

Collagen VI, an ECM protein up-regulated in triple-negative breast cancer and obese individuals, drives breast cancer metastasis.
Abstract
The extracellular matrix (ECM), a major component of the tumor microenvironment, promotes local invasion to drive metastasis. Here, we describe a method to study whole-tissue ECM effects from disease states associated with metastasis on tumor cell phenotypes and identify the individual ECM proteins and signaling pathways that are driving these effects. We show that decellularized ECM from tumor-bearing and obese mammary glands drives TNBC cell invasion. Proteomics of the ECM from the obese mammary gland led us to identify full-length collagen VI as a novel driver of TNBC cell invasion whose abundance in tumor stroma increases with body mass index in human TNBC patients. Last, we describe the mechanism by which collagen VI contributes to TNBC cell invasion via NG2-EGFR cross-talk and MAPK signaling. Overall, these studies demonstrate the value of decellularized ECM scaffolds obtained from tissues to identify novel functions of the ECM.
INTRODUCTION
One in two men and one in three women will be diagnosed with cancer within their lifetime, and the leading cause of death in cancer patients is metastasis, the dissemination of tumor cells from the primary tumor to secondary sites in the body. Triple-negative breast cancer (TNBC), which represents approximately 15 to 20% of breast cancer cases, is aggressive and remains difficult to treat because of its heterogeneous nature and limited targetable driver mutations (1). TNBC also has a recurrence rate of more than 30%, which is highest in the first 3 years after diagnosis (2). Chemotherapy with anthracyclines and taxane-based drugs remains the primary treatment for patients with TNBC (3), commonly administered in the neoadjuvant setting before surgery. Pathologic complete response rates vary between 15 and 40%, with higher rates of residual disease and shorter disease-free and overall survival for patients with TNBC (4). Unfortunately, there is no single mutation that drives tumor invasion, which occurs as result of a continual feedforward loop between cancer cells and the local tumor microenvironment (TME), making the development of biomarkers challenging (5). Understanding the features of the TME that promote local invasion will increase our ability to predict and track metastatic recurrence and ultimately reduce cancer deaths.
In particular, we and others (6) have shown that the extracellular matrix (ECM), which provides structure and support to our tissues, can regulate many hallmarks of cancer, such as invasion and metastasis (7). Recent advances in the field of ECM proteomics have increased our understanding of the complexity and heterogeneity of the tumor matrisome (8). Specifically, Mayorca-Guiliani et al. (9) describe the matrisome of breast tumor tissue generated via syngeneic 4T1 xenografts relative to normal mammary gland, while Naba et al. (10) characterize the ECM of highly metastatic LM2 tumor xenografts versus the ECM of poorly metastatic breast tumors from MDA-MB-231 in immunocompromised mice. While these studies revealed the presence of well-studied ECM proteins such as fibronectin (FN) and collagen I, they also highlighted the diversity of individual cues in ECM proteins in tumors, many of which whose function has been poorly studied until now. There is a clear need for strategies to identify and study individual ECM proteins and the signaling pathways they activate, which drive invasion and ultimately promote metastasis.
Obesity is a systemic disease that affects the composition and physiological properties of the brain and peripheral tissues such as the adipose tissue. Obesity is a global pandemic: According to the U.S. Centers for Disease Control and Prevention (CDC), 70% of U.S. adults are overweight or obese (11). Obesity contributes to 20% of cancer-related deaths, with a higher incidence of breast cancer in obese individuals. Epidemiological studies reveal that the risk of TNBC is associated with an increase in body mass index (BMI) and that a higher proportion of obese patients suffer from TNBC (12, 13). Furthermore, individuals with a high BMI have increased rates of metastasis (14) and decreased response to chemotherapy (15). Given the increasing rates of obesity and the poor outcomes associated with TNBC, it is important to gain a detailed understanding of the mechanisms of obesity-driven breast cancer progression. Obesity is thought to drive cancer cell proliferation because of increased hormone, growth factor, and cytokine secretion by adipocytes and/or macrophages (16). Obesity is also associated with fibrosis, the modification of both the amount and composition of ECM proteins. Obesity-associated macrophages promote myofibroblast activation, leading to ECM deposition and ultimately fibrosis in adipose tissue (17). As a consequence of these complex physiological changes, the local microenvironment of tumors in obese patients is drastically different than in lean patients. A recent study explored the role of the ECM in obesity-induced breast cancer invasion (18). Adipose stromal cells isolated from mammary fat pads of obese and lean mice were plated in vitro for 2 weeks, where they secreted their own ECM. ECM generated from cells from obese mice showed increased abundance of FN and collagen cross-linking, leading to increased breast cancer cell invasion via mechanosignaling. These studies demonstrated that obesity affects the ability of stromal cells isolated from the mammary gland to secrete ECM in vitro. However, these experiments focused on ECM generated in vitro and did not characterize the landscape of ECM changes associated with obesity.
Studying the role of ECM and the contribution of physiological states to the ECM composition in cancer progression remains challenging. Cell-ECM responses are studied in vitro using recombinant proteins, evaluating cell adhesion to, migration on, and invasion in individual ECM proteins. We have recently shown that cell shape when a cell is adhering to ECM protein is more predictive of three-dimensional (3D) invasion in response to that same protein than migration on the same 2D substrate (19). While these assays are informative and can be used to dissect the signaling pathways involved in ECM-driven responses, they also only evaluate response to a single cue. Studying responses to combinations of ECM proteins is more challenging. Matrigel, which is composed of basement membrane components and growth factors, is commonly used; however, this is only representative of a small subset of ECM proteins tumor cells interact with in the mammary gland. Last, cell-derived matrices generated by fibroblasts have been used to generate more physiologically relevant diverse ECM matrices. However, this method studies cell responses to ECM generated in vitro by single cell type in the TME. Proteomics studies using mammary tumors generated with human tumor cells in a mouse host demonstrate that ECM peptide fragments originate from both the tumor and stromal compartment (10). Further, various studies show that ECM is secreted by multiple cell types within the TME: Tumor cells, fibroblasts, macrophages, adipocytes, and endothelial cells have all been shown to secrete ECM proteins and contribute to ECM remodeling. Novel methods that allow us to study cell response to native ECM are needed.
Here, we describe a method and experimental pipeline to identify and study ECM proteins implicated in driving breast cancer metastasis that involves isolation of decellularized ECM (dECM) scaffolds from lean, obese, and tumor-bearing mammary glands and reseeding of cells to study whole-tissue ECM-driven effects on tumor cell proliferation and invasion via live imaging. Coupled to proteomics and mechanistic signaling analysis, our method can be used to study the effect of the ECM on tumor cell behaviors, identify ECM proteins that contribute to local invasion, and determine the mechanism by which they affect tumor cell behaviors. We use our pipeline to identify collagen VI as an ECM protein up-regulated in both obese and tumor-bearing mammary glands and a major driver of TNBC cell invasion. We show that inhibition of collagen VI–driven invasion via NG2–epidermal growth factor receptor (EGFR) cross-talk and mitogen-activated protein kinase (MAPK) signaling decreases TNBC cell response to both tumor and obese ECM, identifying targets that can be leveraged for detection and targeting of cancer.
RESULTS
dECM scaffolds obtained from tumor models can be used to study ECM-driven cell invasion
To investigate how tissue-level changes in ECM composition affect breast tumor cells, we used two models of metastatic TNBC: a xenograft model where MDA-MB-231 cells are injected into the mammary gland of immunocompromised mice and left to grow for 8 weeks, and PyMT-MMTV mice, an autochthonous model where mice develop tumors in all mammary glands, which were grown for 10 weeks (Fig. 1A). At these time points, all tumors have metastases to the lungs. We developed a novel experimental pipeline that involves isolation of the ECM fraction of mammary tissue via decellularization, reseeding of fluorescently labeled tumor cells into the isolated ECM scaffolds, and live microscopy of cells to quantify effects on cell proliferation and cell invasion (Fig. 1B). Decellularization with low-dose SDS removed all cell nuclei in healthy mammary gland, 231-tumors, and PyMT-tumors, as shown by hematoxylin and eosin (H&E) staining (Fig. 1C), while maintaining overall tissue structure. We confirmed removal of soluble intracellular proteins from different compartments by Western blot (Fig. 1D) and the removal of adipocytes by staining for the lipid droplet marker perilipin (fig. S1A). We also verified that decellularization did not affect individual ECM abundance and organization, by staining for Picrosirius red to visualize collagen I and III fibers and using an antibody to stain for collagen IV, a major component of the basement membrane (fig. S1, B and C). We also performed structural analysis of collagen I fibers as visualized by second-harmonic generation (SHG) and find that decellularization does not alter fiber angle, straightness, length, or width (fig. S2). These data demonstrate that decellularization with low-dose SDS does not affect ECM protein abundance and organization. The dECM scaffolds were then prepared for cell seeding by cutting into 5-mm pieces and incubating overnight in the appropriate cell culture medium. MDA-MB-231 cells labeled with green fluorescent protein (231-GFP) were seeded into the stabilized ECM scaffolds and imaged for more than 16 hours to evaluate effects of intact healthy and tumor ECM on 3D cell proliferation and migration using live imaging. We find that ECM isolated from xenograft 231-tumors significantly increased 231-GFP cell migration and cell proliferation (Fig. 1, F to H, and movies S1 and S2). We seeded GFP-labeled PyMT tumor–derived cells in the ECM isolated from primary MMTV-PyMT tumors, which also increased tumor cell invasion without affecting proliferation (Fig. 1, F to J, and movies S3 and 4). These data demonstrate that whole-tissue ECM can be isolated from healthy and tumor-containing mammary glands and used to study ECM-driven effects on individual cell phenotypes. In addition, these findings functionally validate that whole-tissue ECM changes in ECM composition coordinately drive pro-proliferation and invasive phenotypes in TNBC tumor cells.
