

Abstract
Herein we present a comparative study of the effects of isoquinoline alkaloids belonging to benzo[c]phenanthridine and berberine families on β-amyloid aggregation. Results obtained using a Thioflavine T (ThT) fluorescence assay and circular dichroism (CD) spectroscopy suggested that the benzo[c]phenanthridine nucleus, present in both sanguinarine and chelerythrine molecules, was directly involved in an inhibitory effect of Aβ1–42 aggregation. Conversely, coralyne, that contains the isomeric berberine nucleus, significantly increased propensity for Aβ1–42 to aggregate. Surface Plasmon Resonance (SPR) experiments provided quantitative estimation of these interactions: coralyne bound to Aβ1–42 with an affinity (KD = 11.6 μM) higher than benzo[c]phenanthridines. Molecular docking studies confirmed that all three compounds are able to recognize Aβ1–42 in different aggregation forms suggesting their effective capacity to modulate the Aβ1–42 self-recognition mechanism. Molecular dynamics simulations indicated that coralyne increased the β-content of Aβ1–42, in early stages of aggregation, consistent with fluorescence-based promotion of the Aβ1–42 self-recognition mechanism by this alkaloid. At the same time, sanguinarine induced Aβ1–42 helical conformation corroborating its ability to delay aggregation as experimentally proved in vitro. The investigated compounds were shown to interfere with aggregation of Aβ1–42 demonstrating their potential as starting leads for the development of therapeutic strategies in neurodegenerative diseases.
Keywords: Amyloid beta, Neurodrug, Amyloid aggregation, Alzheimer's disease, Chelerythrine, Sanguinarine, Coralyne, Berberine
Graphical abstract
Highlights
-
•
We compared the effects of three isoquinoline alkaloids on β-amyloid aggregation.
-
•
Sanguinarine and chelerythrine showed inhibitory effects on Aβ1–42 aggregation.
-
•
Coralyne significantly increased propensity for Aβ1–42 to aggregate.
-
•
Molecular dynamics suggested the alkaloid ability to affect β-content of Aβ1–42.
1. Introduction
Several neurodegenerative disorders, including Alzheimer's (AD), Parkinson's (PD) and Huntington's (HD) diseases are associated with aggregation of misfolded proteins [1,2]. Among these, AD, a predominant cause of dementia worldwide [3,4], is characterized by extracellular amyloid deposits, whose main component is the 42-amino acid amyloid β peptide (Aβ1-42), and by intracellular neurofibrillary tangles composed of tau [5,6].
Aβ1–42 is a peptide cleaved from the amyloid precursor protein (APP), comprised of a charged N-terminal segment (amino acids 1–22), a hydrophobic central region (KLVFFA, amino acids 16–21), which alone is able to aggregate into insoluble fibrils, and a hydrophobic C-terminal region (residues 23–42). Once released as a monomer from APP into the extracellular space, Aβ1-42 undergoes a structural transition gaining β-sheet content, and tends to aggregate into oligomeric, protofibrillar and fibrillar species [7]. Aβ1–42 oligomeric assemblies have been related to AD pathogenesis for their role in neuronal damage and neurotoxicity following Aβ1–42 aggregation [8]. In this context, preventing Aβ1–42 aggregation with small molecules is one of the prominent strategies for the development of new therapies for AD [[9], [10], [11]]. To this scope, several plant extracts and natural products, such as curcumin, epigallocatechin-3-gallate, and resveratrol, were evaluated with promising results [[12], [13], [14]].
Isoquinoline alkaloids (Fig. 1 ) belong to one of the most complex families of plant alkaloids. They are nitrogenous metabolites distributed in many botanical families investigated nowadays for their significant biomedical importance [[15], [16], [17]]. Among these, benzo[c]phenanthridines and protoberberines are found in various vegetal sources belonging to the Rutaceae family (in particular from the Zanthoxylum genus [18]), with berberine (Fig. 1) being an interesting candidate for PD and AD thanks to multi-faceted defensive mechanisms and bio-molecular pathways involving this alkaloid [19,20]. However, its use as a neurodrug is hampered by its cytotoxic effects at relatively high concentration [21]. Hence, a structurally modified version of berberine that results in the nontoxic, free hydroxyl-bearing Ber-D was prepared, which was found to inhibit the aggregation and cell toxicity of Aβ1-42 in vitro [22]. The berberine nucleus in Ber-D comprises four rings, of which three aromatic, whereas the anti-leukemic berberine-like drug coralyne (here indicated as CO, Fig. 1) contains all four aromatic rings [23,24].
Fig. 1.

The isoquinoline alkaloids of synthetic (CO) and plant (CH and SA) origin investigated in this work. All share an isoquinoline core (up, left) but are based on two different polycycle rearrangements (bottom, left).
Other examples of plant isoquinoline alkaloids are sanguinarine (SA) and chelerythrine (CH, Fig. 1), two tetracyclic aromatic compounds isolated from Macleaya cordata belonging to the family of benzo[c]phenanthridines, and also classifiable as azachrysenes [25,26]. In particular, SA is endowed with several properties of therapeutic relevance, including the reduction of levels of stress hormone as shown in studies carried out in animal models [27], as well as of serum haptoglobin, and serum amyloid A (SAA) [27,28]. This latter is mainly produced in the liver but also expressed extrahepatically in the central nervous system (CNS) [29], with increased levels in AD patients [29], and it was recently recognized as a biomarker for COVID-19 [30], that is a recently-emerged viral disease causing severe acute respiratory syndrome and diverse injuries in other systems [[31], [32], [33], [34]]. SA and CH are believed to possess potential as neurodrugs for AD due to their ability to inhibit several neuropathologically-relevant enzymes [35]. However, clues of neuroprotective properties were found experimentally only for CH which inhibited in vitro amyloid aggregation [36], whereas the same inhibitory activity, predicted in silico for SA by some of us [37], had not been validated before on an experimental basis.
Thus, the scope of this work was to investigate the interaction between tetracyclic aromatic structures endowed with benzo[c]phenanthridine (SA, CH) and berberine (CO, Fig. 1) nuclei, respectively, with Aβ1–42 peptide, by means of ThT fluorescence and CD spectroscopy to evaluate their effects on the aggregation of Aβ1–42, and by surface plasmon resonance (SPR) assays to characterize these interactions.
Experimental data were further corroborated by in silico studies, through molecular docking simulations, to unveil preferential binding modes of ligands to different aggregated forms of Aβ1–42, and by molecular dynamics simulations to explore the effects of these compounds in early aggregation stages of Aβ1–42.
2. Materials and methods
2.1. Chemicals
Aβ1-42 peptide (for CD and SPR), SA, CH, SA isoquinoline alkaloids and all other chemicals and solvents were purchased from Sigma-Aldrich (Amsterdam, The Netherlands). Aβ1-42 peptide for ThT assay was purchased from rPeptide (GA, USA).
2.2. Aβ1-42 peptide solubilization
Solutions of recombinant Aβ1-42 peptide were prepared according to a previously published procedure [38]. In short, Aβ1-42 was sequentially dissolved in hexafluoroisopropanol (HFIP) and DMSO. The DMSO was removed from the Aβ1-42 solution by using a HiTrap™ desalting column (GE Healthcare, Zwijndrecht, The Netherlands) and elution with PBS at pH 7.4. We measured the Aβ1-42concentration by the Coomassie (Bradford, UK) Protein Assay Kit (ThermoFisher, Landsmeer, The Netherlands) and, afterwards, the final concentration required for the subsequent experiments was achieved by dilution. Aβ peptide aggregation, in the presence or absence of SA, CH and CO, was evaluated at 37 °C under quiescent conditions.
2.3. Thioflavin-T assay
Aggregation was measured by a ThT fluorescence assay. The Aβ1-42 concentration was adjusted to 25 μM using PBS buffer (pH 7.4), while a final ThT concentration of 12 μM was realized in a 96-well plate (Greiner flat bottom transparent black, Sigma–cat. M9685). Fluorescence intensity was measured at 37 °C using an automated well-plate reader (Tecan Infinite 200 PRO) at an excitation wavelength of 450 nm and emission detection from 480 to 600 nm. The fluorescence intensity from ThT at its maximum value (485 nm) was reported in a graph for the three complexes with the ligands (C = 25 μM). Measurements were performed in triplicate, the values recorded were averaged and background measurements that corresponded to buffer containing 12 μM ThT and the tested isoquinoline alkaloids subtracted. Measurements were performed after incubation for 2 h to allow Aβ to aggregate.
