

Abstract
In 1965 Dr. Harry Angelman reported a neurodevelopmental disorder affecting three unrelated children who had similar symptoms: brachycephaly, mental retardation, ataxia, seizures, protruding tongues, and remarkable paroxysms of laughter. Over the past 50 years, the disorder became Angelman’s namesake and symptomology was expanded to include hyper-activity, stereotypies, and severe sleep disturbances. The sleep disorders in many Angelman syndrome (AS) patients are broadly characterized by difficulty falling and staying asleep at night. Some of these patients sleep less than 4 hours a night and, in most cases, do not make up this lost-sleep during the day—leading to the speculation that AS patients may “need” less sleep. Most AS patients also have severely reduced levels of melatonin, a hormone produced by the pineal gland exclusively at night. This nightly pattern of melatonin production is thought to help synchronize internal circadian rhythms and promote nighttime sleep in humans and other diurnal species. It has been proposed that reduced melatonin levels contribute to the sleep problems in AS patients. Indeed, emerging evidence suggests melatonin replacement therapy can improve sleep in many AS patients. However, AS mice show sleep problems that are arguably similar to those in humans despite being on genetic backgrounds that do not make melatonin. This suggests the hypothesis that the change in nighttime melatonin may be a secondary factor rather than the root cause of the sleeping disorder. The goals of this review article are to revisit the sleep and melatonin findings in both AS patients and animal models of AS and discuss what AS may tell us about the underlying mechanisms of, and interplay between, melatonin and sleep.
Keywords: Melatonin, sleep, Angelman syndrome, Ube3a
Introduction
Melatonin has become a very popular over-the-counter pharmaceutical and is frequently recommended by clinicians for adults and children with sleep disturbances1. Melatonin’s role in the regulation of sleep and its frequent use as a somnogen have led to the investigation of melatonin as both a cause of, and treatment for, sleep disturbances in neurological disorders. This review is intended to highlight the relationship between melatonin, sleep, and Angelman syndrome (AS).
Sleep and Melatonin
The need for sleep is universal; all animals do it (for a review see2). And while the answers to the question “why sleep?” are hotly debated, it is clear that sleep is very tightly regulated. The consensus view of sleep regulation, the two-process model, proposes that sleep is regulated by the interaction of two processes that separately control timing and amount of sleep (see Figure 1). Although several variations of the two-process model have been proposed3,4, the basic tenets described here remain consistent.
Figure 1: Two-process model depictions of how sleep might be altered in Angelman syndrome.
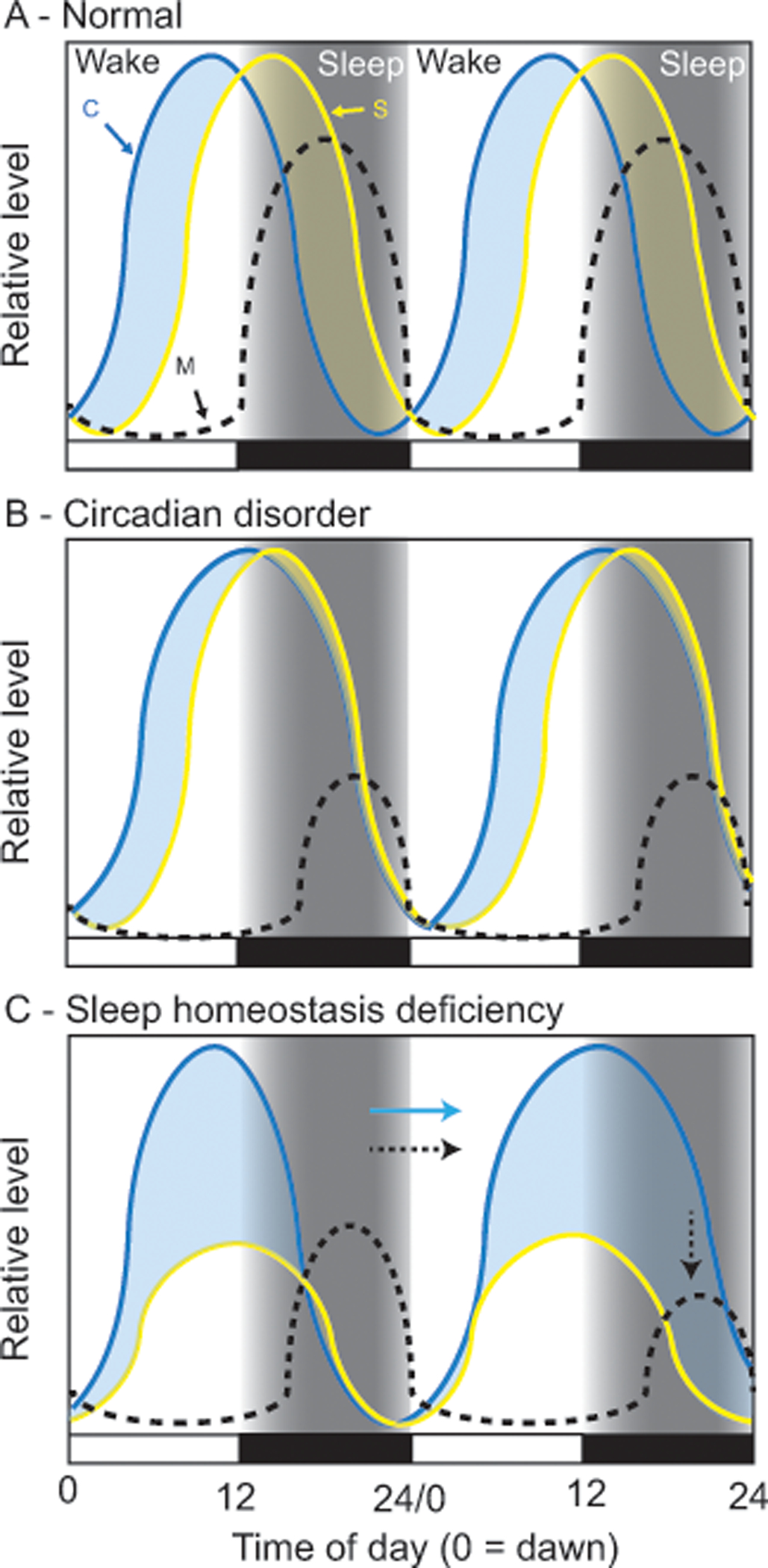
In these simplified models, Process C (C; circadian clock) provides drive for wakefulness and Process S (S; sleep homoeostasis) builds pressure to sleep. At any time of day, whichever drive is higher dictates sleep-state (light blue shading = wake, yellow shading = sleep). Under normal conditions (A) Wake drive (blue curve), driven by Process C, dominates during the day keeping the subject awake; sleep-drive (yellow curve) is building simultaneously, just at a slower rate. At night, melatonin (M) is produced, the circadian clock stops providing wake drive, and the accumulation of sleep drive causes the subject to fall asleep; sleep causes sleep drive to dissipate. In Angelman syndrome, sleep disorders could be caused by changes to circadian control (B), sleep homeostatic control (C), or both (not shown). Data suggest that AS patients may have a long circadian period and/or a delayed and suppressed nighttime melatonin rhythm, as depicted by Process C and melatonin curves (B). If Process S is normal, then Process C may compete against it during the night, delaying and suppressing both sleep and melatonin production. Alternatively, data also suggest the sleep homeostatic process is blunted (C) and may accumulate less efficiently. In this scenario, overall sleep drive is reduced, leading to decreases in sleep. In either B or C, not sleeping may also lead to an increase of light exposure at night (i.e. turning on lights or TV) that may further contribute to circadian-like deficits in sleep regulation (indicated by the arrows in C).