Fig. 1. Reseeding tumor cells onto dECM scaffolds reveals whole-tissue ECM effects on tumor cell proliferation and invasion.
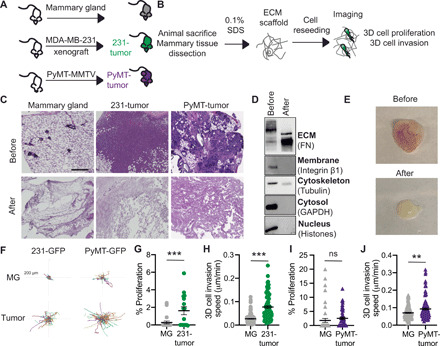
(A) MDA-MB-231 xenografts (231-tumor) and tumors from PyMT-MMTV (PyMT-tumor) mice were used. (B) Schematic of experimental pipeline. (C) H&E of mammary gland, 231-tumors, and PyMT-tumors before and after decellularization (scale bar, 200 μm). (D) Representative Western blot of 231-tumor before and after decellularization for cellular compartments. (E) Photo of PyMT-tumor before and after decellularization Photo credit: Jackson Fatherree, Tufts University. (F) Representative tracks of 231-GFP or PyMT-GFP cells seeded on dECM from mammary gland (MG), 231-tumors, or PyMT-tumor. Each line represents the track of a cell over 16 hours. (G) Percentage of 231-GFP cells proliferating on healthy mammary gland or 231-tumor. (H) Cell migration speed of 231-GFP seeded on dECM from mammary gland and 231-tumors. (I) Percentage of PyMT-GFP cells proliferating on mammary gland or PyMT-tumor scaffolds. (J) Cell migration speed of PyMT-GFP seeded on dECM from mammary gland and PyMT-tumors. For proliferation, data point is a field of view. For cell migration, each point is the average speed of a cell over 16 hours. Data obtained from at least three dECM scaffolds from three mice show mean ± SEM. Significance was determined by unpaired two-tailed Mann Whitney t test. **P < 0.01 and ***P < 0.005. ns, not significant.
Obesity is associated with changes in ECM composition of mammary gland tissue
To investigate whether our method could be used to identify novel drivers of breast cancer metastasis, we focused on obesity, which is associated with increased metastasis in breast cancer and has been shown to affect breast cancer ECM properties. We set out to understand whether obesity-driven changes in ECM could contribute to TNBC invasion and how these effects compared to tumor ECM. We used a diet-induced model of obesity (Fig. 2A). We isolated the mammary tissues of female C57BL/6 mice fed on chow or 60% kcal high-fat diet (HFD) for 16 weeks. Mice fed an HFD diet weighed twice as much as those fed with a chow diet and had 2 to 5 g of subcutaneous fat (fig. S3, A and B). H&E staining of mammary gland tissue sections confirmed adipocyte hypertrophy in the tissue obtained from obese mice (fig. S3C). Decellularization of the obese mammary gland was performed using the same protocol as used in Fig. 1 and confirmed by H&E, perilipin, ECM protein, and Picrosirius red staining (fig. S3, D and E). The dECM scaffolds from lean and obese mice were used to evaluate the effect of obesity on whole-tissue ECM-driven tumor cell phenotypes and characterize the ECM composition of the obese mammary gland (Fig. 2B). We seeded 231-GFP cells on the ECM scaffolds from lean and obese mammary glands, imaged them overnight, and analyzed cell movement (Fig. 2C and movie S5). dECM isolated from obese mammary glands significantly increased cell invasion speed relative to ECM from lean mammary glands (Fig. 2D); however, it did not have an effect on tumor cell proliferation (Fig. 2E). These data demonstrate that obesity induces whole-tissue changes in ECM composition that drive TNBC tumor cell invasion.
Fig. 2. ECM from the mammary gland of obese mice increases cell invasion and overlaps with breast tumor ECM composition.
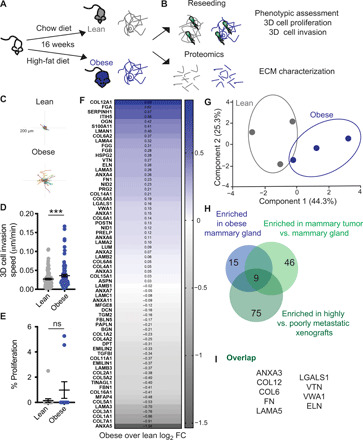
(A) Diet-induced obesity model in female C57BL/6 mice. (B) dECM of lean and obese mammary glands by decellularization was used for reseeding and proteomics. Representative tracks (C) and quantification (D) of cell migration speed of 231-GFP cells seeded on dECM from lean and obese mammary gland. Each point represents the average speed of a cell over 16 hours. (E) Percentage of proliferating 231-GFP cells on ECM scaffolds from lean and obese mammary gland. Data show mean ± SEM, obtained from at least three dECM scaffolds from three mice. Significance was determined by unpaired two-tailed Mann-Whitney t test. ***P ≤ 0.005. (F) Average log2 fold change (FC) differences of 68 ECM proteins identified by proteomics of the ECM fraction and principal components analysis (G). (H) ECM proteins found enriched in obese mammary gland were overlapped with proteins enriched in 4T1 orthotopic syngeneic mammary (9) tumor and in MDA-MB-231-LM2 highly metastatic (10) xenografts in a Venn diagram. (I) List of nine matrisome proteins present in obese mammary gland ECM, 4T1 tumor ECM, and metastatic LM2 tumors.
To comprehensively characterize the changes in ECM associated with obesity, we performed proteomics analysis. To directly compare the abundance of each peptide within the different samples, we used label-based quantification with unique isobaric tandem mass tags (TMTs) to label three samples per group. We used the existing in silico murine matrisome data to annotate the data and identify proteins with differential abundance in obese versus lean mammary tissues. We first assessed reproducibility between biological replicates by plotting median-centered reporter ion intensities and found significant correlation between all combinations of individual samples within each group (fig. S4). In the matrisome of the obese mammary gland, we identified 53 core matrisome proteins (27 glycoproteins, 18 collagens, and 8 proteoglycans) and 15 ECM-associated proteins (10 ECM-affiliated proteins, 3 ECM regulators, and 2 secreted factors) (fig. S5), providing the first characterization of the matrisome of obese mammary tissues. These data demonstrate that the ECM composition of lean and obese mammary tissues is distinct (Fig. 2, F and G) with a number of proteins found to up-regulated in these obese mammary glands relative to lean. We then compared the obese matrisome to two previously published mammary gland tumor proteomics (Fig. 2H) (9, 10). We identify a common signature of nine matrisome proteins, which are up-regulated in both tumor and obese ECM, which includes collagen VI, collagen XII, FN, LAMA5, VTN, ELN, VWA1, LGALS1, and ANXA3 (Fig. 2I). These data demonstrate obesity-induced tissue-level changes in ECM composition that overlap with those seen during tumorigenesis and that also drive TNBC tumor cell invasion.
