

Coarse-grained modeling, machine learning, and optimization enable efficient design of polymers with target properties.
Abstract
The chemical design of polymers with target structural and/or functional properties represents a grand challenge in materials science. While data-driven design approaches are promising, success with polymers has been limited, largely due to limitations in data availability. Here, we demonstrate the targeted sequence design of single-chain structure in polymers by combining coarse-grained modeling, machine learning, and model optimization. Nearly 2000 unique coarse-grained polymers are simulated to construct and analyze machine learning models. We find that deep neural networks inexpensively and reliably predict structural properties with limited sequence information as input. By coupling trained ML models with sequential model-based optimization, polymer sequences are proposed to exhibit globular, swollen, or rod-like behaviors, which are verified by explicit simulations. This work highlights the promising integration of coarse-grained modeling with data-driven design and represents a necessary and crucial step toward more complex polymer design efforts.
INTRODUCTION
Machine learning (ML) algorithms, enabled by preexisting experimental and computational data, have emerged as powerful tools for molecular property prediction and design (1–5). For example, synthetic protocols have been optimized via the training of ML models on experimental reaction databases (USPTO, Reaxsys, and SciFinder) (6), while generative design strategies have enabled targeted small-molecule design (7). However, materials science often presents problems where substantially less data are available, thereby necessitating the development of creative approaches for navigating data-scarce regimes (8, 9).
One major impediment for the application of ML to soft materials concerns the chemical, topological, and morphological complexity of macromolecular systems, which precludes facile generation and/or integration of requisite data (10–12). These concerns have limited the success of ML in soft materials to a few notable cases (13–15). Although combinatorial and high-throughput polymer synthesis and characterization techniques are now emerging (16, 17), some applications will require advanced or nuanced synthetic approaches that will further limit the number of well-defined systems that can be characterized. Moreover, the proper representation or description of soft materials (18–20) remains an outstanding challenge that inhibits the integration of related databases. Some difficulties might be mitigated by advanced data selection techniques (9) and/or augmentation with in silico datasets (8), presuming that extracting useful data from simulations is feasible.
To date, most computational data for ML on polymers are derived from density functional theory calculations of monomeric or small oligomeric species (21–23). Polymers, however, owe much of their structural and conformational complexity to their large molecular weight. From a simulation standpoint, first-principles characterization of macromolecular systems is challenging due to the span of relevant spatiotemporal scales that dictates material functionality. Consequently, computation of macromolecular properties is often the realm of coarse-grained (CG) classical modeling (24), where reduced representations of the system that retain essential physics are developed to make the calculations computationally tractable. Although ML has been recently used to develop CG force fields (25, 26) and even as a means to predict optoelectronic properties directly from CG models of conjugated polymer systems (11, 27), there are so far very few polymer-based simulations that use ML to make surrogate predictions for CG simulations themselves (14, 15). If available, such data-driven workflows could be extremely helpful in materials design efforts (10, 11).
The directed design of polymers with tailored composition or sequence has considerable potential in numerous application areas (28, 29). This work demonstrates the targeted design of polymer sequences in a portion of the CG polymer genome. It is enabled by (i) generating data in a reduced genome space with molecular dynamics (MD) simulations, (ii) training an ML model that accurately captures the statistical information contained in the generated data, and (iii) making predictions in regions external to the reduced genome space. Coupling the ML model to an optimization framework facilitates the design of specific polymer sequences to achieve a target property. While this strategy is demonstrated for CG polymer models of single-chain structural properties, the efficacy of the approach is promising for future endeavors that go beyond the cases demonstrated here.
RESULTS
Scope of CG polymer genome and calculated properties
Polymers can feature a vast array of monomeric chemistries and topologies that obfuscate design efforts. Because complete coverage of properties across the polymer genome (22) is unrealistic, we first study a specific region of CG chemical space, as denoted by the associated polymer classes summarized in Fig. 1 (A and B). Here, we briefly review some of the more salient features of the CG chemical space; additional details are provided in Materials and Methods.
Fig. 1. Schematic of the scope of the CG polymer genome and the approach to property prediction.
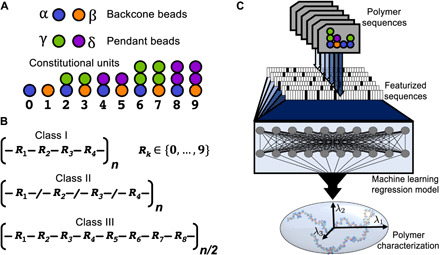
(A) CG bead types and topologies that comprise the CG polymers. Backbone bead types are denoted as either α or β; pendant bead types are denoted as either γ or δ; and allowable combinations of backbone and pendant beads yields 10 unique CUs, which are labeled from 0 to 9. (B) Structural representations and labels for the classes of polymers studied; Rk denotes the kth CU. Class I polymers correspond to regular copolymers with a repeat pattern of four CUs, class II polymers correspond to random polymers constructed from four CUs, and class III polymers correspond to regular copolymers constructed with a repeat pattern of eight CUs. (C) General workflow for predicting CG polymer properties. The polymer sequence (or a repeat unit thereof) is featurized and provided as input to an ML algorithm, which maps the input onto structural characterizations of the polymer.
As shown Fig. 1A, the CG polymers are composed of four different bead types, two that can be found within the backbone of the polymer and two found in pendant groups; for ease of reference, the backbone bead types are denoted as α and β, and the pendant bead types are denoted as γ and δ. Note that these CG bead types do not reflect specific chemistries; however, the CG polymer interactions are formulated to provide a test case using potential energy functions that are typical for CG simulations. All polymer beads have characteristic size, σ. However, the bead types (α, β, γ, and δ) are distinguished by the strength of their nonbonded interactions as dictated by the parameter εii; because beads with large εii exhibit stronger attractive forces, εii functions as a proxy for relative solvophobicity. Furthermore, unique two-, three-, and four-body intramolecular interactions based on local composition lend additional complexity to the conformational characteristics of the polymers.
Within this CG chemical space, Fig. 1A also shows how backbone and pendant bead types can be connected such that 10 distinct constitutional units (CUs) (30) are observed across all polymer sequences. We consider three polymer classes, which are distinguished by the number of distinct CUs and their arrangement in a given polymer sequence (Fig. 1B). The sequence of any class I or class II polymer contains up to four distinct CUs, while a class III polymer can feature as many as eight distinct CUs. A repeating pattern of CUs is referred to as a constitutional repeat unit (CRU) (30) such that all class I and III polymers can be described by CRUs of four and eight CUs, respectively; the class II polymers have stochastically generated sequences and thus cannot be described using a CRU.