The circadian clock (Process C) is responsible for regulating sleep/wake to the appropriate time of day, by driving a daily rhythm in a sleep-wake threshold or set-point by which sleep or wake is initiated (depending on time of day)5. Behavioral circadian rhythms are controlled by intracellular clocks localized to the 20,000 neurons within suprachiasmatic nucleus (SCN) of the hypothalamus. This timekeeping mechanism is a coupled set of transcriptional negative feedback loops that ultimately drive rhythms in firing rates of SCN neurons6. SCN neurons project through the ventral and dorsal subparaventricular zone (SPZ) to the dorsal medial hypothalamus (DMH) to regulate sleep/wake and other rhythmic outputs7 like melatonin synthesis.
Sleep is also regulated by a homeostatic process (Process S) that is responsible for “sleep pressure” and the regulation of total daily sleep amount. Sleep pressure (i.e. sleep drive, sleep load) is homeostatically regulated and accumulates with increasing duration of wakefulness: The longer the duration of wakefulness, the higher the drive to sleep and the longer and deeper the subsequent sleep episode. When sleep pressure reaches the upper threshold set by the circadian clock (Process C), sleep is initiated. Most organisms require a species-specific duration of sleep; in humans this is ~ 7 hours/day8. Not reaching this minimum threshold leads to the feelings and symptoms of sleepiness, including neurocognitive impairment9. Despite popular anecdotes, empirical evidence has repeatedly shown that one cannot “learn” or “train” themselves to need less sleep10. Once asleep, accumulated sleep pressure dissipates in a duration-specific manner (the more you sleep, the more sleep pressure is reduced). When sleep pressure reaches the lower limit set by the circadian clock (Process C), wake is initiated (Figure 1A). While we have a good understanding of circadian clock mechanisms (see below), and extensive information of the neural circuitry promoting both sleep and arousal, the nature of the sleep homeostatic process is a major gap in our understanding of how sleep is regulated5,7,11–13.
Projections from the SCN ultimately send time of day information to the pineal gland to regulate melatonin production. Melatonin is involved in regulating multiple physiological processes in mammals14 including the sleep-wake cycle15,16. The pineal gland produces melatonin exclusively during the night; light at night quickly shuts down melatonin synthesis. In this way, plasma melatonin levels transduce information about light/dark cycle as well as the presence of light at night17. The Dim Light Melatonin Onset/Offset (DLMO on/off) is among the most accurate measures for assessing the circadian pacemaker in human subjects18. The robustness or amplitude of the melatonin rhythm is directly related to overall strength and health of the circadian system, and flattened nighttime melatonin production signifies a potential problem in the underlying circadian clock19. When combined, melatonin onset and amplitude are commonly used to provide key insights into the timing and functioning of the circadian system in humans and other mammals18.
Melatonin administration can also help induce sleep, but it seems to do this at least in part by resetting the circadian clock. Evidence from rodent studies suggests that the actions of melatonin in the SCN are mediated primarily by two G-protein coupled melatonin receptors, MT1 and MT2. Melatonin feeds back into the SCN to modulate circadian clock timing as a ‘nighttime’ signal (as opposed to just a ‘dark’ environment)20,21.
When taken at night, melatonin and melatonin receptor agonists (i.e. Agomelatin, Ramelteon, Tasimelteon)22 shift the circadian system towards night, improving sleep latency. Exogenous melatonin is not affected by light-at-night and, therefore, can be used to offset the effects of light exposure at night. Because derivative compounds of melatonin appear to shift the circadian clock, they are effective in treating some Circadian Rhythm Sleep Disorders (CRSDs), such as Non-24-Hour Sleep-Wake Disorder23. This, combined with the fact that CRSDs are often accompanied by an alteration in the melatonin rhythm, suggests that melatonin may act directly as a ‘somnolescent’ and regulate sleep. However, melatonin and its derivative compounds are not efficacious for treating primary (i.e. non-circadian) sleep disorders24; this argues against actions as a somnolescent independent of the circadian clock. This may explain why melatonin therapy has mixed results in treating the sleep disorders associated with Angelman syndrome, as discussed below.
Angelman Syndrome
AS is a rare but severe neurodevelopmental disorder with a prevalence of 1 in 12,000–20,000. Its broad, complex presentation is characterized by severe developmental delay and speech impairment resulting in the use of few if any words; disordered balance and/or movement, ranging from ataxia and unsteadiness, to quick jerky movements; and a ‘behavioral uniqueness’ described as a happy, easily excitable demeanor accompanied by hypermotoric behaviors (typically hand-flapping/waving motions)25–28. More than 80% of individuals with AS display microencephaly, abnormal electroencephalography (EEG), and epilepsy29,30. An estimated 70–80% of those with AS also exhibit sleep disturbances31–33, described in detail below.
AS is caused by a lack of ubiquitin protein E3A ligase gene (UBE3A) expression in the brain. Both UBE3A alleles are expressed in peripheral cells and tissues. But in the vast majority of neurons, the paternal UBE3A allele is imprinted, preventing its expression. Thus, the maternally-inherited allele is responsible for the expression of UBE3A protein in neurons. The imprinting mechanism is not fully understood, though it appears to be due to expression of a large, non-coding, antisense transcript (LNCAT) that selectively blocks expression of the paternal allele, which has been nicely reviewed elsewhere34,35. There are a few regions in the brain where UBE3A expression is biallelic. Interestingly, the SCN is the site with the highest concentration of neurons that express UBE3A from the paternal allele, at least in mice. UBE3A levels are reduced by only ~50% in the SCN of a mouse model of AS compared to wild type controls36,37, whereas UBE3A is absent most everywhere else37,38. On the surface, this would suggest a relatively intact circadian rhythmicity in AS; however, this is not the case and circadian impairments, which are discussed later, do exist.