Collagen VI, which is up-regulated in obese and tumor ECM, drives migration of TNBC cancer cells
Our next goal was to identify which of the ECM proteins up-regulated in both tumor and obese ECM contributes to TNBC cell invasion. From the nine ECM proteins identified above, we chose to focus on four: FN, collagen VI, elastin, and laminin; each of these proteins is commercially available and has published antibodies for staining. We used three main assays commonly used to study cell-ECM interactions: an adhesion assay, 2D cell migration, and 3D invasion. For adhesion assays, wells were coated with recombinant ECM, washed, and then seeded with MDA-MB-231 cells that were left to adhere for 2 hours (Fig. 3A). Collagen VI increased both cell area and cell eccentricity (fig. S6, A and B). FN increased cell area but did not alter cell shape. Neither laminin nor elastin had any significant effect on cell shape. Similar results were obtained with another TNBC cell line, MDA-MB-468 (fig. S6, C to E). We then plated the cells overnight on the four ECM proteins to evaluate their effects on cell migration speed. Here, we find that collagen VI significantly increases cell migration of both MDA-MB-231 and MDA-MB-468 cells when compared to no ECM control (Fig. 3, C and D, and movies S6 and S7). To investigate whether these effects on 2D cell migration are specific to TNBC tumor cells, we also performed the same assay on the epithelial premalignant breast cell line MCF-10A. We find that collagen VI also increases cell migration of MCF-10A cells (fig. S6F). We confirmed that collagen VI could induce cell migration of TNBC cells at a range of concentrations (fig. S6G). We then investigated whether collagen VI could also drive 3D invasion of TNBC cells. We generated spheroids embedded in collagen I, the most abundant ECM protein present in normal breast and breast tumor tissue. We find that addition of collagen VI to the matrix significantly increases MDA-MB-231 invasion in a 3D spheroid model (Fig. 3, E and F). Overall, these data identify collagen VI, an ECM protein up-regulated in both tumor and obese ECM, as a major driver of TNBC cell migration.
Fig. 3. Collagen VI, ECM protein up-regulated in obese and tumor ECM, drives TNBC cell adhesion, migration, and invasion.
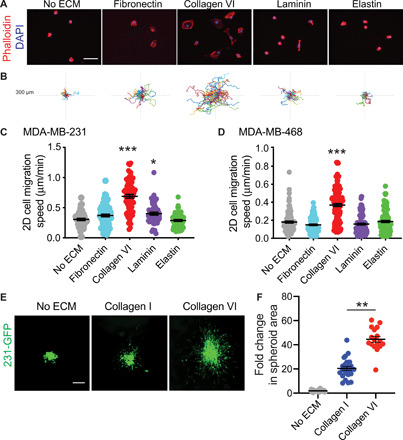
(A) Representative images from an adhesion assay of MDA-MB-231 cells seeded onto wells coated with FN (20 μg/ml), collagen VI, laminin, or elastin (scale bar, 50 μm). (B) Representative rose plots of cell migration tracks of MDA-MB-231 cells on ECM substrates, with each colored line representing a single cell track over 16-hour time course. (C) 2D migration speed of MDA-MB-231 cells seeded on glass, FN (20 μg/ml), collagen VI, laminin, or elastin. (D) 2D migration speed of MDA-MB-468 cells seeded on wells coated with PBS (n = 111), FN (20 μg/ml) (n = 133), collagen VI, laminin, or elastin. Each point represents the average speed of a cell over 16 hours. (E) Representative images of spheroid 231-GFP cells after 5 days within medium without ECM, in collagen I (1 mg/ml) and in a collagen I + collagen VI (50 μg/ml) matrix (scale bar, 300 μm). (F) Quantification of fold change in 231-GFP spheroid area after 5 days. Each dot represents an individual spheroid. For all graphs in this figure, data were pooled from at least three independent experiments and show mean ± SEM. Significance was determined by a nonparametric Kruskal-Wallis test with Dunn’s multiple testing correction. *P < 0.05, **P < 0.01, and ***P < 0.005.
Collagen VI is up-regulated in obese and tumor ECM
We next wanted to validate the proteomics findings within mouse and human samples. Collagen VI is secreted by stromal adipocytes in breast tissue (20) and is present in the stromal compartment of tumors. We performed immunostaining for collagen VI in the lean and obese mammary glands obtained from mice described in Fig. 2. We found increased accumulation of collagen VI in obese mammary gland relative to lean, with high levels surrounding mammary glands and in the stromal areas (Fig. 4, A and B). Collagen VI is encoded by six different genes (COL6A1 to COL6A6) that code for an α chain. Collagen VI assembles into a triple-helix composed of α1-α2-αX chains (21), where αX is either α3, α4, α5, or α6 (22). COLA3 is the most abundant chain, and the COL6A4 gene is not functional owing to a large chromosome inversion (22). We mined a publicly available dataset and found that patients with high levels of COL6A1/COL6A2/COL6A3 chains have worse outcomes than those with low levels (Fig. 4C and fig. S7, A to C). We then investigated whether collagen VI was associated with BMI in human patients with TNBC. We stained biopsy tissue samples from a cohort of nine women with BMI ranging from 20 to 36 diagnosed with TNBC for collagen VI and perilipin, a marker for adipocytes (Fig. 4D). Within the tumor compartment, there is no correlation between BMI and collagen VI expression levels (Fig. 4E). However, in the stromal compartment and peritumoral area, there is a positive correlation between BMI and collagen VI expression in TNBC patients (Fig. 4, F and G). These data demonstrate that our experimental platform can identify ECM drivers of metastasis that are relevant to human breast cancer.
Fig. 4. Collagen VI is enriched in obese mammary gland ECM, and TNBC patients with high tumor collagen VI have lower overall survival.
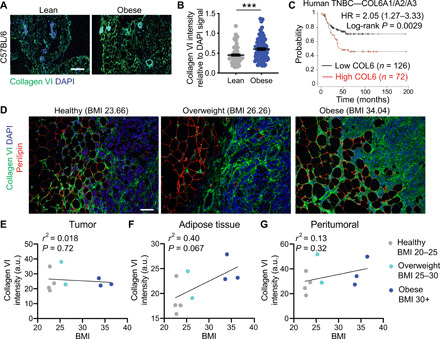
(A) Immunostaining of mammary gland sections from lean and obese C57BL/6 mice stained for collagen VI (green) and nuclei (blue) (scale bar, 100 μm). (B) Collagen VI intensity relative to DAPI signal shows increased abundance in obese ECM. Data are from more than 75 fields of view from at least three mammary gland tissues from three lean and obese mice. Unpaired two-tailed Mann-Whitney test was performed. Data represent mean ± SEM. ***P ≤ 0.005. (C) Kaplan-Meier curve of human TNBC breast cancer patients with low versus high mRNA of the most abundant form of collagen VI (COL6A1/COL6A2/COL6A3); data from (38). (D) Collagen VI (green), perilipin (red), and DAPI (blue) staining of a human TNBC biopsy samples from an average weight TNBC patient (BMI = 23.66), an overweight TNBC patient (BMI = 26.26), and an obese TNBC patient (BMI = 34.04) (scale bar, 100 μm). Correlation between BMI and collagen VI intensity relative to region area for the tumor area (as identified by DAPI staining) (E), adipose tissue (as identified by perilipin) (F), and peritumoral region (G) for nine patients of ranging BMI. a.u., arbitrary units.
Collagen VI drives migration of TNBC cancer cells via NG2 and EGFR cross-talk and MAPK signaling
We next set out to dissect the mechanism by which collagen VI promotes breast cancer invasion. In breast cancer, collagen VI has been suggested to drive breast cancer cell migration via its C-terminal fragment, endotrophin (ETP). ETP is a cleavage product of the COL6α3 chain (fig. S7A) (21). Previous studies have found that ETP is abundant in tumor tissues in PyMT mice and that ETP enhances fibrosis, angiogenesis, inflammation, and the epithelial to mesenchymal transition, thereby promoting primary tumor growth and metastasis. ETP is thought to serve as the major mediator of the tumor-promoting effects of collagen VI in breast cancer (23). We compared effects of collagen VI and ETP in our assays, by both embedding ETP in the matrix and including it as a soluble factor in the solution, alone or in combination with collagen I. ETP did not have an effect on the cell migration of MDA-MB-231 cells (fig. S7, D and E). In other contexts, collagen VI can signal through the proteoglycan NG2 and β1 integrins, both of which can cross-talk with receptor tyrosine kinases (RTKs) (Fig. 5A) (21). We find that collagen VI–driven MDA-MB-231 breast cancer cell migration is partially inhibited by an NG2-specific antibody (NG2ab) but not by β1 inhibitory antibody (β1ab) (Fig. 5B). Neither antibody affected proliferation at the doses used (fig. S8A). We confirmed these findings in MDA-MB-468 cells (fig. S8B). Because NG2 binds to the central portion of collagen VI and not the C-terminal ETP fragment, these data demonstrate that collagen VI drives TNBC invasion through an ETP-independent mechanism. NG2 has been shown to cross-talk with RTKs such as EGFR (24) to drive intracellular signaling. Treatment with NG2ab and the EGFR inhibitor lapatinib, individually or in combination, reduced cell migration on collagen VI relative to their own vehicle controls (Fig. 5C) at a dose that did not affect cell viability (fig. S6D). We also used a small interfering RNA (siRNA) to knock down NG2 in MDA-MB-231 cells. We obtained a 55% knockdown of NG2 mRNA levels and found that cells treated with siNG2 showed a significant decrease in collagen VI–driven cell migration (fig. S8, D and E). Further, we find that cells treated with siNG2 and lapatinib also decreased collagen VI–driven cell migration relative to lapatinib or siNG2 alone (fig. S8E). Given our recent findings that cell adhesion in response to ECM is more predictive of 3D ECM, we also examined the role of these pathways in collagen VI–driven cell adhesion. Treatment with NG2ab and the EGFR inhibitor lapatinib, individually or in combination, reduced cell adhesion to collagen VI relative to their own vehicle controls (Fig. 5, D and E). Together, these studies suggest that NG2 and EGFR synergize to mediate collagen VI–driven TNBC cell adhesion and migration.