In contrast to previous work, in which ML models have been trained on available (and possibly sparse) experimental data or quantum chemistry calculations, our design approach relies on data systematically generated from CG MD simulations. To effectively gauge efficacy and data requirements, we focus on the simple yet nontrivial single-chain property of the average square radius of gyration 〈Rg2〉, for which a high-quality dataset can be reasonably generated using MD. 〈Rg2〉 also has practical relevance to the rheological behavior of polymers in solution, because it sets an overlap concentration (c*∝ 〈Rg2〉−3/2) that relates to the onset of chain entanglements and gelation (31). Furthermore, polymer compactness, as expressed through the radius of gyration and hydrodynamic radius determined from small-angle x-ray scattering, was also the focus of a recent combinatorial and high-throughput experimental approach toward the design of single-chain polymer nanoparticles (17, 32).
To isolate the effects of polymer chemistry and sequence design, rather than that due to chain molecular weight, we compare polymers with the same degree of polymerization, as given by the number of backbone beads Nbb. All polymers are first constructed with Nbb = 400 and simulated in implicit solvent using CG MD; 〈Rg2〉 is then computed from the simulation trajectory (see Materials and Methods for details on simulation methodology and property computation). Figure 2 (A and B) illustrates the range of values obtained from explicit simulation of class I and class II polymers. In Fig. 2A, the polymers are rank-ordered from smallest to largest 〈Rg2〉 and further delineated by class; Fig. 2B shows histograms of the same data. Because class I polymers are regular polymers, in the limit of a large Nbb, cyclic and inverse sequence permutations are expected to yield identical results such that our dataset includes all 1540 unique polymers. Meanwhile, the number of unique polymers in class II is extraordinarily large, as they are stochastically constructed from up to four unique CUs; therefore, the dataset comprises 200 representative polymers. Other than the dataset size, the distribution of 〈Rg2〉 is similar between class I and II polymers in terms of mean and SD across the dataset. Although the apparent complexity of the CG chemical space appears small, Fig. 2A indicates that the CG space spans the breadth of anticipated polymer behavior in solution, ranging from collapsed globules to rod-like polymers, and thus provides a nontrivial testing ground for ML-enhanced design.
Fig. 2. Summary of square radius of gyration (〈Rg2〉) datasets.
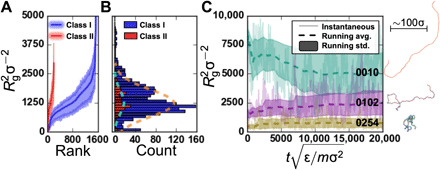
(A) 〈Rg2〉 for class I (blue) and class II (red) polymers, rank-ordered by 〈Rg2〉. The value for each polymer is given by the solid line, while the shaded region spans the 25th and 75th percentile values observed during production. (B) Histogram of 〈Rg2〉 data illustrating the distribution of mean values for class I (blue) and class II (red) polymers. For reference, fits to a Gaussian distribution are shown by the dashed lines and have means of 1234σ2 and 1281σ2 and SDs of 577σ2 and 492σ2 for class I and II polymers, respectively. (C) 〈Rg2〉 variability and convergence for class I polymers with labels 0010, 0102, and 0254 as representative examples of polymers with large, intermediate, and small 〈Rg2〉. Note that (A) and (B) share the vertical axis, and a small set of polymers is not shown with 〈Rg2〉 > 5000σ2.
Along with the calculated values (solid line), Fig. 2A also provides a measure of the width of the underlying conformational distribution with the shaded region spanning the 25th to 75th percentile values. The difference between these percentile values is approximately 50 to 70% of 〈Rg2〉 for the bulk of the data, indicating broad distributions. This highlights an underlying challenge with predicting polymer behavior, which is that properties arise from conformational distributions with substantial heterogeneity. When evaluating the prediction errors of surrogate ML models for 〈Rg2〉, it will be important to compare these errors to (i) the distribution of 〈Rg2〉 values in the dataset resulting from different polymer sequences and (ii) the distribution of simulated Rg2 values for a single polymer sequence due to conformational heterogeneity. Figure 2C illustrates the variation in polymer conformations for three representative class I polymers by tracking Rg2 over the course of their simulation (post-equilibration). From Fig. 2C, it is clear that Rg2 fluctuates considerably; however, estimates of 〈Rg2〉 (dashed lines) and its SD (bounded shaded regions) begin to stabilize for all three polymers after about 104 reduced time units (Materials and Methods). Consequently, the 〈Rg2〉 values are expected to be well converged across all polymer sequences.
Predictions within the space of regular polymer sequences
In a first application, we consider whether an ML model can predict 〈Rg2〉 for polymer sequences with regular patterns, in lieu of explicitly running an MD simulation. To do so, we train a deep neural network (DNN) to take featurized class I polymer sequences as input and output a corresponding 〈Rg2〉. Then, the ML model is tasked to predict 〈Rg2〉 for class I polymer sequences that are not part of the training set. Because the polymer sequence is a regular pattern for class I polymers, each polymer is uniquely described by a CRU, which is represented to the DNN as a one-hot vector, with each bit indicating one of the 10 possible CUs; because the CRU has four CUs, the CRU is a 40-bit vector (see Fig. 1C for a schematic representation). The DNN consists of two hidden, fully connected layers with 20 neurons that precede a single output neuron that yields a value of 〈Rg2〉 for a given input vector; further details of the polymer featurization and DNN are provided in Materials and Methods. Figure 3A compares the values predicted using the ML model to those obtained from explicit simulation; in this case, 80% of the class I polymer dataset, or 1232 polymers, is used for training (with 20% of the 1232 used for training validation), and the remaining 20%, or 308 polymers, is used for testing.
Fig. 3. Performance of ML model for prediction of mean square radius of gyration, 〈Rg2〉, for class I and class II polymers.