2.4. CD experiments
The CD experiments were conducted as previously described [[39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53]]. The spectra were obtained using a JascoJ-715 spectropolarimeter coupled to a PTC-348WI temperature control system, and a quartz cell with a path length of 1 cm, at 37 °C with a response of 1 s, a scanning speed of 100 nm/min and a 2.0 nm bandwidth. All the spectra were averaged over three scans. Experiments were carried out using a 5 μM concentration of Aβ1-42 in PBS (overall volume = 2 ml, pH 7.2) and a twofold concentration of ligands. Spectra were collected after incubation at 37 °C for 0.5, 24 and 48 h.
2.5. Surface plasmon resonance (SPR) experiments
Surface plasmon resonance (SPR) binding assays were performed on a Biacore 3000 (GE Healthcare). Aβ1-42 peptide was immobilized on a CM5 chip through an amine coupling procedure at 100 μg/mL in 10 mM sodium acetate (pH 4) at 2 μL/min until reaching an immobilization level of ∼400 RU. Binding assays were carried out by injecting 90 μL of analyte, at 20 μL/min-1. Experiments were carried out using PBS as running buffer. The association phase (kon) was followed for 270 s, whereas the dissociation phase (koff) was followed for 300 s. The reference chip sensorgrams were subtracted from sample sensorgrams. After each cycle, the sensor chip surface was regenerated with a 10 mM NaOH solution for 30 s. Analyte concentrations were for cheletrine 20, 40, 80 and 100 μM, sanguinarine 100, 300, 500, 700, 900 and 1100 μM and for coralyne 5, 20, 30, 40, 50, 70 μM. Experiments were carried out in duplicates. Kinetic parameters were estimated assuming a 1:1 binding model and using version 4.1 Evaluation Software (GE Healthcare).
2.6. In silico studies
In all computational studies, as initial Aβ1-42 conformations we utilized S-shape and U-shape fibril models (PDB codes: 2LMN and 2MXU) and three of the most representative monomeric models from previous extensive computational studies [54].
2.7. Ligand parameterization
Fully-protonated structures of the three compounds (CO, SA, CH) were optimized by gaussian 09 software [55], utilizing Hartree-Fock method and 6-31G∗ basis set. AM1-BCC method [56] implemented in the AmberTools 19 package was used to derive charges of all atoms. Parameters for bonds, valence and dihedral angles were adapted from General Amber Force Field [57] based on structural similarity.
2.8. Docking
Global molecular docking of compounds to the monomeric, tetrameric, and fibrillar structures of Aβ1-42 was performed using AutoDock 4.2.6 software [58] allowing flexibility of the ligand with rigid conformation of the receptor due to computational limitations. The algorithm was set to generate 100 initial docking positions and subsequently perform clustering using 10, 15, and 15 Å criteria for monomeric, tetrameric, and fibril structures, respectively, to obtain most probable docking positions (modes) of the compounds. Two different cutoff values were used due to large size differences between monomeric and other systems. AutoDock 4.2 was selected for docking, because it was found to provide more reliable binding energies than AutoDock Vina in recent studies [59]. In general, AutoDock 4.2.6 should provide reliable docking poses and estimated binding energies [60]. It should be mentioned that in all computational methods using approximate system representation, such as molecular docking or MD simulations, relative energies, rather than absolute should be analyzed, treating the latter with large possible error [61], however, usually binding energy stronger than −9 kcal/mol is treated as strong binding [62].
2.9. Molecular dynamics simulations
Two series of molecular dynamics (MD) simulations were performed: (i) fibrillar structures with the compounds bound to them, obtained through docking procedure, and (ii) 16 non-bound semi-extended Aβ1-42 chains in the presence and absence of compounds. MD simulations of fibrillar Aβ1-42 with compounds were performed using Amber ff14sb [63] force field with TIP3P water model [64], which should provide reliable results for these systems. Due to computational restrictions, MD simulations were performed for top 2 binding modes of each system, each of 10 separate trajectories, reaching in total 1 μs for each of the binding modes.
For MD simulations of 16 chains, we used an in-house algorithm to put pre-generated semi-extended Aβ1-42 chains of random conformations as close to each other as possible, with the restriction to keep minimum distance of 8 Å between any heavy atoms of different chains to avoid possible bias coming from initial orientation of the chains. Such system was hydrated by adding approximately 47500 water molecules and charge was neutralized by inserting counterions, resulting in truncated octahedron boxes of total volume of approximately 1800 nm3 resulting in total Aβ1-42 concentration of approximately 1 cM, which is order of magnitude higher than in other studies [65,66], yet still not in glass phase [67]. In simulations with compounds, small molecules were placed between Aβ1-42 chains using the same criterion. In all simulations, initial orientations of Aβ1-42 chains and compounds were identical.
Obtained systems were energy minimized, using steepest descent and conjugate gradient algorithm and equilibrated for 1ns. For each type of system, two trajectories were run, each of 800ns and then recorded 20,000 snapshots from the last 200 ns (600–800ns) were analyzed. To better capture aggregation effects in simulations of systems containing 16 chains, we utilized state-of-the-art Amber ff19sb force field [68] coupled with OPC water model [69], which should provide reliable results, especially for binding-dissociation process. Analysis of these simulations included root-mean-square deviation (RMDd) using initial structure as a reference, radius of gyration (Rg), solvent-accessible surface area (SASA) using LCPO method [70] and secondary structure determinations with DSSP [71] algorithm implemented into Amber19 package and various distance calculations. Distance criterion of 6.5 Å between centers of mass of two side-chains was used to determine a contact between chains, and a criterion of 5 contacts was used to determine the size of the oligomer (e.g. two chains have to form at least 5 contacts to be named as dimer), as in our previous work [66] to discard structures forming weak interaction due to accidental proximity of the chains.
2.10. Molecular mechanics - Poisson Boltzmann Surface Area (MM/PBSA) method
MM-PBSA is a post-processing method which was used to calculate the free energy difference, ΔGbind, between the free and bound states of a molecule complex: receptor and ligand. ΔGbind is calculated for a set of selective snapshots from simulation trajectory and is defined as follows:
| ΔGbind = ΔEelec + ΔEvdW + ΔESUR + ΔEPB – TΔS, | (1) |
where ΔEelec and ΔEvdW are differences in electrostatic and van der Waals energy components, respectively, ΔESUR and ΔEPB describe differences in non-polar and polar solvation free energies, respectively, and TΔS represents the entropic contribution.
In this study, MM/PBSA methods implemented into the AmberTools 19 package was used to estimate ΔGbind of compounds to fibrillar models using second halves of performed MD simulations. As a standard procedure, for energy calculation in MM/PBSA procedure we used the same force field adopted to perform the simulations, however, without cutoff for electrostatic and van der Waals interactions. The entropic term, TΔS, was estimated by normal mode approximation method, where ΔEPB was obtained by solving numerically linearized Poisson-Boltzmann equation and ΔESUR was calculated from the following equation:
| ΔESUR = α x SASA + β, | (2) |
where SASA was calculated using LCPO method [66], regression coefficient α was set to 0.005 and the regression offset β was set to 0.
3. Results and discussion
3.1. Modulation of Aβ1–42 aggregation
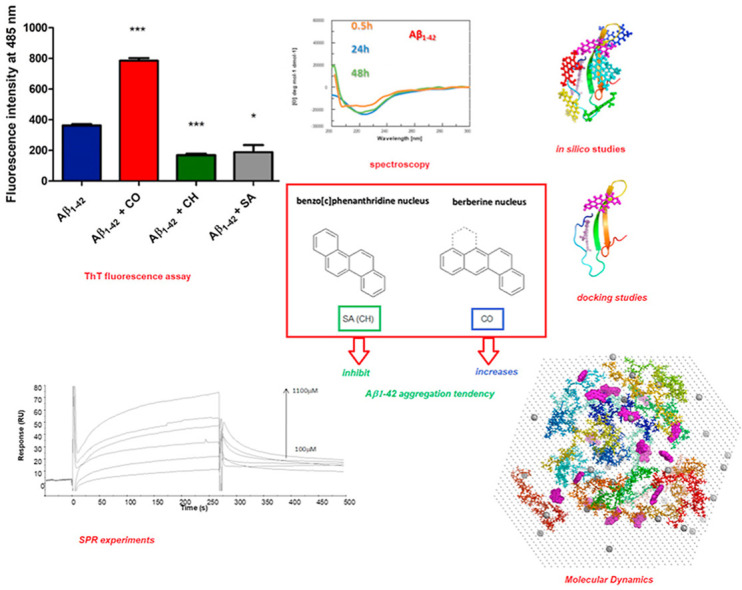
Toxicity of Aβ and related Alzheimer's disease-associated neuronal loss have been clinically associated with the accumulation of oligomeric forms of the peptide which generally are known to precede amyloid fibril formation [72,73]. In vitro assays have shown that short incubation times, of 1.5–6 h, result in the formation of ThT positive oligomeric Aβ1-42 assemblies that significantly associate with apoptotic neurons and cognitive dysfunction in a mouse model [74]. To obtain preliminary insights into the ability of isoquinoline alkaloids to modulate the accumulation of Aβ1-42 oligomers we evaluated herein thioflavin (ThT) fluorescence intensity after 2 h incubation [75]. First of all, the Aβ1-42 monomer (25 μM) was incubated with SA, CH or CO (25 μM). The extent of ThT-positive aggregation of Aβ1-42 within this incubation time was then assessed by recording the fluorescence emission of ThT (12 μM, λex = 450 nm, λem = 485 nm) (Fig. 2 ).