Fig. 5. Collagen VI–driven TNBC cell migration is mediated by NG2/EGFR cross-talk and MAPK signaling.
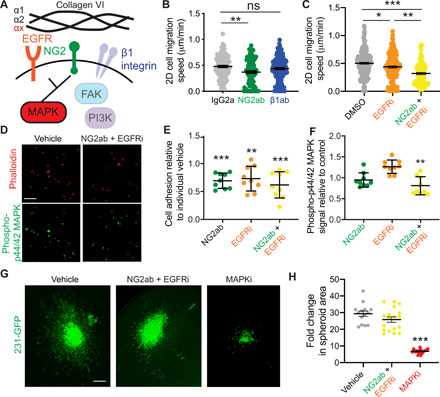
(A) Schematic of collagen VI signaling. (B) Migration speed of 231-GFP cells on collagen VI (20 μg/ml) treated with IgG2a, NG2 targeting antibody (NG2ab), or β1 integrin inhibitory antibody (β1ab). (C) Migration speed of 231-GFP cells on collagen VI (20 μg/ml) treated with DMSO, lapatinib (EGFRi, 10 μM), and lapatinib/NG2ab (NG2ab + EGFRi) combination. Significance was determined by a Kruskal-Wallis test with Dunn’s multiple testing correction, with *P < 0.05, **P < 0.01, and ***P < 0.005. (D) Immunostaining for phalloidin and phospho-p44/42ERK (Thr202/Tyr204) in MDA-MB-231 cells seeded on collagen VI (20 μg/ml) and treated with IgG and DMSO or NG2ab + EGFRi. Quantification of MDA-MB-231 cell adhesion (E) and phospho-p44/42 MAPK (F) in cells treated with NG2ab, EGFRi, or combination, relative to vehicle control. (G) Representative images of spheroid 231-GFP cells after 5 days in collagen I + collagen VI (50 μg/ml) matrix treated with DMSO, NG2ab/EGFRi, or trametinib (MAPKi) (scale bar, 300 μm). (H) Quantification of fold change in 231-GFP spheroid area after 5 days. Each dot represents an individual spheroid. For all graphs, data were pooled from at least three independent experiments and show mean ± SEM. Significance was determined by Kruskal-Wallis test with Dunn’s multiple testing correction. *P < 0.05, **P < 0.01, and ***P < 0.005.
Next, we investigated the downstream signaling pathways that drive collagen VI–mediated responses. In neuronal tissues, collagen VI acts via Akt, c-Jun N-terminal kinase (JNK), extracellular signal–regulated kinase (ERK), and p38 MAPKs (25). In sarcoma, collagen VI activates phosphatidylinositol 3-kinase (PI3K) signaling to drive cell migration (26), whereas in breast cancer collagen VI activates Akt and β-catenin to drive cell proliferation (20). We investigated the effects of alpelisib (a PI3K inhibitor), defactinib [a focal adhesion kinase (FAK) inhibitor], and trametinib (a MAPK kinase inhibitor) on cell migration at concentrations of 10, 1, and 0.1 μM, respectively. Cell viability assays were used to determine the appropriate dosage of the drugs investigated (fig. S9, A to C). Trametinib, but not alpelisib or defactinib, resulted in a significant decrease in collagen VI–driven cell migration (fig. S9D), indicating that MAPK signaling downstream of EGFR and NG2 may be mediating collagen VI–driven cell migration. Consistent with this observation, inhibition of EGFR and NG2 decreased phospho-p44/42 MAPK signal in MDA-MB-231 cells seeded on collagen VI (Fig. 5, D to F), without affecting pFAK397 (fig. S9E). Last, we investigated the ability of NG2, EGFR, and MAPK signaling to inhibit collagen VI–driven invasion in our 3D spheroid model. Inhibition of MAPK signaling significantly decreased invasion of cells seeded in our collagen VI 3D spheroid model, and while inhibition of both NG2 and EGFR slightly decreased invasion, the result was not statistically significant (Fig. 5, G and H). These data demonstrate that collagen VI drives migration in TNBC cells via NG2/EGFR cross-talk and MAPK signaling.
Blocking collagen VI signaling reduces obese and tumor ECM-driven invasion
Last, we investigated whether collagen VI–driven signaling was responsible for the increase in TNBC cell invasion seen on dECM from tumor-bearing and obese mice (Figs. 1 and 2). We seeded MDA-MB-231 cells on dECM from lean, obese, and tumor-bearing mice and treated them with NG2ab. We found that inhibition of NG2 decreased migration in dECM obtained from both obese and tumor-bearing mice (Fig. 6, A and B, and movies S8 and S9). We then investigated whether the NG2-EGFR cross-talk and MAPK signaling were driving tumor cell ECM-driven migration of PyMT mouse tumor cells. We find that inhibition of NG2/EGFR together and MAPK decreased PyMT-tumor ECM-driven invasion (Fig. 6, C and D). These studies demonstrate that our platform can be used to study the contribution of individual signaling pathways to ECM-driven invasion. Further, they confirm that collagen VI–driven signaling contributes to ECM-mediated TNBC cell invasion in the context of obesity and tumor progression.
Fig. 6. Inhibition of collagen VI–driven signaling reduces TNBC cell migration on dECM derived from the mammary gland of obese and tumor-bearing mice.
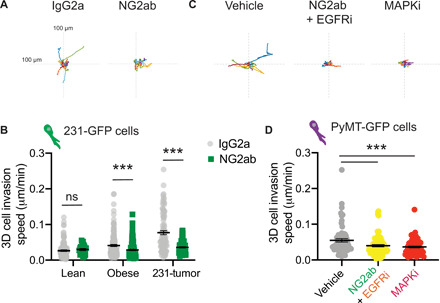
(A) Representative rose plots of 231-GFP cell migration seeded on dECM from obese mammary gland and treated with IgG2a or NG2ab, where each colored line represents the tracked movement of a cell over the time course (16 hours). (B) Cell migration speed of 231-GFP cells seeded on mammary gland ECM derived from lean mice, obese mice, and tumor-bearing mice treated with IgG2a or NG2ab. Graphs show mean ± SEM, with dot representing data from a single cell. Unpaired two-tailed Mann-Whitney tests were performed between respective IgG2a control and NG2ab conditions. Data were obtained from at least three different ECM scaffolds from three different mice, with an average of 108 total cells per condition. Significance by t test, with ***P < 0.005. (C) Representative rose plots of PyMT-GFP cell migration seeded on dECM from PyMT-tumors and treated with vehicle, NG2ab+ EGFRi or MAPKi, where each colored line represents the tracked movement of a cell over the time course (16 hrs) (D) Cell migration speed of PyMT-GFP cells seeded on mammary gland ECM derived from PyMT-mice treated with vehicle, NG2ab+ EGFRi or MAPKi. Data obtained from at least 3 different ECM scaffolds from 3 different mice, statistic by one-way Anova, with ***P < 0.005.
DISCUSSION
It is well established that the ECM can regulate many hallmarks of cancer and is a major driver of metastasis, the leading cause of death in cancer patients. Recent advances in proteomics have revealed an abundance of individual ECM proteins within breast tumor tissues, and novel methods are needed to investigate which ECM proteins regulate proliferation and invasion within this complex mesh of proteins. Here, using a combination of dECM scaffolds, proteomics, and live imaging, we identified collagen VI as a driver of TNBC cell invasion. Specifically, we leveraged the fact that obesity, which is associated with increased breast cancer incidence, metastasis, and chemoresistance, can also induce compositional changes in the mammary gland ECM via fibrosis. Using our model, we find that ECM from obese and tumor-bearing mammary glands drive TNBC cell invasion and find overlap of ECM proteins in the obese mammary gland with published mammary tumor ECM datasets. Collagen VI is up-regulated in both tumor and obese ECM, and we show that collagen VI promotes TNBC cell adhesion, 2D migration, and 3D invasion via NG2 binding, activating an EGFR/MAPK signaling axis. Overall, these studies define a novel mechanism by which obesity may contribute to TNBC progression and identify collagen VI as a driver of TNBC cell invasion.