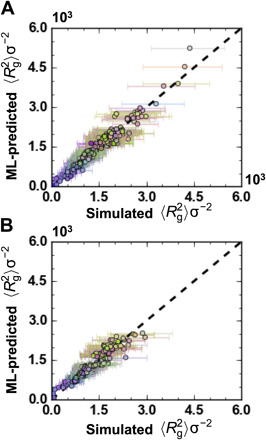
In (A), an ML model is trained on 80% of the sequences, and the predictions for 〈Rg2〉 versus simulated 〈Rg2〉 are shown for the 308 held-out sequences; the r2 is 0.953, and the MAE is 111.32σ2. In (B), a model trained on class I polymers is applied to make predictions on the class II polymers; the r2 is 0.895, and the MAE is 130.34σ2. The coloring of the markers reflects the polymer composition as described in the “Data coloring by composition” section in Materials and Methods.
Overall, the simple DNN model exhibits good predictive capabilities within the space of class I polymers; for this test set, the coefficient of determination, r2, exceeds 0.95, the mean absolute error (MAE) is ~111σ2, and the SD of absolute errors (SDAE) is ~110σ2. Both the MAE and SDAE are considerably smaller than the SD of 〈Rg2〉 across all polymers, which is 577σ2 (Fig. 2B), as well as the SD of Rg2 observed in a simulation for a given polymer (Fig. 2, A and C). This suggests that DNNs provide a viable surrogate for explicit CG MD simulations without substantial loss in accuracy and at considerably reduced computational cost.
The DNN performance in Fig. 3A is achieved without providing any chemically specific input information. Specifically, because the 4-CU repeat pattern is represented as a one-hot vector, each CU is categorically different despite any common chemical motifs. To provide insight into predictive accuracy as a function of chemical composition, the data in Fig. 3A are colored according to the chemical composition of the sequence as described in Materials and Methods. Qualitatively, in Fig. 3A, blue/purple shades trend toward small 〈Rg2〉 and green shades trend toward large 〈Rg2〉. Intuitively, this is expected because more solvophobic polymers exhibit smaller 〈Rg2〉, and here, blue and purple are assigned to α and δ beads, which have the largest nonbonded interaction parameters (εii) and thus the greatest solvophobicity. This suggests that some chemical similarity among CUs is learned during training to reflect the correlation between polymer composition and size. Alternative featurization approaches such as chemical fingerprinting might directly encode this chemical similarity at the input level; some additional possibilities are explored later.
Transferability of ML models from the space of regular to random polymers
The restriction of a regular, repeating pattern made it possible to enumerate and simulate all class I polymers, but complete enumeration of all sequences is generally unlikely. For design, a relevant consideration is whether an ML model trained on data in one region of the polymer genome can predict polymer properties in a related region of the genome. To this end, we next examine the viability of applying a model trained on class I polymers, which have regular sequences, to predict the properties of class II polymers, which have stochastically generated sequences.
Because the class II polymers do not have a fixed-length repeat pattern of CUs, the input featurization approach must be generalized to account for variable sequence lengths. Although the size of the one-hot input vector could be increased to accommodate the entire length of chain, the increased dimensionality and number of fitting parameters associated with this strategy makes this undesirable. Instead, we use a long short-term memory (LSTM) recurrent neural network as a convenient way to consider sequence variability along a polymer chain. Because the polymer sequences do not have inherent directionality, a bidirectional LSTM processes the one-hot encoding (OHE) polymer sequence both forward and backward to produce a new feature vector that is fed into the DNN architecture; further details regarding the featurization and LSTM implementation are provided in Materials and Methods.
Using a model trained on class I polymer sequences, Fig. 3B compares predicted 〈Rg2〉 values to those obtained from explicit CG MD simulation for class II sequences. In this case, r2 remains high (∼0.9), while both the MAE (~130σ2) and SDAE (~114σ2) remain low by comparison to the SD across the dataset (492σ2). The distribution of colors in Fig. 3B is also similar to that in Fig. 3A, again indicating that a higher concentration of solvophobic beads leads to smaller 〈Rg2〉. Because the class I dataset contains information on all possible 4-CU combinations, it is reasonable to expect that the trained ML model would be transferable to the space of random sequences.
To further investigate this transferability, we examine the predictive performance of several simple linear mixing models for treating the class II polymer sequences (see the “Description of simple linear mixing models” section in Materials and Methods). Specifically, we consider predictions derived from weighted averages from homopolymers (model A), from polymers with a CRU of two CUs (model B), and from polymers with a CRU of four CUs (model C). Table 1 summarizes the performance of these models as applied to the class II polymers. The ML model outperforms the simpler surrogate models in all cases; however, model C performs comparably to the ML, albeit with a somewhat larger MAE. This may suggest that the transferability observed between class I and class II polymers in the ML is mostly a linear mapping. The fact that model A and model B yield inferior results indicates that the sequence information on the 1-CU or 2-CU scale is insufficient to predict the global properties of the polymer sequence. Inclusion of sequence information at larger length scales can be expected to enhance surrogate model performance.
Table 1. Comparison of model regression performance metrics for predicting 〈Rg2〉 of class II polymers using data from class I polymers.
| Model | r2 | MAE/σ2 |
| A | 0.595 | 764 |
| B | 0.745 | 306 |
| C | 0.886 | 174 |
| ML | 0.895 | 130 |
Performance of regression models with dataset size
Given the computational cost and potential complexity of soft matter simulations, it is important to gauge the quantity of data necessary to train an effective ML regression model. Figure 4 assesses the performance of ML regression models, as quantified by r2 (blue circles, left axis) and MAE (red diamonds, right axis), on predicting 〈Rg2〉 for held-out class I polymers as a function of dataset size. The figure shows that the quality of the model improves as the training set size increases. However, the quality of the models does not improve significantly after 20% of the available data (300 polymers) have been incorporated into the training set, and only a rough sampling over the CG polymer genome is necessary to build predictive models (r2 > 0.9). The fact that only hundreds of data points may be required for models of this quality is promising, although this likely depends on both the complexity of the chemical space and the property considered. It also highlights a potential benefit of using ML versus the simple models proposed in the previous section, because the ML models might be constructed from more limited data without significant detriment to accuracy, whereas the linear mixing models depend on the data for all sequences for a given CRU length to be available for evaluation. Nevertheless, it is also worth noting that the polymers used in training here are randomly selected, and some of the expected data augmentation requirements could be offset with better training strategies, such as active learning (9, 11).
Fig. 4. r2 and MAE associated with ML regression models when predicting 〈Rg2〉 for class I polymers as a function of the number of polymers in the training set.
