protecting them from natural killer cell-mediated lysis. Immunol Lett 2001;76:37–41. [DOI] [PubMed] [Google Scholar]
- 27.Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med 2002;195:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tha-In T, Metselaar HJ, Tilanus HW, et al. Intravenous immunoglobulins suppress T-cell priming by modulating the bidirectional interaction between dendritic cells and natural killer cells. Blood 2007;110:3253–3262. [DOI] [PubMed] [Google Scholar]
- 29.Schneider-Hohendorf T, Schwab N, Uceyler N, Gobel K, Sommer C, Wiendl H. CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2012;78:402–408. [DOI] [PubMed] [Google Scholar]
- 30.Chong WP, Ling MT, Liu Y, et al. Essential role of NK cells in IgG therapy for experimental autoimmune encephalomyelitis. PLoS One 2013;8:e60862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heming M, Schulte-Mecklenbeck A, Brix T, et al. Immune cell profiling of the cerebrospinal fluid provides pathogenetic insights into inflammatory neuropathies. Front Immunol 2019;10:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nedvetzki S, Sowinski S, Eagle RA, et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 2007;109:3776–3785. [DOI] [PubMed] [Google Scholar]
- 33.Crouse J, Xu HC, Lang PA, Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol 2015;36:49–58. [DOI] [PubMed] [Google Scholar]
- 34.Berger M, Allen JA. Optimizing IgG therapy in chronic autoimmune neuropathies: a hypothesis driven approach. Muscle Nerve 2015;51:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.