Fig. 2.

SA and CH inhibit ThT-positive oligomer formation of Aβ1-42, whereas CO induces ThT-positive assemblies. Solutions containing Aβ1-42 at a concentration of 25 μM were incubated in the presence and absence of SA, CH and CO (at 1:1 ratio) at 37 °C for 2 h. ThT-positive aggregate formation was detected using ThT fluorescence intensity measurements at a fluorescence emission wavelength of 485 nm upon excitation at 450 nm. The reported values represent the results obtained from three independent experiments. The statistical significance of the replicates was assessed by p-values using paired two-tailed t-tests (GraphPad Prism).
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared with the control (‘Aβ1-42’).
Data show that SA and CH reduce the ThT fluorescence signal by ∼40% compared with Aβ1-42 in the absence of these compounds. On the other hand, the berberine-like CO increased the aggregation level of Aβ1-42 as indicated by a strong two-fold increase in ThT fluorescence intensity compared to untreated Aβ1-42. These results show that berberine-like and benzo[c]phenanthridine alkaloids differently modulate Aβ1-42 aggregation.
3.2. Aβ1-42 conformational response to isoquinoline alkaloids
To investigate if the observed effects of isoquinoline alkaloids on Aβ1-42 aggregation were accompanied by conformational variations, we performed circular dichroism (CD) time-dependent studies. The aggregation of Aβ1-42, which reportedly coincides with increasing β-sheet content [76], was monitored using CD at different time points of incubation (0.5, 24 and 48 h, in PBS at 37 °C; Fig. 3 ). The obtained time-dependent CD profiles of Aβ1-42 showed spectral changes in agreement with those reported in literature [11,77] with a progressive transition towards a β-sheet conformation at 24 h indicated by a broad band centered at ∼225 nm, that is a spectral element previously assigned to this secondary structure in many amyloid systems [[78], [79], [80], [81], [82], [83]].
Fig. 3.

Conformational response of Aβ1-42peptide to SA, CH and CO. Circular dichroism spectra of Aβ1-42 (5 μM concentration in PBS, black line) and Aβ1-42 in the presence of isoquinoline alkaloids (1:2 M ratio, peptide: small molecule) after 0.5 (orange), 24 (blue), and 48 (green) h of incubation at 37 °C.
At longer incubation times CD intensity at 225 nm showed a tendency to decrease (Fig. 3) suggestive of amyloid aggregation/precipitation as previously observed under similar conditions [77]. In parallel, Aβ1-42 was incubated, under the same experimental conditions, with the isoquinoline alkaloids (which did not contribute to the observed CD signal).
Remarkably, the presence of CO, already at t = 0.5 h of analysis, (Fig. 3) favors a β-like structure as indicated by a minimum at ∼225 nm that, in the following 24 h, slightly shifts toward higher wavelengths (Fig. 3). The observed increase of Cotton effect for Aβ1-42 in the presence of CO at 24 h (Fig. 3) can be ascribed to a stabilization of these secondary structures [[84], [85], [86], [87], [87], [88], [89]]. When comparing the CD spectra of Aβ1-42 in the presence of all three compounds after 24 h, it became apparent that the presence of the three isoquinoline alkaloids induced differences in the structural organization of Aβ1-42 (Fig. 3). The observed changes, impacting on both intensity and shape of spectra, were already described by Guo et al. [80], suggesting that benzo[c]phenanthridines partly limited β-sheet content of Aβ1-42 leading to new structural elements. The effect is more appreciable for CH while the main significant variations are evident in the 210–220 nm range for SA.
3.3. Isoquinoline alkaloids interact with Aβ1-42
To further evaluate the ability of isoquinoline alkaloids to interact with Aβ1-42 we carried out SPR assays [90]. Binding profiles for all three molecules (Fig. 4 ) suggested the formation of complexes, in a concentration-dependent manner. Freshly dissolved Aβ1-42, after HFIP treatment, was covalently immobilized on Sensor chip [91]. Kinetic parameters, reported in Table 1 , allowed the estimation of thermodynamic dissociation constant values that appear in the low, for CO, high, for SA, and very high, for CH, micromolar range. The higher affinity exhibited by CO compared to CH and SA can be due to the faster association phase. Our data are in agreement with a previous study [91] that showed the ability of berberine-like inhibitors of Aβ1-42 to interact with the polypeptide at low micromolar KD values [91].
Fig. 4.

Overlay of sensorgrams for the binding to immobilized Aβ1-42 of (A) SA, (B) CH and (C) CO.
Table 1.
SPR based equilibrium dissociation constants (KD) and kinetic parameters for the interaction of Aβ1-42 with SA, CH and CO using the BIA evaluation v.4.1 software. Data reported were obtained through SPR analyses using small molecules as analyte on immobilized Aβ1-42.
| kon (M−1s−1 × 104) | koff (s−1 × 10−3) | KD (μM) | |
|---|---|---|---|
| Sanguinarine (SA) | 13.1 | 6.07 | 463 |
| Cheletrine (CH) | 5.14 | 19.7 | 3.83∗10 3 |
| Coralyne (CO) | 983 | 11.4 | 11.6 |
3.4. Computational study of the interaction of SA, CH and CO with monomeric and fibrillar Aβ1-42
To further explore the molecular-level interactions responsible for the observed modulating effects of Aβ1-42 aggregation displayed by small molecules we performed in silico studies as described below.
3.5. Binding energies
3.5.1. Docking of ligands to monomers
The binding energies of the three ligands were estimated by means of Molecular Docking. Since Aβ peptides are intrinsically disordered, their native structures are transient and cannot be resolved experimentally.
Therefore, for our simulations we adopted three most representative Aβ1-42 monomeric models obtained by clustering ensembles of monomeric Aβ1-42 conformations at 300K from extensive all-atom Replica-Exchange and conventional MD simulations with explicit water model performed with various Amber and CHARMM force fields [54], as targets (Fig. 5 ). Because of the disordered character of monomeric Aβ1-42 there is no possibility to treat properly flexibility of the receptor during docking procedure, therefore, we utilized three various Aβ1-42 conformations to better sample the possible binding modes and which should minimize the impact of conformational selection. It should be noted that the use of multiple targets can significantly enhance the quality of docking results as shown by the McCammon group [92] and this approach is known as ensemble-based virtual screening.
Fig. 5.

Representations of docking positions of CO (left column: A, D, G), SA (middle column: B, E, H), and CH (right column: C, F, I) to three models of monomeric Aβ1-42 (presented as rainbow-colored cartoons).
As expected for similar small compounds, their modes of interactions appeared quite similar, but significant differences were observed in the number of possible binding modes (Table 2 ), which is higher for CO for all three monomeric structures. Conversely, the lowest number of binding modes was found for SA suggesting a more selective binding mechanism toward Aβ1-42 with respect to the other compounds. The drug-amyloid interactions are stabilized by both hydrophobic and hydrogen bonds (three for SA and CH and one for CO, Fig. 6 ). Interestingly, CH and SA, contrary to CO, form hydrogen bonds with two histidine residue (His13 and His14), that are reported as responsible of the binding of ions, e.g. Cu2+, which impacts Aβ1-42 aggregation [93].
Table 2.
AutoDock-predicted binding energies (kcal/mol) for the binding of the compounds CO, SA, and CH to three representative amyloid monomeric models obtained in the previous simulation study [50].
|
Binding Mode |
Aβ1-42 Model 1 |
Aβ1-42 Model 2 |
Aβ1-42 Model 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| CO | SA | CH | CO | SA | CH | CO | SA | CH | |
| 1 | −8.03 | −9.10 | −8.59 | −10.17 | −10.26 | −10.07 | −9.44 | −9.07 | −8.82 |
| 2 | −6.68 | −7.24 | −7.39 | −7.53 | −8.58 | −8.36 | −8.11 | −8.99 | −8.31 |
| 3 | −6.36 | −6.05 | −7.52 | −6.68 | −8.69 | −7.05 | |||
| 4 | −6.24 | −6.03 | −7.07 | −6.58 | −6.87 | −6.86 | |||
| 5 | −5.57 | −7.00 | −5.84 | ||||||
| 6 | −5.42 | −5.30 | |||||||
| 7 | −5.34 | ||||||||
| 8 | −5.28 | ||||||||
Fig. 6.