An absence of maternal UBE3A expression can be caused by four different molecular mechanisms39–44. The most common cause (70–80%) is a chromosomal deletion from 15q11-q13 on the maternal allele, containing the UBE3A gene45. This deletion usually occurs as a de novo event but it can be due to structural rearrangement in the mother46. Mutations within the UBE3A gene account for 5–10% of AS cases47, with the remaining cases due to paternal uniparental disomy (UPD) of chromosome 15 (1–2%)48 or defects in the imprinting mechanism ( 1–3% )49. The severity of some aspects of AS correlates with the underlying genetic anomaly. For example, 15q11-q13 deletions tend to cause more severe phenotypes compared to other mechanisms, suggesting potential contribution of nearby genes in AS. However, mutations that only affect the maternal UBE3A allele cause all of the core AS disabilities suggesting this is the primary underlying cause of AS.
Mouse lines in which maternal UBE3A is disrupted provide additional evidence of a causal role of UBE3A in AS. Mice with a maternal deletion of Ube3a (Ube3am-/p+ mice) have phenotypes that resemble many of the clinical symptoms of AS, including motor deficits, impaired spatial learning, seizures, and deficiencies in synaptic plasticity. Ube3am-/p+ mice have an imprinting mechanism - homologous to humans - that silences the paternal Ube3a allele in neurons. Thus, Ube3am-/p+ mice are widely used as a preclinical model to understand how UBE3A loss leads to AS-like pathologies and to evaluate potential treatments. Mice, however, are nocturnal, and AS-like behavioral phenotypes can vary by genetic background, strain, and age. All of these factors should be considered when using Ube3am-/p+ mice as an animal model for AS50.
Sleep disorders in Angelman Syndrome
The widespread sleep disturbances reported in AS are based on two broad categories of evidence: 1) survey-based studies relying on the reports of parents and caregivers and 2) polysomnographic (PSG) studies based on EEG data. Both evidence categories provide important information, with surveys providing subjective information regarding mood, sleep patterns, sleep habits and behavioral consequences, whereas PSG provides objective metrics of sleep quantity, quality and potential diagnostic criteria. In addition to clinical studies, animal models also provide detailed sleep metrics and analysis that are not available in the human literature. Overall, these data converge to indicate that AS is associated with a debilitating sleep disorder that appears to have its roots in the altered accumulation of sleep-pressure.
The first consensus guidelines for the diagnostic criteria of AS included sleep disturbances as an associated feature of the disorder51; this was later updated in the guidelines to include abnormal sleep-wake cycles and a diminished need for sleep52. The latter feature, a diminished need for sleep, is of high interest, as it appears AS patients do not suffer the negative consequences of sleep deprivation. Currently, “sleep need” cannot be objectively measured, and its biological basis represents a critical knowledge-gap in sleep research. Indeed, evidence for altered sleep-need is rare in the literature and mostly based on evidence from natural short-sleepers; individuals reporting significantly less daily sleep than the population average with seemingly little effect on health and performance53,54. Thus, the study of sleep in AS may provide important information on the nature of sleep need and related health problems, as well as uncover novel sleep-regulatory mechanisms.
EEG studies provide a potential means for AS diagnosis, and when combined with other PSG measures they can provide objective analysis of sleep in AS patients. Studies focused on AS diagnosis have found widespread EEG disturbances in these patients55–62. Two of the most commonly reported abnormalities in the EEG are intervals of high-amplitude slow waves (in the EEG delta/theta band) and interictal epileptiform spike-wave discharges55,63. The degree to which these commonly reported abnormalities are present during sleep is not clear. This is because, although these recordings were made during both wake and spontaneous sleep56,59–61,63, the prevalence of anomalies during specific sleep/wake states have not been reported. State-specific analysis is critical for determining the degree to which EEG anomalies are related to the sleep disturbances in AS; several studies have used PSG to accomplish this goal.
PSG uses EEG and additional measures (e.g., electrooculography, electromyography) to objectively assess sleep and wakefulness with a level of precision far superior to behavioral analysis. PSG studies of AS patients provide direct evidence that multiple sleep parameters indicative of decreased sleep efficiency exist. AS patients have nearly twice the number of transitions between sleep states, a four-fold increase in the frequency of awakenings (i.e., sleep is fragmented), and a 50% reduction in the deepest stages of NREM sleep – all indications that AS patients have reduced sleep quality and sleep efficiency during the night64,65.
Recently, differences in the number and duration of NREM sleep spindles were also reported66. Approximately half the number of sleep spindles of a significantly decreased length were found in the EEG of children with AS. This spindle activity in the 11–16 Hz range occurs during NREM sleep and is associated with memory consolidation66, suggesting a potential direct relationship between poor sleep quality and memory difficulties in AS patients. Despite the aforementioned deficits in NREM sleep quality, there are no indications that the total amount of NREM sleep is reduced in AS patients. This is somewhat at odds with subjective reports from parents/caregivers regarding overall amount of sleep. Nevertheless, though the degree to which AS patients sleep less may be unclear, there is no question that the quality of their sleep is significantly impaired. Studies report a significant decrease in rapid-eye movement (REM) sleep in AS patients compared to healthy controls—especially in patients younger than eight years old64,65. This could be directly due to poor NREM sleep in AS patients, as they may not be efficiently progressing through the 3 stages of NREM in order to reach REM sleep. Thus, the emerging picture from objective PSG studies is consistent with caregiver reports that AS patients sleep poorly during the night. However, the subtle inconsistencies in overall sleep time make it difficult to predict the possible underlying causes of this disturbed sleep.
Origin of the sleep phenotypes in AS
A -. Evidence of circadian dysfunction
One potential cause of the sleep disturbances observed in AS (and other neurodevelopmental disabilities) is an inability to synchronize the sleep–wake cycle with the light-dark cycle, resulting in abnormal melatonin secretion and, consequently, CRSDs67,68. Indeed, emerging evidence from studies in model organisms36,69–71 provide some support for this hypothesis (more on this below). Although few studies have examined the melatonin secretion profile of AS patients, the available data indicate that it varies broadly between individuals with a tendency towards reduced levels and/or altered timing of nighttime melatonin secretion. Additionally, melatonin administration in the early evening has been shown to consistently improve sleep outcomes in individuals with AS.
Some of the first evidence indicating that AS patients exhibit altered nighttime melatonin levels came from studies that compared nighttime melatonin secretion patterns in AS children to published reports of healthy children. A study of 13 AS children demonstrated that both the average hourly rate of production across the 24-hour period and peak nighttime levels of melatonin production were highly variable between individuals72. In a majority of these children, peak nighttime melatonin levels were noticeably lower compared to a published report of unaffected children of a similar age73. In addition, the nighttime surge in melatonin secretion was significantly delayed in 3 AS children (relative to habitual bedtime and sleep onset). Two of these children exhibited prolonged morning sleepiness that was correlated with low sleep quality and quantity. A separate study of 8 AS patients with idiopathic insomnia (4–9yo, N=5; 12–20yo, N=3) also reported low levels of endogenous melatonin74. In this case, sampling times were restricted to 5–11pm, making it impossible to distinguish between low melatonin per se vs. a delayed onset in nighttime melatonin secretion (see below).
