

Impairment in gut microbial detoxification is correlated with mitochondrial dysfunction and disease severity in autistic children.
Abstract
Growing evidence suggests that autism spectrum disorder (ASD) is strongly associated with dysbiosis in the gut microbiome, with the exact mechanisms still unclear. We have proposed a novel analytic strategy—quasi-paired cohort—and applied it to a metagenomic study of the ASD microbiome. By comparing paired samples of ASD and neurotypical subjects, we have identified significant deficiencies in ASD children in detoxifying enzymes and pathways, which show a strong correlation with biomarkers of mitochondrial dysfunction. Diagnostic models based on these detoxifying enzymes accurately distinguished ASD individuals from controls, and the dysfunction score inferred from the model increased with the clinical rating scores of ASD. In summary, our results suggest a previously undiscovered potential role of impaired intestinal microbial detoxification in toxin accumulation and mitochondrial dysfunction, a core component of ASD pathogenesis. These findings pave the way for designing future therapeutic strategies to restore microbial detoxification capabilities for patients with ASD.
INTRODUCTION
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder characterized by impaired social communication and repetitive and stereotyped behavior, interests, or activities (1). The prevalence of ASD has been increasing worldwide and was estimated by the Autism and Developmental Disabilities Monitoring Network in 2014 to affect approximately 1.6% of American 8-year old children (2). The etiology of ASD remains unknown and likely involves a wide range of environmental factors that affect many physiological processes in genetically susceptible individuals (3). Recently, lines of evidence have shown possible contributions of the intestinal microbiome to ASD pathogenesis, as in many other diseases (4). First, gastrointestinal comorbidity is common in ASD children (5). Second, patients with ASD are mostly picky eaters (6) and are often deficient in digestive enzymes (7), which unavoidably alters the nutrients or media of the microbes inhabiting their gut and influences the growth of component species, leading to dysbiosis. Last, many studies have found obvious dysbiosis in the gut microbiome of patients with ASD, such as a deficiency in Bifidobacterium longum and overgrowth of Clostridium spp. and Candida albicans (8, 9). Dysbiosis of the ASD microbiome is believed to be associated with inflammation of intestinal epithelia and increased permeability of the gut-blood barrier (10). However, how the intestinal microbiome affects ASD pathogenesis is not fully understood.
Metagenome research based on shotgun sequencing is widely applied to exploring microbe-host interactions and microbiome-associated pathogenesis in many diseases. Compared to targeted sequencing of 16S ribosomal DNA (rDNA), which only presents species profiles, shotgun sequencing is more informative, as it provides comprehensive information for the inference of concrete metabolic pathways and physiological functions of the microbiome (11). Although few studies of shotgun-based metagenome analysis have been reported for the ASD intestinal microbiome (12, 13), obstacles remain because of high interindividual diversity, which is influenced by a wide range of factors such as genetics, age, diet, and healthy conditions (14). The individual diversity is often so great and difficult to control that it even overwhelms disease-associated alterations and profoundly affects the identification of disease-associated microbiome features. Thus, the results of metagenomic studies are highly dependent on the samples collected and often include stochastic false positives or negatives (15, 16).
It is well known that microbiome composition varies widely, even among healthy people (17). Moreover, microbial components and their abundance are greatly influenced by complex metabolic interactions and are strictly constrained by the entire metabolic network in the microbiome (18, 19). In this way, the relative activity of a specific pathway can be compared between samples of similar metabolic backgrounds (16, 20). On the basis of this concept, we developed a novel strategy for metagenomic analysis, “quasi-paired cohort,” in which we paired ASD samples with control samples of similar metabolic backgrounds—that is, the profile of metabolic pathways inferred from the metagenomic data. This approach allowed us to transform the original group cohort into a paired cohort, which not only controls for individual diversity but also increases statistical power. We then performed shotgun-based metagenome sequencing of 79 fecal samples from ASD individuals and healthy controls; this analytic strategy enabled us to identify apparent deficiencies in deoxidation and toxicant-degradation pathways in ASD microbiome. As toxin exposure has been epidemiologically demonstrated as one of the main etiological factors of ASD (21), the impaired detoxification capability of the intestinal microbiome suggests a previously unknown mechanism to explain why patients with ASD are more vulnerable to toxicant exposure and how the intestinal microbiome contributes to the pathogenesis of ASD.
MATERIALS AND METHODS
Study design
ASD children and gender- and age-matched neurotypical controls were consecutively recruited from the Autism Research Center of Peking University Health Science Center and surrounding communities. Fecal and morning urine samples were collected from each participant for metagenome sequencing and urine metabolite measurements, respectively. Following the protocols of quasi-paired cohort (see below), a paired cohort was constructed where individuals from ASD and control groups were cross paired to identify ASD-associated microbiome features (see below and Fig. 1). Microbiome features, such as the abundance of metabolic pathways, were compared between samples in each pair, and those significantly overrepresented or deficient in ASD samples were suggested as ASD associated. Urine biomarkers and clinical rating scores were used to confirm the role of the identified ASD-associated microbiome features. Last, the identified features were used to construct a diagnostic model, and its performance was evaluated with receiver operating characteristic (ROC) analysis.
Fig. 1. Principle of quasi-paired cohort analysis.