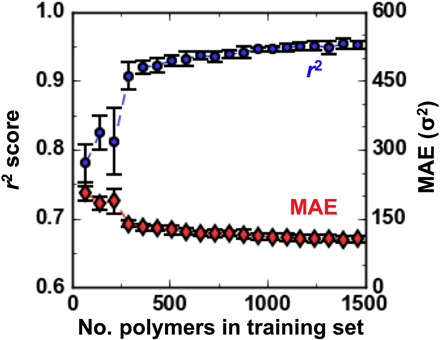
The error bars reflect the SD of scores obtained from 20-fold cross-validation, in which each fold is used as a test set for regression models trained using between 5 and 100% of the data from the remaining 19 folds.
Targeted sequence design using sequential model-based optimization
In a final application, we aim to design new polymers with specific 〈Rg2〉 by leveraging ML models trained on our previously generated class I polymer data. For concreteness, we target class III polymers (Fig. 1B). Like class I polymers, they are regular polymers, but their constitution is considerably more complex. Thus, this task probes the viability of collecting data in a relatively simple and manageable chemical space and applying that knowledge to design in a much broader and complex space. We use a sequential model-based optimization technique (SMBO)—namely, the tree-structured Parzen estimator (TPE) algorithm (33)—coupled to a DNN trained on class I polymers for the prediction of 〈Rg2〉. For a target value of 〈Rg2〉, the TPE algorithm generates a candidate sequence, compares the estimated 〈Rg2〉 from the ML model to the target, and then proposes a new sequence based on historical performance. Although it is possible to use the OHE featurization approaches described earlier for this task, we transition to a new featurization approach, referred to as property coloring, with the aim of enhancing chemical flexibility and transferability. In this scheme, the polymer is encoded as an image wherein each CG bead in the polymer is assigned a color based on local characteristics, such as size and solvophobicity. The resulting “image” is then further processed by a two-dimensional convolutional neural network (2D-CNN), and the flattened output of the convolutional layer is used as input for training the DNN (Materials and Methods). When tested on the same 308 held-out class I polymer sequences, a DNN trained using the property coloring scheme yielded r2 = 0.958, MAE = 106σ2, and SDAE = 103σ2 versus 0.947, 120σ2, and 115σ2 for a DNN trained using OHE vectors, suggesting that property coloring would perform at least as well as OHE as a representation for this task (fig. S1).
For demonstration, we generate sequences according to target values of 〈Rg2〉 = 250, 2000, and 3800σ2, which we refer to as globular, swollen, and rod-like targets. In addition to spanning observed behaviors of polymers in solvents of varying quality, the numerical values are greater than 1 SD outside of the mean values of the training data (Fig. 2A) to curtail the likelihood of generating viable candidates by chance. For each target, 20 candidate sequences of the class III type are generated via the SMBO-TPE approach, and their behavior is subsequently simulated using CG MD to benchmark the predictions.
Figure 5A demonstrates that the combination of CG modeling with ML and SMBO enables targeted sequence design with high fidelity. The figure displays the statistical distribution of Rg2 obtained from explicit simulations (in the form a violin plot with a notch at the median value and a bar extending from the 25th to the 75th percentile values) and the 〈Rg2〉 (white dot) for all candidate polymers; these results are compared to the target values for globular, swollen, and rod-like targets (horizontal lines) as well as Rg2 distribution widths that are typical of polymers of that size (rectangular shaded regions). Overall, the bulk of simulated values compares quite favorably to their intended targets. While the distribution of values underlying 〈Rg2〉 can span many thousand σ2 for some polymer sequences, as shown by the extent of the violin plots, nearly all of the simulated values lie within the distribution bands of their intended targets.
Fig. 5. Targeted sequence design of size-specific polymers.
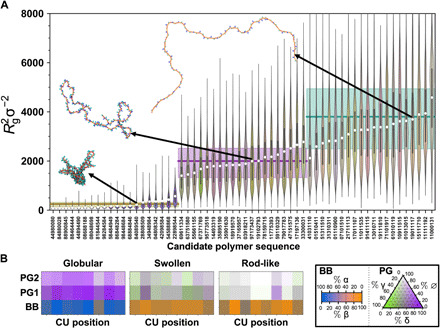
(A) Statistical comparison of 〈Rg2〉 distributions obtained from explicit MD simulations of all candidate polymers. (B) Average composition maps of the CUs for candidate rod-like, swollen, and globular targets with 〈Rg2〉σ−2 = 3800 (top), 2000 (middle), and 250 (bottom). In (A), from left to right, the first 20 sequences are the globular targets, the next 20 are the swollen targets, and the remaining 20 are the rod-like targets; within each set, the sequences are ordered by ascending 〈Rg2〉, given by the white dots. The violin plots indicate the distribution of values underlying the mean, with a notch at the median value and a bar extending from the 25th to the 75th percentile values. For reference, the target value is indicated by the horizontal line, and the shaded region indicates the average spread between 25th and 75th percentiles for class I polymers of similar size. The color of each violin is based on the average composition of the total polymer sequence. In (B), the colors are resolved by CU and backbone/pendant group but averaged over all sequences for each specific target size. The color contributions for each bead type are shown in the boxed legend, with ∅ indicating no bead present.
Even where exact correspondence to the target is not observed, Fig. 5A presents three sets of distinguishable predictions, with the globular targets distinct from the swollen targets, which are largely separated from the rod-like targets. Figure 5B examines the average sequence composition of proposed targets to qualitatively assess the characteristics. As suggested by the color of the violin distributions in Fig. 5A, the proposed targets tend toward specific constitutional characteristics. The globular targets feature a high density of δ pendant beads atop α backbones. Meanwhile, the rod-like targets have predominantly β backbones with sparse pendant groups, which tend toward γ beads, if present. The swollen targets have significant β-type backbone character, but the composition of the pendant groups appears fairly diverse. While some of this behavior might be qualitatively expected, such as polymers tending toward rods with increasing β character, the tandem ML/SMBO approach yields complex sequences with quantitative accuracy beyond that granted by simple intuition.