More recent investigations compare melatonin secretion profiles between AS patients and unaffected age-matched controls within the same study. A direct comparison of 15 AS patients (including children, adolescents, and young adults) and age-matched controls revealed that individuals with AS exhibit significantly lower nighttime melatonin levels67. Surprisingly, total sleep time and ratio of nocturnal to total sleep time did not differ from controls. AS patients with a diagnosed CRSD, however, exhibited less total sleep than AS patients without CRSD. Patients with CRSD also had a lower percentage of nocturnal sleep to total sleep compared to both AS patients without CRSD and unaffected age-matched controls. AS patients with CRSD also exhibited a significantly smaller nighttime peak in melatonin secretion compared to AS patients without CRSD. AS patients with free-running type and irregular sleep-wake type CRSDs exhibited low serum levels of melatonin throughout the 24-hour day with virtually no nighttime surge in melatonin. Peak melatonin levels in AS patients with delayed sleep phase type CRSD were similar to controls, but the peak was significantly delayed.
Another study75, in which AS children were directly compared to both non-AS/epilepsy and non-AS/non-epilepsy age-matched controls, found differences in the pattern, but not levels, of melatonin secretion in AS patients. Although these AS children — all of whom were diagnosed with a CRSD — had melatonin levels similar to levels reported in the literature for both age-matched control groups and healthy children73, their individual melatonin secretion curves were highly variable. The duration of nighttime melatonin secretion was elongated, and both the phase and the offset of nighttime melatonin secretion were significantly delayed in AS children. Rather than the typical bell-shaped curve, these children exhibited a ‘triangular” melatonin secretion profile that may explain the sleep onset insomnia and sleep maintenance problems so common to AS.
The shortage of studies, small sample sizes, high individual variability and differences in patient characteristics make it difficult to draw conclusions from most of these human studies. The fact that most AS patients take anti-seizure medications containing sodium valproate - known to suppress plasma melatonin levels76 — further complicates interpretation of these data. Braam et al.,74 reported no differences in melatonin levels between AS children taking and not taking sodium valproate; furthermore, Takaesu et al.67 found no relationship between daily dose of sodium valproate and peak melatonin levels in AS patients. These data indicate that reduced melatonin production is a characteristic of AS and not a result of anti-seizure medication per se. On the whole, it appears that in AS patients nighttime melatonin levels are low and/or its pattern disturbed.
Melatonin is commonly used by caregivers and routinely recommended by physicians as a treatment for sleep disruption in AS patients1,74. Despite the limited number and inconsistent methodologies of investigations into the effectiveness of melatonin in AS children, the available data consistently indicate that melatonin administration improves sleep latency, efficiency, duration, and nighttime awakenings. In an open label study, a physiological dose of melatonin (0.3mg) just before bedtime decreased sleep latency, reduced motor activity during sleep, and increased sleep duration in children with AS72. Parents of these children reported improved sleep consolidation and quality. This treatment also resulted in a 2 to 3-hour phase advance in the onset of melatonin secretion in children that otherwise exhibited a significant phase delay in nighttime melatonin secretion72. In a double-blind, randomized placebo-controlled trial, evening melatonin treatment (<6yo: 2.5mg at 6pm; ≥6yo: 5mg at 7p) advanced sleep onset, decreased sleep latency, reduced nighttime awakening, and increased total sleep time in children with AS74. Parents indicated that their children were less sleepy and more attentive during the day, with easier to manage behavior. In a case study of a 9-year old boy with AS that exhibited prolonged mid-night awakenings, treatment with sustained-release melatonin (3mg) 30 minutes before bedtime improved sleep efficiency, decreased nighttime arousals, and increased total sleep time77. Furthermore, melatonin treatment (1mg between 6–7pm) increased the percentage of nocturnal sleep in four of six individuals with AS (mostly children) that also exhibited a recognized CRSD67.
All of these studies indicate that improving nighttime melatonin can improve sleep and appears to be doing it by realigning AS patients’ circadian timing with the day-night schedule. This suggests that perhaps the sleep disorders in AS patients could be due to a defect in the circadian system – either with its overall functionality or in how it is aligned with the day-night cycle. The data also suggest that if this is indeed the case, the effects on the circadian system vary quite broadly from patient to patient. It is not yet clear if CRSDs fully explain all of the idiosyncratic sleeping disorders in AS patients. However, the data do suggest that melatonin can improve sleep in those with CRSDs and those that exhibit reduced melatonin levels, suggesting that circadian dysfunction is a leading contributor to the sleep disturbances described in this patient population.
The cause-effect relationship between circadian dysfunction and sleep disorders is not yet clear. While it is possible that circadian dysfunction (Process C) could be the root cause of the sleeping disorders, it is also possible that a defect in sleep homeostasis (Process S) is the root cause of the sleeping disorders in AS, including CRSDs. Insomnia can lead to circadian misalignment even though the circadian system is functioning normally, simply by increasing an individual’s exposure to light at night (watching TV, reading, playing on a cellphone, etc.) while struggling to go to sleep. This would cause the circadian system to shift out of alignment with the day-night cycle (Fig 1C). While melatonin therapy does improve sleep in many AS patients, it’s not clear that it restores sleep back to normal levels, durations, and consolidation. Thus, more work is needed to understand the etiology of the sleep disorders in AS, improve the treatment of these sleep disorders, and potentially uncover new aspects of sleep regulation in general.
In order to determine if the sleep disorders in AS are a possible consequence of a defect in Process C, several groups have begun examining interactions of UBE3A with core circadian clock proteins, as well as examining circadian rhythmicity in AS model Drosophila and mice. The first evidence that circadian dysfunction may be involved comes from findings that removing UBE3A protein in fruit flies disrupts circadian patterns of locomotor activity69,70; however effects on the molecular circadian clockwork have not yet been reported for this species. In human cells, the UBE3A protein (also known as E6-AP) interacts with and regulates the stability of the BMAL1 protein – a protein essential for mammalian clock function70. UBE3A is an E3 ubiquitin ligase that targets substrate proteins for proteasomal degradation78. Thus, the hypothesis was that UBE3A loss dysregulated BMAL1 protein levels by impairing its normal degradation. Indeed, depleting UBE3A using RNAi in cell-based circadian clock models results in a slight lengthening of circadian periods36,70, providing support for the circadian-based sleep disorder hypothesis.