Our study provides the first description of using dECM from tumors to identify novel ECM drivers of cancer cell invasion pipeline that involves isolation of dECM scaffolds and subsequent cellular reseeding. Our method provides several advantages. First, we show that decellularization using low-dose SDS maintains the ECM composition of tumor and obese tissues while removing most of the cells. Using various methods including immunostaining and Western blotting, we show that the abundance of several ECM proteins and fibril structure of collagens is maintained in the decellularized tissues. Second, the composition of the dECM scaffolds is independent of the cell type that secreted it. Proteomics studies have shown that ECM of breast tissue is derived from both tumor and stromal compartments (10). Most studies that attempt to recreate the ECM composition of tumors use fibroblasts or other stromal cells isolated from tumors but cultured in vitro (18). Our method allows us to isolate ECM directly from tissues from different disease states without any bias toward proteins secreted by stroma or the tumor. Last, we have found that our method is compatible with mouse and human cell types, tissues of different composition, live imaging, and mechanistic analysis. Our method can therefore be used to dissect the role of signaling pathways in driving ECM responses and to assay multiple cell phenotypes. Overall, this method will help provide a novel platform to study ECM-driven responses.
Collagen VI is expressed in a range of tissues and has been shown to be involved in cartilage, bone, and skeletal muscle function; nervous system regeneration and myelination; and immune cell recruitment and polarization (21). Previous studies have found that collagen VI plays an important role in stimulating mammary tumor growth and breast cancer progression (20). In MMTV-PyMT collagen VI−/− mice, the lack of collagen VI reduced hyperplasia and primary tumor growth. In this study, the authors state that collagen VI has no effect on metastasis. However, the only method used to assess metastasis in this study was by injecting Met1 cells into the tail vein of wild-type or collagen VI−/− mice and quantifying homing to the lungs. While this assay evaluates whether collagen VI is important for extravasation and metastatic outgrowth, it does not address the role of collagen VI in driving local invasion in the primary tumor. Here, we demonstrate that collagen VI directly drives cell migration of TNBC cells. A subsequent study demonstrated that ETP, a cleavage product of the COL6α3 chain, serves as the major mediator of the collagen VI–driven effects on cell migration and metastasis (23) by enhancing transforming growth factor β (TGFβ) signaling to promote epithelial-mesenchymal transition, although the cell surface receptors and signaling pathways activated downstream of ETP are not known. Here, we do not see an effect of ETP on TNBC cell migration, although this may be because MDA-MB-231 cells are mesenchymal. We find that collagen VI–driven cell migration is partially inhibited by an NG2-specific antibody. Because NG2 binds to the central portion of collagen VI and not the C-terminal ETP fragment, these data demonstrate that collagen VI drives breast cancer invasion independently of ETP. NG2 has been suggested to cross-talk with RTKs, consistent with our data showing synergistic suppression of cancer cell migration by NG2/EGFR coinhibition. Several ECM proteins are known to signal via adhesion receptor/RTK cross-talk, and it will be interesting to further dissect the mechanisms that mediate this, particularly whether collagen VI synergizes with growth factors to promote invasion in breast cancer.
While our results demonstrate how obesity-induced changes in ECM can contribute to tumor progression, we still do not know what cells secrete ECM in the adipose mammary gland. Both the tumor and stroma compartment are known to contribute to ECM in mouse models of metastasis, demonstrated in species-specific peptide identification studies (10). Collagen VI is secreted by adipocytes, and collagen VI secretion increases during adipose differentiation (27). Although adipocytes can secrete ECM proteins directly, other factors such as TGFβ can activate other cells such as myofibroblasts or macrophages to synthesize ECM proteins (28). In the Seo et al. (18) study, adipose stromal cells were isolated and those isolated from obese tissue were found to have myofibroblast-like characteristics, as indicated by α-SMA (smooth muscle actin) staining. It will be important to dissect which cell types are contributing to the changes in ECM associated with obesity in the mammary gland, which may reveal therapeutic strategies for targeting ECM-driven signaling that promotes tumor progression and metastasis. Last, obese breast cancer patients have been shown to be more resistant to chemotherapy, the main standard of care for metastatic breast cancer. Obesity has been shown to contribute to chemoresistance by altering drug pharmacokinetics, inducing chronic inflammation, as well as altering tumor-associated adipocyte adipokine secretion (29). The role of obesity-driven ECM production in chemoresistance remains unknown. Collagen VI has been shown to drive resistance to chemotherapeutic drug cisplatin in ovarian cancer (30). Future studies investigating the role of obesity-induced changes in ECM in drug resistance may also shed light on strategies to treat this growing population of breast cancer patients.
Overall, these findings will facilitate the translation of novel ECM-based tools to both detect and target local invasion and metastasis. Recent tools using ECM-targeting antibodies (31) or mimetic peptides (32) have been developed to visualize and leverage ECM presence for drug targeting or cargo delivery. The ECM is an attractive target for development of blood-based biomarkers, substances whose presence in the blood can inform on how responsive a tumor is to a given drug and how aggressive the tumor is. The ECM is cleaved, and ECM fragments shed into the circulation and can be detected by proteomics (33). We have previously shown that peptides shed by proteases in the tumor can be detected in the blood and help predict drug response in human patients. ECM fragments can therefore inform on protease activity in the blood (34). The ECM also lends itself well to being imaged using clinical modalities such as photoacoustic imaging or magnetic resonance imaging (MRI) (35), which could help with detection and monitoring of tumors throughout treatment. Last, obesity disproportionately affects certain minorities, with the highest prevalence among Blacks and Hispanics (32). Understanding how obesity affects cancer metastasis may provide insight into how cancer develops in different ethnic populations and novel treatment options for these subpopulations, which are highly affected by cancer.
MATERIALS AND METHODS
Reagents, antibodies, ECM substrates, growth factors, peptides, and inhibitors
Reagents were purchased from Fisher Scientific (Hampton, NH) or Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Antibodies used were anti–immunoglobulin 2a (IgG2a) isotype control (ab18414; Abcam, Cambridge, MA), anti–collagen VI (ab199720; Abcam, Cambridge, MA), anti-FN (ab2413; Abcam, Cambridge, MA), anti–integrin β1 (12594-1-AP; Proteintech, Rosemont, IL), anti–α-tubulin (T9026; Sigma-Aldrich, St. Louis, MO), anti–histone H3 (ab1791; Abcam, Cambridge, MA), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 14C10; Cell Signaling Technology, Danvers, MA), anti–phospho-p44/42 ERK (Thr202/Tyr204; 4370; Cell Signaling Technology, Danvers, MA), anti-pFAK397 (3283; Cell Signaling Technology, Danvers, MA), and anti-perilipin (ab61682, Abcam, Cambridge, MA). ECM substrates used were collagen VI (ab7538; Abcam, Cambridge, MA), collagen I (CB-40236; Fisher Scientific, Hampton, NH), FN (F1141; Sigma-Aldrich, St. Louis, MO), elastin (E7277; Sigma-Aldrich, St. Louis, MO), and laminin (L6274; Sigma-Aldrich, St. Louis, MO). ETP peptide, a 77–amino acid length peptide with >98% purity as determined by analytical high-performance liquid chromatography (HPLC), was synthesized by Thermo Fisher Scientific (Waltham, MA). Inhibitors used were anti-NG2 (NG2ab) (LS-C22113; Lifespan Biosciences, Seattle, WA), anti–integrin β1 (β1ab; P4C10; MilliporeSigma, Burlington, MA), lapatinib (S2111; Selleckchem, Houston, TX), alpelisib (HY-15244; MedChemExpress, Monmouth Junction, NJ), defactinib (VS-6063; Selleckchem, Houston, TX), and trametinib (S2673; Selleckchem, Houston, TX).
Cell culture
MDA-MB-231, MDA-MB-468, and MCF-10A cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). MDA-MB-231 and MDA-MB-468 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% serum and penicillin-streptomycin-glutamine, while MCF-10A cells were cultured as per ATCC recommendations in MEGM bullet kit growth medium (CC-3150; Lonza, Walkersville, MD) without gentamicin–amphotericin B mix but with additional cholera toxin (100 ng/ml; C8052; Sigma-Aldrich, St. Louis, MO). PyMT cells were cultured in 1:1 ratio of DMEM and Ham’s F-12 Nutrient Mixture containing 2% serum and penicillin-streptomycin-glutamine, with additional 1% (w/v) bovine serum albumin lyophilized powder (A2153-50G; Sigma-Aldrich, St. Louis, MO), EGF (10 ng/ml; PHG0313; Fisher Scientific, Hampton, NH), and insulin (10 μg/ml; 12585014; Fisher Scientific, Hampton, NH). Cells were routinely checked for the presence of mycoplasma by a polymerase chain reaction (PCR)–based method using the Universal Mycoplasma Detection Kit (30-1012 K; ATCC, Manassas, VA). Only mycoplasma-negative cells were used in this study.