Schematic representation of the strongest binding mode of monomeric Aβ1-42 to compounds (monomeric model 2, binding mode 1; see Table 2 for more details) showed in 2D form for: A) CH, B) CO, C) SA. Aβ1-42 residues involved in hydrophobic interactions with compounds are showed by red lines and black three-letter residue codes, hydrogen bonds are represented by cyan dashed lines and green three-letter residue codes. For clarity, hydrogens are not presented on the plot.
Averaging over all target structures in the best docking mode (mode 1) of the monomer, from Table 2 we obtain the binding energy ΔE bind = −9.21, −9.48 and −9.16 kcal/mol for CO, SA and CH, respectively
The highest interaction energy was observed for the least structured model 2, due to the disordered and extended character of this conformation allowing compounds to maximize the number of hydrogen bonds between molecules maintaining a high number of hydrophobic contacts (Table 3 ).
Table 3.
Number of hydrogen bonds (HB) and hydrophobic interactions (HI) between monomeric Aβ1-42 models and the ligands CO, SA, and CH in the strongest binding mode (mode 1).
| Aβ1-42 Model 1 |
Aβ1-42 Model 2 |
Aβ1-42 Model 3 |
||||
|---|---|---|---|---|---|---|
| HI | HB | HI | HB | HI | HB | |
| CO | 10 | 1 | 11 | 1 | 9 | 1 |
| SA | 10 | 1 | 9 | 3 | 7 | 1 |
| CH | 9 | 1 | 8 | 3 | 7 | 1 |
3.5.2. Docking of ligands to Aβ1-42 tetramers
As oligomeric states are a bridging step between monomers and fibrils, we decided to study the impact of the three ligands on tetramers, that are considered crucial in Aβ1-42 aggregation [94], by using models obtained in previous multi-scale MD simulations [66]. Similar to the monomeric Aβ1-42, SA exhibited minor binding modes for all three tetrameric models (Table 4 and Fig. S1) confirming major selectivity of interaction. Averaging over three models of the tetramer and using data shown in Table 4, in the best docking mode we obtained ΔE bind = −8.88, −9.98, and −9.60 kcal/mol for CO, SA and CH, respectively. Which these values of the binding energy, IC50 of the three compounds is of order of μM. Moreover, the small differences in binding affinity of compounds to monomeric and tetrameric forms are probably due to compact forms of Aβ1-42 tetramers, which did not allow many interactions with drugs even when more chains and possible binding sites are available. Overall, the high binding affinity of the studied compounds to oligomers indicates they can alter the fibril formation kinetics and pathways.
Table 4.
AutoDock-predicted binding energies (kcal/mol) for the binding of the CO, SA, and CH to three representative amyloid tetrameric models obtained in the previous simulation study [62] (models 1, 2, and 3 correspond to tetramers 1, 3, and 5 from the mentioned work, respectively).
|
Binding Mode |
Aβ1-42 tetramer 1 |
Aβ1-42 tetramer 2 |
Aβ1-42 tetramer 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| CO | SA | CH | CO | SA | CH | CO | SA | CH | |
| 1 | −9.06 | −9.97 | −9.72 | −9.36 | −9.52 | −9.26 | −8.21 | −10.45 | −9.81 |
| 2 | −8.48 | −9.89 | −8.93 | −8.84 | −9.46 | −9.05 | −7.94 | −8.44 | −8.43 |
| 3 | −7.56 | −9.14 | −8.78 | −8.80 | −9.13 | −7.91 | −7.24 | −8.26 | −8.32 |
| 4 | −7.51 | −7.19 | −7.28 | −8.34 | −7.42 | −7.02 | −7.96 | −7.57 | |
| 5 | −6.43 | −7.13 | −7.25 | −7.52 | −7.33 | −7.00 | −7.68 | −7.50 | |
| 6 | −5.77 | −6.55 | −7.04 | −6.96 | −6.80 | −6.88 | |||
| 7 | −6.55 | −6.50 | |||||||
| 8 | −6.01 | −6.48 | |||||||
3.5.3. Docking of ligands to Aβ1-42 protofibrils
The structure of Aβ1-42 fibrils is still under debate. Old solid state NMR experiments showed that the monomer structure has the U-shape in the fibril state (here we call protofibril because we deal with a small number of chains) [95], but the S- and LS-shapes have been recently reported [96,97]. Assuming that protofibrils and fibrils have similar structures [98], we used the fibrillar structures deposited in PDB databank, for further docking simulation. Namely, we have chosen two experimental structures with U-shape (PDB ID 2LMN) [99] and LS-shape (PDB ID 2MXU) [96].
In both 2LMN and 2MXU models, the three ligands can bind in different regions depending on the docking mode (Figs. S2 and S3 in Supporting Information). In the docking mode with the lowest energy, they are all preferentially located in the loop region of 2LMN, while for 2MXU CO and CH seem to prefer the terminal part, while SA is mainly located in the middle of the structure. In analogy to the monomeric case, SA is endowed with the poorest variety in docking positions compared to the other two ligands (Figs. S2 and S3, and Table 5 ).
Table 5.
AutoDock-predicted binding energies (kcal/mol) of the clustered orientations with 2LMN and 2MXU fibril models.
|
Mode |
2LMN |
2MXU |
|||||
|---|---|---|---|---|---|---|---|
| CO | SA | CH | CO | SA | CH | Color | |
| 1 | −12.12 | −12.16 | −10.93 | −10.41 | −11.76 | −10.89 | Purple |
| 2 | −11.90 | −10.71 | −10.83 | −9.70 | −10.92 | −10.36 | Magenta |
| 3 | −10.07 | −10.50 | −9.83 | −8.71 | −10.09 | −9.73 | Red |
| 4 | −10.00 | −10.37 | −9.71 | −7.14 | −9.98 | −7.96 | Yellow |
| 5 | −9.97 | −9.94 | −9.60 | −6.34 | −7.87 | Cyan | |
| 6 | −8.84 | −9.56 | −7.80 | Teal | |||
| 7 | −8.78 | −9.42 | Blue | ||||
| 8 | −8.37 | −8.15 | Green | ||||
| 9 | −6.72 | −6.95 | Darkgrey | ||||
| 10 | −6.21 | Lightgrey | |||||
With the binding energy of about −12 kcal/mol (Table 5), IC50 of CO and SA with 2LMN and SA with 2MXU is in the range of nM. Overall, all ligands are more strongly associated with protofibrils than with monomers and tetramers. The identified potential for the ligands to interact with both monomeric, oligomeric and protofibrillar Aβ1-42 suggests ample means for the ligands to modulate the subsequent aggregation process. Molecular Mechanics - Poisson Boltzmann Surface Area (MM-PBSA) docking assays on two compounds provided similar results (Figs. S4 and S5 and Table S1).
3.5.4. Binding affinity of ligands to Aβ1-42 protofibrils: MM-PBSA results
Because in general docking results are not sufficiently reliable we performed molecular mechanics - Poisson Boltzmann surface area (MM-PBSA) assays on two compounds CO and SA. For each protofibril-ligand complex, the binding free energy was calculated for two binding sites obtained in modes 1 and 2 of the docking simulations. The details of simulations and the results are described in SI (Figs. S4 and S5 and Table S1), which show that, in agreement with the docking results, the ligands strongly bind to protofibrils with IC50 ∼ nM.
3.5.5. Molecular dynamics simulations
In silico prediction of binding of the alkaloids to Aβ1-42 indicated that the presence of a ligand can alter the rate of Aβ1-42 aggregation, but it is unclear if it accelerates or slows down aggregation. On the other hand, our in vitro experiment demonstrated that CO speeds up fibril formation, while SA retards it. Thus, to clarify this issue, we performed MD simulations with 16 Aβ1-42 chains in the absence or presence of CO and SA to mimic the first stages of Aβ1-42 aggregation from semi-extended non-interacting chains. Studies of early aggregations stages of Aβ are believed to be key to understand the whole process and are commonly performed [100], even though the computational studies of it on all-atom level cannot reach equilibration, which would require probably minutes of real time [101].The simulation started from the initial configuration of the 16 non-interacting randomly generated Aβ1-42 chains in the presence of ligands in a 1:1 ratio (Fig. 7 ). For each set, we carried out two trajectories of 800 ns: this short interval even if it does not allow reaching equilibrium provides insights the initial steps of the aggregation.
Fig. 7.

Initial structure of the 16 Aβ1-42 chains with SA in 1:1 ratio. Aβ1-42 is represented by ball-and-sticks, SA by magenta spheres, counter ions by light-grey sphere, water by black dots.
