

The FAM171A2 gene is a key regulator of progranulin expression and modifies the risk of multiple neurodegenerative diseases.
Abstract
Progranulin (PGRN) is a secreted pleiotropic glycoprotein associated with the development of common neurodegenerative diseases. Understanding the pathophysiological role of PGRN may help uncover biological underpinnings. We performed a genome-wide association study to determine the genetic regulators of cerebrospinal fluid (CSF) PGRN levels. Common variants in region of FAM171A2 were associated with lower CSF PGRN levels (rs708384, P = 3.95 × 10−12). This was replicated in another independent cohort. The rs708384 was associated with increased risk of Alzheimer’s disease, Parkinson’s disease, and frontotemporal dementia and could modify the expression of the FAM171A2 gene. FAM171A2 was considerably expressed in the vascular endothelium and microglia, which are rich in PGRN. The in vitro study further confirmed that the rs708384 mutation up-regulated the expression of FAM171A2, which caused a decrease in the PGRN level. Collectively, genetic, molecular, and bioinformatic findings suggested that FAM171A2 is a key player in regulating PGRN production.
INTRODUCTION
Progranulin (PGRN) is a secreted pleiotropic glycoprotein expressed in the central nervous system (CNS) and peripheral tissues. Its deficiency has been associated with the development of frontotemporal dementia (FTD) (1), Alzheimer’s disease (AD) (2), and Parkinson’s disease (PD) (3). Loss-of-function animal models revealed that potential pathogenesis mechanisms may involve neuroinflammation (4), autophagy (5), and cell signaling. PGRN overexpression provided protection against pathological protein deposition and toxicity and inhibited phenotypic progression in AD- and PD-like disease models (3, 6). Recently, PGRN in cerebrospinal fluid (CSF) is found to be increased during the course of AD (7). Together, these lines of evidence indicate that PGRN dysregulation might be involved in the pathogenesis of neurodegenerative diseases and could be a promising therapeutic target. Mounting evidence illustrates that PGRN levels might be modified by genetic variants (8, 9), including the encoding gene GRN, which could partially explain variability of the protein (10). Previous genome-wide screens have identified multiple loci outside the GRN [SORT1 (8) and PSRC1 (9)] as regulators of blood PGRN levels. Nonetheless, to date, no genome-wide association study (GWAS) has been conducted to reveal the genetic modulators of PGRN in CNS. The search for CNS PGRN regulators is of tremendous importance, especially considering that (i) PGRN levels might be regulated differently in the peripheral blood and CNS (11) and (ii) increasing PGRN levels as a neuroprotective approach might be hindered by its peripheral effects of promoting carcinogenesis (12) and obesity (13). This study aimed to identify genetic modifiers of CSF PGRN levels. We first conducted a GWAS of CSF PGRN levels from 1362 adults without dementia and successfully identified an independent locus within the FAM171A2 gene region that showed genome-wide significant association with CSF PGRN levels. Then, we used bioinformatics and cellular approaches to annotate the functionality of the identified variant and investigate its potential mechanisms underlying neurodegenerative diseases. On the basis of these findings, we propose that the FAM171A2 gene is a key regulator of CSF PGRN expression and modifies risk of neurodegenerative diseases.
RESULTS
This study aimed to detect genetic modifiers of CSF PGRN levels and better understand the role of PGRN in biological pathways relevant to neurodegenerative diseases. To achieve this goal, we analyzed genetic data and CSF PGRN levels from 432 individuals without dementia from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) in a GWAS. Then, we sought to replicate our findings in an independent cohort composed of 930 Chinese adults without dementia.
CSF PRGN levels were influenced by sex, educational level, and APOE ε4
In the GWAS cohort, CSF PGRN levels were significantly influenced by sex (P = 3.87 × 10−5, b = 0.003) and potentially influenced by APOE ε4 allele (P = 0.08, b = −8.73 × 10−4) and educational level (P = 0.12, b = −1.77 × 10−4). CSF PGRN levels showed no association with either age (P = 0.48) or clinical diagnosis [healthy controls (HC) versus subjects with mild cognitive impairment (MCI) subjects, P = 0.21].
Common variants in FAM171A2 gene were associated with CSF PGRN levels
A total of 432 individuals without dementia (157 HC and 275 subjects with MCI) participated in the GWAS. Age (P = 1.38 × 10−6) and APOE ε4 carrier percentage (P = 1.37 × 10−6) differed between the two diagnostic groups (Table 1). After adjusting for sex, educational level, APOE ε4 allele, and the first three principal components, no inflation due to population stratification was identified (genomic inflation factor λ = 1.012). A total of 18 single-nucleotide polymorphisms (SNPs) (5 in FAM171A2, 12 in ITGA2B, and rs5848 in GRN) on chromosome 17 were significantly associated with CSF PGRN levels, with rs708384 (genotyped) in FAM171A2 gene yielding the most significant signal (P = 5.09 × 10−12) (Table 2 and Fig. 1A). Located in the transcription factor (TF) binding site (intron 1) of FAM171A2, rs708384 also survived the permutation-based empiric corrections for multiple testing (empiric P = 0.0002; permutation-based corrected empiric family-wise error rate controlled at 0.0002) (Fig. 1B).
Table 1. Characteristics of discovery and replication samples.
CV, coefficient of variation.
| ADNI (discovery sample) | CABLE (replication sample) | ||||||
| Baseline diagnosis | Total | HC | MCI | Total | AA | AC | CC |
| n | 432 | 157 | 275 | 930 | 93 | 392 | 445 |
| Age, years, mean ± SD | 72.8 ± 6.8 | 74.7 ± 5.5 | 71.8 ± 7.3 | 62.6 ± 10.4 | 62.1 ± 9.9 | 62.9 ± 10.2 | 62.4 ± 10.7 |
| Male/female, n | 252/180 | 85/72 | 167/108 | 380/550 | 46/47 | 157/235 | 177/268 |
| Education, years | 16.2 ± 2.8 | 16.4 ± 2.8 | 16.0 ± 2.8 | 9.7 ± 5.2 | 10.6 ± 10.1 | 9.5 ± 4.5 | 9.7 ± 4.2 |
| APOE Ɛ4 carrier, % | 37.5% | 22.3% | 46.5% | 16.3% | 15.05% | 17.98% | 16.34% |
| CSF PGRN, mean ± SD, pg/ml | 1600.3 ± 612 | 1551.2 ± 368.6 | 1628.4 ± 713.9 | 1739 ± 372 | 1687 ± 453 | 1696 ± 335 | 1787 ± 380 |
| CV of CSF PGRN, mean ± SD, % | 2.3 ± 1.7 | 2.4 ± 1.9 | 2.2 ± 1.7 | 2.95 ± 2.69 | 2.74 ± 2.40 | 2.90 ± 2.71 | 3.04 ± 2.72 |
Table 2. SNPs significantly (P < 5 × 10−8) associated with CSF PGRN levels.
CHR, chromosome; BP, base pair; Alt, altered allele; UTR, untranslated region; NA, not available.
| CHR | SNP |
LD (r2) |
BP (GRCh37) |
Alt |
Observed MAF |
Source |
Closest gene |
SNP location |
Regulome DB score* |
P values† | |||
| P1 | P2 | P3 | P4 | ||||||||||
| 17 | rs708384‡ | 1 | 42437682 | A | 0.4074 | Genotyping | FAM171A2 |
Intron (TF binding site) |
5 | 5.09 × 10−12 | 0.0002 | 0.0002 | … |
| 17 | rs708383‡ | 1 | 42437754 | A | 0.4074 | Imputation | FAM171A2 | Intron | 5 | 5.09 × 10−12 | 0.0002 | 0.0002 | … |
| 17 | rs34603233 | NA | 42455022 | T | 0.4201 | Imputation | ITGA2B | Intron | 5 | 6.48 × 10−12 | 0.0002 | 0.0002 | 0.4561 |
| 17 | rs850730 | 0.89 | 42454463 | C | 0.4213 | Imputation | ITGA2B | Intron | 5 | 1.90 × 10−11 | 0.0002 | 0.0002 | 0.8554 |
| 17 | rs850729 | 0.88 | 42454806 | T | 0.4213 | Imputation | ITGA2B | Intron | NA | 1.90 × 10−11 | 0.0002 | 0.0002 | 0.8554 |
| 17 | rs850736 | 0.93 | 42446420 | C | 0.4225 | Imputation | ITGA2B | NA | NA | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs850734 | 0.91 | 42449025 | G | 0.4225 | Imputation | ITGA2B | NA | 5 | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs5910 | 0.90 | 42449789 | A | 0.4225 | Genotyping | ITGA2B | Synonymous | 5 | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs35905006 | NA | 42451281 | C | 0.4225 | Imputation | ITGA2B | Intron | 6 | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs850733 | 0.89 | 42451305 | A | 0.4225 | Imputation | ITGA2B | Intron | NA | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs5911 | 0.89 | 42453065 | C | 0.4225 | Imputation | ITGA2B | Missense | 2b | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs850732 | 0.89 | 42454243 | T | 0.4225 | Imputation | ITGA2B | Intron | NA | 2.47 × 10−11 | 0.0002 | 0.0002 | 0.9572 |
| 17 | rs708382 | 0.86 | 42442344 | C | 0.4340 | Genotyping | FAM171A2 | NA | 6 | 2.91 × 10−11 | 0.0002 | 0.0002 | 0.7185 |
| 17 | rs850737 | 0.86 | 42443784 | C | 0.4340 | Imputation | FAM171A2 | NA | 5 | 2.91 × 10−11 | 0.0002 | 0.0002 | 0.7185 |
| 17 | rs850738 | 0.73 | 42434630 | G | 0.4317 | Imputation | FAM171A2 | Intron | NA | 4.89 × 10−11 | 0.0002 | 0.0004 | 0.2513 |
| 17 | rs11389756 | NA | 42448210 | AG | 0.4699 | Imputation | ITGA2B | NA | 6 | 3.62 × 10−11 | 0.0002 | 0.0022 | 0.6897 |
| 17 | rs850731 | 0.71 | 42454270 | C | 0.4699 | Imputation | ITGA2B | Intron | 5 | 3.62 × 10−11 | 0.0002 | 0.0022 | 0.6897 |
| 17 | rs5848 | 0.60 | 42430244 | T | 0.3275 | Imputation | GRN | 3′-UTR | 5 | 1.22 × 10−11 | 0.0002 | 0.0074 | 0.6925 |
*Lower scores indicate increasing evidence for a variant to be located in a functional region (www.regulomedb.org).