Animal experiments
Institutional Animal Care and Use Committee guidelines were followed for all animal experiments. We used two mouse models of metastatic breast cancer. For xenograft tumors, MDA-MB-231 cells [2 million per mouse in phosphate-buffered saline (PBS) and 20% collagen I] were injected into the fourth right mammary fat pad of three 6-week-old female nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice (Taconic, Hudson, NY). Once the tumors had all reached 1 cm in diameter, mice were euthanized by CO2 and tumors were excised. PyMT-MMTV mice (The Jackson Laboratory) were used for a genetic mouse model of breast cancer. Female mice were left to grow for 12 weeks until tumor burden reached 2 cm3. Obese mice were generated with a diet. Female C57BL/6 mice were fed on chow diet Teklad 2016S or HFD (Research Diets D12492, 60% kcal fat) for 18 weeks, after which mice were euthanized by CO2 and mammary fat pads were excised.
Decellularization and protein isolation
Tumors dissected from NOD-SCID mice and PyMT-MMTV mice and mammary fat pads from C57BL/6 mice were submerged in 0.1% SDS solution and stirred for several days, replacing the solution twice daily. Once the tissue turned completely white (visual sign of successful decellularization), the solution was replaced with a 0.05% Triton X-100 solution for 3 hours. The tissues were then washed for 4 days by stirring in dH2O to remove any residual detergent. Decellularized tissue was sectioned and H&E-stained, along with Western blot, to assess decellularization. The dECM or intact tissues were lysed in 25 mM tris, 150 mM NaCl, 10% glycerol, 1% NP-40, and 0.5 M EDTA with 1× protease Mini-complete protease inhibitor (04693124001; Roche, Indianapolis, IN) and 1× phosphatase inhibitor cocktail (4906845001; Roche, Indianapolis, IN) at 4°C. In addition, for dECM, tissue homogenization was performed using the BeadBug 3 Place Microtube Homogenizer (D1030; Benchmark Scientific, Sayreville, NJ). The dECM or cell homogenate was then spun down at 21,000g for 10 min at 4°C, and the supernatant was stored at −20°C until used for Western blot or proteomics analysis.
Sample preparation for mass spectrometry
The dECM homogenate (from ±5-mm piece of tissue) was denatured in 8 M urea and 10 mM dithiothreitol and incubated on a shaker at 37°C for 2 hours. The samples were then alkylated with 25 mM iodoacetamide and incubated at room temperature in the dark for 30 min. Following dilution to 2 M urea in 11 mM ammonium bicarbonate (pH 8.0), deglycosylation was performed by adding 1000 U of peptide N-glycosidase F (PNGaseF) (P0704S; New England Biolabs, Ipswich, MA), whereafter the samples were incubated on a shaker at 37°C for 2 hours. Samples were digested first using 1 μg of endoproteinase LysC (125-05061; Wako Chemicals USA, Richmond, VA), shaking at 37°C for 2 hours, then adding 3 μg of trypsin (PR-V5113; Promega, Madison, WI), and shaking at 37°C overnight. The next day, additional 1.5 μg of trypsin was added to each sample and incubated on a shaker at 37°C for a further 2 hours. Samples were acidified with 50% trifluoroacetic acid and pH-tested to <2.0 pH. Samples were centrifuged, and the supernatant was collected. Peptide labeling with TMT10plex (90110; Thermo Fisher Scientific, Waltham, MA) was performed per the manufacturer’s instructions. Briefly, lyophilized samples were dissolved in ethanol and triethylammonium bicarbonate (TEAB; pH 8.5) and mixed with the TMT reagent. The resulting solution was vortexed and incubated at room temperature for 1 hour. Samples labeled with the 10 different isotopic TMT reagents were combined and concentrated to completion in a vacuum centrifuge. The peptides were fractionated via high-pH reversed-phase HPLC. Peptides were resuspended in 100 μl of buffer A [10 mM TEAB (pH 8)] and separated on a 4.6 mm by 250 mm 300 Extend-C18, 5-μm column (Agilent, Santa Clara, CA) using a 90-min gradient with buffer B [90% acetonitrile (ACN) and 10 mM TEAB (pH 8)] at a flow rate of 1 ml/min. The gradient was as follows: 1 to 5% B (0 to 10 min), 5 to 35% B (10 to 70 min), 35 to 70% B (70 to 80 min), and 70% B (80 to 90 min). Fractions were collected over 75 min at 1-min intervals from 10 to 85 min. The fractions were concatenated into 15 fractions noncontiguously (1 + 16 + 31 + 46 + 61, 2 + 17 + 32 + 47 + 62). The fractions were concentrated in a vacuum centrifuge and then lyophilized. Lyophilized peptides were resuspended in 100 μl of 0.1% formic acid, and 10 μl was injected into the liquid chromatography–mass spectrometry (LC-MS) system.
LC-MS/MS analysis
Peptides were separated by reversed-phase HPLC (Thermo Easy nLC1000, Thermo Fisher Scientific, Waltham, MA) using a precolumn (made in house, 6 cm of 10-μm C18) and a self-pack 5-μm tip analytical column (12 cm of 5-μm C18; New Objective, Woburn, MA) over a 140-min gradient before nanoelectrospray using a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Solvent A was 0.1% formic acid, and solvent B was 80% ACN/0.1% formic acid. The gradient conditions were 2 to 10% B (0 to 3 min), 10 to 30% B (3 to 107 min), 30 to 40% B (107 to 121 min), 40 to 60% B (121 to 126 min), 60 to 100% B (126 to 127 min), 100% B (127 to 137 min), 100 to 0% B (137 to 138 min), and 0% B (138 to 140 min), and the mass spectrometer was operated in a data-dependent mode. The parameters for the full-scan MS were resolution of 60,000 across 350 to 2000 m/z (mass/charge ratio), automatic gain control 3 x 106, and maximum ion injection time of 50 ms. The full-scan MS was followed by tandem mass spectrometry (MS/MS) for the top 15 precursor ions in each cycle with a normalized collision energy of 34 and dynamic exclusion of 30 s.
Proteomics data processing and visualization
Raw mass spectral data files (.raw) were searched using Proteome Discoverer (Thermo Fisher Scientific, Waltham, MA) and Mascot version 2.4.1 (Matrix Science, Boston, MA). Mascot search parameters were 10 ppm (parts per million) mass tolerance for precursor ions, 15 mmu for fragment ion mass tolerance, and two missed cleavages of trypsin; fixed modification was carbamidomethylation of cysteine and TMT 10-plex modification of lysine and peptide N termini; variable modifications were methionine, lysine and proline oxidation, asparagine and glutamine deamination, Gln>pyro-flu(N-Term Q), carbamylation of N terminus, and tyrosine, serine, and threonine phosphorylation. Only peptides with a Mascot score greater than or equal to 25 and an isolation interference less than or equal to 30 were included in the data analysis. Reporter ion intensities were median-centered and log2-transformed. Data were filtered for ECM protein groups with more than one unique peptide, and the resulting groups were imported into Perseus data visualization and statistics software v1.6.1.1. Data visualization included scatterplot generation with coefficient of determination (R2) values to assess reproducibility between biological replicates and principal components analysis to assess differences between groups.
Western blot
Standard procedures were used for protein electrophoresis and Western blotting. Protein lysates were separated by SDS–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, blocked with 5% nonfat dry milk solution, and incubated in primary antibody overnight at 4°C. Proteins were detected using horseradish peroxidase–conjugated secondary antibodies. Imaging was performed using a ChemiDoc MP imaging system (12003154; Bio-Rad, Hercules, CA).