DISCUSSION
In this work, we have examined the efficacy of a design paradigm wherein (i) a manageable number of simulations are run to generate an in silico dataset; (ii) the dataset is used to train an ML model, which functions as a surrogate for additional simulations; and (iii) the surrogate model is exploited with optimization techniques to propose sequence-defined polymers that exhibit target property values. The paradigm was specifically explored in consideration of how chemistry and sequence dictate the characteristic size of polymers within a specified region of the polymer genome (22). The chemical and topological complexity of the polymers studied is commensurate with experimental literature examples of using ML for polymer properties (9, 21) and sufficient to observe a broad spectrum of polymer behaviors, from collapsed globules to extended rods. We find that ML provides a viable surrogate for mapping sequence to structure in these polymers, yielding predictions with errors that are generally much smaller than the variability in polymer size itself and the variability of data obtained across polymer sequences. The ML models trained in one region of the polymer genome can be sufficiently transferable so as to usefully direct design efforts, with the aid of optimization techniques, at significantly reduced computational cost.
While primarily illustrative, the specific case of manipulating polymer size/conformation could have direct implications in a number of application areas. For example, polymer sequence could provide another means to control or understand the rheology of polymer solutions by exploiting the link between viscosity and polymer size (31). Similar workflows could also facilitate the design of single-chain polymeric nanoparticles for which intramolecular interactions drive targeted polymer collapse (17, 32). Furthermore, the proposed framework might be repurposed to interpret chemical sequence and structure underlying experimental data obtained from techniques that report on macromolecular structure (e.g., small-angle x-ray scattering, small-angle neutron scattering, or Förster resonance energy transfer). Such ML models, if trained on biological systems, could quantitatively interrogate structure-function relationships in systems like intrinsically disordered proteins, whose conformational properties have been empirically correlated to sequence-level information (34). The combination of CG modeling, ML, and optimization is not itself limited to the interrogation of single-chain or structural properties. Provided that a target property or figure of merit can be tractably computed using simulations within the desired chemical space, it should be possible to design sequences around dynamic properties, complex phenomena such as self-assembly, or even multi-objective properties (14, 15).
In addition to targeting other physicochemical properties of polymeric materials, future work may build on the technical foundation of the design strategy outlined here. For example, the use of DNNs as the surrogate model was motivated by their simplicity of implementation and their flexibility for applications, particularly as it relates to the exploration of featurization approaches. However, future design problems may be better served by other ML algorithms, like Gaussian process regression (GPR) or even simpler surrogate models; our own preliminary data suggested that GPR exhibits similar accuracies as DNN, with the added benefit of providing measures of uncertainty that can be exploited by active learning strategies. Featurization is also likely to play a more prominent role in future applications. Here, the use of OHE was convenient for a finite set of CUs and simple to understand. However, more flexible representation methods, such as the property coloring scheme used in our 2D convolution or the use of graph convolutional networks, may be better when chemical complexity is increased. The property coloring scheme could be easily adapted to use more conventional chemical descriptors, instead of scalar CG variables in cases where the chemistry of the CG beads is known. In addition, methods to handle polymer sequences of varying sizes will also find great utility (27, 35). Last, we relied on the SMBO/TPE algorithm as a means to propose CG polymer sequences using a finite, discrete set of CG polymer beads; this is a sensible, inexpensive, and facile approach for situations where underlying chemical units are discrete and the design space can be structured around a countable set of possible synthetic modifications. While it is both possible and intriguing to allow our CG bead properties to adjust on a continuum rather than a discrete scale, back-mapping or chemical inversion methods would be required and are thus of great interest. Meanwhile, other optimization strategies, such as those based on genetic algorithms, generative adversarial networks, or variational autoencoders, are reasonable alternatives that are already in use (1, 11, 12).
Irrespective of the choice of ML architecture, featurization scheme, or optimization strategy, the quality of available CG models will be integral to successful utilization of this paradigm. In this work, the CG polymers were chemically distinct but did not reflect specific chemical structures. A similar approach may be sufficient to investigate generic phenomena, such as the effect of polymer topology on self-assembly; however, applications that emphasize chemical design will need to rely on existing CG force fields or include model parameterization as part of the design workflow. In either scenario, the viability of a hybrid CG modeling/ML design paradigm will benefit from continued development of systematic coarse-graining methodologies (11, 36–38) and approaches to enhance the capabilities and accuracy of CG models (26, 27, 39).
In conclusion, we have presented a new practical paradigm for soft materials design that combines CG modeling, ML, and model optimization. This unique combination addresses technical challenges related to experimental synthesis and characterization as well as soft materials modeling. The approach is exemplified through the mapping of sequence to structure relationships in a nontrivial region of the CG polymer genome. Although this paradigm only relies on simulation data, we anticipate that integration with experimental data will be both possible and highly effective in certain applications. Overall, the results reported here highlight significant potential for enhancing efforts to design polymer-based materials via the combination of CG modeling and ML.
MATERIALS AND METHODS
Definition of CG chemical space
A summary of the polymers considered in this study is shown in Fig. 1 (A and B). All polymers are constructed from four possible CG beads (α, β, γ, and δ); the different beads are distinguished by the self, nonbonded interaction parameter, εii, which functions as a proxy for relative solvophobicity. Two bead types (α and β) form the backbone of the polymer, while the remaining two (γ and δ) can form pendant groups that adorn the backbone. In addition to εii changing with bead type, certain combinations of beads yield specific stretching, angle bending, and torsional interaction constants (Supplementary Materials) to add additional complexity to the CG chemical space. Furthermore, the pendant groups can be either one bead or two beads of the same type. Given these restrictions, if a CU is considered as a backbone bead plus its pendant group, if any, then there are 10 unique CUs or building blocks (based on combined composition and topology) that can be found in a given polymer sequence; these CUs are assigned numerical labels from 0 to 9 for easy association. All polymers are composed of 400 CUs such that the number of backbone beads is Nbb = 400 and the total number of pendant group beads is Npg ≤ 800.
Within this CG chemical space, three classes of polymers are constructed (Fig. 1B). The first class, class I, includes regular polymers with a CRU (30) containing equal to or fewer than four CUs. In the limit of large Nbb, inverted sequences or cyclic permutations of sequences should yield identical properties. Consequently, class I is limited to 1540 unique polymers, rather than 104. The second class, class II, includes random copolymers composed of up to four unique CUs in the polymer sequence. The third class, class III, includes regular polymers like class I, except that the CRU contains equal to or fewer than eight CUs.