†P1 = P value for GWAS; P2 = empiric P value; P3 = permutation-based corrected empiric familywise error rate; P4 = empiric P value after controlling for rs708384
‡rs708383 is in complete LD with rs708384 (r2 = 1) according to the HaploRegv4.1 online database.
Fig. 1. GWAS results and regional plots for associations with CSF PGRN levels.
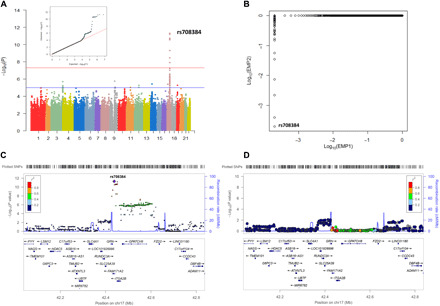
(A) Manhattan plots [showing the −log10 (P value) for individual SNP] and q-q plot. (B) Association results after permutation test. EMP1, empiric P value; EMP2, permutation-based corrected empiric family-wise error rate. (C) Regional association results for the GRN-FAM171A2-ITGA2B region. (D) Regional association results after controlling for rs708384.
In addition to the loci in FAM171A2, 13 SNPs in ITGA2B and GRN also showed significant associations with CSF PGRN levels (empiric P = 0.0002; permutation-based corrected empiric family-wise error rate controlled at <0.005). Notably, eight loci showed high linkage disequilibrium (LD) (r2 ≥ 0.8) with rs708384. After controlling for the rs708384 genotype, the association signals for all loci were markedly attenuated (P > 0.1), indicating that the relationships in these loci were driven by rs708384 (Table 2 and Fig. 1, C and D). The analysis also identified 149 suggestive loci; 92% were located in FAM171A2, ITGA2B, and GPATCH8 genes of chromosome 17. However, they did not survive after permutation-based empiric corrections (table S1). Accordingly, rs708384 in FAM171A2 gene was selected as the top SNP, for which the minor allele (A allele, MAF = 0.4074) was significantly linked to lower CSF PGRN levels in a dose-dependent manner (P = 3.95 × 10−12) (Fig. 2A).
Fig. 2. CSF PGRN levels as a function of rs708384 genotype in two samples.
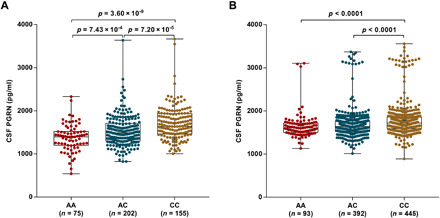
(A) The minor allele (A allele, MAF = 0.41) of rs708384 was significantly associated with lower CSF levels of PGRN in a dose-dependent manner. (B) CSF PGRN levels were compared across the AA, AC, and CC genotypes of rs708384 in a larger independent cohort of 930 nondemented Chinese participants to validate the initial observed top signal. A significant association of decreasing CSF PGRN levels with increased minor allele (A) dose of rs708384 was observed, independent of age, gender, education, APOE ε4 genotype, MMSE score at baseline, CV, and rs5848 genotype.
Subgroup and sensitivity analyses
In the subgroup analysis, the minor allele (A) of rs708384 was associated with lower CSF PGRN levels in different strata according to sex (male, P = 2.67 × 10−6; female, P = 1.76 × 10−6) and baseline diagnosis (HC, P = 0.017; MCI, P = 1.36 × 10−11). Notably, the effect size drastically increased in the HC population (beta for HC = 100.9; beta for MCI = 8.77 × 10−5) (fig. S2A). As CSF PGRN levels were reported to be influenced by age, diagnosis, and rs5848 in GRN gene (11), sensitivity analysis was performed to further adjust these confounders. Modifying for age and baseline diagnosis did not significantly change the results (P = 3.51 × 10−12). After adjusting for rs5848, a strong, although less significant, association for rs708384 with CSF PGRN levels was found (P = 8.28 × 10−5) (fig. S2B). Excluding those who developed AD within 3 years from baseline did not change the result, and rs708384 remained the top SNP (n = 359, P = 4.73 × 10−10) (fig. S2C). According to Aβ pathology status, subgroup analysis showed the same dose-response relationship for both groups (P < 5.00 × 10−4 for Aβ-negative group, P = 1.07 × 10−7 for Aβ-positive group). Furthermore, we added the rs5848 genotype, which did not pass the GWAS quality control (QC) stage, as a covariate in the regression model. After adjusting for the rs5848 genotype and other covariates mentioned above, the association of PGRN levels with rs708384 remained significant (n = 337; P = 0.0009).
Specificity of the association with PGRN
We tested whether the observed association may be influenced by other candidate proteins related to neurodegeneration. First, the significance remained unchanged after including CSF levels of Aβ42, T-tau, P-tau, α-synuclein (α-SYN), neurofilament light chain (NFL), and secreted triggering receptor expressed on myeloid cells 2 (sTREM2) as covariates. Next, no correlations with Aβ42 or P-tau biomarkers were identified (r2 ranging from 0.04 to 0.1) although weak correlations were observed of CSF PGRN with T-tau (P = 0.04, r = 0.1), α-SYN (P = 0.0003, r = 0.36), NFL (P = 0.03, r = 0.22), and sTREM2 (P = 1.134 × 10−8, r = 0.27) levels. Furthermore, no associations of rs708384 were revealed with any of the abovementioned proteins when they were separately used as the endophenotype. Together, these results supported the specificity of our results (table S2).
Variability in CSF PGRN levels explained by genetic variants
We used a genome-partitioning analysis to estimate the proportion of variance in CSF PGRN levels explained by chromosomes, significant genes, and loci. All genotyped and imputed variants on chromosome 17 can explain approximately 17.4% of the variability in the CSF PGRN levels (P < 0.05) (Fig. 3A). Most significant loci were in high LD with rs708384 (r2 ≥ 0.8) (Fig. 3B). The SNPs in the GRN region explained 13.3% of the variability in CSF PGRN levels (P = 8.13 × 10−8), suggesting that genetic variants other than the GRN gene also played significant roles. Rs708384 was identified to explain 9.1% of the variability with the smallest P value (P = 2.60 × 10−10) (Fig. 3C). Although rs5848 was not genotyped in ADNI, the 1000 Genomes Project data indicated that rs5848 has only low-to-moderate LD (r2 = 0.6), with rs708384 in the CEU (Utah residents with Northern and Western European ancestry) population (Fig. 3D) and no loci showed LD with rs5848. Thus, it can be reasonably implied that the observed associations for other significant loci were more likely to be influenced by rs708384.
Fig. 3. Variability in CSF PGRN levels explained by genetic variants.
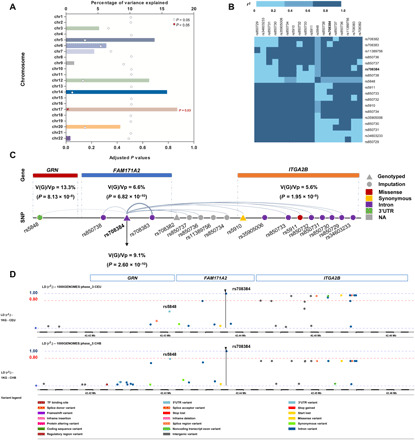
(A) Chromosome 17 explained approximately 17.4% of the variability in the CSF levels of PGRN. (B) Most of the significant loci were in LD with rs708384. (C) SNPs in the GRN region explained the most but not all of the variability in CSF PGRN. Analysis of FAM171A2 and ITGA2B region showed that these two regions explained 9.1 and 5.6% of the variability in CSF levels of PGRN, respectively. rs708384 was shown to explain 9.1% of the variability. (D) rs5848 was only in low-to-moderate LD with rs708384 in CEU population (r2 = 0.6), and no loci were in LD with rs5848. Nonetheless, rs5848 was in high LD with rs708384 in CHB population (r2 ≈ 0.8).
Replication in an independent cohort and meta-analysis
To validate our finding that rs708384, as the top signal, was associated with CSF PGRN levels, we measured CSF PGRN levels and genotyped rs708384 in a larger, independent cohort of 930 Northern Han Chinese individuals without dementia (Table 1). In this cohort, the participants’ age ranged from 40 to 88 years (mean = 62.6 years, SD = 10.4 years) at the time of CSF extraction. Both sexes were well-represented, including 380 men and 550 women. The mean CSF PGRN levels was 1739 pg/ml (SD = 372 pg/ml), with mean intrabatch coefficient of variation (CV) of 2.95% and mean interbatch CV of 3.82%. Consistent with the result of the discovery cohort, the A allele of rs708384 was significantly associated with lower CSF PGRN levels in a linear regression model accounting for age, sex, educational level, APOE ε4 genotype, MMSE (mini-mental state examination) score at baseline, and CV (P = 7.47 × 10−9, Fig. 2B).