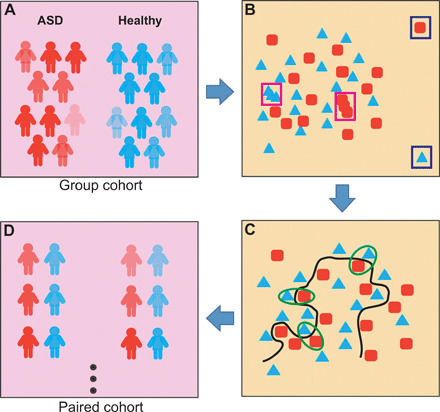
(A) Group cohort of ASD individuals and healthy control participants. (B) A high-dimensional space with each microbiome feature representing a dimension was constructed, and samples were positioned. Samples with abnormally far distances to others (outliners, labeled with blue rectangles) or that were too close (redundancies, labeled with pink rectangles) were removed. (C) Boundary samples near the decision boundary (the black line) were selected for both groups and paired with their nearest samples from the opposite group. (D) Quasi-paired cohort was constructed and used to test for the significance of the difference in each dimension (microbial feature) between case and control samples.
Participants
Children with ASD and typically developing matched control subjects from 3 to 8 years old were recruited. The diagnoses of ASD were confirmed with the Autism Diagnostic Interview–Revised (22) and Autism Diagnostic Observation Schedule (ADOS) (23) according to Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) criteria. Rating or scoring instruments of Childhood Autism Rating Scale (CARS) (24), Repetitive Behavior Scale–Revised (RBS-R) (25), Autism Behavior Checklist (ABC) (26), and Gastrointestinal Severity Index (27) were also performed for autistic children. For the control subjects, medical examinations and parent interviews were performed to exclude anyone with psychiatric disorders. Subjects were also excluded if they experienced an infection or took psychoactive medications, antibiotics, probiotics, prebiotics, or a special diet (such as ketogenic diet) 3 months before enrollment.
Metagenome sequencing and annotation
Fecal specimens of all participants were collected and immediately frozen at −20°C in sample tubes and stored at −80°C for subsequent metagenome sequencing. The total DNA of each fecal sample was extracted using the QIAamp PowerFecal Pro DNA Kit (QIAGEN), and paired-end sequencing was performed on the Illumina HiSeq X10 platform (150 base pairs × 2). Raw reads were first applied to quality control, where ambiguous sequences and adapters were filtered out using FastQC (version 0.11.5; www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality bases and reads were trimmed using the FASTX-Toolkit (version 0.0.13; http://hannonlab.cshl.edu/fastx_toolkit/). Next, reads of human origin that mapped to Hs37 human genomes by Burrows-Wheeler Aligner (mem module with default parameters; http://bio-bwa.sourceforge.net/) were removed, and polymerase chain reaction duplicates were removed using PRINSEQ (http://prinseq.sourceforge.net/). The final clean reads were applied to taxonomy and metabolic function annotation. For each sample, MetaPhlAn2 (www.huttenhower.org/metaphlan2) was used to perform microbial classification and calculate the relative abundance of each species, and HUMAnN2 (http://huttenhower.sph.harvard.edu/humann2) was used to annotate microbial pathways according to the BioCyc Database Collection (https://biocyc.org/) and to determine the abundance of each pathway. Alpha diversity of each sample was indicated by the Shannon index, which was calculated using the R vegan package (https://cran.r-project.org/web/packages/vegan/index.html), and principal components analysis (PCA) was used to evaluate the beta diversity among samples.
Construction of quasi-paired cohort
To construct the “paired cohort” from the original group cohort, we first constructed a high-dimensional space where the abundance of each metabolic pathway represented a dimension, and all subjects were positioned in the space according to their profiles of metabolic pathways. Our goal is to delineate the boundary between the case and control samples described as significant differences in ASD-associated pathways. First, the similarity of each sample to its nearest k neighbors was presented as KNN (the average Bray-Curtis distance to k nearest neighbors), where k was the square root of the sample size. Next, outliers that were too far (KNN > mean KNN + SD) and redundancies that were too near (KNN < mean KNN − SD) to their neighbors were removed to avoid stochastic impacts from these samples on the selection of paired sample and statistics. To determine the boundary between ASD and control, we identified boundary samples for both groups using samples that were more similar to neighbors in the opposite group than their own group, i.e., intragroup KNN > intergroup KNN. These boundary samples, which bear similar backgrounds to neighbors of the opposite group, are more valuable than others in the control of metabolic background, leaving the different metabolic pathways between paired samples more likely to be ASD associated. Then, comparable numbers of boundary samples were selected for each group, and ASD-control pairs were constructed with one boundary sample and one of its k nearest neighbors of the opposite side. Thus, the pairs are composed of samples of similar metabolic backgrounds (nearest neighbors in the high-dimensional space) but in opposite phenotype groups. Last, the quasi-paired cohort was constructed with these ASD-control pairs after removing redundancy (Fig. 1 and fig. S1).
Measurement of urine organic acid
Morning urine of ASD and control children was collected and immediately frozen at −20°C and then transferred to a −80°C freezer. For each urine sample, a total of 75 metabolites were measured using Agilent 7890A gas chromatograph–mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions and analyzed with MSD ChemStation (E.02.02.1431) to calculate the concentration of each metabolite normalized by the concentration of creatine in the same sample.