Immunohistochemistry
Fixation, processing, and staining of tissue sections from tumors and mammary fat pads were carried out as previously described (7). Mammary fat pads dissected from C57BL/6 mice and tumors dissected from NOD-SCID mice were fixed in 10% buffered formalin and embedded in paraffin. Human formalin-fixed, paraffin-embedded tissue sections (5 μm) of TNBC were obtained from Tufts Medical Center as deidentified samples. For H&E staining, standard procedures were followed for H&E, including deparaffinizing, hydration, staining with hematoxylin (GHS280; Sigma-Aldrich, St. Louis, MO), and counterstaining with eosin (HT110180; Sigma-Aldrich, St. Louis, MO). For Picrosirius red staining, tissue sections (10 μm) were deparaffinized and staining was performed using the Picro Sirius Red Stain Kit (ab150681; Abcam, Cambridge, MA) according to the manufacturer’s instructions, including staining with Picrosirius red solution for 1 hour and washing with 0.5% acetic acid. Sections were mounted with Permount mounting medium (SP15-500, Fisher Scientific, Hampton, NH). For immunofluorescence, tissue sections (10 μm thick) were deparaffinized followed by antigen retrieval using Citra Plus solution (HK057; Biogenex, Fremont, CA). After blocking in PBS–0.5% Tween 20 and 10% serum, sections were incubated with primary antibodies overnight at 4°C and fluorescently labeled secondary antibodies at room temperature for 2 hours. 4′,6-Diamidino-2-phenylindole (DAPI; D1306; Thermo Fisher Scientific, Waltham, MA) was used to stain cell nuclei, and fluorochromes on secondary antibodies included Alexa Flour 488, Alexa Fluor 534, and Alexa Fluor 647 (Jackson ImmunoResearch, West Grove, PA). Sections were mounted in Fluoromount mounting medium (00-4958-02; Thermo Fisher Scientific, Waltham, MA) and imaged using a Keyence BZ-X710 microscope (Keyence, Elmwood Park, NJ). For the collagen imaging, microscopy images were collected using a Leica TCS SP8 confocal microscope system equipped with an Insight DS+ Ti:Sapphire laser (Spectra-Physics) for multiphoton near-infrared excitation. SHG images were obtained at an excitation wavelength of 920 nm, with an average power of 26 mW at the sample. The SHG emission channel was recorded on a nondescanned Leica HyD hybrid detector, using an emission bandpass filter at 460 ± 25 nm. A Leica DMi8 inverted microscope was used with a Leica HC PL APO 40×/1.10 water-immersion objective. Individual images were sampled using eight-bit dynamic range and 512 × 512 pixels per frame (corresponding to a 291 μm × 29 μm field of view or 0.57 μm per pixel). Macroscopic images were generated by stitching multiple frames acquired using the Leica LAS X TileScan modality. In addition to SHG imaging, images were also collected in transmission and reflection mode using 552-nm laser illumination and photomultiplier tube detection, with 10-nm bandpass filters centered around the 552-nm illumination wavelength. Images were analyzed using CT-FIRE (36).
Adhesion assay
Depending on the experimental condition, cells were suspended in medium containing control antibody, vehicle [dimethyl sulfoxide (DMSO)], inhibitory antibody, or drug and plated on glass-bottomed dishes (2500 cells per well; MatTek, Ashland, MA) coated with ECM protein (20 μg/ml) and allowed to adhere for 2 hours. Cells were then fixed for 15 min in 4% paraformaldehyde, then permeabilized with 0.2% Triton X-100, blocked with 3% bovine serum albumin, and incubated with primary antibodies overnight at 4°C. Cells were stained with DAPI (D1306; Thermo Fisher Scientific, Waltham, MA) and phalloidin (A12390; Thermo Fisher Scientific, Waltham, MA) along with incubation with fluorescently labeled secondary antibodies at room temperature for 2 hours. Imaging was performed using a Keyence BZ-X710 microscope (Keyence, Elmwood Park, NJ), and CellProfiler (37) v3.1.8 was used for imaging analysis using a custom pipeline. ImageJ (National Institutes of Health, Bethesda, MD) was used for pFAK foci counting. Data are the result of three independent experiments with three technical replicates per experiment.
2D migration assay and reseeding in dECM
For 2D migration, cells were plated on glass-bottomed dishes (7500 cells per well; MatTek, Ashland, MA) coated with recombinant ECM protein (20 μg/ml) and allowed to adhere for 1 hour. For reseeding on dECM, 5-mm-diameter pieces of decellularized tissue were cut and 250,000 cells per tissue were seeded and allowed to adhere for 6 hours. Depending on the experimental condition, the medium was then replaced with medium containing control antibody, vehicle (DMSO), inhibitory antibody, drug, growth factor, or peptide. Cells were imaged overnight with images acquired every 10 min (for 2D migration assay) or 20 min (for reseeding in dECM) for 16 hours in an environmentally controlled chamber within a Keyence BZ-X710 microscope (Keyence, Elmwood Park, NJ). Cells were then tracked using VW-9000 Video Editing/Analysis Software (Keyence, Elmwood Park, NJ), and both cell speed and distance migrated were calculated using a custom MATLAB script vR2018a (MathWorks, Natick, MA). Data are the result of three independent experiments with six fields of view per experiment and an average of six cells tracked per field of view (at least 100 cells per condition).
Spheroid invasion assay
Cells (2000 cells per well) were seeded in low-attachment plates in medium, followed by centrifugation to form spheroids. Spheroids were grown for 3 days, after which the matrix was added to each well, which included (depending on the condition) collagen I protein, collagen VI protein, ETP, 10 mM NaOH, 7.5% 10× DMEM, and 50% 1× DMEM. The spheroids in the matrix were then spun down, and a further 50 μl of medium was added to each well, which included (depending on the condition) vehicle (DMSO), inhibitory antibody, or drug at four times the concentration to diffuse through the gel. Following another 4 days of growth, spheroids were imaged as a Z-stack using a Keyence BZ-X710 microscope (Keyence, Elmwood Park, NJ), and Z-projection images were analyzed using a Hybrid Cell Count feature within the BZ-X Analyzer software v1.3.1.1 (Keyence, Elmwood Park, NJ). Data are the result of three independent experiments with six technical replicates per experiment.
Cell viability assay
Cells were seeded in plates coated with ECM protein (20 μg/ml) and allowed to adhere for 6 hours. The medium was then changed to medium containing control antibody, vehicle (DMSO), inhibitory antibody, or drug and incubated for 16 hours. PrestoBlue Cell Viability Reagent (A13261, Invitrogen, Carlsbad, CA) was added to each well according to the manufacturer’s instructions and incubated for 25 min at 37°C. Fluorescence was then read on a plate reader at 562 nm. Background was corrected to control wells containing only cell culture medium (no cells). Data are the result of three independent experiments with three technical replicates per experiment.
RNA extraction and real-time quantitative PCR
Total RNA was isolated using the Quick-RNA MiniPrep Plus Kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. To quantify gene expression, complementary DNA (cDNA) was synthesized from 500 ng of total RNA using SuperScript IV reverse transcriptase and an oligo-d(T) primer (Invitrogen, Carlsbad, CA). PCRs were conducted by mixing cDNA with 2× Power Up SYBR Green PCR master mix (Applied Biosystems, Carlsbad, CA) and 0.3 μM forward and reverse gene-specific primers. Primers used include NG2-forward (5′-GGCAAACCCAGAGCCCTGCC-3′), NG2-reverse (5′-GCTGGAGCTGACAGCGGGTG-3′), SDHA-forward (5′-TGGGAACAAGAGGGCATCTG-3′), SDHA-reverse (5′-CCACCACTGCATCAAATTCATG-3′), HPRT-forward (5′-TCAGGCAGTATAATCCAAAGATGGT-3′), HPRT-reverse (5′-AGTCTGGCTTATATCCAACACTTCG-3′), TBP-forward (5′-CACGAACCACGGCACTGATT-3′), and TBP-reverse (5′-TTTTCTTGCTGCCAGTCTGGAC-3′). SDHA, HPRT, and TBP were used as internal controls. Ct values for NG2 were normalized to the internal controls and then quantified relative to their corresponding negative control within the experiment. Experiments were performed using a QuantStudio 5 Real-Time PCR machine (Applied Biosystems).
siRNA transfection
Human breast cancer cells (100,000) were seeded in each well of a six-well plate. Cells were transfected with 30 nM of either a nontargeting control siRNA or an NG2-specific siRNA (Ambion Inc., Austin, TX). Transfections were performed using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions. Seventy-two hours after delivery of siRNA, cells were either trypsinized for use in 2D migration experiments or lysed for RNA isolation.