To exclude the potential influence of rs5848 in the GRN gene, we genotyped and included it as a covariate. Single regression analysis indicated that rs5848 was significantly associated with CSF PGRN levels (P < 0.005). However, the significance of rs5848 was no longer present (P = 0.88) when rs708384 was added as a covariate. In contrast, the association of rs708384 with the CSF PGRN level still reached borderline significance when controlling for rs5848 (P = 0.10). This may be because rs5848 showed relatively high but not complete LD with rs708384 in the Chinese population (r2 ≈ 0.8, Fig. 3D). The additive model (AA versus CC) indicated that the association for rs708384 remained significant (P < 0.05) after controlling for rs5848 and other covariates (table S3). These results demonstrated that the influence of rs708384 on the CSF PGRN level was not determined by rs5848. The meta-analysis of the two-stage findings further strengthened the association of rs708384 with the CSF PGRN level (P = 1.74 × 10−18). We identified that the loci previously associated with blood PGRN levels had no significant relationships with CSF PGRN level, suggesting that the genetic mechanisms of PGRN regulation may be different in the peripheral system and CNS (table S3).
Functional annotation of the top signal in the FAM171A2 gene region
Enhancer enrichment analysis showed that the tagged variants by rs708384 were significantly enriched in specific brain regions (e.g., middle hippocampus, inferior temporal lobe, and prefrontal lobe) (table S4). This suggests that the variants might be linked to the regulation of gene expression in these sites. We also found significant differential expression of FAM171A2 in the temporal cortex (P = 0.004) in AD (fig. S3). Moreover, the expression quantitative trait loci (eQTL) analyses showed that rs708384 (z = 4.74; P = 2.5 × 10−6) and some of its tagged variants can potentially influence GRN expression in both the blood (table S5) and specific brain regions (anterior cingulate cortex and frontal cortex) (table S6). In addition to GRN, rs708384 was also associated with the expression of FAM171A2 and ITGA2B in multiple sites, such as the frontal lobe (table S7). Together, the evidence points to the hypothesis that rs708384, a TF binding site, may functionally modulate the expression of GRN and its downstream gene FAM171A2. We identified that the expression of GRN was highly correlated with that of FAM171A2 (P = 1.1 × 10−17), in either the brain (P = 5.3 × 10−10) or blood (P = 3.1 × 10−6).
FAM171A2 and GRN genes were clustered in common pathways
There was a significant excess of enriched categories: 96 categories in the Protein Analysis Through Evolutionary Relationships (PANTHER) analysis and 107 categories in the Consensus PathDB (CPDB) analysis. There were 34 significant categories that overlapped in the two analyses (table S8). These categories were primarily related to regulation of nervous system development, molecular transport, signal transduction, cell-to-cell adhesion, and response to stimuli (Fig. 4A). The gene network analysis revealed a total of 31 significant categories (P < 0.001) forming four clusters of genes (Fig. 4B). As expected, the most significant genes (GRN, FAM171A2, and ITGA2B) were clustered together (Fig. 4B). The Gene Ontology (GO) items for this specific cluster included all the abovementioned categories (P < 0.001) and another two items: regulation of blood pressure (P = 3.84 × 10−4) and interleukin-8 secretion (P = 4.20 × 10−4).
Fig. 4. GO and pathway analysis.
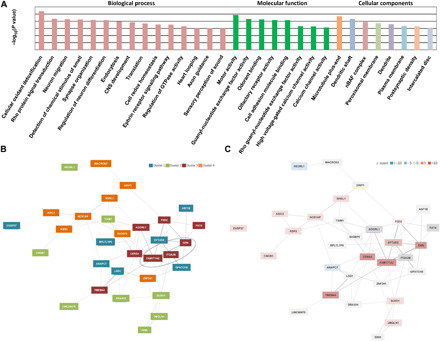
(A) Functional categories were identified that were significantly enriched (P < 0.001), primarily including those involving regulation of nervous system development, molecular transport, signal transduction, cell-cell adhesion, and response to stimuli. GTPase, guanosine triphosphatase. (B) Four clusters were identified in the gene network analysis. The most significant genes (FAM171A2 and GRN) were clustered together. (C) The highest z-scores were achieved by FAM171A2 and GRN. z-score was used by GeneNetwork Assisted Diagnostic Optimization to prioritize the candidate genes: Gene with a higher z-score is more likely to explain the phenotype.
Among 16 pathways [12 from REACTOME and 4 from Kyoto Encyclopedia of Genes and Genomes (KEGG)] that achieved the statistical significance level (P < 0.005), the L1 cell adhesion molecule (L1CAM) interaction pathway was the most significant (REACTOME, P = 2.49 × 10−6), for which the highest z-scores were achieved by FAM171A2 and GRN gene (Fig. 4C). Most other significant pathways were also primarily related to cell-to-cell interactions (table S9).
FAM171A2 considerably expressed in the cerebral vascular endothelium and microglia
It was essential to determine the location of FAM171A2 in the brain before investigating its functions. Therefore, we conducted immunohistochemistry (IHC) and immunofluorescence (IF) staining on brain sections (cortex and hippocampus) of 10-week-old male C57BL/6 mice. Evident DAB (Diaminobenzidine) staining was observed around the vessels (Fig. 5, A1 and B1, tilted arrows) and on cells in a similar form to microglia (Fig. 5, A2 and B2, horizontal arrows). The IF staining of FAM171A2 colocalized with CD31 and IBA1 on the brain sections (Fig. 5, C1, C2, D1, and D2). Five independent experiments were conducted, and the abovementioned results indicate that FAM171A2 was considerably expressed in the cerebral vascular endothelium and microglia.
Fig. 5. FAM171A2 high expression on cerebral vascular endothelium and microglia.
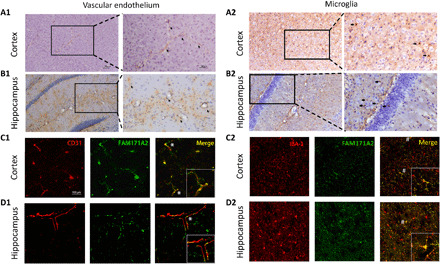
(A and B) The IHC staining of FAM171A2 on mouse cortex and hippocampi. The DAB staining along and around the cerebral vascular (A1 and B1) was marked by tilted arrows. The DAB staining on the cells in a similar form of microglia (A2 and B2) was marked by horizontal arrows. (C and D) The IF staining of FAM171A2 with the CD31 and IBA1 antibodies on mouse cortex and hippocampus. Their colocalization was marked by “*” and “#.” n = 5 mice in these experiments.
Targeting rs708384 regulates expression of FAM171A2 and GRN genes in vitro
First, we conducted dual-luciferase reporter assay in human embryonic kidney 293 (HEK293) cells to evaluate the regulation of rs708384 to FAM171A2 (Fig. 6A). A total of four independent experiments were conducted. We observed that the c > a mutation significantly improved the ratio of firefly/Renilla luciferase reporter gene expression compared to the wild type. This indicates that c > a stimulated the expression of FAM171A2 (Fig. 6B).
Fig. 6. rs708384 stimulates FAM171A2 expression and subsequently inhibits GRN/PGRN level.
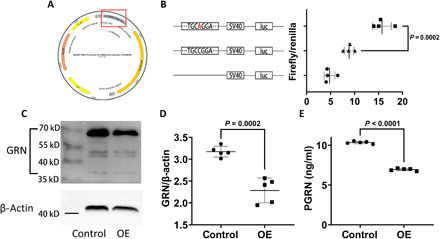
(A) The structure of firefly luciferase reporter plasmid. The sequence containing rs708384 was labeled by a red square. (B) The plasmids with or without rs708384 (c > a), including the empty control, had different expressing levels of firefly luciferase after transfection into the HEK293 cells. The luminous intensity was calibrated by Renilla luciferase, and the result was presented by ratio of firefly/Renilla luciferase. n = 4 per group, the data were analyzed by the one-way analysis of variance (ANOVA) (P < 0.0001) followed by the Tukey post hoc test (P = 0.0002 c > a versus wild type). (C and D) The change of intracellular GRN after FAM171A2 overexpression. n = 5 per group, the data were analyzed by t test (P = 0.0002). (E) The change of the supernatant PGRN levels by FAM171A2 overexpression (OE). n = 5 per group, the data were analyzed by t test (P < 0.0001).
Next, we examined the influences of FAM171A2 overexpression on GRN/PGRN levels. Cultured human umbilical vein endothelial cells were used, and five independent experiments were conducted. A significant decrease in the intracellular GRN level (Fig. 6, C and D) and the supernatant PGRN level (Fig. 6E) was observed after FAM171A2 overexpression, which was consistent with the GWAS data. Meanwhile, FAM171A2 was knocked down by small interfering RNA (siRNA), and we observed a significant increase in the intracellular GRN level (fig. S4, A and B). The efficiency of FAM171A2 siRNA and overexpression were verified by Western blot analysis (fig. S4, C and D). These results showed that FAM171A2 overexpression led to decreased GRN/PGRN levels, while knockdown had the opposite effect.
Impact of rs708384 on phenotypes of neurodegenerative disease
Last, we tested whether rs708384 had relationships with Aβ pathologic features, CSF biomarkers of neuronal injury, cognitive functions, brain structure/metabolism using the ADNI database, and the risk of neurodegenerative diseases [including AD, PD, amyotrophic lateral sclerosis (ALS), and FTD] using previous published GWAS summary statistics. The “AA” genotype of the rs708384 was significantly linked to higher CSF tau levels (fig. S5A) and poorer global cognition (fig. S5, B and C). There were no significant influences of rs708384 genotypes on Aβ pathologic features, CSF P-tau levels, and brain structure/metabolism of the regions of interest. Ten variants were tagged by rs708384 with r2 ≥ 0.8. In the large-scale GWAS dataset, these loci were significantly related to AD, PD, and FTD risk, with effect allele (A) of rs708384 associated with increased risks [P = 0.01 for AD (14), P = 1.42 × 10−5 for PD (15), and P = 0.04 for FTD (16)]. No significant relationships of rs708384 or its tagged loci were found for ALS (table S10).
DISCUSSION
It has previously been shown that loci on genes other than GRN could modify blood PGRN levels (8, 9). However, no previous GWAS has been conducted to report the genetic modifiers of PGRN in CSF. In the present study, we conducted a genome-wide analysis of CSF PGRN levels and observed an independent signal in the FAM171A2 gene. Collectively, evidence from genetic, molecular, and bioinformatic findings suggests that the FAM171A2 gene is a key regulator of PGRN production.