Simulations showed that the flexibility of the chains was unaffected by the presence of the ligands (Table 6 ), as RMSD, gyration radius Rg, solvent accessible surface area (SASA), and end-to-end (N–C) distance did not vary significantly in absence or presence of the ligand. This was expected due to the semi-extended nature of the initial Aβ1-42 chains, which in the early aggregation steps firstly try to hide hydrophobic residues from the solvent and only then form stable interactions with other chains forming oligomeric structures [102,103]. Decrease of the number of interchain contacts indicates that both compounds are interacting with Aβ1-42 replacing some of the interaction which normally would form between chains. In general, calculated properties are quite dispersed, which is visible as high standard deviation values in Table 6, a feature caused by averaging over 16 chains, 2 trajectories and snapshots from the second halves of the simulations which are not fully equilibrated, and by the fact that Aβ1-42 chains are subjected to large conformational changes. However, even relatively small changes at early aggregation steps caused e.g. by the presence of external compounds, can significantly impact aggregation pathways and fibrilization process [104,105]. It was also previously reported that the beta content of Aβ1-42 monomers exponentially affects the aggregation rate [106], therefore we believe that these small changes may have significant impact on the behavior of the Aβ1-42 taking into account its disordered nature in low-mass forms, which increase its susceptibility to external factors.
Table 6.
Calculated average properties of the Aβ1-42chains from simulations of 16 chains with standard deviations. Other details are given in Fig. S6.
| Aβ1-42 | Aβ1-42+ CO | Aβ1-42+ SA | |
|---|---|---|---|
| RMSD [Å] | 10.97 ± 0.49 | 11.30 ± 0.36 | 11.52 ± 0.40 |
| Rg [Å] | 14.04 ± 0.28 | 14.45 ± 0.63 | 14.21 ± 0.41 |
| SASA [nm2] | 553.4 ± 21.1 | 576.6 ± 24.2 | 569.0 ± 20.3 |
| N–C distance [Å] | 33.64 ± 1.98 | 32.40 ± 2.29 | 34.24 ± 1.29 |
| Number of contacts between chains | 3.76 ± 0.25 | 3.06 ± 0.33 | 3.02 ± 0.19 |
| Alpha content [%] | 3.12 ± 1.11 | 2.92 ± 1.83 | 3.72 ± 0.50 |
| Beta content [%] | 4.04 ± 0.72 | 5.01 ± 0.81 | 2.33 ± 0.66 |
Both ligands reduced the population of monomers: a remarkable variation in the population of tetramers, heptamers, 14- and 15-mers (Fig. S7) was observed, which means that aggregation pathways to the fibril state are extensively modified by the ligands. In addition to size of the oligomers, our data confirm the secondary content is the prevalent factor governing aggregation rate of Aβ1-42 [107], suggesting that it could be in relation with the opposite effects by the SA and CO on the aggregation.
4. Conclusion
Herein we studied early Aβ1-42 aggregation stages in the presence of three alkaloids and our preliminary findings suggested that aromatic tetracycles with benzo[c]phenanthridine and berberine nuclei and similar functionalization of the aromatic core may oppositely affect the aggregation of Aβ1-42 peptide.
While benzo[c]phenanthridines SA and CH seemed to inhibit aggregation, the berberine-like CO increased propensity for Aβ1-42 to aggregate, showing also the highest affinity for monomeric Aβ1-42, as revealed by SPR experiments, and it displayed the highest variety of binding modes (as found in silico). These observations suggest that, different from benzo[c]phenanthridines, the bent berberine-like structure of CO can be accommodated in a higher number of diverse Aβ1-42 conformations. The presence of CO also led to increased Aβ1-42 β-content as revealed by CD experiments and MD calculations: this effect appears in perfect agreement with the promotion of Aβ1-42 aggregation observed in the ThT assay. Both docking and MM-PBSA simulations showed that all three studied alkaloids interact with monomeric, oligomeric and protofibrillar Aβ1-42. Our in silico study revealed that SA inhibits the assembly of Aβ1-42 into aggregates as a result of helix stabilization in the Aβ1-42 amyloid structure. On the contrary, the aggregation promoting effect caused by CO possibly occurs through enhancement of the β structures, which are predominantly reported in the fibril state. Interestingly, both benzo[c]phenanthridine and berberine derivatives are able to modulate the amyloid aggregation pathways by showing differences in the population of different oligomeric states, and in particular the Aβ1-42 oligomer assembly state undergoes significant changes upon ligand binding.
Finally, since berberine and Ber-D (Fig. S8), compounds differing from CO by carrying one non-aromatic ring (berberine) or free hydroxyl-groups besides the non-aromatic ring (Ber-D), both inhibit Aβ1-42 aggregation [22], future synthetic efforts and, biological studies should be carried out on chelerythrine-derived compounds CH-D1 and CH-D2 (Fig. S8) as promising candidates as neurodrugs in the family of the benzo[c]phenanthridine alkaloids [22].
Funding
We are grateful to the financial support received from Campania Region, Italy [POR-FESR 2014–2020 project PON03PE_0060_4], “Combattere la resistenza tumorale: piattaforma integrata multidisciplinare per un approccio tecnologico innovativo alle oncoterapie-Campania Oncoterapie” (Project N. B61G18000470007) and ZonMw for Memorabel project “Exploring the potential of multi-target treatment for Alzheimer's disease” (Project N. 733050304). Mai Suan Li was supported by Department of Science and Technology, Ho Chi Minh city, Vietnam (grant No. 07/2019/HĐ-KHCNTT) and Narodowe Centrum Nauki (NCN) in Poland (grant No 2019/35/B/ST4/02086).
CRediT authorship contribution statement
Daniela Marasco: Conceptualization, Investigation, Data curation, Writing - original draft. Caterina Vicidomini: Investigation, Methodology, Data curation, Validation. Pawel Krupa: Investigation, Methodology, Data curation, Writing - original draft, Writing - review & editing. Federica Cioffi: Investigation, Methodology. Pham Dinh Quoc Huy: Investigation, Methodology. Mai Suan Li: Investigation, Methodology, Writing - original draft. Daniele Florio: Investigation, Methodology. Kerensa Broersen: Investigation, Methodology, Writing - original draft. Maria Francesca De Pandis: Conceptualization, Writing - original draft. Giovanni N. Roviello: Supervision, Conceptualization, Investigation, Methodology, Data curation, Writing - original draft.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cbi.2020.109300.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Rochet J.-C., Lansbury P.T. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 2.Nasica-Labouze J., Nguyen P.H., Sterpone F., Berthoumieu O., Buchete N.-V., Coté S., De Simone A., Doig A.J., Faller P., Garcia A., Laio A., Li M.S., Melchionna S., Mousseau N., Mu Y., Paravastu A., Pasquali S., Rosenman D.J., Strodel B., Tarus B., Viles J.H., Zhang T., Wang C., Derreumaux P. Amyloid β protein and Alzheimer's disease: when computer simulations complement experimental studies. Chem. Rev. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weller J., Budson A. Current understanding of Alzheimer's disease diagnosis and treatment. F1000Research. 2018;7:1161. doi: 10.12688/f1000research.14506.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blennow K., de Leon M.J., Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 5.Gu L., Guo Z. Alzheimer's Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013;126:305–311. doi: 10.1111/jnc.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Helmond Z., Miners J.S., Kehoe P.G., Love S. Oligomeric Aβ in alzheimer's disease: relationship to plaque and tangle Pathology,APOEGenotype and cerebral amyloid angiopathy. Brain Pathol. 2010;20:468–480. doi: 10.1111/j.1750-3639.2009.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jan A., Adolfsson O., Allaman I., Buccarello A.-L., Magistretti P.J., Pfeifer A., Muhs A., Lashuel H.A. Aβ42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Aβ42 species. J. Biol. Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gylys K.H., Fein J.A., Yang F., Miller C.A., Cole G.M. Increased cholesterol in Aβ-positive nerve terminals from Alzheimer's disease cortex. Neurobiol. Aging. 2007;28:8–17. doi: 10.1016/j.neurobiolaging.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Doig A.J., Derreumaux P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015;30:50–56. doi: 10.1016/j.sbi.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Re F., Airoldi C., Zona C., Masserini M., Ferla B.L., Quattrocchi N., Nicotra F. Beta amyloid aggregation inhibitors: small molecules as candidate drugs for therapy of alzheimers disease. Curr. Med. Chem. 2010;17:2990–3006. doi: 10.2174/092986710791959729. [DOI] [PubMed] [Google Scholar]