Our group as well as Carl Johnson’s group at Vanderbilt independently extended these studies to the same in vivo mouse model, and largely came up with conflicting results36,71. The AS model mice used in these studies all inherited a null Ube3a allele from their mothers (i.e. maternally deleted Ube3a or Ube3am-/p+), and show phenotypes very similar to those described in humans, including sleep deficits79–81. Of note here, all of the mice used in these studies are on the C56Bl6 background, a strain that does not produce detectable amounts of melatonin. The Vanderbilt group found that these mice, as well as another model with a larger deletion, displayed slightly longer circadian periods (~20–30 min) and realigned more quickly to phase advances in the light cycle71, consistent with observations in AS patients described above. However, our group, using nearly identical assays, did not find these, or any, differences in circadian function in Ube3am-/p+ mice36. Although subtle differences in experimental design and/or animal environment likely accounts for the discrepancy in the data, the difference in findings between groups suggests that any potential circadian deficiencies are not robust.
Quite surprisingly, we found that UBE3A is still present, albeit at reduced levels, in SCN neurons of Ube3am-/p+ mice37, perhaps explaining why we do not find alterations in circadian behavior36. This relaxation in the imprinting of the paternal Ube3a allele is specific to the SCN, as UBE3A is absent from nearly all other brain regions in these mice36–38. Altogether, although UBE3A may regulate BMAL1 stability and cellular clock function, it’s not clear that its loss in AS simply results in changes in Process C that explain the severe sleep disorders described for these patients.
B-. Evidence of deficits in sleep homeostasis
As previously discussed, sleep disturbances in AS patients—twice the number of sleep-state transitions, four-fold increase in awakenings and a halving of time in the deepest stages of NREM sleep—indicate reduced sleep efficiency64,65. A defect in the homeostatic mechanism(s) regulating sleep (Process S) is a potential cause of this reduced sleep efficiency (see Fig. 1C). Indeed, our group has uncovered evidence in the mouse model that sleep homeostasis, rather than the circadian clock, may be a major underlying source of the sleep deficits in AS36.
As noted above, the maternally-deleted Ube3a mouse model (Ube3am-/p+) exhibits imprinting of the paternal allele and most, if not all, of the core features of AS, including sleep disorders79–81. The sleep features in this AS mouse model include an increased number of transitions between sleep states, decreased depth of NREM sleep, and a trend for decreased REM amount during undisturbed sleep81 – traits reminiscent of humans.
The physiological basis of the homeostatic mechanism regulating sleep remains unknown; therefore, EEG and behavioral measures of sleep pressure are used to identify changes in this homeostatic mechanism. This homeostatic process is responsible for the increases in sleep amount and EEG power in low frequency ranges (i.e. slow-wave power a.k.a NREM delta power, 0.5–4 Hz) during NREM sleep and increases in the number of higher frequency EEG peaks during waking (‘wave-incidence’), especially prolonged durations of waking82,83. NREM delta power is among the most reliable measures of Process S, as it builds with wake, dissipates with sleep, and increases in response to sleep deprivation. In this way, it is a widely accepted as an objective indicator of sleep pressure. Wave-incidence provides similar information on changes in sleep pressure, except that it is measured during wake, when sleep pressure is actively accumulating.
We serendipitously discovered that mice inheriting a null allele of the Ube3a gene from their mothers (Ube3am-/p+ mice) have pronounced deficits in NREM delta power and subsequent NREM sleep patterns36. Mice are nocturnal, with high amounts of wakefulness to start the night that devolve into a late-night ‘siesta’ (i.e. nap) before an increase in wake again before lights-on. During this time, normal mice build wave incidence and NREM delta power that peaks coincident with the timing of the siesta, suggesting a potential causal relationship (though it should be noted that in normal mice, the precise timing and presence of siesta from day to day can be variable). AS model mice, in contrast, show little changes in wave-incidence and display a much slower increase in NREM delta power during the night, with the latter peaking at lights-on. These mice also lack a mid-to-late night siesta (Fig 2). Recently, elevated delta power (2–4 Hz) was reported in both children with AS and a mouse model of AS59. This elevated delta power was found to be an effective biomarker of AS that is present in both sleep and waking; however, based on our findings, this reported overall elevation does not impact changes in delta power (0.5–4 Hz) related to the duration of sleep and wakefulness. Overall, these findings are consistent with reduced accumulation of sleep pressure and suggestive of a deficit in sleep homeostasis.