Random forest model
The random forest model was constructed with the caret (https://cran.r-project.org/web/packages/caret/) and randomForest (https://cran.r-project.org/web/packages/randomForest/index.html) R packages to select the most deviated markers of enzymes. The model was trained by 50% of samples through twofold cross-validation and tested with all samples. Bootstrapping was done 1000 times. For each bootstrap result, each enzyme’s contribution to the model, and the diagnostic score for each subject was recorded. Then, the diagnostic capacity of the model in discriminating the patients with ASD from control subjects was evaluated with AUC (area under the ROC curve) through the R packages pROC (https://cran.r-project.org/web/packages/pROC/index.html) and ROCR (https://cran.r-project.org/web/packages/ROCR/index.html). The mean contributions of each enzyme to the model were calculated to evaluate their deviations between ASD and control. The mean diagnostic score of each subject, which was inferred from the abundance of detoxification enzymes and adjusted by the model according to their deviations between ASD and control, was used to represent the extent of dysfunction in microbial detoxification comprehensively and tested for correlations to clinical rating scores.
Statistical analysis
Wilcoxon rank sum test was used to compare the average between the ASD and control groups in their alpha diversity indicators, the abundance of each species or metabolic pathway, with adjusted P < 0.05 as significant. Differences in the abundance of pathways between paired samples of the quasi-paired cohort were tested with the Wilcoxon signed-rank test for paired samples, with P < 0.05 and false discovery rate (FDR) < 0.1 as significant. Correlations of pathways versus urine organic acids and score in detoxifying dysfunction versus ASD rating scores were evaluated with Spearman’s rank correlation coefficient and regarded as strongly correlated if r < −0.4 or r > 0.4.
RESULTS
Intestinal microbiome showed great individual diversity
The study enrolled 79 participants, including 39 ASD children and 40 age- and gender-matched neurotypical control subjects (mean age, 5.59 years; range, 3 to 8 years; male percentage, 82%; table S1). Metagenome sequencing and analysis of stool samples identified a total of 209 species in all samples with each sample of 101 ± 14 species. The alpha diversity or richness of the microbiome was similar between ASD and control subjects (fig. S2A). Among the 209 species, 18 showed significant differences between groups (fig. S2B), which included species of Veillonella parvula and Lactobacillus rhamnosus enriched in ASD and species of B. longum and Prevotella copri enriched in controls (Wilcoxon rank-sum test, P < 0.05, FDR < 0.3). These findings are partially consistent with previous studies on 16S rDNA sequencing–based species profiling (8, 28, 29). However, each of these studies, including ours, identified a list of study-specific differential species for ASD, and these species do not provide precise mechanisms for understanding ASD pathogenesis.
PCA of species profiles did not obviously separate samples of ASD from those of control (fig. S2C) when plotted with the top two PCs, and the samples of each group were widely scattered, indicating great individual diversity even among samples of the same group. Next, we calculated the pairwise Bray-Curtis distances between each pair of samples and found that intergroup distances were not longer than intragroup differences (fig. S2D), implying that the individual diversity was so great that it overwhelmed ASD-associated alterations. After functional annotation of the metabolic pathways present in the microbiome, we inferred the abundance of each pathway for each sample. PCA of the metabolic profiles showed that samples of the same group did not cluster (fig. S2E), and the pathway-based Bray-Curtis distances of paired samples were similar between intergroup and intragroup (fig. S2F), suggesting great metabolic diversity among samples as well. Direct comparison of the abundance of each metabolic pathway across case and control groups did not identify many relevant pathways; only three pathways showed significant enrichment in ASD samples compared to control samples (Wilcoxon rank-sum test, P < 0.05). The three pathways are NAD.NADP.NADH.NADPH mitochondrial interconversion (yeast), superpathway of acetyl–coenzyme A biosynthesis, and superpathway of thiamin diphosphate biosynthesis.
The quasi-paired cohort strategy identified deficient microbial detoxification in ASD
Since conventional analysis did not provide clear clues on how microbiome contributes to ASD pathogenesis, we next conducted the analysis based on quasi-paired cohort strategy. In doing so, we constructed a cohort of 65 ASD-control pairs, which ultimately included 20 ASD subjects and 18 control subjects from the original groups based on the metabolic profiles of samples. Comparison between the paired samples identified a total of 96 ASD-associated pathways (Wilcoxon signed-rank test, P < 0.05, FDR < 0.1; table S2) with 39 overrepresented and 57 deficient pathways involved in many metabolic categories (table S3).
Among the list of the ASD-associated pathways, we identified a conspicuous trend in the deficiencies in the metabolic category of detoxification in ASD samples. A total of five complete pathways in this category exhibited obviously decreased abundance in ASD subjects when compared to their control counterparts (Fig. 2). Two of the impaired detoxification pathways in ASD were involved in the generation of glutathione (GSH): pathways of the γ-glutamyl cycle and biosynthesis of l-glutamate and l-glutamine, the precursor of GSH (fig. S3). The three other pathways functioned in degrading organic toxicants of methylphosphonate, 3-phenylpropanoate or 3-(3-hydroxyphenyl)-propanoate, and methylglyoxal (fig. S4). Almost all the enzymes in the pathways exhibit significant deficiency in ASD samples including the following key enzymes (Wilcoxon signed-rank test, P < 0.05): glutamate-cysteine ligase (gshA), GSH synthase (gshB), 𝛾-glutamyltransferase (ggt), and aminopeptidase B (pepB) in GSH biosynthesis; C-P lyase (phnJ), which removes the phosphate group in degrading methylphosphonate; dehydrogenase (hcaB) and oxidase (mhpB), which break the benzene ring; and glyoxalase (gloA) and hydrolase (gloB/gloC), which degrade the methylglyoxal to lactate (Fig. 2).