- 11.Vicidomini C., Cioffi F., Broersen K., Roviello V., Riccardi C., Montesarchio D., Capasso D., Gaetano S.D., Musumeci D., Roviello G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019;11:285–302. doi: 10.4155/fmc-2018-0473. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y., Latshaw D.C., Hall C.K. Aggregation of aβ(17–36) in the presence of naturally occurring phenolic inhibitors using coarse-grained simulations. J. Mol. Biol. 2017;429:3893–3908. doi: 10.1016/j.jmb.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Permyakov E.A., Viet M.H., Chen C.-Y., Hu C.-K., Chen Y.-R., Li M.S. Discovery of dihydrochalcone as potential lead for Alzheimer's disease: in silico and in vitro study. PloS One. 2013;8 doi: 10.1371/journal.pone.0079151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ngo S.T., Li M.S. Curcumin binds to aβ1–40 peptides and fibrils stronger than ibuprofen and naproxen. J. Phys. Chem. B. 2012;116:10165–10175. doi: 10.1021/jp302506a. [DOI] [PubMed] [Google Scholar]
- 15.Bentley K.W. 1975. The Isoquinoline Alkaloids; pp. 259–348. [Google Scholar]
- 16.Orhan I., Özçelik B., Karaoğlu T., Şener B. Antiviral and antimicrobial profiles of selected isoquinoline alkaloids from fumaria and corydalis species. Z. Naturforsch. C Biosci. 2007;62:19–26. doi: 10.1515/znc-2007-1-204. [DOI] [PubMed] [Google Scholar]
- 17.Mehrzadi S., Fatemi I., Esmaeilizadeh M., Ghaznavi H., Kalantar H., Goudarzi M. Hepatoprotective effect of berberine against methotrexate induced liver toxicity in rats. Biomed. Pharmacother. 2018;97:233–239. doi: 10.1016/j.biopha.2017.10.113. [DOI] [PubMed] [Google Scholar]
- 18.Patiño Ladino O.J., Cuca Suárez L.E. Isoquinoline alkaloids of Zanthoxylum quinduense (Rutaceae) Biochem. Systemat. Ecol. 2010;38:853–856. [Google Scholar]
- 19.Kim M.I.A., Cho K.-H., Shin M.-S., Lee J.-M., Cho H.-S., Kim C.-J., Shin D.-H., Yang H.J. Berberine prevents nigrostriatal dopaminergic neuronal loss and suppresses hippocampal apoptosis in mice with Parkinson's disease. Int. J. Mol. Med. 2014;33:870–878. doi: 10.3892/ijmm.2014.1656. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed T., Gilani A.-u.-H., Abdollahi M., Daglia M., Nabavi S.F., Nabavi S.M. Berberine and neurodegeneration: a review of literature. Pharmacol. Rep. 2015;67:970–979. doi: 10.1016/j.pharep.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Shin K.S., Choi H.S., Zhao T.T., Suh K.H., Kwon I.H., Choi S.O., Lee M.K. Neurotoxic effects of berberine on long-term l-DOPA administration in 6-hydroxydopamine-lesioned rat model of Parkinson's disease. Arch Pharm. Res. (Seoul) 2013;36:759–767. doi: 10.1007/s12272-013-0051-4. [DOI] [PubMed] [Google Scholar]
- 22.Rajasekhar K., Samanta S., Bagoband V., Murugan N.A., Govindaraju T. Antioxidant berberine-derivative inhibits multifaceted amyloid toxicity. iScience. 2020;23:101005. doi: 10.1016/j.isci.2020.101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zee-Cheng K.-Y., Paull K.D., Cheng C.C. Experimental antileukemic agents. Coralyne, analogs, and related compounds. J. Med. Chem. 1974;17:347–351. doi: 10.1021/jm00249a020. [DOI] [PubMed] [Google Scholar]
- 24.Xing F., Song G., Ren J., Chaires J.B., Qu X. Molecular recognition of nucleic acids: coralyne binds strongly to poly(A) FEBS (Fed. Eur. Biochem. Soc.) Lett. 2005;579:5035–5039. doi: 10.1016/j.febslet.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 25.Pi G., Ren P., Yu J., Shi R., Yuan Z., Wang C. Separation of sanguinarine and chelerythrine in Macleaya cordata (Willd) R. Br. based on methyl acrylate-co-divinylbenzene macroporous adsorbents. J. Chromatogr. A. 2008;1192:17–24. doi: 10.1016/j.chroma.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 26.Dostál J., Potáček M. Quaternary benzo[c]phenanthridine alkaloids. Collect. Czech Chem. Commun. 1990;55:2840–2873. [Google Scholar]
- 27.Zhang R., Wang X.W., Zhu J.Y., Liu L.L., Liu Y.C., Zhu H. Dietary sanguinarine affected immune response, digestive enzyme activity and intestinal microbiota of Koi carp (cryprinus carpiod) Aquaculture. 2019;502:72–79. [Google Scholar]
- 28.Uhlar C.M., Whitehead A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 29.Miida T., Yamada T., Seino U., Ito M., Fueki Y., Takahashi A., Kosuge K., Soda S., Hanyu O., Obayashi K., Miyazaki O., Okada M. Serum amyloid A (SAA)-induced remodeling of CSF-HDL. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2006;1761:424–433. doi: 10.1016/j.bbalip.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Li H., Xiang X., Ren H., Xu L., Zhao L., Chen X., Long H., Wang Q., Wu Q. Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J. Infect. 2020 Jun;80(6):646–655. doi: 10.1016/j.jinf.2020.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang T., Du Z., Zhu F., Cao Z., An Y., Gao Y., Jiang B. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet. 2020;395:e52. doi: 10.1016/S0140-6736(20)30558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothan H.A., Byrareddy S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020;109:102433. doi: 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costanzo M., De Giglio M.A.R., Roviello G.N. SARS CoV-2: recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020;27 doi: 10.2174/0929867327666200416131117. [DOI] [PubMed] [Google Scholar]
- 34.Roviello V., Roviello G.N. Lower COVID-19 mortality in Italian forested areas suggests immunoprotection by Mediterranean plants. Environ. Chem. Lett. 2020 Aug 14:1–12. doi: 10.1007/s10311-020-01063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiart C. Academic Press; London: 2014. Lead Compounds from Medicinal Plants for the Treatment of Neurodegenerative Diseases. [Google Scholar]
- 36.Brunhofer G., Fallarero A., Karlsson D., Batista-Gonzalez A., Shinde P., Gopi Mohan C., Vuorela P. Exploration of natural compounds as sources of new bifunctional scaffolds targeting cholinesterases and beta amyloid aggregation: the case of chelerythrine. Bioorg. Med. Chem. 2012;20:6669–6679. doi: 10.1016/j.bmc.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 37.Ngo S.T., Li M.S. Top-leads from natural products for treatment of Alzheimer's disease: docking and molecular dynamics study. Mol. Simulat. 2013;39:279–291. [Google Scholar]
- 38.Broersen K., Jonckheere W., Rozenski J., Vandersteen A., Pauwels K., Pastore A., Rousseau F., Schymkowitz J. A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer's disease. Protein Eng. Des. Sel. 2011;24:743–750. doi: 10.1093/protein/gzr020. [DOI] [PubMed] [Google Scholar]
- 39.Moccia M., Roviello G.N., Bucci E.M., Pedone C., Saviano M. Synthesis of a l-lysine-based alternate alpha,epsilon-peptide: a novel linear polycation with nucleic acids-binding ability. Int. J. Pharm. 2010;397:179–183. doi: 10.1016/j.ijpharm.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 40.Roviello G.N., Gaetano S.D., Capasso D., Cesarani A., Bucci E.M., Pedone C. Synthesis, spectroscopic studies and biological activity of a novel nucleopeptide with Moloney murine leukemia virus reverse transcriptase inhibitory activity. Amino Acids. 2010;38:1489–1496. doi: 10.1007/s00726-009-0361-5. [DOI] [PubMed] [Google Scholar]
- 41.Roviello G.N., Roviello V., Autiero I., Saviano M. Solid phase synthesis of TyrT, a thymine–tyrosine conjugate with poly(A) RNA-binding ability. RSC Adv. 2016;6:27607–27613. doi: 10.1039/c6ra00294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roviello G.N., Crescenzo C., Capasso D., Di Gaetano S., Franco S., Bucci E.M., Pedone C. Synthesis of a novel Fmoc-protected nucleoaminoacid for the solid phase assembly of 4-piperidyl glycine/l-arginine-containing nucleopeptides and preliminary RNA interaction studies. Amino Acids. 2010;39:795–800. doi: 10.1007/s00726-010-0532-4. [DOI] [PubMed] [Google Scholar]