Calculation of polymer properties
We consider the structural properties of a single polymer chain. For each polymer, simulation trajectories are used to compute the gyration tensor S defined as
| (1) |
where ri is a column vector of the position of the ith bead, rcm is the center-of-mass position of the polymer, and T denotes the transpose. Subsequently, diagonalization of Eq. 1 yields S = diag(λ12, λ22, λ32), where the diagonal elements λ12 ≤ λ22 ≤ λ32 are known as the principal moments of the gyration tensor. The square radius of gyration is conveniently computed from the principal moments as
| (2) |
and measures the size of a particular conformation. The target of the ML regression model is then given as the ensemble average over all sampled configurations, 〈Rg2〉.
Details of MD simulations
MD simulations are used to generate polymer configurations for computing 〈Rg2〉, and the resulting dataset is used to train and evaluate all ML models. All MD simulations are performed using the LAMMPS simulation package (40) in reduced units with characteristic quantities of m, σ, and ε for mass, distance, and energy, respectively; the reduced time unit is (mσ2/ε)1/2. Simulations correspond to a single CG polymer chain in implicit solvent such that the polymer dynamics are evolved according to the Langevin equation using the velocity-Verlet integration scheme with a 0.001 timestep; the solvent friction is set to be ς = 0.1. After initializing the polymer chain in an extended configuration, simulations are run for 109 simulation steps, with the first half used as equilibration. During the second half of the trajectory, configurations are recorded every 5 × 104 timesteps for analysis.
Polymer interactions are described by summation of typical bonded and nonbonded potential energy functions such that the total potential energy of the system with configuration rN is given by
| (3) |
where rij, θijk, and φijkl are internal distances, angles, and dihedrals, respectively, derived from the coordinates rN; the functional forms for the various interaction types are described below. All pairs of beads have a nonbonded energy contribution given by
| (4) |
Directly bonded beads have the stretching energy
| (5) |
beads connected by two bonds have an angle bending energy
| (6) |
and beads connected through three bonds have a torsional interaction
| (7) |
The constants appearing in Eqs. 4 to 7 depend on the specific bead types involved in the interaction and are provided in the Supplementary Materials. Constants involving different bead types in Eq. 4 are computed using Lorentz-Berthelot combination rules.
Featurization of polymer sequences
A critical problem in chemistry-inspired ML applications is the appropriate featurization of chemical inputs. Here, we use two approaches: simple OHE and property coloring. In the OHE approach, each CU is represented as a 10-bit vector with a single high element that corresponds to a particular CU type. Because this approach is categorical, the various CU types are simply recognized as distinct, and the featurization vectors do not specifically encode any chemical or topological information. A further limitation is that OHE requires explicit enumeration of all possible categorical features, which here corresponds to the set of CU types.
We also consider a more flexible featurization approach, which we refer to as property coloring; the use of property coloring is schematically depicted in Fig. 6. In effect, the polymer is encoded as an image, with each bead of the polymer represented by a pixel with coloring determined by local properties. In this application, a CU is depicted as a 3 × 1 × M array, where the first dimension, which is 3 long, encodes bead properties for the backbone and any associated pendant groups, while the last dimension, which is M long, is the number of property channels. Then, the polymer sequence (or CRU) is mapped to an N × 3 × M array, with N being the number of backbone beads in the input structure (e.g., Nbb if the entire polymer sequence is used). As an image, the polymer representation is well suited as input to CNNs. Using a number of filters (Fig. 6), a convolved image is produced and flattened to produce a featurized vector for the polymer sequence.
Fig. 6. A schematic of property coloring featurization.
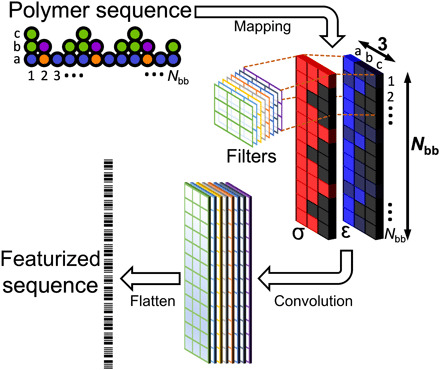
The polymer sequence is mapped to a number of property channel layers (here, denoted as σ and ε); filters are used to produce a convolved image; and the result is flattened to produce the feature vector.
Here, we use M = 2, where one channel provides the σi for each bead and the second channel provides the εii for each bead. Because all σi are equivalent, this channel indicates whether a CG bead exists at the given location. Because all the properties are already of order one, no normalization was applied. In principle, additional properties could be provided as additional channels.
In all cases, featurized inputs are based on either a repeating subunit of the polymer or the entire polymer sequence. For example, class I polymers can be defined using a CRU of four CUs, which is represented as a 40-bit OHE vector. Alternatively, featurizing the entire sequence would yield a 400-bit OHE vector. Handling the stochastic sequences of the class II polymers requires the latter approach.
Details of ML architectures, hyperparameters, and training
The regression models in this study all have the same basic architecture and differ only based on the input featurization. Little effort was expended on hyperparameter optimization, because our emphasis is on evaluating the overall viability of the design approach and not on the development of the best regression model. Hence, all regression models have the same final three layers: two hidden, fully connected layers with 20 neurons preceding a single output neuron for the predictions of 〈Rg2〉.
The input to the final DNN just described is a vector with dimensionality and origin determined based on the featurization technique. Three cases are considered. In the first, a 40-bit OHE vector representing a 4-CU CRU is supplied directly to the DNN; this is the case for the results in Fig. 3A. In the second, a 400-bit OHE vector representing the entire polymer sequence is supplied to a bidirectional LSTM recurrent neural network, the result of which is an 80-bit vector that feeds into the DNN; this is the approach to obtain the results in Fig. 3B. In the third, an Nbb × 3 × 2 array representing the entire polymer sequence is supplied to a 2D-CNN with eight 3 × 3 filters and unit stride length, the result of which is an Nbb × 3 × 8 array that is further flattened to a 9600-bit feature vector that feeds into the DNN; this is the approach for the regression model used to propose the candidates in Fig. 5.
The DNN weights are initially set using LeCun normal initialization, and the network is trained using the Nesterov-accelerated adaptive moment estimation algorithm (41) (learning rate = 0.001, β1 = 0.9, β2 = 0.999) to minimize the MAE of the training set predictions on 〈Rg2〉; the batch size is set to 32. Exponential linear units (42) are used as activation functions in all DNN layers except the final layer. For training, the output is standard-normalized. To limit potential overfitting, an early stopping procedure was implemented to halt training if the MAE on a validation set (20% of the training data) ceased to improve over an interval of 50 training epochs; after training is halted, the weights of the model corresponding to the smallest observed MAE are used. When training on class I polymers, the dataset is augmented to also include cyclic and inverse permutations as inputs with the same output data.