Fig. 2. ASD-associated detoxifying enzymes and pathways.
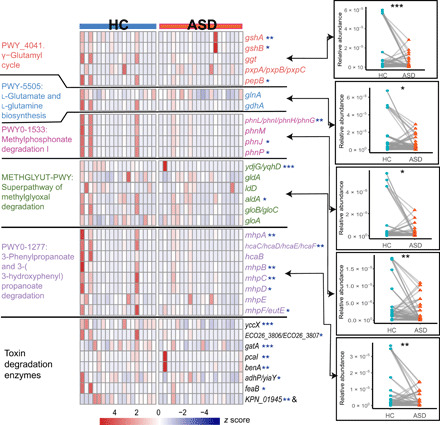
The heatmap shows the relative abundance of each detoxifying enzyme in each sample involved in the paired cohort. Colors indicate the relative abundance that is normalized with z score. Names of pathway modules and representative genes in Enterobacteriaceae of the enzyme are labeled on the left and right sides of the heatmap, respectively. Comparisons of the relative abundance of pathways between controls and ASD subjects are shown to the right side of the heatmap and linked to corresponding pathways with arrows. Significance was tested with Wilcoxon signed-rank test; *P < 0.05, **P < 0.01, and ***P < 0.001. HC, healthy control.
Apart from the above detoxification pathways, we further scrutinized all enzymes participating in the degradation of various kinds of toxins and compared their abundance between subjects of ASD and their control counterparts (Wilcoxon signed-rank test, P < 0.05). None of the enzymes exhibit significant overrepresentation, while eight were significantly deficient in ASD and participated in the degradation of a wide range of toxicants, including chloroalkane/chloroalkene, aminobenzoate, benzamide, styrene, naphthalene, xylene, and benzoate (Fig. 2 and table S4). These toxicants are widely used as insecticides and food additives. The deficiency in these critical enzymes suggests a wider range of impairment in detoxification in ASD, although the corresponding pathways that contained these enzymes were not significantly different between the paired samples.
The other ASD-associated pathways that we identified were largely consistent with previous knowledge about ASD, including many involved in the biosynthesis of pyrimidine, purine, and tetrahydrofolate (table S3) exhibited deficiency in ASD children. It is well known that the metabolism of tetrahydrofolate is often defective in patients with ASD (30). Tetrahydrofolate is the one-carbon donor for the biosynthesis of nucleotides and homocysteine remethylation to form methionine and subsequently S-adenosylmethionine. The primary source of folate, the precursor of tetrahydrofolate, is food, which implies a common deficiency in folate supply for both microbial and host metabolism of tetrahydrofolate in ASD. In addition, among the 39 ASD-enriched pathways, 17 are from yeast. One of these yeast pathways synthesizes 2-amino-3-carboxymuconate semialdehyde (table S3), the intermediate to generate an excitotoxin, quinolinic acid. These results are consistent with previous findings of overproduction of quinolinic acid (31) and overgrowth of yeast in the intestine in ASD patients (32). The consistency of the ASD-associated pathways that we identified with previously reported metabolic alterations in ASD supported the reliability of the quasi-paired cohort method and further confirmed the contributions of gut microbial metabolism to hosts.
Impaired microbial detoxification is associated with mitochondrial dysfunction and the extent of ASD severity
Because of the evident metabolic disturbances seen in ASD, we quantified the absolute concentrations of urine metabolites to evaluate the metabolic alterations in our subjects. The results showed that most ASD children exhibited metabolic abnormalities when compared with controls, in accordance with previous findings (table S5) (33). The most significantly abnormal metabolites in our ASD subjects included aconitic acid, suberic acid, 2-hydroxyhippuric acid, and fumaric acid. All these organic acids are metabolites involved in the Krebs cycle and might be released into circulation and discharged from urine when mitochondria are damaged, thus serving as biomarkers of damage or dysfunction in mitochondria. The measurements of these biomarkers were negatively correlated with the abundance of most of the detoxification enzymes deficient in ASD subjects (Fig. 3). The correlations imply a potential protective role of these microbial detoxification enzymes for mitochondria: The ingested environmental toxicants that may cause damage to mitochondria are substantially detoxified by intestinal microbes. The loss of such protection in ASD may contribute to mitochondrial dysfunction, one of the major pathological alterations in various tissues of ASD children, including the brain.
Fig. 3. The correlation of detoxifying enzymes to urine biomarkers of mitochondrial dysfunction.

(Top) Average concentrations of each urine biomarker of mitochondrial dysfunction (labeled below the heatmap) in ASD and control samples. The y axis indicates their concentrations (μmol normalized by the concentration of creatine of the same sample). Significance was labeled with *P < 0.05 and ***P < 0.001 on the basis of a Wilcoxon signed-sum test. (Bottom) Heatmap of correlations between each detoxifying enzyme and urine biomarker. The color indicates the ρ value of the Spearman rank test, *ρ ≥ 0.4 or ρ ≤ −0.4.
To further corroborate the role of these detoxification enzymes in the pathogenesis of ASD, we constructed a random forest classifier based on the abundance of the ASD-associated detoxification enzymes that we identified (as listed in Fig. 2) and evaluated its performance in discriminating ASD from control subjects. ROC evaluation with 1000 bootstrap showed that the model based on the detoxification enzyme panel accurately described the deviations between ASD and control subjects and achieved a diagnostic power as high as 88% AUC (Fig. 4A). The diagnostic model also outputs the contributions of each enzyme to the model with the top five as key enzymes in the biosynthesis of GSH and l-glutamate/l-glutamine and degradation of aminobenzoate, chloroalkane/chloroalkene/naphthalene, and methylglyoxal, which means these enzymes mostly deviate between ASD and controls (Fig. 4B).