Figure 2: Sleep patterns are disrupted in AS model mice.
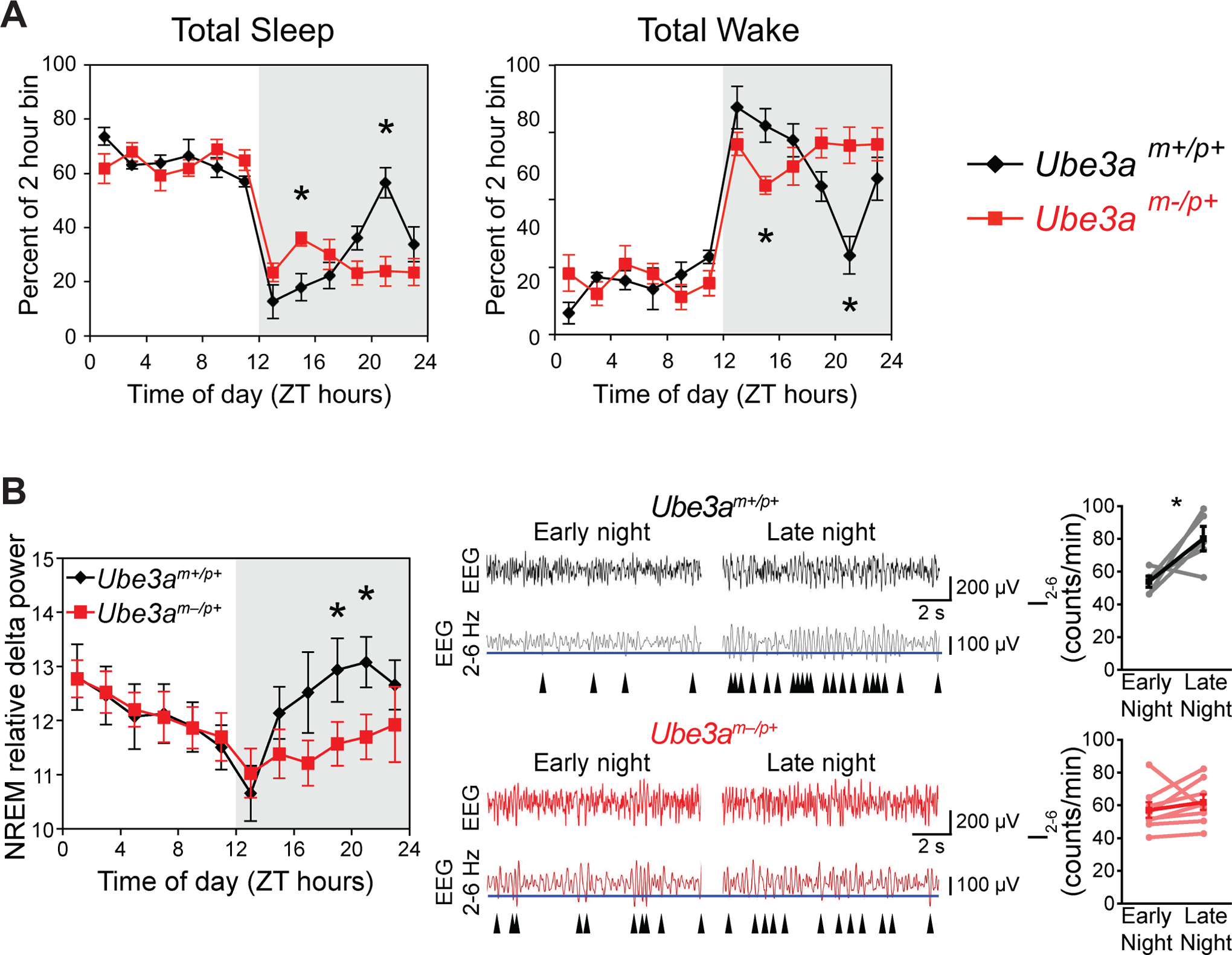
A. Total sleep or wake in wild type (Ube3am+/p+) or AS (Ube3am-/p+) mice recorded over an uninterrupted 24 h period in 12 h light/dark cycle. Data are shown as percentage of each 2 h block spent asleep or awake within each mouse, averaged within genotypes (mean+/−SEM; n=6 wild type or 8 AS mice). Note the difference in sleep/wake amounts between genotypes across the night (indicated by the gray shading). B. AS model mice exhibit reduced nighttime sleep-pressure accumulation. Left - NREM sleep intensity, as measured by delta power, is significantly reduced in Ube3am-/p+ mice during the latter portion of the night. * = p<0.05 between genotypes, n= 6–8 mice. Right - The increase in the wave-incidence (I2– 6) across the night (wake period) is abolished in Ube3am-/p+ mice. Representative EEG recordings (black and red) are shown above bandpass-filtered versions (gray and dark red) of the same EEG recording. The incidence of peaks in the upper 30% by amplitude (blue line) in the filtered signal were counted in epochs scored as waking are indicated by arrowheads. I2– 6 has been previously validated as a measure of sleep-pressure accumulation in awake mice82. I2– 6 significantly increased across the active period in wild type (* = p<0.05) but not Ube3am-/p+mice. All data are replotted from Ehlen et al, 201536.
To test this hypothesis more directly, we forcibly deprived mice of sleep during the early daytime – when mice are sleeping most intensely, a common paradigm used to probe sleep homeostasis. Indeed, AS mice displayed blunted changes in wave-incidence and NREM delta power accumulation, as well as less recovery sleep after sleep deprivation36. Taken together, our findings demonstrate that neuronal loss of UBE3A in mice eliminates the homeostatic sleep responses to both ad libitum wakefulness and forced wakefulness; thus, indicating a deficit in the accumulation of sleep pressure.
Overall, the sleep studies in mice provide direct evidence that UBE3A-loss reduces sleep pressure. However, direct comparisons to humans with AS are complicated by differences in nocturnality/diurnality, experimental paradigm (i.e. forced sleep deprivation), and by the fact that most clinical sleep studies focused on times when AS patients typically sleep. Furthermore, when studied in a controlled environment, animal models are not subject to the disruptive nighttime influences of light. Nevertheless, this animal model demonstrates that undisturbed, spontaneous sleep is altered by neuronal loss of UBE3A in ways that are at least superficially similar to AS patients. Both display similar NREM sleep characteristics, an increase in the number of transitions between sleep states, and decreased depth of NREM sleep. Both effects are indicative of reduced sleep need. Detailed analysis of EEG waveforms from AS patients are needed to confirm that markers of sleep pressure are reduced similarly in both mice and humans. These studies in humans are technically challenging but are critically important as they have the potential to confirm a core mechanism for sleep disturbances associated with UBE3A-related developmental disorders.
Conclusion and final considerations
In Figure 1, we depict two possible causes of sleep problems in AS that are both suggested by the data and consistent with the Two-Process model11. Sleep in AS patients has thus far been most successfully improved by nighttime melatonin therapy, suggesting that some aspects/portions of the sleep disorders may be due to changes in circadian timing. In mouse models, however, neuronal loss of UBE3A does not produce robust circadian deficits but results in marked disruption in sleep homeostasis. So, the truth may lie somewhere in-between. Mouse models, because of the varied genetic tools available (i.e. floxed Ube3a alleles, melatonin deficient/proficient strains, strains with very diverse sleep characteristics), are going to be extremely valuable for identifying the fundamental relationships between AS and circadian and sleep dysfunction. One intriguing possibility is that UBE3A loss impairs clock function in a brain region outside of the SCN to cause some of these effects. It’s also possible that there are interactions between clocks/SCN and an unknown site responsible for maintaining sleep homeostasis. In that regard, although its loss causes a wide array of phenotypes, UBE3A appears to be a “sleep gene.” Determining how and where in the brain it functions to regulate sleep could prove pivotal in understanding the mechanism(s) underlying sleep homeostasis regardless of the exact mechanism that is impaired by neuronal loss of UBE3A.
Funding information:
This work was supported by grants from: NSF IOS grant 1832069 to JPD, DLH, and JCE, NIH NIGMS grants GM127260 to JCE, and GM109861 and GM127044 to JPD. This work was also supported in part by NIH NINDS grant U54 NS083932 and NIH NIMHD grant U54MD007602 to Morehouse School of Medicine.