There were several sources of evidence that support the identified genome-wide locus as a real and specific signal and not an artificial type I error. First, rs708384 showed a highly significant P value and survived the permutation-based empiric corrections for multiple testing (Fig. 1). Moreover, the locus was directly genotyped, eliminating the possibility that the signal was a result of an imputation error. Second, the association was successfully replicated in a larger, independent sample. Third, both bioinformatics and in vitro evidence confirmed that rs708384 could modulate the expression of the GRN and FAM171A2 genes. Fourth, FAM171A2 and GRN were closely connected and may functionally act together in the human brain. Last, the genotype of rs708384 was associated with the CSF biomarker and risk of neurodegenerative diseases. The finding was consistent with that of a recent study in which PGRN deficiency was reported to increase AD risk by influencing tau rather than Aβ pathology (17).
It has recently been found that PGRN expression was up-regulated in patients with AD compared to controls (18). Thus, participants with dementia were excluded from the present analysis to preclude the potential selection bias due to case ascertainment. Moreover, sensitivity or subgroup analysis showed that the results were significant in both Aβ-positive and Aβ-negative groups. Subgroup analysis indicated that the influence of rs708384 on CSF PGRN levels was vastly greater in HCs than in patients with MCI.
GRN and FAM171A2 are clustered in common pathways that are potentially involved in regulating CSF PGRN levels. The most significant is L1CAM interactions. LICAM is an axonal glycoprotein that is important for neurite outgrowth and neuronal survival (19). Although previous animal studies depicted this molecule to reduce AD pathology (20), it is still unclear how it contributes to the development of neurodegenerative diseases via interaction with PGRN. In addition, our GO analyses included categories related to oxidative stress, transport system of neurons, sensory perception of smell and sound, and endocytosis. Previous studies demonstrated that all these biological processes were associated with neurodegenerative diseases, especially AD, PD, and FTD. Future research is necessary to explore the roles of PGRN in these processes and how these may influence the progression of neurodegenerative diseases.
Several limitations should also be noted. The validation cohort is based on Han Chinese individuals recruited from the hospital, which leads to constrained generalizability. Future studies are required to replicate the association in community-based cohorts of other races. As a critical covariate in the GWAS stage, rs5848, which did not pass the QC filter, was imputed but not genotyped. We provided multiple forms of evidence to lower this risk: (i) rs5848 was in low-to-moderate LD with rs708384 in the CEU population, suggesting that the potential influences were limited; (ii) the consistency test showed a high imputation accuracy (n = 427, k = 0.874) for the rs5848 locus; and (iii) we found that the association of rs708384 with the CSF PGRN level was only slightly influenced by genotyped rs5848, in either ADNI or Chinese Alzheimer’s Biomarker and LifestylE (CABLE). The in vitro evidence suggests that rs708384 could influence expression of FAM171A2, which showed close relationships with expression of the GRN gene. However, the specific mechanisms have yet to be studied. In the following research, we plan to further investigate the specific mechanisms of FAM171A2 to regulate PGRN. We will also focus on the role of FAM171A2 in the expression of PGRN within microglia and various models of neurodegeneration diseases. Further investigation is needed to determine whether the effect size of the top locus on CSF PGRN varies by the diagnostic group, although the present study focused on individuals without dementia.
Therefore, we found evidence to show that FAM171A2, downstream of GRN, is a novel genetic regulator of PGRN production. Future studies are warranted to fully understand FAM171A2 function in the brain and the mechanisms for which FAM171A2 affects PGRN levels. Targeting FAM171A2 may potentially modify neurodegenerative disease risk by regulating brain PGRN levels.
MATERIALS AND METHODS
Participants for GWAS
In the discovery cohort, 432 (HC = 157, MCI = 275) non-Hispanic white nondemented individuals were included from the Alzheimer’s Disease Neuroimaging Initiative GO/2 (ADNI-GO/2). Table 1 showed the summarized characteristics of this sample. Data used in the preparation for GWAS were obtained from the ADNI database (adni.loni.usc.edu). The original cohort with CSF PGRN and GWAS data comprised 508 participants. We confined our analysis to non-Hispanic white individuals (n = 434) to constrain the risk of bias from population stratification. We tested unanticipated duplicates and cryptic relatedness among samples using pairwise genome-wide estimates of proportion identity-by-descent by the PLINK software (beta 6.4) (21). The genome-wide complex trait analysis (GCTA) was used to calculate principal component and confirm the ethnicity of the samples (22). Two outlier participants were removed, and 432 were finally included (fig. S1). The study was approved by institutional review boards of all participating institutions, and written informed consent was obtained from all participants or authorized representatives.
CSF PGRN measurements
CSF PGRN levels were measured by a previously reported sandwich immunoassay (7) using the Meso Scale Discovery platform (23). All CSF samples were distributed randomly across plates and measured in duplicate. All the antibodies and plates were from a single lot to exclude variability between batches. Experiments were performed by experienced operators blinded to clinical information. The mean intrabatch CV was 2.2% (with all duplicate measures presented CV < 15%) and the mean interbatch CV was 4.21%. PGRN levels were corrected by interbatch variation, and corrected values were used for analyses.
Genotyping and imputation
The ADNI-GO/2 samples were genotyped by the Human OmniExpress BeadChip (Illumina Inc., San Diego, CA). Before association analysis, all samples and genotypes underwent stringent QC with the following criteria: call rates for SNPs > 95%, call rates for individuals > 95%, minor allele frequencies > 0.2, and Hardy-Weinberg equilibrium test P > 0.001. The original dataset included a total of 710,618 genotyped variants, including rs7412 and rs429358, which were genotyped separately by an APOE genotyping kit to define the APOE ε2/ε3/ε4 isoforms, as previously described (24). Following the standard procedure, imputation was conducted using Beagle software (version 5.0) with the HapMap GRCh37 as reference. SNPs with a Beagle R2 < 0.8, a call rate < 95%, a minor allele frequency < 0.2, and Hardy-Weinberg equilibrium test P < 0.001 were removed. Last, the filters and QC produced a total of 3,262,988 imputed and genotyped SNPs for analyses.
Statistical methods
Because the CSF PGRN values were skewed (Shapiro-Wilk test P < 0.05), data transformation was performed to achieve a normal distribution using “car” package of R. A linear regression model was used to determine the associations of CSF PGRN levels with genetic polymorphisms using the PLINK v1.9 with an additive genetic model. Stepwise linear regression analysis was used to determine whether CSF levels of PGRN were influenced by diagnosis, age, gender, education, APOE ε4 allele, and CV of PGRN measurements. Those with P < 0.2 were included as covariates. To correct for confounding by genetic ancestry that could lead to population stratification, the first three principal components of a genetic relationship matrix between pairs of individuals were further included as covariates. Given that 30% of nondemented elders met the biomarker criteria for AD (25), subgroup analyses were first conducted for Aβ pathological status (A positive versus A negative) according to A-T-N criteria, for which A positive was defined as positive evidence of cerebral Aβ deposition as detected by PET (AV45 > 1.11) or CSF (Aβ < 192 pg/ml) (26). We further excluded those who progressed to dementia within 3 years since baseline. We also performed sensitivity analyses including rs5848 (GRN, imputed), age, and diagnosis as covariates because it was previously shown that levels of PGRN might be influenced by these factors (11, 18). Analyses stratified according to gender and diagnoses were conducted to examine the strata effect.
The specificity of the association with top signal (the SNP with the smallest P value) was explored to examine whether the association was influenced by other CSF proteins linked with neurodegenerative diseases, including Aβ, p-tau181 (P-tau), total tau (T-tau) (27), α-SYN (28), NFL (29, 30), and sTREM2 (31, 32). The measurement data of all these proteins were downloaded from ADNI: (i) The associations of top signal with these proteins were separately explored via single variable linear regression; (ii) the relationships between normalized levels of CSF PGRN and other CSF proteins were estimated using Pearson’s product moment correlation coefficient; (iii) these CSF proteins were respectively included as covariates in the model. Statistical analyses and data visualization were performed in R v3.5.1, PLINK v1.9 (21), GCTA v1.91.6beta (33), and LocusZoom v1.3 (34).
Bonferroni-corrected statistical significance was defined as P < 5 × 10−8, and P < 1 × 10−5 was considered suggestive association. To identify additional independent genetic signals, conditional analyses were performed by adding the top SNP as a covariate and testing all remaining regional SNPs for association. As an additional alternative to exclude possible false-positive results, the PLINK max (T) permutation test with 5000 permutations was used to generate empiric P values for multiple testing correction. Genome-wide associations were visualized with the R package “qqman”; genome partitioning was conducted using the algorithm GCTA to estimate the proportion of phenotypic variance explained (35).
Replication cohort
We used an independent replication cohort from nondemented northern Han Chinese elders who were derived from CABLE study. The study has been approved by the Institutional Review Board of Qingdao Municipal Hospital. Since 2017, CABLE is an ongoing large-scale study majorly focused on AD’s risk factors and biomarkers in Chinese Han population. CABLE is aimed to determine the genetic and environmental modifiers of AD biomarkers and their utility in early diagnosis. Individuals were recruited at Qingdao Municipal Hospital, Shandong Province, China.
All enrolled participants were Han Chinese aged between 40 and 90 years. The exclusion criteria include the following: (i) CNS infection, head trauma, epilepsy, multiple sclerosis, or other major neurological disorders; (ii) major psychological disorders; (iii) severe systemic diseases (e.g., malignant tumors); (iv) family history of genetic diseases. All participants underwent clinical and neuropsychological assessments, biochemical testing, as well as blood and CSF sample collection. Demographic information and medical history were collected via a structured questionnaire and an electronic medical record system.