Fig. 4. Diagnostic model based on detoxifying enzymes for ASD status.
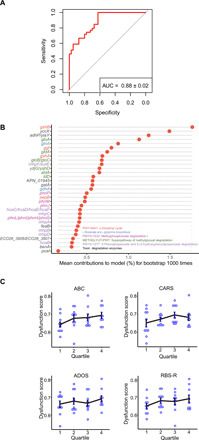
All identified detoxifying enzymes were used to construct a diagnostic model for prediction of ASD status by random forest classifiers with 1000 bootstrap replicates. (A) ROC analysis of the performance of the diagnostic model. (B) Mean contributions of each detoxifying enzyme based on 1000 bootstrap replicates. (C) Dysfunction scores of the microbial detoxification inferred from the diagnostic model for samples in each quartile of patients divided according to their clinical ASD rating scores.
Furthermore, we investigated the correlations between the mean diagnostic score of each sample inferred from the model and the severity of ASD. As the diagnostic score was inferred from the abundance of detoxification enzymes and adjusted by their respective deviation between ASD and control, the score can be regarded as an overall score for the extent of impairment in microbial detoxification and is hereinafter named the dysfunction score. As there are no objective biomarkers for the disease severity or even diagnosis of ASD, we applied clinical rating scores for ASD, i.e., ADOS, ABC, CARS, and RBS-R, to divide patients into respective quartiles according to each of the scores. The dysfunction scores of patients in quartiles showed a trend of increasing with the rating scores (Fig. 4C). Logistic regression between the dysfunction score and clinical rating scores largely confirmed their positive correlations, although they were not significant, possibly because of the subjectivity in the clinical rating of ASD (fig. S5). These correlations between impaired microbial detoxification and the extent of severity of ASD further demonstrated the involvement of the intestinal microbiome in the pathogenesis of ASD due to its dysfunction in detoxification.
DISCUSSION
Supported by the power of the quasi-paired cohort strategy, our study revealed a previously unidentified ASD-associated deficiency in microbial detoxification, which exhibited a strong correlation with the degree of mitochondrial dysfunction, as well as the severity of clinical ASD manifestations. Toxicant exposure has been epidemiologically confirmed as an important etiological factor of ASD, and patients often show some clinical manifestations of intoxication. Mammals are consistently exposed to toxicants from the external environment like glyphosate or from the internal metabolic process of host or microbes, such as methylglyoxal. Detoxification ability is thus essential for life. In addition to the host detoxification system such as hepatic enzymes like P450, the intestinal microbiome may function as the first defense toxicants by degrading or expelling them, as the digestive tract is the major route by which we ingest toxins. In this way, microbial detoxification serves as an essential constituent of the host’s detoxification system.
One of the main pathological manifestations of ASD is the dysfunction in mitochondria, major targets of organic toxicants due to their lipophilic properties. When the intestinal microbial detoxification is severely impaired in ASD, more toxicants of external and internal origins might enter circulation and injure the mitochondria of various tissues. Thus, our finding of impaired microbial detoxification helps explain why ASD children are so vulnerable to environmental toxins and suggests that impairment in microbial detoxification might be involved in the pathogenesis of ASD (Fig. 5). However, the reasons for deficiencies in microbial detoxification are not clear. One possibility is that microbial detoxification deficiency is the consequence of microbiome dysbiosis caused by various genetic and environmental factors such as altered diet and defects in the digestive system, which change the nutrients provided to the microbial inhabitants.
Fig. 5. Role of impaired microbial detoxification in the pathogenesis of ASD.
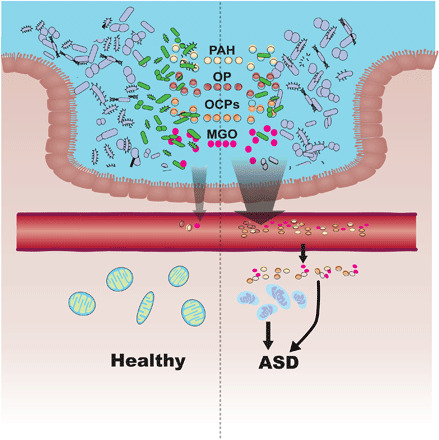
Colored dots in the intestinal lumen indicate common toxicants indigested. PAH, polycyclic aromatic hydrocarbon; OP, organophosphorus; OCPs, organochlorine pesticides; and MGO, methylglyoxal. Intestinal bacteria in green indicate microbes able to detoxify the toxicants, which are deficient in ASD. More toxicants may enter circulation if the function of intestinal microbial detoxification is impaired, and accumulation of toxicants in tissues potentially causes injury in mitochondria or other structure/function integrity, representing the featured manifestations of ASD.
Toxicants implicated in ASD include organochlorine pesticides, polycyclic aromatic hydrocarbons, automotive exhaust, and heavy metals, most of which are organic compounds, especially aromatic and halogenated compounds. Organic toxicants are often lipophilic and prone to accumulate in fatty tissue such as the brain following chronic intake and then damage membrane structures such as those of mitochondria. Glyphosate (N-phosphonomethyl-glycine), the most widely used herbicide in the world, is a methylphosphonate well known for its nuclear and mitochondrial toxicity. Microbial degradation of methylphosphonate is very common in bacteria, and many species use this pathway as the sole source of phosphorus (34). This important detoxification function of microbes is obviously deficient in ASD in our results.