Keras (43) and scikit-learn (44) are used to implement all ML methods. The Hyperopt package (45) is used for implementation of the TPE optimization algorithm (33). Python scripts demonstrating the construction and training of the regression models are provided in the Supplementary Materials.
Data coloring by composition
To facilitate visual identification of possible compositional trends underlying the organization of the data, we devised a data coloring scheme, which is used in Figs. 3 and 5. In short, the marker colors in Fig. 3 and the violin colors in Fig. 5 are obtained as a weighted average of five colors assigned to the bead types (α, β, γ, and δ) or the lack of a bead. With RGB values ranging from 0 to 255 expressed as the triple [R,G,B], we assign the color cα ≡ [3,95,220] to α, cβ ≡ [255,145,3] to β, cγ ≡ [112,255,3] to γ, cδ ≡ [145,3,255] to δ, and c∅ [255,255,255] to no bead present (∅). The colors for the four bead types approximate a rectangular tetrad, and the scheme as a whole is shown at the bottom right of Fig. 5. Therefore, for a sequence with Nbb backbone beads, the color is given by
| (8) |
where Ni denotes the number of beads (or lack thereof) of the ith type in the polymer sequence.
Description of simple linear mixing models
Three simple linear mixing models are proposed as an alternative to ML for predicting the properties of stochastic polymer sequences. Let a given polymer sequence with N CUs be defined as PN = (R1, R2, ..., RN), with Rk indicating one of the 10 CUs in Fig. 1A. The sequences of regular polymers can be specified by replication of a CRU of length m, Pm, such that its full sequence is given by (Pm)N/m, presuming that N mod n = 0. Then, the predictions for linear mixing models are obtained as
| (9) |
where m is an integer specific to the model, the CRU Pim is given by (Ri, ..., Ri + m−1) with Rk taken from the sequence PN, and the notation 〈Rg2|PN〉 indicates the 〈Rg2〉 for the polymer with sequence PN. In Table 1, m = 1 for model A, m = 2 for model B, and m = 4 for model C.
Supplementary Material
Acknowledgments
Funding: This work is supported by the Department of Energy, Basic Energy Sciences, Materials Science and Engineering Division. The development of models and simulation strategies such as those described here for high–molecular weight biopolymers is supported by Solvay. The computational resources required for this work were provided by the LCRC of Argonne National Laboratory and the GM4 cluster at the University of Chicago; the GM4 cluster is supported by the National Science Foundation’s Division of Materials Research under the Major Research Instrumentation (MRI) program award #1828629. The development of software was supported by the Midwest Center for Computational Materials (MICCOM), which is funded by the Department of Energy, Basic Energy Sciences, Materials Science and Engineering Division. N.E.J. thanks the Maria Goeppert Mayer Named Fellowship from Argonne National Laboratory for support. Author contributions: M.A.W. and J.J.d.P. conceived and designed the approach for generating and using the simulation dataset for ML-enhanced design. M.A.W. prepared, executed, and analyzed the simulations and ML models with input from N.E.J. and J.J.d.P. M.A.W. and P.S.G. implemented and analyzed the linear mixing models. M.A.W., N.E.J., and J.J.d.P. prepared the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All datasets used in this work are available as Supplementary Materials along with example scripts demonstrating the construction of the ML models. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc6216/DC1
REFERENCES AND NOTES
- 1.Sanchez-Lengeling B., Aspuru-Guzik A., Inverse molecular design using machine learning: Generative models for matter engineering. Science 361, 360–365 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Butler K. T., Davies D. W., Cartwright H., Isayev O., Walsh A., Machine learning for molecular and materials science. Nature 559, 547–555 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Meredig B., Agrawal A., Kirklin S., Saal J. E., Doak J. W., Thompson A., Zhang K., Choudhary A., Wolverton C., Combinatorial screening for new materials in unconstrained composition space with machine learning. Phys. Rev. B. 89, 094104 (2014). [Google Scholar]
- 4.Piñeros W. D., Lindquist B. A., Jadrich R. B., Truskett T. M., Inverse design of multicomponent assemblies. J. Chem. Phys. 148, 104509 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Gómez-Bombarelli R., Aguilera-Iparraguirre J., Hirzel T. D., Duvenaud D., Maclaurin D., Blood-Forsythe M. A., Chae H. S., Einzinger M., Ha D.-G., Wu T., Markopoulos G., Jeon S., Kang H., Miyazaki H., Numata M., Kim S., Huang W., Hong S. I., Baldo M., Adams R. P., Aspuru-Guzik A., Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 15, 1120–1127 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Coley C. W., Green W. H., Jensen K. F., Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 51, 1281–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Gómez-Bombarelli R., Wei J. N., Duvenaud D., Hernández-Lobato J. M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T. D., Adams R. P., Aspuru-Guzik A., Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iovanac N. C., Savoie B. M., Improved chemical prediction from scarce data sets via latent space enrichment. J. Phys. Chem. A 123, 4295–4302 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Kim C., Chandrasekaran A., Jha A., Ramprasad R., Active-learning and materials design: The example of high glass transition temperature polymers. MRS Commun. 9, 860–866 (2019). [Google Scholar]
- 10.Ferguson A. L., Machine learning and data science in soft materials engineering. J. Phys. Condens. Matter 30, 043002 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Jackson N. E., Webb M. A., de Pablo J. J., Recent advances in machine learning towards multiscale soft materials design. Curr. Opin. Chem. Eng. 23, 106–114 (2019). [Google Scholar]
- 12.Sherman Z. M., Howard M. P., Lindquist B. A., Jadrich R. B., Truskett T. M., Inverse methods for design of soft materials. J. Chem. Phys. 152, 140902 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Afzal M. A. F., Haghighatlari M., Ganesh S. P., Cheng C., Hachmann J., Accelerated discovery of high-refractive-index polyimides via First-Principles molecular modeling, virtual high-throughput screening, and data mining. J. Phys. Chem. C 123, 14610–14618 (2019). [Google Scholar]
