

Abstract
Albocycline (ALB) is a unique macrolactone natural product with potent, narrow-spectrum activity against methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-intermediate (VISA), and vancomycin-resistant S. aureus (VRSA) strains (MIC = 0.5–1.0 μg/mL). Described herein is the synthesis and evaluation of a novel series analogs derived from albocycline by functionalization at three specific sites: the C2-C3 enone, the tertiary carbinol at C4, and the allylic C16 methyl group. Exploration of the structure-activity relationships (SAR) by means of minimum inhibitory concentration assays (MICs) revealed that C4 ester analog 6 was twice as potent as ALB, which represents a class of lead compound that can be further studied to address multi-drug resistant pathogens.
Keywords: albocycline, MIC, SAR, antibiotic
Antibacterial drug discovery has benefited greatly from the systematic investigation of natural products (NPs). In fact, approximately 60% of all the FDA-approved antibiotics are either NPs, NP-derived, or NP-inspired small molecules.1,2 However, the rapid development of bacterial resistance to antibiotics coupled with marked changes in the Pharmaceutical Sector has resulted in a global health crisis.3 The structural modification of natural product derived antibiotics (e.g., β-lactams, aminoglycosides, macrolides, tetracyclines) by either semi- or total synthesis is a highly effective tactic for exploring novel drugs to address bacterial resistance. As part of a program directed at bioactive natural products synthesis, we were drawn to albocycline (ALB, 1) due to its unique structure—a 14-membered macrolactone with four stereogenic centers and three alkenes—in addition to its antibacterial activity (Figure 1).4
Figure 1.
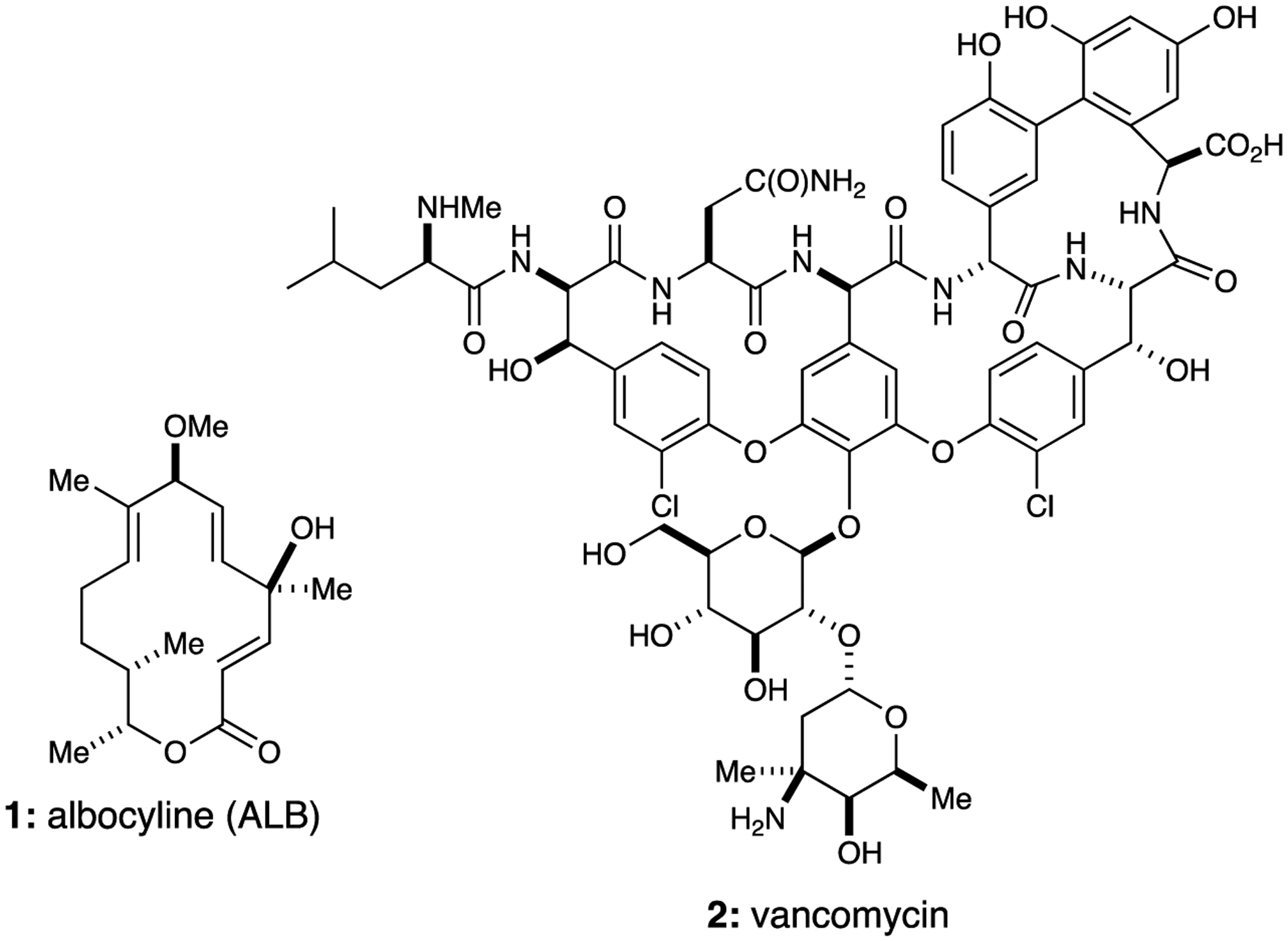
Structures of albocycline (ALB, 1) and vancomycin (2).