References
- 1.Schwichtenberg AJ, Malow BA. Melatonin Treatment in Children with Developmental Disabilities. Sleep Med Clin. 2015;10(2):181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cirelli C, Tononi G. Is sleep essential? PLoS Biol. 2008;6(8):e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boivin DB, Czeisler CA, Dijk DJ, et al. Complex interaction of the sleep-wake cycle and circadian phase modulates mood in healthy subjects. Arch Gen Psychiatry. 1997;54(2):145–152. [DOI] [PubMed] [Google Scholar]
- 4.Achermann P, Dijk DJ, Brunner DP, Borbely AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull. 1993;31(1–2):97–113. [DOI] [PubMed] [Google Scholar]
- 5.Borbely AA, Daan S, Wirz-Justice A, Deboer T. The two-process model of sleep regulation: a reappraisal. J Sleep Res. 2016;25(2):131–143. [DOI] [PubMed] [Google Scholar]
- 6.Herzog ED, Hermanstyne T, Smyllie NJ, Hastings MH. Regulating the Suprachiasmatic Nucleus (SCN) Circadian Clockwork: Interplay between Cell-Autonomous and Circuit-Level Mechanisms. Cold Spring Harb Perspect Biol. 2017;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437(7063):1257–1263. [DOI] [PubMed] [Google Scholar]
- 8.Zepelin H, Siegel JM and Tobler I Mammalian sleep In: Kryger MH, Roth T and Dement WC (Eds) Principles and Practice of Sleep Medicine , 4th edn Elsevier Saunders, Philadelphia, 2005: 91–100. [Google Scholar]
- 9.Liu Y, Wheaton AG, Chapman DP, Cunningham TJ, Lu H, Croft JB. Prevalence of Healthy Sleep Duration among Adults--United States, 2014. MMWR Morb Mortal Wkly Rep. 2016;65(6):137–141. [DOI] [PubMed] [Google Scholar]
- 10.Robbins R, Grandner MA, Buxton OM, et al. Sleep myths: an expert-led study to identify false beliefs about sleep that impinge upon population sleep health practices. Sleep Health. 2019;5(4):409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1(3):195–204. [PubMed] [Google Scholar]
- 12.Donlea JM, Alam MN, Szymusiak R. Neuronal substrates of sleep homeostasis; lessons from flies, rats and mice. Curr Opin Neurobiol. 2017;44:228–235. [DOI] [PubMed] [Google Scholar]
- 13.Jones BE. Principal cell types of sleep-wake regulatory circuits. Curr Opin Neurobiol. 2017;44:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zisapel N New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br J Pharmacol. 2018;175(16):3190–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson D, Encel N. Melatonin and sleep in humans. J Pineal Res. 1993;15(1):1–12. [DOI] [PubMed] [Google Scholar]
- 16.Scheer FA, Czeisler CA. Melatonin, sleep, and circadian rhythms. Sleep medicine reviews. 2005;9(1):5–9. [DOI] [PubMed] [Google Scholar]
- 17.Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep medicine reviews. 2005;9(1):25–39. [DOI] [PubMed] [Google Scholar]
- 18.Pandi-Perumal SR, Smits M, Spence W, et al. Dim light melatonin onset (DLMO): a tool for the analysis of circadian phase in human sleep and chronobiological disorders. Progress in neuro-psychopharmacology & biological psychiatry. 2007;31(1):1–11. [DOI] [PubMed] [Google Scholar]
- 19.Hood S, Amir S. The aging clock: circadian rhythms and later life. J Clin Invest. 2017;127(2):437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubocovich ML, Rivera-Bermudez MA, Gerdin MJ, Masana MI. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front Biosci. 2003;8:d1093–1108. [DOI] [PubMed] [Google Scholar]
- 21.Dubocovich ML. Melatonin receptors: role on sleep and circadian rhythm regulation. Sleep Med. 2007;8 Suppl 3:34–42. [DOI] [PubMed] [Google Scholar]
- 22.Hardeland R, Poeggeler B. Melatonin and synthetic melatonergic agonists: actions and metabolism in the central nervous system. Cent Nerv Syst Agents Med Chem. 2012;12(3):189–216. [DOI] [PubMed] [Google Scholar]
- 23.Williams WP 3rd, McLin DE 3rd, Dressman MA, Neubauer DN. Comparative Review of Approved Melatonin Agonists for the Treatment of Circadian Rhythm Sleep-Wake Disorders. Pharmacotherapy. 2016;36(9):1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buscemi N, Vandermeer B, Hooton N, et al. The efficacy and safety of exogenous melatonin for primary sleep disorders. A meta-analysis. J Gen Intern Med. 2005;20(12):1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laan LA, v Haeringen A, Brouwer OF. Angelman syndrome: a review of clinical and genetic aspects. Clin Neurol Neurosurg. 1999;101(3):161–170. [DOI] [PubMed] [Google Scholar]
- 26.Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40(2):87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–395. [DOI] [PubMed] [Google Scholar]
- 28.Duca DG, Craiu D, Boer M, et al. Diagnostic approach of angelman syndrome. Maedica (Buchar). 2013;8(4):321–327. [PMC free article] [PubMed] [Google Scholar]
- 29.Sidorov MS, Deck GM, Dolatshahi M, et al. Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. Journal of Neurodevelopmental Disorders. 2017;9(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frohlich J, Miller MT, Bird LM, et al. Electrophysiological Phenotype in Angelman Syndrome Differs Between Genotypes. Biological Psychiatry. 2019;85(9):752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira JA, Ravichandran CT, Mullett J, McDougle CJ, Keary CJ. Characterization of sleep habits and medication outcomes for sleep disturbance in children and adults with Angelman syndrome. Am J Med Genet A. 2020. [DOI] [PubMed] [Google Scholar]
- 32.Larson AM, Shinnick JE, Shaaya EA, Thiele EA, Thibert RL. Angelman syndrome in adulthood. Am J Med Genet A. 2015;167A(2):331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spruyt K, Braam W, Curfs LM. Sleep in Angelman syndrome: A review of evidence. Sleep medicine reviews. 2018;37:69–84. [DOI] [PubMed] [Google Scholar]
- 34.Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21(13):3001–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7(7):1213–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehlen JC, Jones KA, Pinckney L, et al. Maternal Ube3a Loss Disrupts Sleep Homeostasis But Leaves Circadian Rhythmicity Largely Intact. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35(40):13587–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones KA, Han JE, DeBruyne JP, Philpot BD. Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci Rep. 2016;6:28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. The Journal of comparative neurology. 2014;522(8):1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knoll JH, Nicholls RD, Lalande M. On the parental origin of the deletion in Angelman syndrome. Hum Genet. 1989;83(2):205–207. [DOI] [PubMed] [Google Scholar]
- 40.Malcolm S, Clayton-Smith J, Nichols M, et al. Uniparental paternal disomy in Angelman’s syndrome. Lancet. 1991;337(8743):694–697. [DOI] [PubMed] [Google Scholar]
- 41.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(1):70–73. [DOI] [PubMed] [Google Scholar]
- 42.Matsuura T, Sutcliffe JS, Fang P, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15(1):74–77. [DOI] [PubMed] [Google Scholar]
- 43.Glenn CC, Nicholls RD, Robinson WP, et al. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993;2(9):1377–1382. [DOI] [PubMed] [Google Scholar]
- 44.Buiting K, Williams C, Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12(10):584–593. [DOI] [PubMed] [Google Scholar]