CSF PGRN levels in CABLE were determined with the Human PGRN Enzyme-Linked Immunosorbent Assay (ELISA) kit (Biovendor-Laboratornimedicina A.s. Czech Republic) on the microplate reader (Thermo Fisher Scientific Multiskan MK3). Samples were diluted 10-fold and run in duplicate. The association for the most significant locus (top SNP) in the discovery stage was chosen for validation. Several loci were selected for genotyping with restriction fragment length polymorphism technology, including rs708384 (top SNP for validation), rs5848 (important confounder in GRN), two loci related to APOE ε4 status (rs7412 and rs429358), and three loci (rs660240, rs4747197, and rs646776) associated with blood PGRN by previous GWAS. Until October 2019, eligible measurements of CSF PGRN and genotyping have been available for 930 nondemented individuals. Data analysis was performed with linear regression via R, adjusting for age, sex, education, APOE ε4 status, MMSE at baseline, and CV of PGRN measurements. Sensitivity analysis was conducted by including rs5848 as covariate. Meta-analysis was performed to pool the P values of the two-stage findings using “metap” in Stata/SE 12.0 software. In addition, we also examined whether the loci associated with blood PGRN also influenced the levels in CSF.
Bioinformatics analyses
SNP annotations were conducted using the NCBI Database of SNPs (dbSNP, GRCh37/hg19 assembly, 105 release) (www.ncbi.nlm.nih.gov/SNP/) (36), SNPnexus (www.snp-nexus.org) (37), and SNP and CNV Annotation Database (SCAN) (www.scandb.org/newinterface/index_v1.html). The potential regulatory functions were examined using HaploReg v4.1 (38), RegulomeDBv1.1(build 141 of dbSNP) (39), and 1000 Genomes Project (www.internationalgenome.org/). LD analyses were conducted on the basis of data from the 1000 Genomes Project [EUR and Han Chinese in Beijing, China (CHB)]. Variants tagged by the top SNP were used to perform the following enhancer enrichment analysis and expression analysis.
Enhancer enrichment analysis was conducted via HaploReg v4.1 to evaluate in which cell types the tag variants were significantly enriched. Enhancers were defined using Roadmap Epigenomics data with four different methods (including the 15-state core model, the 25-state model incorporating imputed epigenomes, the H3K4me1/H3K4me3 peaks, and the H3K27ac/H3K9ac peaks) (40). A binomial test was performed using all 1000 Genomes variants with a MAF > 5% in any population as the background set. A total of 28 blood cells and 13 brain cells were selected for the present analysis.
To validate the result of enhancer enrichment analysis and characterize these associations, expression analyses were performed to determine (i) whether the expression levels of genes where the identified significant loci are located are associated with AD case-control status. The differential expression of identified genes in AD blood (41) and brain (42) (fig. S3) was investigated by analyzing the gene expression profiles from Gene Expression Omnibus (GEO) via NCBI web tool GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r/) and AlzData web server (www.alzdata.org) (43). P < 0.05 after adjustment for multiple testing was regarded as statistically significant (44); (ii) whether SNPs associated with CSF PGRN also affect the protein-encoding gene (GRN) expression levels in brain and blood; (iii) whether the significant SNPs were associated with expression levels of the candidate genes within each locus. To these end, eQTL analyses were conducted using multiple publicly available datasets in human brain tissues [including brain expression GWAS eQTL datasets (45) and Genotype-Tissue Expression (GTEx) (46)] and the whole blood [including Blood eQTL browser (47), Consortium for the Architecture of Gene Expression browser (48), NCBI Molecular QTL Browser Search database (49), and Framingham Heart Study eQTL database (49)]. The eQTL results were also searched from 1000 Genomes Project. (iv) Pair-wise gene expression correlation analysis was performed using public RNA sequencing data of 31,499 samples (https://genenetwork.nl) and GTEx expression data via GEPIA web server (http://gepia.cancer-pku.cn/index.html) (50).
GO overrepresentation analyses and pathway analyses
The Association List GO Annotator method (51) was used to search for the GO terms and KEGG pathways. SNPs were clumped if they were within 1 Mb and in high LD (r2 ≥ 0.8) with the index SNP (defined as P < 1.0 × 10−4). These SNPs were mapped to genes using SNP annotation tools mentioned above. SNPs were mapped to a gene if they were situated within 20 kb of that gene; genes were only counted once irrespective of how many SNPs were mapped to the gene. Last, 34 genes were included.
GO overrepresentation analyses were performed using the PANTHER statistical overrepresentation test v9.0 (www.pantherdb.org/) (52) and the CPDB overrepresentation gene set analysis Release 33 (http://cpdb.molgen.mpg.de/) (53). Both used data from the GO Consortium (www.geneontology.org) and calculated overrepresentation of candidate genes, relative to different background (18,043 genes for CPDB and 20,814 genes for PANTHER). Of the 34 genes in the analysis, CPDB recognized all and 29 were assigned to at least one GO term by PANTHER. The resulting P values are corrected for multiple testing using the false discovery rate method in CPDB (P < 0.05). As for PANTHER, Bonferroni-adjusted (six separate tests) P values were used (P < 8.33 × 10−3).
In addition, multiple sources of pathway sets were used, including KEGG (1 August 2018, www.genome.jp/kegg/) (54), PANTHER v8.1 (52), Reactome pathways, v65 (13 June 2018, https://reactome.org/) (55), WikiPathways (www.wikipathways.org/index.php/WikiPathways), and Prioritization And Functional Assessment (http://159.226.67.237:8080/pafa) (56). Last, the function of gene network was predicted on the basis of the functional enrichment of its coregulatory partners using the GeneNetwork Assisted Diagnostic Optimization tool (www.genenetwork.nl).
In vitro and in vivo experiments
Luciferase reporter plasmid and antibodies
The plasmid was purchased from Genomeditech Co. Ltd. Shanghai, China. Briefly, we cloned an about 500-bp region of the FAM171A2 intron 1 containing SNP rs708384 or wild-type site and inserted the cloned fragment into a pGL3-promoter vector, which contained the SV40 promoter upstream of the firefly luciferase reporter gene. Empty pGL3-promoter vector was transferred as a systemic control. Antibodies used were as follows: anti-FAM171A2 antibody (1:20 for IHC and IF; ATLAS, HPA019770); anti-FAM171A2 antibody (1:500 for Western blot; Proteintech, 20836-1-AP); anti-CD31 antibody (1:250 for IF; ab24590), anti-granulin antibody (1:1000 for Western bolt; ab108608); anti–β-actin antibody (1:1000 for Western bolt; Affinity, AF7018).
Animals
Eight-week-old C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd. and housed in 23° ± 1°C, 55 ± 5% humidity and 12-hour light/dark cycles (lights on between 07:00 and 19:00) with libitum food and water. Mice were euthanized for IHC and IF staining (n = 5) after adaptation for 2 weeks. The study was performed with the approval of the Institutional Animal Care and Use Committee of Fudan university (approval number: JS-256). The experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory animals.
FAM171A2 knockdown and overexpression
The in vitro FAM171A2 knockdown was achieved by siRNA, purchased from GenePharma Co. Ltd, Shanghai, China. Sequence: 5′-GCAAUGGCACUGGUGUAAUTT, AUUACACCAGUGCCAUUGCTT-3′. The FAM171A2 overexpression plasmid was purchased from GenePharma. The pEX-3 vector was used for plasmid construction. Both FAM171A2 siRNA and plasmid were transferred into human umbilical vein endothelial cells (HUVECs) by Lipofectamine2000. The effect of knockdown and overexpression was confirmed 48 hours after transfection.
Cell culture
The HUVECs we used were purchased from iCell, Shanghai, China. This cell line was immortalized by lentiviral transformation in 2018 from human primary umbilical vein endothelial cells, which was confirmed by CD31 IF staining. HUVECs was cultured in extracellular matrix + 5% fetal bovine serum (FBS) + 1% endothelial growth factor + 1% penicillin/streptomycin (P/S). The HEK293 cell was purchased from Chinese Academy of Sciences Cell Bank. The cells were cultured in Dulbecco’s modified Eagle’s medium + 5% FBS + 1% P/S. The incubator maintained an environment of 37°C and 5% CO2. Cells were subcultured by 1:3 until a convergence of 80%.
Dual-luciferase reporter assay
The assay was conducted according to the technical manual of Promega Dual-Luciferase Reporter Assay System. The firefly and Renilla luciferase reporter plasmid (5:1) was cotransferred into HEK293 cells by Lipofectamine2000. After 24 hours, cells were homogenized by the passive lysate buffer provided by the manufacture. Luciferase Assay Reagent II and Stop and Glo Reagent were added to the lysates sequentially. Luminous intensity was detected by the Synergy H1 hybrid reader using the luminescence model.
Immunohistochemistry
Mice was injected by pentobarbital sodium into deep anesthesia and perfused with saline solution followed by 4% (w/v) paraformaldehyde. Hippocampi and cortex were frontally sectioned using a freezing microtome, and 30-μm serial sections were collected. Sections were incubated in blocking buffer [0.01% Triton X-100 and 20% normal goat serum in phosphate-buffered saline, (pH 7.4)] for 1 hour and were subsequently incubated at 4°C overnight in rabbit anti-FAM171A2 primary antibody, followed by 2 hours in secondary biotinylated antibody, at room temperature. After three washes, sections were incubated in avidin-biotin complex (ABC Standard, Vector Laboratories) and color developed by using 0.025% 3,3′-diaminobenzidine and 0.1% H2O2. Then, the sections were stuck on microslides and immersed into hematite dyeing solution for 1 min followed by deionized water washes. After treated with ethanol and dimethylbenzene, the sections were covered by neutral gum and slips. An Olympus camera (DP72; Olympus) was used to take images.
IF staining
The process was similar to IHC. The brain sections were incubated by the rabbit anti-FAM171A2 antibody together with the mouse anti-CD31 antibody and shaken overnight. After washing, sections were immersed into goat anti-rabbit Alex Fluor 488 and goat anti-mouse Alex Fluor 647 (FAM171A2 and CD31) or donkey anti-rabbit Alex Fluor 488 and donkey anti-goat Alex Fluor 594 (FAM171A2 and IBA1) for 2 hours, followed by washing and imaged using the Olympus FV10 laser scanning confocal microscope.