- 43.Roviello G.N. Novel insights into nucleoamino acids: biomolecular recognition and aggregation studies of a thymine-conjugated l-phenyl alanine. Amino Acids. 2018;50:933–941. doi: 10.1007/s00726-018-2562-2. [DOI] [PubMed] [Google Scholar]
- 44.Fik-Jaskółka M.A., Mkrtchyan A.F., Saghyan A.S., Palumbo R., Belter A., Hayriyan L.A., Simonyan H., Roviello V., Roviello G.N. Spectroscopic and SEM evidences for G4-DNA binding by a synthetic alkyne-containing amino acid with anticancer activity. Spectrochim. Acta Mol. Biomol. Spectrosc. 2020;229:117884. doi: 10.1016/j.saa.2019.117884. [DOI] [PubMed] [Google Scholar]
- 45.Musumeci D., Mokhir A., Roviello G.N. Synthesis and nucleic acid binding evaluation of a thyminyl L-diaminobutanoic acid-based nucleopeptide. Bioorg. Chem. 2020:103862. doi: 10.1016/j.bioorg.2020.103862. [DOI] [PubMed] [Google Scholar]
- 46.Oliviero G., Borbone N., Amato J., D'Errico S., Galeone A., Piccialli G., Varra M., Mayol L. Synthesis of quadruplex-forming tetra-end-linked oligonucleotides: effects of the linker size on quadruplex topology and stability. Biopolymers. 2009;91:466–477. doi: 10.1002/bip.21153. [DOI] [PubMed] [Google Scholar]
- 47.Saghyan A.S., Simonyan H.M., Petrosyan S.G., Geolchanyan A.V., Roviello G.N., Musumeci D., Roviello V. Thiophenyl-substituted triazolyl-thione l-alanine: asymmetric synthesis, aggregation and biological properties. Amino Acids. 2014;46:2325–2332. doi: 10.1007/s00726-014-1782-3. [DOI] [PubMed] [Google Scholar]
- 48.Roviello G.N., Moccia M., Sapio R., Valente M., Bucci E.M., Castiglione M., Pedone C., Perretta G., Benedetti E., Musumeci D. Synthesis, characterization and hybridization studies of new nucleo-gamma-peptides based on diaminobutyric acid. J. Pept. Sci. 2006;12:829–835. doi: 10.1002/psc.819. [DOI] [PubMed] [Google Scholar]
- 49.Carella A., Roviello V., Iannitti R., Palumbo R., La Manna S., Marasco D., Trifuoggi M., Diana R., Roviello G.N. Evaluating the biological properties of synthetic 4-nitrophenyl functionalized benzofuran derivatives with telomeric DNA binding and antiproliferative activities. Int. J. Biol. Macromol. 2019;121:77–88. doi: 10.1016/j.ijbiomac.2018.09.153. [DOI] [PubMed] [Google Scholar]
- 50.Musumeci D., Roviello G.N., Rigione G., Capasso D., Di Gaetano S., Riccardi C., Roviello V., Montesarchio D. Benzodifuran derivatives as potential antiproliferative agents: Possible correlation between their bioactivity and aggregation properties. Chempluschem. 2017 Feb;82(2):251–260. doi: 10.1002/cplu.201600547. [DOI] [PubMed] [Google Scholar]
- 51.D'Atri V., Oliviero G., Amato J., Borbone N., D'Errico S., Mayol L., Piccialli V., Haider S., Hoorelbeke B., Balzarini J., Piccialli G. New anti-HIV aptamers based on tetra-end-linked DNA G-quadruplexes: effect of the base sequence on anti-HIV activity. Chem Commun (Camb) 2012 Oct 4;48(76):9516–9518. doi: 10.1039/c2cc34399a. [DOI] [PubMed] [Google Scholar]
- 52.Roviello G.N., Musumeci D., Castiglione M., Bucci E.M., Pedone C., Benedetti E. Solid phase synthesis and RNA‐binding studies of a serum‐resistant nucleo‐ε‐peptide. J. Pept. Scie. Off. Publ. Eur. Pept. Soc. 2009;15(3):155–160. doi: 10.1002/psc.1072. [DOI] [PubMed] [Google Scholar]
- 53.Roviello G.N., Roviello G., Musumeci D., Bucci E.M., Pedone C. Dakin–West reaction on 1-thyminyl acetic acid for the synthesis of 1, 3-bis (1-thyminyl)-2-propanone, a heteroaromatic compound with nucleopeptide-binding properties. Amino Acids. 2012;43(4):1615–1623. doi: 10.1007/s00726-012-1237-7. [DOI] [PubMed] [Google Scholar]
- 54.Krupa P., Quoc Huy P.D., Li M.S. Properties of monomeric Aβ42 probed by different sampling methods and force fields: role of energy components. J. Chem. Phys. 2019;151 [Google Scholar]
- 55.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., Li X., Caricato M., Marenich A.V., Bloino J., Janesko B.G., Gomperts R., Mennucci B., Hratchian H.P., Ortiz J.V., Izmaylov A.F., Sonnenberg J.L., Williams, Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J.J., Brothers E.N., Kudin K.N., Staroverov V.N., Keith T.A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Foresman J.B., Fox D.J. Rev. C.01; Wallingford, CT: 2016. Gaussian 16. [Google Scholar]
- 56.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 57.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 58.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen N.T., Nguyen T.H., Pham T.N.H., Huy N.T., Bay M.V., Pham M.Q., Nam P.C., Vu V.V., Ngo S.T. Autodock Vina adopts more accurate binding poses but Autodock4 forms better binding affinity. J. Chem. Inf. Model. 2019;60:204–211. doi: 10.1021/acs.jcim.9b00778. [DOI] [PubMed] [Google Scholar]
- 60.Castro-Alvarez A., Costa A.M., Vilarrasa J. The performance of several docking programs at reproducing protein-macrolide-like crystal structures. Molecules. 2017:22. doi: 10.3390/molecules22010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreira L., dos Santos R., Oliva G., Andricopulo A. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adinarayana K.P.S., Devi R.K. Protein-Ligand interaction studies on 2, 4, 6-trisubstituted triazine derivatives as anti-malarial DHFR agents using AutoDock. Bioinformation. 2011;6:74–77. doi: 10.6026/97320630006074. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 65.Barz B., Liao Q., Strodel B. Pathways of amyloid-β aggregation depend on oligomer shape. J. Am. Chem. Soc. 2017;140:319–327. doi: 10.1021/jacs.7b10343. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen H.L., Krupa P., Hai N.M., Linh H.Q., Li M.S. Structure and physicochemical properties of the Aβ42 tetramer: multiscale molecular dynamics simulations. J. Phys. Chem. B. 2019;123:7253–7269. doi: 10.1021/acs.jpcb.9b04208. [DOI] [PubMed] [Google Scholar]
- 67.Mioduszewski Ł., Cieplak M. Protein droplets in systems of disordered homopeptides and the amyloid glass phase. Phys. Chem. Chem. Phys. 2020;22:15592–15599. doi: 10.1039/d0cp01635g. [DOI] [PubMed] [Google Scholar]
- 68.Tian C., Kasavajhala K., Belfon K.A.A., Raguette L., Huang H., Migues A.N., Bickel J., Wang Y., Pincay J., Wu Q., Simmerling C. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theor. Comput. 2019;16:528–552. doi: 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- 69.Izadi S., Anandakrishnan R., Onufriev A.V. Building water models: a different approach. J. Phys. Chem. Lett. 2014;5:3863–3871. doi: 10.1021/jz501780a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weiser J.r., Shenkin P.S., Still W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO) J. Comput. Chem. 1999;20:217–230. [Google Scholar]
- 71.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 72.Walsh D.M., Selkoe D.J. A? Oligomers ? a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 73.Hayden E.Y., Teplow D.B. Amyloid β-protein oligomers and Alzheimer's disease. Alzheimer's Res. Ther. 2013;5:60. doi: 10.1186/alzrt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuperstein I., Broersen K., Benilova I., Rozenski J., Jonckheere W., Debulpaep M., Vandersteen A., Segers-Nolten I., Van Der Werf K., Subramaniam V., Braeken D., Callewaert G., Bartic C., D'Hooge R., Martins I.C., Rousseau F., Schymkowitz J., De Strooper B. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Picken M.M., Herrera G.A. 2012. Thioflavin T Stain: an Easier and More Sensitive Method for Amyloid Detection; pp. 187–189. [Google Scholar]
- 76.Di Natale C., Scognamiglio P.L., Cascella R., Cecchi C., Russo A., Leone M., Penco A., Relini A., Federici L., Di Matteo A., Chiti F., Vitagliano L., Marasco D. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. Faseb. J. 2015;29:3689–3701. doi: 10.1096/fj.14-269522. [DOI] [PubMed] [Google Scholar]
- 77.Monji A., Utsumi H., Ueda T., Imoto T., Yoshida I., Hashioka S., Tashiro K.-i., Tashiro N. The relationship between the aggregational state of the amyloid-β peptides and free radical generation by the peptides. J. Neurochem. 2001;77:1425–1432. doi: 10.1046/j.1471-4159.2001.00392.x. [DOI] [PubMed] [Google Scholar]