Statistical analysis
GraphPad Prism v7.04 was used for generation of graphs and statistical analysis. For data with normal distribution: For comparison between two groups, an unpaired two-tailed Student’s t test was used, and a P value of ≤0.05 was considered significant, and for comparison between multiple groups, a one-way analysis of variance (ANOVA) with Bonferroni multiple testing correction was used, with a corrected P value of ≤0.05 considered significant. For data with nonnormal distribution: For comparison between two groups, an unpaired two-tailed Mann-Whitney test was used, and a P value of ≤0.05 was considered significant, and for comparison between multiple groups, a Kruskal-Wallis test with Dunn’s multiple testing correction was used, with a corrected P value of ≤0.05 considered significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the NIH (R00-CA207866-04 to M.J.O.) and Tufts University (Start-up funds from the School of Engineering to M.J.O., Collaborative Cancer Biology Award to J.P.F., Tufts Summer Scholars Award to Y.P., and Laidlaw Foundation Funds to R.A.M.). A.S.G. was supported by ARS Project 1950-51000-071-02S, P30 DK046200-23, P30 DK046200-27S2, DK108722 01A1, and 1R21HD098056-01 and the Robert C and Veronica Atkins Foundation. M.H. was supported by NIH Research Infrastructure grant S10 OD021624. This study was also supported by the Breast Cancer Alliance Young Investigator Award to M.J.O. Author contributions: M.J.O. and A.L.W. designed the experiments. A.L.W., S.J.C., J.R.G., J.P.F., Y.P., R.A.M., and M.H. completed experiments in the manuscript. S.P.N. provided human tissue samples. R.C. and A.S.G. provided lean and obese adipose tissue. A.L.W., S.J.C., J.R.G., J.P.F., and M.J.O. wrote the manuscript. All authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc3175/DC1
REFERENCES AND NOTES
- 1.Bianchini G., Balko J. M., Mayer I. A., Sanders M. E., Gianni L., Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anders C. K., Zagar T. M., Carey L. A., The management of early-stage and metastatic triple-negative breast cancer: A review. Hematol. Oncol. Clin. North Am. 27, 737–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J. H., Ahn J.-H., Kim S.-B., How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 3( suppl. 1), e000357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liedtke C., Mazouni C., Hess K. R., André F., Tordai A., Mejia J. A., Symmans W. F., Gonzalez-Angulo A. M., Hennessy B., Green M., Cristofanilli M., Hortobagyi G. N., Pusztai L., Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–12781 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Steeg P. S., Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Socovich A. M., Naba A., The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 89, 157–166 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Oudin M. J., Jonas O., Kosciuk T., Broye L. C., Guido B. C., Wyckoff J., Riquelme D., Lamar J. M., Asokan S. B., Whittaker C., Ma D., Langer R., Cima M. J., Wisinski K. B., Hynes R. O., Lauffenburger D. A., Keely P. J., Bear J. E., Gertler F. B., Tumor cell-driven extracellular matrix remodeling drives haptotaxis during metastatic progression. Cancer Discov. 6, 516–531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naba A., Clauser K. R., Ding H., Whittaker C. A., Carr S. A., Hynes R. O., The extracellular matrix: Tools and insights for the "omics" era. Matrix Biol. 49, 10–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayorca-Guiliani A. E., Madsen C. D., Cox T. R., Horton E. R., Venning F. A., Erler J. T., ISDoT: In situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 23, 890–898 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Naba A., Clauser K. R., Lamar J. M., Carr S. A., Hynes R. O., Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 3, e01308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell S., Shaw D., The worldwide epidemic of female obesity. Best Pract. Res. 29, 289–299 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Choi Y., Park S. K., Ahn K. J., Cho H., Kim T. H., Yoon H. K., Lee Y.-H., Being overweight or obese increases the risk of progression in triple-negative breast cancer after surgical resection. J. Korean Med. Sci. 31, 886–891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarroudi O. A., Abda N., Seddik Y., Brahmi S. A., Afqir S., Overweight: Is it a prognostic factor in women with triple-negative breast cancer? Asian Pac. J. Cancer Prev. 18, 1519–1523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osman M. A., Hennessy B. T., Obesity correlation with metastases development and response to first-line metastatic chemotherapy in breast cancer. Clin. Med. Insights 9, 105–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontanella C., Lederer B., Gade S., Vanoppen M., Blohmer J. U., Costa S. D., Denkert C., Eidtmann H., Gerber B., Hanusch C., Hilfrich J., Huober J., Schneeweiss A., Paepke S., Jackisch C., Mehta K., Nekljudova V., Untch M., Neven P., von Minckwitz G., Loibl S., Impact of body mass index on neoadjuvant treatment outcome: A pooled analysis of eight prospective neoadjuvant breast cancer trials. Breast Cancer Res. Treat. 150, 127–139 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Quail D. F., Dannenberg A. J., The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 15, 139–154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka M., Ikeda K., Suganami T., Komiya C., Ochi K., Shirakawa I., Hamaguchi M., Nishimura S., Manabe I., Matsuda T., Kimura K., Inoue H., Inagaki Y., Aoe S., Yamasaki S., Ogawa Y., Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nat. Commun. 5, 4982 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Seo B. R., Bhardwaj P., Choi S., Gonzalez J., Andresen Eguiluz R. C., Wang K., Mohanan S., Morris P. G., Du B., Zhou X. K., Vahdat L. T., Verma A., Elemento O., Hudis C. A., Williams R. M., Gourdon D., Dannenberg A. J., Fischbach C., Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 7, 301ra130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baskaran J. P., Weldy A., Guarin J., Munoz G., Shpilker P. H., Kotlik M., Subbiah N., Wishart A., Peng Y., Miller M. A., Cowen L., Oudin M. J., Cell shape, and not 2D migration, predicts extracellular matrix-driven 3D cell invasion in breast cancer. APL Bioeng. 4, 026105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyengar P., Espina V., Williams T. W., Lin Y., Berry D., Jelicks L. A., Lee H., Temple K., Graves R., Pollard J., Chopra N., Russell R. G., Sasisekharan R., Trock B. J., Lippman M., Calvert V. S., Petricoin E. F. III, Liotta L., Dadachova E., Pestell R. G., Lisanti M. P., Bonaldo P., Scherer P. E., Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 115, 1163–1176 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cescon M., Gattazzo F., Chen P., Bonaldo P., Collagen VI at a glance. J. Cell Sci. 128, 3525–3531 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Gara S. K., Grumati P., Urciuolo A., Bonaldo P., Kobbe B., Koch M., Paulsson M., Wagener R., Three novel collagen VI chains with high homology to the α3 chain. J. Biol. Chem. 283, 10658–10670 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Park J., Scherer P. E., Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Investig. 122, 4243–4256 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price M. A., Colvin Wanshura L. E., Yang J., Carlson J., Xiang B., Li G., Ferrone S., Dudek A. Z., Turley E. A., Mc Carthy J. B., CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 24, 1148–1157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng I. H., Lin Y.-C., Hwang E., Huang H.-T., Chang W.-H., Liu Y.-L., Chao C.-Y., Collagen VI protects against neuronal apoptosis elicited by ultraviolet irradiation via an Akt/Phosphatidylinositol 3-kinase signaling pathway. Neuroscience 183, 178–188 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Cattaruzza S., Nicolosi P. A., Braghetta P., Pazzaglia L., Benassi M. S., Picci P., Lacrima K., Zanocco D., Rizzo E., Stallcup W. B., Colombatti A., Perris R., NG2/CSPG4-collagen type VI interplays putatively involved in the microenvironmental control of tumour engraftment and local expansion. J. Mol. Cell Biol. 5, 176–193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ojima K., Oe M., Nakajima I., Muroya S., Nishimura T., Dynamics of protein secretion during adipocyte differentiation. FEBS Open Bio 6, 816–826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcelin G., Silveira A. L. M., Martins L. B., Ferreira A. V., Clément K., Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J. Clin. Invest. 129, 4032–4040 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mentoor I., Engelbrecht A.-M., van Jaarsveld P. J., Nell T., Chemoresistance: Intricate interplay between breast tumor cells and adipocytes in the tumor microenvironment. Front. Endocrinol. 9, 758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherman-Baust C. A., Weeraratna A. T., Rangel L. B. A., Pizer E. S., Cho K. R., Schwartz D. R., Shock T., Morin P. J., Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell 3, 377–386 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Jailkhani N., Ingram J. R., Rashidian M., Rickelt S., Tian C., Mak H., Jiang Z., Ploegh H. L., Hynes R. O., Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 116, 14181–14190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urello M. A., Kiick K. L., Sullivan M. O., ECM turnover-stimulated gene delivery through collagen-mimetic peptide-plasmid integration in collagen. Acta Biomater. 62, 167–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pitteri S. J., Kelly-Spratt K. S., Gurley K. E., Kennedy J., Buson T. B., Chin A., Wang H., Zhang Q., Wong C.-H., Chodosh L. A., Nelson P. S., Hanash S. M., Kemp C. J., Tumor microenvironment-derived proteins dominate the plasma proteome response during breast cancer induction and progression. Cancer Res. 71, 5090–5100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller M. A., Oudin M. J., Sullivan R. J., Wang S. J., Meyer A. S., Im H., Frederick D. T., Tadros J., Griffith L. G., Lee H., Weissleder R., Flaherty K. T., Gertler F. B., Lauffenburger D. A., Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 6, 382–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heidari P., Esfahani S. A., Turker N. S., Wong G., Wang T. C., Rustgi A. K., Mahmood U., Imaging of secreted extracellular periostin, an important marker of invasion in the tumor microenvironment in esophageal cancer. J. Nucl. Med. 56, 1246–1251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y., Keikhosravi A., Mehta G. S., Drifka C. R., Eliceiri K. W., Methods for quantifying fibrillar collagen alignment. Methods Mol. Biol. 1627, 429–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., Sabatini D. M., CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Györffy B., Lanczky A., Eklund A. C., Denkert C., Budczies J., Li Q., Szallasi Z., An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eabc3175/DC1