Western blotting analysis
Thirty-microgram protein for each sample was added to a lane of 10% SDS–polyacrylamide gel electrophoresis gels. After electrophoresis, polyvinylidene difluoride membranes were transferred by constant current of 250 mA for 90 min, and then blocked with 5% bovine serum albumin for 1 hour, and incubated in the rabbit anti-granulin antibody overnight. After washing, membranes were incubated with horseradish peroxidase–conjugated secondary antibodies for 2 hours. The blots were visualized using the Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific Inc.). The grayscale was measured by ImageJ. All experiments were performed in triplicate. The final data were expressed as a ratio of the relative optical density (OD) of the protein of interest to that of β-actin.
Enzyme-linked immunosorbent assay
We used the R&D PGRN Quantikine ELISA kit to detect the supernatant PGRN content. The experiment was conducted according to the manual of the ELISA kit. Briefly, 50 μl of cell lysate was added per well and incubated for 2 hours at room temperature. A 200-μl human PGRN conjugate was added to each well after washing for four times with 400 μl of wash buffer per well and incubated for 2 hours at room temperature. A 200-μl substrate solution was added to each well subsequently and incubated for 30 min at room temperature. Last, the reaction was ended by 50 μl of stop solution per well. Within 30 min, the OD of each sample was detected at 450 nm and corrected by wavelength at 540 nm. PGRN contents were calculated from OD according to protein standards.
Statistics
The data were presented in the means ± SEM. GraphPad Prism 8 software (version 8.0.2, GraphPad Software Inc., La Jolla, CA, USA) was used to test the difference via the one-way analysis of variance (ANOVA) followed by the Tukey post hoc analysis and unpaired t test. P < 0.01 was considered statistically significant.
Associations of the top signal with neurodegenerative diseases
The influences of the top SNP on cognition, brain Aβ deposition, FDG, and volume/thickness of brain regions of interest (hippocampus, parahippocampus, posterior cingulate, precuneus, cuneus, entorhinal cortex, and middle temporal region) were further explored using ADNI. Those who developed AD in the first 3 years were excluded. Linear regression models were conducted to determine the associations with the above indexes at baseline. Subgroup analysis based on the diagnosis (HC versus MCI) was conducted. R packages “lm” was used to perform the above analyses. In addition, previous GWAS for AD (14), PD (15, 57) (www.pdgene.org/), ALS (58), and FTD (16, 59–62) was searched for associations of the top signal with risk of neurodegenerative diseases.
Supplementary Material
Acknowledgments
We thank all the participants and their families, as well as many institutions and their staff that provided support for all studies involved in this collaboration. We also thank the participants, researchers, ADNI, and staff associated with many other studies from which we used data for this report. This work was made possible by the generous sharing of GWAS summary statistics. We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. The in vitro and in vivo experiments were conducted in the center laboratory of Jing’an District Central Hospital. Funding: This study was supported by grants from the National Natural Science Foundation of China (91849126), the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (no.2018SHZDZX03) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. The i-Select chips were funded by the French National Foundation on Alzheimer’s disease and related disorders. EADI was supported by the LABEX (laboratory of excellence program investment for the future) DISTALZ grant, Inserm, Institut Pasteur de Lille, Université de Lille 2, and the Lille University Hospital. GERAD was supported by the Medical Research Council (grant 503480), Alzheimer’s Research UK (grant 503176), the Wellcome Trust (grant 082604/2/07/Z), and German Federal Ministry of Education and Research (BMBF): Competence Network Dementia (CND) grants 01GI0102, 01GI0711, and 01GI0420. CHARGE was partly supported by the NIH/NIA grant R01 AG033193 and the NIA AG081220 and AGES contract N01-AG-12100, the NHLBI grant R01 HL105756, the Icelandic Heart Association, and the Erasmus Medical Center and Erasmus University. ADGC was supported by the NIH/NIA grants U01 AG032984, U24 AG021886, and U01 AG016976, and the Alzheimer’s Association grant ADGC-10-196728. Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co. Inc.; Meso Scale Diagnostics LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Author contributions: W.X. and J.-T.Y. analyzed the GWAS data, performed bioinformatics analyses, and wrote the manuscript. S.-D.H. and C.M. designed and performed cell-based studies. C.Z. helped design in vitro studies and draft the manuscript. W.X., J.-Q.L., C.-C.T., and H.-Q.L. performed the QC for the GWAS data. W.X., J.-Q.L., C.-C.T., Q.D., L.T., and J.-T.Y. provided CSF PGRN and genetic material for the replication analyses. W.X., J.-Q.L., and C.-C.T. measured and QCed the CSF PGRN. ADNI provided data. Q.D., L.T., and J.-T.Y. supervised and wrote the project. All authors read and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All ADNI data are available through the LONI Image and Data Archive (IDA) and interested scientists may apply for access on the ADNI website (http://adni.loni.usc.edu/). IGAP database URL: http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php. PDGene database URL: www.pdgene.org/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb3063/DC1
REFERENCES AND NOTES
- 1.Zhou X., Sun L., Brady O. A., Murphy K. A., Hu F., Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol. Commun. 5, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minami S. S., Min S.-W., Krabbe G., Wang C., Zhou Y., Asgarov R., Li Y., Martens L. H., Elia L. P., Ward M. E., Mucke L., Farese R. V. Jr., Gan L., Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20, 1157–1164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Kampen J. M., Baranowski D., Kay D. G., Progranulin gene delivery protects dopaminergic neurons in a mouse model of Parkinson’s disease. PLOS ONE 9, e97032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martens L. H., Zhang J., Barmada S. J., Zhou P., Kamiya S., Sun B., Min S.-W., Gan L., Finkbeiner S., Huang E. J., Farese R. V. Jr., Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 122, 3955–3959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang M. C., Srinivasan K., Friedman B. A., Suto E., Modrusan Z., Lee W. P., Kaminker J. S., Hansen D. V., Sheng M., Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 214, 2611–2628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Kampen J. M., Kay D. G., Progranulin gene delivery reduces plaque burden and synaptic atrophy in a mouse model of Alzheimer’s disease. PLOS ONE 12, e0182896 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suárez-Calvet M., Capell A., Caballero M. Á. A., Morenas-Rodríguez E., Fellerer K., Franzmeier N., Kleinberger G., Eren E., Deming Y., Piccio L., Karch C. M., Cruchaga C., Paumier K., Bateman R. J., Fagan A. M., Morris J. C., Levin J., Danek A., Jucker M., Masters C. L., Rossor M. N., Ringman J. M., Shaw L. M., Trojanowski J. Q., Weiner M., Ewers M., Haass C.; Dominantly Inherited Alzheimer Network; Alzheimer’s Disease Neuroimaging Initiative , CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol. Med. 10, e9712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrasquillo M. M., Nicholson A. M., Finch N. C., Gibbs J. R., Baker M., Rutherford N. J., Hunter T. A., De Jesus-Hernandez M., Bisceglio G. D., Mackenzie I. R., Singleton A., Cookson M. R., Crook J. E., Dillman A., Hernandez D., Petersen R. C., Graff-Radford N. R., Younkin S. G., Rademakers R., Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am. J. Hum. Genet. 87, 890–897 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tönjes A., Scholz M., Krüger J., Krause K., Schleinitz D., Kirsten H., Gebhardt C., Marzi C., Grallert H., Ladenvall C., Heyne H., Laurila E., Kriebel J., Meisinger C., Rathmann W., Gieger C., Groop L., Prokopenko I., Isomaa B., Beutner F., Kratzsch J., Fischer-Rosinsky A., Pfeiffer A., Krohn K., Spranger J., Thiery J., Blüher M., Stumvoll M., Kovacs P., Genome-wide meta-analysis identifies novel determinants of circulating serum progranulin. Hum. Mol. Genet. 27, 546–558 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Meeter L. H. H., Patzke H., Loewen G., Dopper E. G. P., Pijnenburg Y. A. L., van Minkelen R., van Swieten J. C., Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement. Geriatr. Cogn. Dis. Extra 6, 330–340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson A. M., Finch N. C. A., Thomas C. S., Wojtas A., Rutherford N. J., Mielke M. M., Roberts R. O., Boeve B. F., Knopman D. S., Petersen R. C., Rademakers R., Progranulin protein levels are differently regulated in plasma and CSF. Neurology 82, 1871–1878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Y., Zheng L., Zhang W., Feng T., Liu J., Wang X., Yu Y., Qi M., Zhao W., Yu X., Tang W., Growth factor progranulin contributes to cervical cancer cell proliferation and transformation in vivo and in vitro. Gynecol. Oncol. 134, 364–371 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Qu H., Deng H., Hu Z., Plasma progranulin concentrations are increased in patients with type 2 diabetes and obesity and correlated with insulin resistance. Mediators Inflamm. 