Aromatic hydrocarbons are compounds containing one benzene ring or more than one fused ring (polyaromatic hydrocarbons). Many bacterial species, especially in the phyla Proteobacteria and Actinobacteria, can break down aromatic hydrocarbons through the pathway of 3-phenylpropanoate and 3-(3-hydroxyphenyl)-propanoate degradation, which is also defective in ASD. Methylglyoxal is an inevitable by-product of the processes of glycolysis and metabolism of fatty acid and protein. Methylglyoxal can permeate cells and is deleterious as a highly potent glycating agent reacting with protein, lipids, and nucleic acids to form advanced glycation end products, which are implicated in various degenerative processes including lesions in the brain (35). One study found that methylglyoxal changes the function and oxidative state of mitochondria and causes damage to mitochondria in the brain of ASD individuals (36). Therefore, gut microbes may quickly degrade the local methylglyoxal in situ to keep the gut concentration of methylglyoxal at a low, nontoxic level.
Apart from direct toxin degradation, the ASD microbiome is also deficient in the biosynthesis of GSH. GSH is one of the body’s major antioxidants and is the key cofactor for many detoxifying enzymes. GSH is essential in degrading organic toxicants and expelling heavy metals, both of which contribute to the maintenance of normal mitochondrial functions (37). As the precursor of GSH, l-glutamine is not only involved in organic toxin degradation but also is beneficial to the healing of ulcers. Glutamine helps maintain the integrity and function of the intestinal barrier to stop organic toxicants such as lipopolysaccharides into circulation (38, 39). Thus, the GSH generated by intestinal microbes adds an essential contribution to local detoxification.
Growing evidence has suggested the importance of mitochondrial dysfunction in the pathogenesis of ASD with both inborn defects in mitochondria and acquired damage caused by environmental toxins reported in ASD (33). Studies have found that damaged mitochondria may release mitochondrial DNA and other damage-associated molecular patterns, which activate a low-grade inflammatory response in various tissues, including the brain (40). Such systemic low-grade inflammation with elevated inflammatory cytokines, chemokines, and cellular immune activity is well documented in patients with ASD (41), and the function of neurocytes and brain development may be thus impaired. Recently, cross-talk between the microbiome and mitochondrion has been reported (42), as specific microbial products may inhibit or fine-tune the function of mitochondria (43–45). Here, we reported a protective effect of the microbiome on mitochondrial structural and functional integrity, which is illustrated by the close correlation between impaired microbial detoxification and mitochondrial dysfunction.
Although this study has discovered a deficiency in the detoxification pathways of the intestinal microbiome in patients with ASD from metagenomic data, evidence of toxicant accumulation is not easy to obtain. First, many toxicants and their reductive antioxidants that we identified are very chemically active, such as methylglyoxal and GSH, and they rapidly react with surrounding biomolecules, making it difficult to detect these compounds in frozen samples. Second, the amount of toxicant exposure often fluctuates, and toxicants are possibly undetectable in feces once absorbed. However, the chronic impairment that they cause may last for a long time, which uncouples the toxicant concentration in samples from clinical manifestations of intoxication. Third, the substances catalyzed by the detoxification enzymes that we identified constitute a long list of organic toxicants, and exposure to them varies from person to person, which makes the study design of any transection cohort difficult for quantitative evaluation of the long-term exposure to various toxicants and its correlations to clinical manifestations of ASD. Last, no suitable method is currently available to simultaneously measure the absolute concentrations of the metabolites. Thus, to clarify the process of pathogenesis related to toxicant exposure and detoxification in ASD, novel methods of toxicant measurement are needed, which would be not only useful in demonstrating toxicant accumulation but also valuable for clinical evaluation of individual etiological factors.
Besides the potential protective role of intestinal microbial detoxification against the development of ASD, the quasi-paired cohort is also a merit of this study, which is powerful in identifying disease-associated microbial features from hundreds of dimensions. The principle of this method is similar to that of twin studies for identifying disease-associated genetic variants in which twin samples are powerful tools for controlling for interindividual diversity in millions of single-nucleotide polymorphism loci, and only those variations that differ in case and control between twins are recognized as causative factors in disease. In constructing the quasi-paired cohort, only boundary samples and their close neighbors of the opposite group are selected, which are more valuable, as in the twin samples applied to genetic analysis. The advantages of the quasi-paired cohort over conventional analysis include the following: (i) better control of the high interindividual diversity in metagenome data; (ii) increased statistical power by transforming a group cohort into a paired cohort, which requires a much smaller sample size to achieve the same level of statistical significance; and (iii) increased sensitivity in identifying causative microbiome features of low abundance.
In conclusion, our study proposes a new strategy for metagenome analysis—quasi-paired cohort—and successfully identifies a conspicuous trend of impairment in detoxification in the ASD gut microbiome. The impaired microbial detoxification is correlated with the clinical rating of ASD and the extent of mitochondrial dysfunction, one of the main pathological alterations of ASD, which strongly suggests that impaired microbial detoxification is deeply involved in the pathogenesis of ASD. Such a previously unknown protective role of intestinal microbes suggests potential future therapeutic strategies of rebuilding the impaired microbial detoxification for patients with ASD. Moreover, the novel quasi-paired cohort strategy is a powerful tool for metagenome analysis and might be applied to studies of other complex diseases or even other -omics datasets with similar characteristics of high dimensionality and complexity.