- 14.Shmilovich K., Mansbach R. A., Sidky H., Dunne O. E., Panda S. S., Tovar J. D., Ferguson A. L., Discovery of self-assembling π-conjugated peptides by active learning-directed coarse-grained molecular simulation. J. Phys. Chem. B 124, 3873–3891 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Xie T., France-Lanord A., Berkley A., Johnson J. A., Shao-Horn Y., Grossman J. C., Toward designing highly conductive polymer electrolytes by machine learning assisted coarse-grained molecular dynamics. Chem. Mater. 32, 4144–4151 (2020). [Google Scholar]
- 16.Oliver S., Zhao L., Gormley A. J., Chapman R., Boyer C., Living in the fast lane—High throughput controlled/living radical polymerization. Macromolecules 52, 3–23 (2019). [Google Scholar]
- 17.Upadhya R., Murthy N. S., Hoop C. L., Kosuri S., Nanda V., Kohn J., Baum J., Gormley A. J., PET-RAFT and SAXS: High throughput tools to study compactness and flexibility of single-chain polymer nanoparticles. Macromolecules 52, 8295–8304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin T.-S., Coley C. W., Mochigase H., Beech H. K., Wang W., Wang Z., Woods E., Craig S. L., Johnson J. A., Kalow J. A., Jensen K. F., Olsen B. D., BigSMILES: A structurally-based line notation for describing macromolecules. ACS Cent. Sci. 5, 1523–1531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peerless J. S., Milliken N. J. B., Oweida T. J., Manning M. D., Yingling Y. G., Soft matter informatics: Current progress and challenges. Adv. Theory Simul. 2, 1800129 (2019). [Google Scholar]
- 20.Audus D. J., de Pablo J. J., Polymer informatics: Opportunities and challenges. ACS Macro Lett. 6, 1078–1082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mannodi-Kanakkithodi A., Pilania G., Huan T. D., Lookman T., Ramprasad R., Machine learning strategy for accelerated design of polymer dielectrics. Sci. Rep. 6, 20952 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim C., Chandrasekaran A., Huan T. D., Das D., Ramprasad R., Polymer genome: A data-powered polymer informatics platform for property predictions. J. Phys. Chem. C 122, 17575–17585 (2018). [Google Scholar]
- 23.John P. C. S., Phillips C., Kemper T. W., Wilson A. N., Guan Y., Crowley M. F., Nimlos M. R., Larsen R. E., Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 150, 234111 (2019). [DOI] [PubMed] [Google Scholar]
- 24.de Pablo J. J., Coarse-grained simulations of macromolecules: From DNA to nanocomposites. Annu. Rev. Phys. Chem. 62, 555–574 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Zhang L., Han J., Wang H., Car R., E W., DeePCG: Constructing coarse-grained models via deep neural networks. J. Chem. Phys. 149, 034101 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Olsson S., Wehmeyer C., Pérez A., Charron N. E., de Fabritiis G., Noé F., Clementi C., Machine learning of coarse-grained molecular dynamics force fields. ACS Cent. Sci. 5, 755–767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson N. E., Bowen A. S., de Pablo J. J., Efficient multiscale optoelectronic prediction for conjugated polymers. Macromolecules 53, 482–490 (2020). [Google Scholar]
- 28.Perry S. L., Sing C. E., 100th anniversary of macromolecular science viewpoint: Opportunities in the physics of sequence-defined polymers. ACS Macro Lett. 9, 216–225 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Panganiban B., Qiao B., Jiang T., DelRe C., Obadia M. M., Nguyen T. D., Smith A. A. A., Hall A., Sit I., Crosby M. G., Dennis P. B., Drockenmuller E., de la Cruz M. O., Xu T., Random heteropolymers preserve protein function in foreign environments. Science 359, 1239–1243 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Kahovec J., Fox R. B., Hatada K., Nomenclature of regular single-strand organic polymers (IUPAC Recommendations 2002). Pure Appl. Chem 74, 1921 (2019). [Google Scholar]
- 31.R. G. Larson, The Structure and Rheology of Complex Fluids (OUP, 1999). [Google Scholar]
- 32.Altintas O., Barner-Kowollik C., Single-chain folding of synthetic polymers: A critical update. Macromol. Rapid Commun. 37, 29–46 (2016). [DOI] [PubMed] [Google Scholar]
- 33.J. Bergstra, R. Bardenet, Y. Bengio, B. Kégl, in Proceedings of the 24th International Conference on Neural Information Processing Systems (NIPS’11) (Curran Associates Inc., 2011), pp. 2546–2554. [Google Scholar]
- 34.Zheng W., Dignon G., Brown M., Kim Y. C., Mittal J., Hydropathy patterning complements charge patterning to describe conformational preferences of disordered proteins. J. Phys. Chem. Lett. 11, 3408–3415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemke T., Peter C., Neural network based prediction of conformational free energies—A new route toward coarse-grained simulation models. J. Chem. Theory Comput. 13, 6213–6221 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Webb M. A., Delannoy J.-Y., de Pablo J. J., Graph-based approach to systematic molecular coarse-graining. J. Chem. Theory Comput. 15, 1199–1208 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Chakraborty M., Xu C., White A. D., Encoding and selecting coarse-grain mapping operators with hierarchical graphs. J. Chem. Phys. 149, 134106 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Wang W., Gómez-Bombarelli R., Coarse-graining auto-encoders for molecular dynamics. npj Comput. Mater. 5, 125 (2019). [Google Scholar]
- 39.Jackson N. E., Bowen A. S., Antony L. W., Webb M. A., Vishwanath V., de Pablo J. J., Electronic structure at coarse-grained resolutions from supervised machine learning. Sci. Adv. 5, eaav1190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plimpton S., Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995). [Google Scholar]
- 41.T. Dozat, Incorporating Nesterov Momentum into Adam (ICLR Workshop, 2016), pp. 2013–2016. [Google Scholar]
- 42.D.-A. Clevert, T. Unterthiner, S. Hochreiter, Fast and accurate deep network learning by exponential linear units (ELUs). arXiv:1511.07289 [cs.LG] (23 November 2015).
- 43.F. Chollet, keras (GitHub, 2015); https://github.com/fchollet/keras.
- 44.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay E., Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 45.J. Bergstra, D. Yamins, D. D. Cox, in Proceedings of the 30th International Conference on International Conference on Machine Learning—Volume 28 (ICML’13) (JMLR.org, 2013), pp. I–115–I–123. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc6216/DC1