2013, 360190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., De Stafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., Russo G., Thorton-Wells T. A., Jones N., Smith A. V., Chouraki V., Thomas C., Ikram M. A., Zelenika D., Vardarajan B. N., Kamatani Y., Lin C. F., Gerrish A., Schmidt H., Kunkle B., Dunstan M. L., Ruiz A., Bihoreau M. T., Choi S. H., Reitz C., Pasquier F., Cruchaga C., Craig D., Amin N., Berr C., Lopez O. L., De Jager P. L., Deramecourt V., Johnston J. A., Evans D., Lovestone S., Letenneur L., Morón F. J., Rubinsztein D. C., Eiriksdottir G., Sleegers K., Goate A. M., Fiévet N., Huentelman M. W., Gill M., Brown K., Kamboh M. I., Keller L., Barberger-Gateau P., McGuiness B., Larson E. B., Green R., Myers A. J., Dufouil C., Todd S., Wallon D., Love S., Rogaeva E., Gallacher J., St George-Hyslop P., Clarimon J., Lleo A., Bayer A., Tsuang D. W., Yu L., Tsolaki M., Bossù P., Spalletta G., Proitsi P., Collinge J., Sorbi S., Sanchez-Garcia F., Fox N. C., Hardy J., Naranjo M. C. D., Bosco P., Clarke R., Brayne C., Galimberti D., Mancuso M., Matthews F.; European Alzheimer’s Disease Initiative (EADI); Genetic, Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart, Aging Research in Genomic Epidemiology, Moebus S., Mecocci P., Del Zompo M., Maier W., Hampel H., Pilotto A., Bullido M., Panza F., Caffarra P., Nacmias B., Gilbert J. R., Mayhaus M., Lannefelt L., Hakonarson H., Pichler S., Carrasquillo M. M., Ingelsson M., Beekly D., Alvarez V., Zou F., Valladares O., Younkin S. G., Coto E., Hamilton-Nelson K. L., Gu W., Razquin C., Pastor P., Mateo I., Owen M. J., Faber K. M., Jonsson P. V., Combarros O., O’Donovan M. C., Cantwell L. B., Soininen H., Blacker D., Mead S., Mosley T. H. Jr., Bennett D. A., Harris T. B., Fratiglioni L., Holmes C., de Bruijn R. F., Passmore P., Montine T. J., Bettens K., Rotter J. I., Brice A., Morgan K., Foroud T. M., Kukull W. A., Hannequin D., Powell J. F., Nalls M. A., Ritchie K., Lunetta K. L., Kauwe J. S., Boerwinkle E., Riemenschneider M., Boada M., Hiltuenen M., Martin E. R., Schmidt R., Rujescu D., Wang L. S., Dartigues J. F., Mayeux R., Tzourio C., Hofman A., Nöthen M. M., Graff C., Psaty B. M., Jones L., Haines J. L., Holmans P. A., Lathrop M., Pericak-Vance M. A., Launer L. J., Farrer L. A., van Duijn C. M., Van Broeckhoven C., Moskvina V., Seshadri S., Williams J., Schellenberg G. D., Amouyel P., Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lill C. M., Lill C. M., Roehr J. T., McQueen M. B., Kavvoura F. K., Bagade S., Schjeide B.-M. M., Schjeide L. M., Meissner E., Zauft U., Allen N. C., Liu T., Schilling M., Anderson K. J., Beecham G., Berg D., Biernacka J. M., Brice A., De Stefano A. L., Do C. B., Eriksson N., Factor S. A., Farrer M. J., Foroud T., Gasser T., Hamza T., Hardy J. A., Heutink P., Hill-Burns E. M., Klein C., Latourelle J. C., Maraganore D. M., Martin E. R., Martinez M., Myers R. H., Nalls M. A., Pankratz N., Payami H., Satake W., Scott W. K., Sharma M., Singleton A. B., Stefansson K., Toda T., Tung J. Y., Vance J., Wood N. W., Zabetian C. P.; 23andMe; The Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium; The International Parkinson’s Disease Genomics Consortium (IPDGC); The Parkinson’s Disease GWAS Consortium; The Wellcome Trust Case Control Consortium (WTCCC), Young P., Tanzi R. E., Khoury M. J., Zipp F., Lehrach H., Ioannidis J. P. A., Bertram L., Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLOS Genet. 8, e1002548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rademakers R., Eriksen J. L., Baker M., Robinson T., Ahmed Z., Lincoln S. J., Finch N., Rutherford N. J., Crook R. J., Josephs K. A., Boeve B. F., Knopman D. S., Petersen R. C., Parisi J. E., Caselli R. J., Wszolek Z. K., Uitti R. J., Feldman H., Hutton M. L., Mackenzie I. R., Graff-Radford N. R., Dickson D. W., Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 17, 3631–3642 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi H., Klein Z. A., Bhagat S. M., Kaufman A. C., Kostylev M. A., Ikezu T., Strittmatter S. M.; Alzheimer’s Disease Neuroimaging Initiative , Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol. 133, 785–807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper Y. A., Nachun D., Dokuru D., Yang Z., Karydas A. M., Serrero G., Yue B.; Alzheimer’s Disease Neuroimaging Initiative, Boxer A. L., Miller B. L., Coppola G., Progranulin levels in blood in Alzheimer’s disease and mild cognitive impairment. Ann. Clin. Transl. Neurol. 5, 616–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutz D., Loers G., Kleene R., Oezen I., Kataria H., Katagihallimath N., Braren I., Harauz G., Schachner M., Myelin basic protein cleaves cell adhesion molecule L1 and promotes neuritogenesis and cell survival. J. Biol. Chem. 289, 13503–13518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djogo N., Jakovcevski I., Müller C., Lee H. J., Xu J.-C., Jakovcevski M., Kügler S., Loers G., Schachner M., Adhesion molecule L1 binds to amyloid beta and reduces Alzheimer’s disease pathology in mice. Neurobiol. Dis. 56, 104–115 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Chang C. C., Chow C. C., Tellier L. C., Vattikuti S., Purcell S. M., Lee J. J., Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price A. L., Patterson N. J., Plenge R. M., Weinblatt M. E., Shadick N. A., Reich D., Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Capell A., Liebscher S., Fellerer K., Brouwers N., Willem M., Lammich S., Gijselinck I., Bittner T., Carlson A. M., Sasse F., Kunze B., Steinmetz H., Jansen R., Dormann D., Sleegers K., Cruts M., Herms J., Van Broeckhoven C., Haass C., Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J. Neurosci. 31, 1885–1894 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S., Swaminathan S., Shen L., Risacher S. L., Nho K., Foroud T., Shaw L. M., Trojanowski J. Q., Potkin S. G., Huentelman M. J., Craig D. W., De Chairo B. M., Aisen P. S., Petersen R. C., Weiner M. W., Saykin A. J.; Alzheimer’s Disease Neuroimaging Initiative , Genome-wide association study of CSF biomarkers Aβ1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 76, 69–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider J. A., Aggarwal N. T., Barnes L., Boyle P., Bennett D. A., The neuropathology of older persons with and without dementia from community versus clinic cohorts. J. Alzheimers Dis. 18, 691–701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaw L. M., Vanderstichele H., Knapik-Czajka M., Clark C. M., Aisen P. S., Petersen R. C., Blennow K., Soares H., Simon A., Lewczuk P., Dean R., Siemers E., Potter W., Lee V. M.-Y., Trojanowski J. Q.; Alzheimer’s Disease Neuroimaging Initiative , Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsson A., Vanderstichele H., Andreasen N., De Meyer G., Wallin A., Holmberg B., Rosengren L., Vanmechelen E., Blennow K., Simultaneous measurement of β-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 51, 336–345 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Hong Z., Shi M., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Zabetian C. P., Leverenz J. B., Baird G., Montine T. J., Hancock A. M., Hwang H., Pan C., Bradner J., Kang U. J., Jensen P. H., Zhang J., DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133, 713–726 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nordlund A., Rolstad S., Hellström P., Sjögren M., Hansen S., Wallin A., The Goteborg MCI study: Mild cognitive impairment is a heterogeneous condition. J. Neurol. Neurosurg. Psychiatry 76, 1485–1490 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zetterberg H., Skillbäck T., Mattsson N., Trojanowski J. Q., Portelius E., Shaw L. M., Weiner M. W., Blennow K.; Alzheimer’s Disease Neuroimaging Initiative , Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 73, 60–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suárez-Calvet M., Kleinberger G., Caballero M. Á. A., Brendel M., Rominger A., Alcolea D., Fortea J., Lleó A., Blesa R., Gispert J. D., Sánchez-Valle R., Antonell A., Rami L., Molinuevo J. L., Brosseron F., Traschütz A., Heneka M. T., Struyfs H., Engelborghs S., Sleegers K., Van Broeckhoven C., Zetterberg H., Nellgård B., Blennow K., Crispin A., Ewers M., Haass C., sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 8, 466–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suárez-Calvet M., Morenas-Rodríguez E., Kleinberger G., Schlepckow K., Caballero M. Á. A., Franzmeier N., Capell A., Fellerer K., Nuscher B., Eren E., Levin J., Deming Y., Piccio L., Karch C. M., Cruchaga C., Shaw L. M., Trojanowski J. Q., Weiner M., Ewers M., Haass C.; Alzheimer’s Disease Neuroimaging Initiative , Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 14, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J., Lee S. H., Goddard M. E., Visscher P. M., GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pruim R. J., Welch R. P., Sanna S., Teslovich T. M., Chines P. S., Gliedt T. P., Boehnke M., Abecasis G. R., Willer C. J., LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J., Manolio T. A., Pasquale L. R., Boerwinkle E., Caporaso N., Cunningham J. M., de Andrade M., Feenstra B., Feingold E., Hayes M. G., Hill W. G., Landi M. T., Alonso A., Lettre G., Lin P., Ling H., Lowe W., Mathias R. A., Melbye M., Pugh E., Cornelis M. C., Weir B. S., Goddard M. E., Visscher P. M., Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 43, 519–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K., dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dayem Ullah A. Z., Oscanoa J., Wang J., Nagano A., Lemoine N. R., Chelala C., SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 46, W109–W113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward L. D., Kellis M., HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyle A. P., Hong E. L., Hariharan M., Cheng Y., Schaub M. A., Kasowski M., Karczewski K. J., Park J., Hitz B. C., Weng S., Cherry J. M., Snyder M., Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward L. D., Kellis M., HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett T., Wilhite S. E., Ledoux P., Evangelista C., Kim I. F., Tomashevsky M., Marshall K. A., Phillippy K. H., Sherman P. M., Holko M., Yefanov A., Lee H., Zhang N., Robertson C. L., Serova N., Davis S., Soboleva A., NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 41, D991–D995 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myers A. J., Gibbs J. R., Webster J. A., Rohrer K., Zhao A., Marlowe L., Kaleem M., Leung D., Bryden L., Nath P., Zismann V. L., Joshipura K., Huentelman M. J., Hu-Lince D., Coon K. D., Craig D. W., Pearson J. V., Holmans P., Heward C. B., Reiman E. M., Stephan D., Hardy J., A survey of genetic human cortical gene expression. Nat. Genet. 