Supplementary Material
Acknowledgments
This study has been approved by the Institutional Review Board of Peking University (Ethical Review Document no: IRB00001052-17100). The study group gained informed consent from the parents/guardians for the collection of stool and urine samples and trial information. We confirmed that all methods were performed in accordance with IRB guidelines and relevant regulations. We are grateful to all the children who participated in this study and their parents for their cooperation. Funding: This work was supported by the Grant of Peking University Health Science Center (BJMU88443Y0306 and BMU2018MX002), the National Science Foundation of China (31671350, 31970568), the National Science and Technology Major Project of China (2018ZX10712001-018-002), and Programs of the Chinese Academy of Sciences (QYZDY-SSW-SMC017 and Y8YZ02E001). Author contributions: J.W. and Y.K. conceived and designed the project. M.Z., Y.H., and R.D. collected information from the participants. Y.C., Q.M., and X.S. processed and analyzed the whole metagenome sequencing data. Y.C. and X.S. performed the statistical analysis. Y.K. and J.Y. conceived and coordinated the project. Z.W. recruited, diagnosed, and examined the recruited participants. Juan Zhang, J.L., and Jie Zhang provided study samples. All authors contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The raw metagenome sequencing data reported in this paper have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA001746 at http://bigd.big.ac.cn/gsa. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaba3760/DC1
REFERENCES AND NOTES
- 1.Posar A., Resca F., Visconti P., Autism according to diagnostic and statistical manual of mental disorders 5th edition: The need for further improvements. J. Pediatr. Neurosci. 10, 146–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baio J., Wiggins L., Christensen D. L., Maenner M. J., Daniels J., Warren Z., Kurzius-Spencer M., Zahorodny W., Rosenberg C. R., White T., Durkin M. S., Imm P., Nikolaou L., Yeargin-Allsopp M., Lee L.-C., Harrington R., Lopez M., Fitzgerald R. T., Hewitt A., Pettygrove S., Constantino J. N., Vehorn A., Shenouda J., Hall-Lande J., Braun K. V. N., Dowling N. F., Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. 67, 1–23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyall K., Croen L., Daniels J., Fallin M. D., Ladd-Acosta C., Lee B. K., Park B. Y., Snyder N. W., Schendel D., Volk H., Windham G. C., Newschaffer C., The changing epidemiology of autism spectrum disorders. Annu. Rev. Public Health 38, 81–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Integrative HMP (iHMP) Research Network Consortium , The integrative human microbiome project. Nature 569, 641–648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vuong H. E., Hsiao E. Y., Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiatry 81, 411–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan Y., Thomas S., Lee B. K., Parent-reported prevalence of food allergies in children with autism spectrum disorder: National health interview survey, 2011-2015. Autism Res. 12, 802–805 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Kushak R. I., Lauwers G. Y., Winter H. S., Buie T. M., Intestinal disaccharidase activity in patients with autism: Effect of age, gender, and intestinal inflammation. Autism 15, 285–294 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Zhang M., Ma W., Zhang J., He Y., Wang J., Analysis of gut microbiota profiles and microbe-disease associations in children with autism spectrum disorders in China. Sci. Rep. 8, 13981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F., Li J., Wu F., Zheng H., Peng Q., Zhou H., Altered composition and function of intestinal microbiota in autism spectrum disorders: A systematic review. Transl. Psychiatry 9, 43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fattorusso A., Di Genova L., Dell’Isola G. B., Mencaroni E., Esposito S., Autism spectrum disorders and the gut microbiota. Nutrients 11, 521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranjan R., Rani A., Metwally A., McGee H. S., Perkins D. L., Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 469, 967–977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dan Z., Mao X., Liu Q., Guo M., Zhuang Y., Liu Z., Chen K., Chen J., Xu R., Tang J., Qin L., Gu B., Liu K., Su C., Zhang F., Xia Y., Hu Z., Liu X., Altered gut microbial profile is associated with abnormal metabolism activity of autism spectrum disorder. Gut Microbes 11, 1246–1267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M., Wan J., Rong H., He F., Wang H., Zhou J., Cai C., Wang Y., Xu R., Yin Z., Zhou W., Alterations in gut glutamate metabolism associated with changes in gut microbiota composition in children with autism spectrum disorder. mSystems 4, e00321-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloyd-Price J., Abu-Ali G., Huttenhower C., The healthy human microbiome. Genome Med. 8, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nayfach S., Pollard K. S., Toward accurate and quantitative comparative metagenomics. Cell 166, 1103–1116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surana N. K., Kasper D. L., Moving beyond microbiome-wide associations to causal microbe identification. Nature 552, 244–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Human Microbiome Project Consortium , Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solden L. M., Naas A. E., Roux S., Daly R. A., Collins W. B., Nicora C. D., Purvine S. O., Hoyt D. W., Schückel J., Jørgensen B., Willats W., Spalinger D. E., Firkins J. L., Lipton M. S., Sullivan M. B., Pope P. B., Wrighton K. C., Interspecies cross-feeding orchestrates carbon degradation in the rumen ecosystem. Nat. Microbiol. 3, 1274–1284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manor O., Levy R., Borenstein E., Mapping the inner workings of the microbiome: Genomic- and metagenomic-based study of metabolism and metabolic interactions in the human microbiome. Cell Metab. 20, 742–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plantinga A. M., Chen J., Jenq R. R., Wu M. C., pldist: Ecological dissimilarities for paired and longitudinal microbiome association analysis. Bioinformatics 35, 3567–3575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Ehrenstein O. S., et al. , Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: Population based case-control study. BMJ 364, l962 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lord C., Rutter M., Le Couteur A., Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24, 659–685 (1994). [DOI] [PubMed] [Google Scholar]
- 23.C. Lord. M. Rutter, P. C. Di Lavore, S. Risi, K. Gotham, S. L. Bishop, R. J. Luyster, W. Guthrie, Autism Diagnostic Observation Schedule, Second Edition: ADOS-2 (Western Psychological Services, 2012).