- 45.Gentile JK, Tan W-H, Horowitz LT, et al. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr. 2010;31(7):592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horsthemke B, Maat-Kievit A, Sleegers E, et al. Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. Journal of medical genetics. 1996;33(10):848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moncla A, Malzac P, Livet MO, et al. Angelman syndrome resulting from UBE3A mutations in 14 patients from eight families: clinical manifestations and genetic counselling. J Med Genet. 1999;36(7):554–560. [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson WP, Christian SL, Kuchinka BD, et al. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clinical Genetics. 2000;57(5):349–358. [DOI] [PubMed] [Google Scholar]
- 49.Le Fevre A, Beygo J, Silveira C, et al. Atypical Angelman syndrome due to a mosaic imprinting defect: Case reports and review of the literature. Am J Med Genet A. 2017;173(3):753–757. [DOI] [PubMed] [Google Scholar]
- 50.Huang H-S, Burns AJ, Nonneman RJ, et al. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams CA, Zori RT, Hendrickson J, et al. Angelman syndrome. Current problems in pediatrics. 1995;25(7):216–231. [DOI] [PubMed] [Google Scholar]
- 52.Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413–418. [DOI] [PubMed] [Google Scholar]
- 53.Hirano A, Hsu PK, Zhang L, et al. DEC2 modulates orexin expression and regulates sleep. Proc Natl Acad Sci U S A. 2018;115(13):3434–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xing L, Shi G, Mostovoy Y, et al. Mutant neuropeptide S receptor reduces sleep duration with preserved memory consolidation. Science translational medicine. 2019;11(514). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyd SG, Harden A, Patton MA. The EEG in early diagnosis of the Angelman (happy puppet) syndrome. Eur J Pediatr. 1988;147(5):508–513. [DOI] [PubMed] [Google Scholar]
- 56.Minassian BA, DeLorey TM, Olsen RW, et al. Angelman syndrome: correlations between epilepsy phenotypes and genotypes. Annals of neurology. 1998;43(4):485–493. [DOI] [PubMed] [Google Scholar]
- 57.Korff CM, Kelley KR, Nordli DR Jr. Notched delta, phenotype, and Angelman syndrome. Journal of clinical neurophysiology : official publication of the American Electroencephalographic Society. 2005;22(4):238–243. [DOI] [PubMed] [Google Scholar]
- 58.Uemura N, Matsumoto A, Nakamura M, et al. Evolution of seizures and electroencephalographical findings in 23 cases of deletion type Angelman syndrome. Brain & development. 2005;27(5):383–388. [DOI] [PubMed] [Google Scholar]
- 59.Sidorov MS, Deck GM, Dolatshahi M, et al. Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. Journal of neurodevelopmental disorders. 2017;9:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buoni S, Grosso S, Pucci L, Fois A. Diagnosis of Angelman syndrome: clinical and EEG criteria. Brain & development. 1999;21(5):296–302. [DOI] [PubMed] [Google Scholar]
- 61.Casara GL, Vecchi M, Boniver C, et al. Electroclinical diagnosis of Angelman syndrome: a study of 7 cases. Brain & development. 1995;17(1):64–68. [DOI] [PubMed] [Google Scholar]
- 62.Valente KD, Andrade JQ, Grossmann RM, et al. Angelman syndrome: difficulties in EEG pattern recognition and possible misinterpretations. Epilepsia. 2003;44(8):1051–1063. [DOI] [PubMed] [Google Scholar]
- 63.Vendrame M, Loddenkemper T, Zarowski M, et al. Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy & behavior : E&B. 2012;23(3):261–265. [DOI] [PubMed] [Google Scholar]
- 64.Miano S, Bruni O, Elia M, Musumeci SA, Verrillo E, Ferri R. Sleep breathing and periodic leg movement pattern in Angelman Syndrome: a polysomnographic study. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2005;116(11):2685–2692. [DOI] [PubMed] [Google Scholar]
- 65.Miano S, Bruni O, Leuzzi V, Elia M, Verrillo E, Ferri R. Sleep polygraphy in Angelman syndrome. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2004;115(4):938–945. [DOI] [PubMed] [Google Scholar]
- 66.den Bakker H, Sidorov MS, Fan Z, et al. Abnormal coherence and sleep composition in children with Angelman syndrome: a retrospective EEG study. Molecular autism. 2018;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takaesu Y, Komada Y, Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012;13(9):1164–1170. [DOI] [PubMed] [Google Scholar]
- 68.Phillips L, Appleton RE. Systematic review of melatonin treatment in children with neurodevelopmental disabilities and sleep impairment. Dev Med Child Neurol. 2004;46(11):771–775. [DOI] [PubMed] [Google Scholar]
- 69.Wu Y, Bolduc FV, Bell K, et al. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci U S A. 2008;105(34):12399–12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gossan NC, Zhang F, Guo B, et al. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic acids research. 2014;42(9):5765–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi SQ, Bichell TJ, Ihrie RA, Johnson CH. Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Current biology : CB. 2015;25(5):537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhdanova IV, Wurtman RJ, Wagstaff J. Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J Pediatr Endocrinol Metab. 1999;12(1):57–67. [DOI] [PubMed] [Google Scholar]
- 73.Cavallo A Plasma melatonin rhythm in normal puberty: interactions of age and pubertal stages. Neuroendocrinology. 1992;55(4):372–379. [DOI] [PubMed] [Google Scholar]
- 74.Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23(6):649–654. [DOI] [PubMed] [Google Scholar]
- 75.Paprocka J, Kijonka M, Wojcieszek P, Pecka M, Emich-Widera E, Sokol M. Melatonin and Angelman Syndrome: Implications and Mathematical Model of Diurnal Secretion. Int J Endocrinol. 2017;2017:5853167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Monteleone P, Tortorella A, Borriello R, Natale M, Cassandro P, Maj M. Suppression of nocturnal plasma melatonin levels by evening administration of sodium valproate in healthy humans. Biological psychiatry. 1997;41(3):336–341. [DOI] [PubMed] [Google Scholar]
- 77.Jain SV, Simakajornboon N, Arthur TM. Central sleep apnea: does stabilizing sleep improve it? J Child Neurol. 2014;29(1):96–98. [DOI] [PubMed] [Google Scholar]
- 78.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. [DOI] [PubMed] [Google Scholar]
- 79.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annual review of genomics and human genetics. 2001;2:153–175. [DOI] [PubMed] [Google Scholar]
- 80.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cellular and molecular life sciences : CMLS. 2007;64(7–8):947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Colas D, Wagstaff J, Fort P, Salvert D, Sarda N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiology of disease. 2005;20(2):471–478. [DOI] [PubMed] [Google Scholar]
- 82.Ehlen JC, Jefferson F, Brager AJ, Benveniste M, Paul KN. Period-amplitude analysis reveals wake-dependent changes in the electroencephalogram during sleep deprivation. Sleep. 2013;36(11):1723–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borbely AA, Baumann F, Brandeis D, Strauch I, Lehmann D. Sleep deprivation: effect on sleep stages and EEG power density in man. Electroencephalogr Clin Neurophysiol. 1981;51(5):483–495. [DOI] [PubMed] [Google Scholar]