39, 1494–1499 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Xu M., Zhang D.-F., Luo R., Wu Y., Zhou H., Kong L.-L., Bi R., Yao Y.-G., A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement. 14, 215–229 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Benjamini Y., Heller R., Screening for partial conjunction hypotheses. Biometrics 64, 1215–1222 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Zou F., Chai H. S., Younkin C. S., Allen M., Crook J., Pankratz V. S., Carrasquillo M. M., Rowley C. N., Nair A. A., Middha S., Maharjan S., Nguyen T., Ma L., Malphrus K. G., Palusak R., Lincoln S., Bisceglio G., Georgescu C., Kouri N., Kolbert C. P., Jen J., Haines J. L., Mayeux R., Pericak-Vance M. A., Farrer L. A., Schellenberg G. D.; Alzheimer’s Disease Genetics Consortium, Petersen R. C., Graff-Radford N. R., Dickson D. W., Younkin S. G., Ertekin-Taner N., Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLOS Genet. 8, e1002707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.GTEx Consortium , Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westra H.-J., Peters M. J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M. W., Fairfax B. P., Schramm K., Powell J. E., Zhernakova A., Zhernakova D. V., Veldink J. H., Van den Berg L. H., Karjalainen J., Withoff S., Uitterlinden A. G., Hofman A., Rivadeneira F., `t Hoen P. A. C., Reinmaa E., Fischer K., Nelis M., Milani L., Melzer D., Ferrucci L., Singleton A. B., Hernandez D. G., Nalls M. A., Homuth G., Nauck M., Radke D., Völker U., Perola M., Salomaa V., Brody J., Suchy-Dicey A., Gharib S. A., Enquobahrie D. A., Lumley T., Montgomery G. W., Makino S., Prokisch H., Herder C., Roden M., Grallert H., Meitinger T., Strauch K., Li Y., Jansen R. C., Visscher P. M., Knight J. C., Psaty B. M., Ripatti S., Teumer A., Frayling T. M., Metspalu A., van Meurs J. B. J., Franke L., Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd-Jones L. R., Holloway A., Rae A. M., Yang J., Small K., Zhao J., Zeng B., Bakshi A., Metspalu A., Dermitzakis M., Gibson G., Spector T., Montgomery G., Esko T., Visscher P. M., Powell J. E., The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 100, 228–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joehanes R., Zhang X., Huan T., Yao C., Ying S.-x., Nguyen Q. T., Demirkale C. Y., Feolo M. L., Sharopova N. R., Sturcke A., Schäffer A. A., Heard-Costa N., Chen H., Liu P.-c., Wang R., Woodhouse K. A., Tanriverdi K., Freedman J. E., Raghavachari N., Dupuis J., Johnson A. D., O’Donnell C. J., Levy D., Munson P. J., Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 18, 16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z., GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holmans P., Green E. K., Pahwa J. S., Ferreira M. A. R., Purcell S. M., Sklar P.; Wellcome Trust Case-Control Consortium, Owen M. J., O’Donovan M. C., Craddock N., Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 85, 13–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mi H., Muruganujan A., Thomas P. D., PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kamburov A., Pentchev K., Galicka H., Wierling C., Lehrach H., Herwig R., ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 39, D712–D717 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M., KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., Kay S. M., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., D’Eustachio P., The Reactome pathway knowledgebase. Nucleic Acids Res. 42, D472–D477 (2016). [Google Scholar]
- 56.Zhou L., Zhao F., Prioritization and functional assessment of noncoding variants associated with complex diseases. Genome Med. 10, 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y., McKay J. D., Rafnar T., Wang Z., Timofeeva M. N., Broderick P., Zong X., Laplana M., Wei Y., Han Y., Lloyd A., Delahaye-Sourdeix M., Chubb D., Gaborieau V., Wheeler W., Chatterjee N., Thorleifsson G., Sulem P., Liu G., Kaaks R., Henrion M., Kinnersley B., Vallée M., Le Calvez-Kelm F., Stevens V. L., Gapstur S. M., Chen W. V., Zaridze D., Szeszenia-Dabrowska N., Lissowska J., Rudnai P., Fabianova E., Mates D., Bencko V., Foretova L., Janout V., Krokan H. E., Gabrielsen M. E., Skorpen F., Vatten L., Njølstad I., Chen C., Goodman G., Benhamou S., Vooder T., Välk K., Nelis M., Metspalu A., Lener M., Lubiński J., Johansson M., Vineis P., Agudo A., Clavel-Chapelon F., Bueno-de-Mesquita H. B., Trichopoulos D., Khaw K.-T., Johansson M., Weiderpass E., Tjønneland A., Riboli E., Lathrop M., Scelo G., Albanes D., Caporaso N. E., Ye Y., Gu J., Wu X., Spitz M. R., Dienemann H., Rosenberger A., Su L., Matakidou A., Eisen T., Stefansson K., Risch A., Chanock S. J., Christiani D. C., Hung R. J., Brennan P., Landi M. T., Houlston R. S., Amos C. I., Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 46, 736–741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lill C. M., Abel O., Bertram L., Al-Chalabi A., Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler. 12, 238–249 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Ferrari R., Grassi M., Salvi E., Borroni B., Palluzzi F., Pepe D., D’Avila F., Padovani A., Archetti S., Rainero I., Rubino E., Pinessi L., Benussi L., Binetti G., Ghidoni R., Galimberti D., Scarpini E., Serpente M., Rossi G., Giaccone G., Tagliavini F., Nacmias B., Piaceri I., Bagnoli S., Bruni A. C., Maletta R. G., Bernardi L., Postiglione A., Milan G., Franceschi M., Puca A. A., Novelli V., Barlassina C., Glorioso N., Manunta P., Singleton A., Cusi D., Hardy J., Momeni P., A genome-wide screening and SNPs-to-genes approach to identify novel genetic risk factors associated with frontotemporal dementia. Neurobiol. Aging 36, 2904.e13–2904.e2926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrari R., Hernandez D. G., Nalls M. A., Rohrer J. D., Ramasamy A., Kwok J. B. J., Dobson-Stone C., Brooks W. S., Schofield P. R., Halliday G. M., Hodges J. R., Piguet O., Bartley L., Thompson E., Haan E., Hernández I., Ruiz A., Boada M., Borroni B., Padovani A., Cruchaga C., Cairns N. J., Benussi L., Binetti G., Ghidoni R., Forloni G., Galimberti D., Fenoglio C., Serpente M., Scarpini E., Clarimón J., Lleó A., Blesa R., Waldö M. L., Nilsson K., Nilsson C., Mackenzie I. R. A., Hsiung G.-Y. R., Mann D. M. A., Grafman J., Morris C. M., Attems J., Griffiths T. D., McKeith I. G., Thomas A. J., Pietrini P., Huey E. D., Wassermann E. M., Baborie A., Jaros E., Tierney M. C., Pastor P., Razquin C., Ortega-Cubero S., Alonso E., Perneczky R., Diehl-Schmid J., Alexopoulos P., Kurz A., Rainero I., Rubino E., Pinessi L., Rogaeva E., St.George-Hyslop P., Rossi G., Tagliavini F., Giaccone G., Rowe J. B., Schlachetzki J. C. M., Uphill J., Collinge J., Mead S., Danek A., Van Deerlin V. M., Grossman M., Trojanowski J. Q., van der Zee J., Deschamps W., Van Langenhove T., Cruts M., Van Broeckhoven C., Cappa S. F., Ber I. L., Hannequin D., Golfier V., Vercelletto M., Brice A., Nacmias B., Sorbi S., Bagnoli S., Piaceri I., Nielsen J. E., Hjermind L. E., Riemenschneider M., Mayhaus M., Ibach B., Gasparoni G., Pichler S., Gu W., Rossor M. N., Fox N. C., Warren J. D., Spillantini M. G., Morris H. R., Rizzu P., Heutink P., Snowden J. S., Rollinson S., Richardson A., Gerhard A., Bruni A. C., Maletta R., Frangipane F., Cupidi C., Bernardi L., Anfossi M., Gallo M., Conidi M. E., Smirne N., Rademakers R., Baker M., Dickson D. W., Graff-Radford N. R., Petersen R. C., Knopman D., Josephs K. A., Boeve B. F., Parisi J. E., Seeley W. W., Miller B. L., Karydas A. M., Rosen H., van Swieten J. C., Dopper E. G. P., Seelaar H., Pijnenburg Y. A. L., Scheltens P., Logroscino G., Capozzo R., Novelli V., Puca A. A., Franceschi M., Postiglione A., Milan G., Sorrentino P., Kristiansen M., Chiang H.-H., Graff C., Pasquier F., Rollin A., Deramecourt V., Lebert F., Kapogiannis D., Ferrucci L., Pickering-Brown S., Singleton A. B., Hardy J., Momeni P., Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurol. 13, 686–699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broce I., Karch C. M., Wen N., Fan C. C., Wang Y., Tan C. H., Kouri N., Ross O. A., Höglinger G. U., Muller U., Hardy J.; International FTD-Genomics Consortium, Momeni P., Hess C. P., Dillon W. P., Miller Z. A., Bonham L. W., Rabinovici G. D., Rosen H. J., Schellenberg G. D., Franke A., Karlsen T. H., Veldink J. H., Ferrari R., Yokoyama J. S., Miller B. L., Andreassen O. A., Dale A. M., Desikan R. S., Sugrue L. P., Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLOS Med. 15, e1002487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mishra A., Ferrari R., Heutink P., Hardy J., Pijnenburg Y., Posthuma D.; International FTD-Genomics Consortium , Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 140, 1437–1446 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabb3063/DC1