- 78.Guivernau B., Bonet J., Valls-Comamala V., Bosch-Morato M., Godoy J.A., Inestrosa N.C., Peralvarez-Marin A., Fernandez-Busquets X., Andreu D., Oliva B., Munoz F.J. Amyloid-beta peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J. Neurosci. 2016;36:11693–11703. doi: 10.1523/JNEUROSCI.1081-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kotler S.A., Brender J.R., Vivekanandan S., Suzuki Y., Yamamoto K., Monette M., Krishnamoorthy J., Walsh P., Cauble M., Holl M.M., Marsh E.N., Ramamoorthy A. High-resolution NMR characterization of low abundance oligomers of amyloid-beta without purification. Sci. Rep. 2015;5:11811. doi: 10.1038/srep11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo J., Sun W., Li L., Liu F., Lu W. Brazilin inhibits fibrillogenesis of human islet amyloid polypeptide, disassembles mature fibrils, and alleviates cytotoxicity. RSC Adv. 2017;7:43491–43501. [Google Scholar]
- 81.Cheng B., Liu X., Gong H., Huang L., Chen H., Zhang X., Li C., Yang M., Ma B., Jiao L., Zheng L., Huang K. Coffee components inhibit amyloid formation of human islet amyloid polypeptide in vitro: possible link between coffee consumption and diabetes mellitus. J. Agric. Food Chem. 2011;59:13147–13155. doi: 10.1021/jf201702h. [DOI] [PubMed] [Google Scholar]
- 82.Cheng B., Gong H., Li X., Sun Y., Zhang X., Chen H., Liu X., Zheng L., Huang K. Silibinin inhibits the toxic aggregation of human islet amyloid polypeptide. Biochem. Biophys. Res. Commun. 2012;419:495–499. doi: 10.1016/j.bbrc.2012.02.042. [DOI] [PubMed] [Google Scholar]
- 83.Palhano F.L., Lee J., Grimster N.P., Kelly J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013;135:7503–7510. doi: 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Natale C., La Manna S., Avitabile C., Florio D., Morelli G., Netti P.A., Marasco D. Engineered β-hairpin scaffolds from human prion protein regions: structural and functional investigations of aggregates. Bioorg. Chem. 2020;96:103594. doi: 10.1016/j.bioorg.2020.103594. [DOI] [PubMed] [Google Scholar]
- 85.Di Natale C., La Manna S., Malfitano A.M., Di Somma S., Florio D., Scognamiglio P.L., Novellino E., Netti P.A., Marasco D. Structural insights into amyloid structures of the C-terminal region of nucleophosmin 1 in type A mutation of acute myeloid leukemia. Biochim. Biophys. Acta Protein Proteonomics. 2019;1867:637–644. doi: 10.1016/j.bbapap.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 86.Florio D., Malfitano A.M., Di Somma S., Mugge C., Weigand W., Ferraro G., Iacobucci I., Monti M., Morelli G., Merlino A., Marasco D. Platinum(II) O,S complexes inhibit the aggregation of amyloid model systems. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20040829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Florio Iacobucci, Ferraro Mansour, Morelli Monti, Merlino Marasco. Role of the metal center in the modulation of the aggregation process of amyloid model systems by square planar complexes bearing 2-(2'-pyridyl)benzimidazole ligands. Pharmaceuticals. 2019;12:154. doi: 10.3390/ph12040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scognamiglio P.L., Di Natale C., Leone M., Cascella R., Cecchi C., Lirussi L., Antoniali G., Riccardi D., Morelli G., Tell G., Chiti F., Marasco D. Destabilisation, aggregation, toxicity and cytosolic mislocalisation of nucleophosmin regions associated with acute myeloid leukemia. Oncotarget. 2016;7:59129–59143. doi: 10.18632/oncotarget.10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.La Manna S., Scognamiglio P.L., Roviello V., Borbone F., Florio D., Di Natale C., Bigi A., Cecchi C., Cascella R., Giannini C., Sibillano T., Novellino E., Marasco D. The acute myeloid leukemia-associated Nucleophosmin 1 gene mutations dictate amyloidogenicity of the C-terminal domain. FEBS J. 2019 Jun;286(12):2311–2328. doi: 10.1111/febs.14815. [DOI] [PubMed] [Google Scholar]
- 90.Poletto M., Malfatti M.C., Dorjsuren D., Scognamiglio P.L., Marasco D., Vascotto C., Jadhav A., Maloney D.J., Wilson D.M., 3rd, Simeonov A., Tell G. Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties. Mol. Carcinog. 2016;55:688–704. doi: 10.1002/mc.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chu M., Chen X., Wang J., Guo L., Wang Q., Gao Z., Kang J., Zhang M., Feng J., Guo Q., Li B., Zhang C., Guo X., Chu Z., Wang Y. Polypharmacology of berberine based on multi-target binding motifs. Front. Pharmacol. 2018;9 doi: 10.3389/fphar.2018.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheng L.S., Amaro R.E., Xu D., Li W.W., Arzberger P.W., McCammon J.A. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J. Med. Chem. 2008;51:3878–3894. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shin B.-k., Saxena S. Substantial contribution of the two imidazole rings of the His13−His14 dyad to Cu(II) binding in amyloid-β(1−16) at physiological pH and its significance. J. Phys. Chem. 2011;115:9590–9602. doi: 10.1021/jp200379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bernstein S.L., Dupuis N.F., Lazo N.D., Wyttenbach T., Condron M.M., Bitan G., Teplow D.B., Shea J.-E., Ruotolo B.T., Robinson C.V., Bowers M.T. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D., Delaglio F., Tycko R. A structural model for Alzheimer's -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. Unit. States Am. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., Ishii Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gremer L., Scholzel D., Schenk C., Reinartz E., Labahn J., Ravelli R.B.G., Tusche M., Lopez-Iglesias C., Hoyer W., Heise H., Willbold D., Schroder G.F. Fibril structure of amyloid-beta(1-42) by cryo-electron microscopy. Science. 2017;358:116–119. doi: 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh D.M., Hartley D.M., Kusumoto Y., Fezoui Y., Condron M.M., Lomakin A., Benedek G.B., Selkoe D.J., Teplow D.B. Amyloid β-protein fibrillogenesis. J. Biol. Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 99.Paravastu A.K., Leapman R.D., Yau W.M., Tycko R. Molecular structural basis for polymorphism in Alzheimer's -amyloid fibrils. Proc. Natl. Acad. Sci. Unit. States Am. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Olsen R.H., DeBusscher G., McCombie W.R. Development of broad-host-range vectors and gene banks: self-cloning of the Pseudomonas aeruginosa PAO chromosome. J. Bacteriol. 1982;150:60–69. doi: 10.1128/jb.150.1.60-69.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Watanabe-Nakayama T., Ono K., Itami M., Takahashi R., Teplow D.B., Yamada M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1–42 aggregates. Proc. Natl. Acad. Sci. Unit. States Am. 2016;113:5835–5840. doi: 10.1073/pnas.1524807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang S., Iwata K., Lachenmann M.J., Peng J.W., Li S., Stimson E.R., Lu Y.a., Felix A.M., Maggio J.E., Lee J.P. The alzheimer's peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 2000;130:130–141. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- 103.Krone M.G., Hua L., Soto P., Zhou R., Berne B.J., Shea J.-E. Role of water in mediating the assembly of alzheimer amyloid-β Aβ16−22 protofilaments. J. Am. Chem. Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu J., Sun H., Hao H., Zheng Q. Prediction of fibril formation by early-stage amyloid peptide aggregation. J. Pharma. Anal. 2020;10:194–199. doi: 10.1016/j.jpha.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giorgetti S., Greco C., Tortora P., Aprile F. Targeting amyloid aggregation: an overview of strategies and mechanisms. Int. J. Mol. Sci. 2018;19:2677. doi: 10.3390/ijms19092677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thu T.T.M., Co N.T., Tu L.A., Li M.S. Aggregation rate of amyloid beta peptide is controlled by beta-content in monomeric state. J. Chem. Phys. 2019;150:225101. doi: 10.1063/1.5096379. [DOI] [PubMed] [Google Scholar]
- 107.Modler A.J., Gast K., Lutsch G., Damaschun G. Assembly of amyloid protofibrils via critical oligomers—a novel pathway of amyloid formation. J. Mol. Biol. 2003;325:135–148. doi: 10.1016/s0022-2836(02)01175-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.