- 24.Ozonoff S., Goodlin-Jones B. L., Solomon M., Evidence-based assessment of autism spectrum disorders in children and adolescents. J. Clin. Child Adolesc. Psychol. 34, 523–540 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Bodfish J. W., Symons F. J., Parker D. E., Lewis M. H., Varieties of repetitive behavior in autism: Comparisons to mental retardation. J. Autism Dev. Disord. 30, 237–243 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Volkmar F. R., Cicchetti D. V., Dykens E., Sparrow S. S., Leckman J. F., Cohen D. J., An evaluation of the autism behavior checklist. J. Autism Dev. Disord. 18, 82–97 (1988). [DOI] [PubMed] [Google Scholar]
- 27.Schneider C. K., Melmed R. D., Barstow L. E., Enriquez F. J., Ranger-Moore J., Ostrem J. A., Oral human immunoglobulin for children with autism and gastrointestinal dysfunction: A prospective, open-label study. J. Autism Dev. Disord. 36, 1053–1064 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Coretti L., Paparo L., Riccio M. P., Amato F., Cuomo M., Natale A., Borrelli L., Corrado G., Comegna M., Buommino E., Castaldo G., Bravaccio C., Chiariotti L., Canani R. B., Lembo F., Gut microbiota features in young children with autism spectrum disorders. Front. Microbiol. 9, 3146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang D.-W., Ilhan Z. E., Isern N. G., Hoyt D. W., Howsmon D. P., Shaffer M., Lozupone C. A., Hahn J., Adams J. B., Krajmalnik-Brown R., Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 49, 121–131 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Shoffner J., Trommer B., Thurm A., Farmer C., Langley W. A. III, Soskey L., Rodriguez A. N., D’Souza P., Spence S. J., Hyland K., Swedo S. E., CSF concentrations of 5-methyltetrahydrofolate in a cohort of young children with autism. Neurology 86, 2258–2263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gevi F., Zolla L., Gabriele S., Persico A. M., Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism. 7, 47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iovene M. R., Bombace F., Maresca R., Sapone A., Iardino P., Picardi A., Marotta R., Schiraldi C., Siniscalco D., Serra N., de Magistris L., Bravaccio C., Intestinal dysbiosis and yeast isolation in stool of subjects with autism spectrum disorders. Mycopathologia 182, 349–363 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Castora F. J., Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuro Psychopharmacol. Biol. Psychiatry 92, 83–108 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Singh B. K., Walker A., Microbial degradation of organophosphorus compounds. FEMS Microbiol. Rev. 30, 428–471 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Allaman I., Bélanger M., Magistretti P. J., Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9, 23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardoso S., Carvalho C., Marinho R., Simões A., Sena C. M., Matafome P., Santos M. S., Seiça R. M., Moreira P. I., Effects of methylglyoxal and pyridoxamine in rat brain mitochondria bioenergetics and oxidative status. J. Bioenerg. Biomembr. 46, 347–355 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Toledano M. B., Huang M. E., The unfinished puzzle of glutathione physiological functions, an old molecule that still retains many enigmas. Antioxid. Redox Signal. 27, 1127–1129 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Achamrah N., Dechelotte P., Coëffier M., Glutamine and the regulation of intestinal permeability: From bench to bedside. Curr. Opin. Clin. Nutr. Metab. Care 20, 86–91 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Emanuele E., Orsi P., Boso M., Broglia D., Brondino N., Barale F., di Nemi S. U., Politi P., Low-grade endotoxemia in patients with severe autism. Neurosci. Lett. 471, 162–165 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Meyer A., Laverny G., Bernardi L., Charles A. L., Alsaleh G., Pottecher J., Sibilia J., Geny B., Mitochondria: An organelle of bacterial origin controlling inflammation. Front. Immunol. 9, 536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pape K., Tamouza R., Leboyer M., Zipp F., Immunoneuropsychiatry—Novel perspectives on brain disorders. Nat. Rev. Neurol. 15, 317–328 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Han B., Lin C.-C. J., Hu G., Wang M. C., ’Inside Out’—A dialogue between mitochondria and bacteria. FEBS J. 286, 630–641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki M., Danilchanka O., Mekalanos J. J., Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 16, 581–591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalghatgi S., Spina C. S., Costello J. C., Liesa M., Morones-Ramirez J. R., Slomovic S., Molina A., Shirihai O. S., Collins J. J., Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 5, 192ra185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi B., Han M., Microbial siderophore enterobactin promotes mitochondrial iron uptake and development of the host via interaction with ATP synthase. Cell 175, 571–582.e11 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaba3760/DC1