Significantly, Tomoda reported in 2013 that ALB was equipotent with vancomycin (2) against methicillin-resistant Staphylococci aureus (MRSA) and more potent against vancomycin-resistant S. aureus (VRSA).4 A recent report on a spike in MRSA incidences, in addition to a pressing need for drugs to address VISA and VRSA, was further motivation to carry SAR studies on this validated antibacterial natural product.5
Studies by Zucchi and co-workers further reported that ALB could be deployed as an anti-mold agent.6 Agricultural losses resulting from “white mold” disease caused by Sclerotinia sclerotiorum in the United States can exceed $200M annually.7 Significantly, in vivo studies suggested further interest in ALB for future therapeutic applications due to the absence of toxicity in mice as well as in humans.8,9 In 2018, we demonstrated ALB was not toxic to human cells at a final concentration ≤64 μg/mL in colorimetric MTT Cell Proliferation Assays using human HepG2 hepatocellular liver carcinoma cells.10 Accordingly, exploring the ALB framework by preparing novel analogs and evaluating them in MIC assays could assist in addressing the aforementioned threats to public health and society.
Two total syntheses of albocycline (i.e., ingramycin) have been reported to date. The first was from Tanner and Somfai in 1987 and required 40 total steps (21 in the longest linear sequence).11 In 2017, we achieved the second synthesis in 14 overall steps.12 Alternatively, ALB can be readily accessed by culturing the producing organism, S. maizeus, as was reported in the Upjohn patent.13 The latter (i.e., semi-synthetic) approach allows access to multi-milligram amounts of material that be employed in the synthesis of novel ALB analogs through judicious modifications that modulate potency, physicochemical, and ADMET properties that have been enormously successful across a wide range of antibiotic classes.14
Previous reports indicated that 2,3-dihydro- (3) and 5,6-dihydroalbocycline (4) did not possess antibacterial activity, thus highlighting the importance of unsaturation at those sites (Figure 2).15 On the other hand, cineromycin B (5) maintains biological activity against MRSA, suggesting that the C7 site is amenable to modification by either semi- or total synthesis.16 Sites that had not been explored included the tertiary C4 carbinol, the O-methyl ether at C7, and the allylic C16 methyl group.
Figure 2.
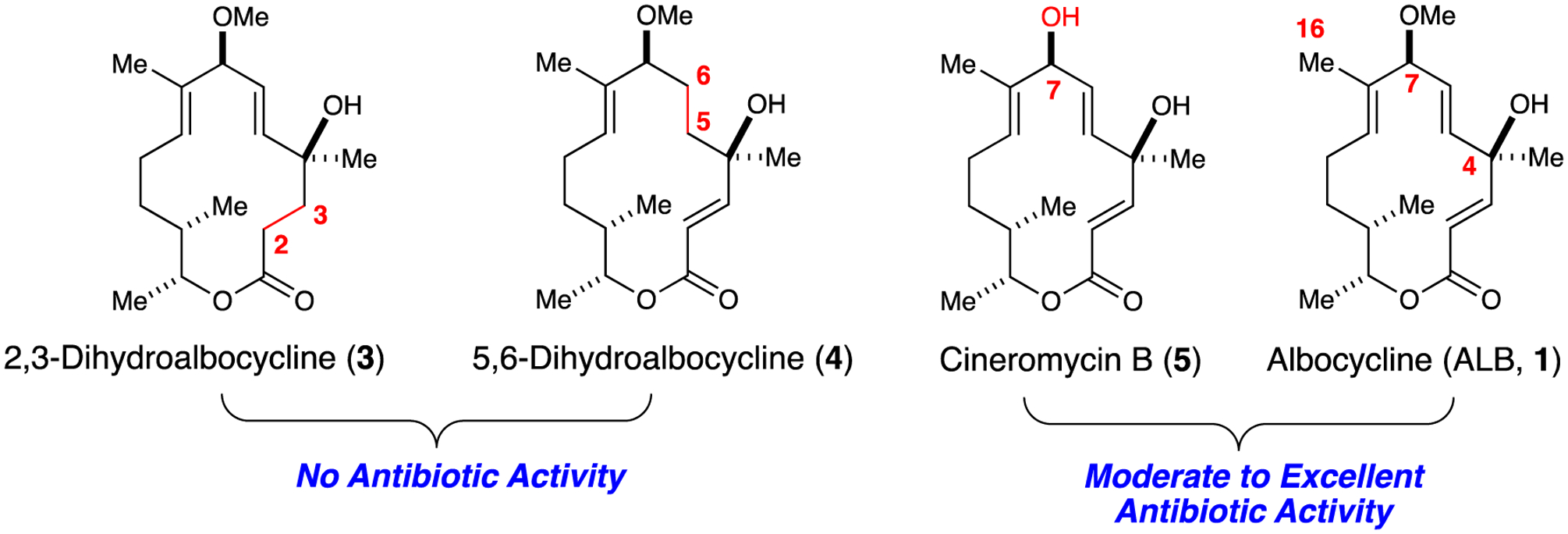
Studying antibiotic activity across albocycline and its analogs.
Straightforward C4 carbinol functionalization includes both esterification and ether formation by acylation and alkylation. With respect to the former, initial attempts based on Yamaguchi or Steglich (i.e., DMAP) conditions did not deliver any ester adduct resulting in recovery of ALB.12,17 However, previous work performed by Upjohn revealed that prolonged heating under pyridine and catalytic DMAP delivered the 4-bromobenzoate adduct, which was subjected to single x-ray crystallography in order to confirm the structure of ALB (1).18 By employing the same reaction conditions, albeit with 4-fluorobenzoyl chloride, which was selected to reflect the increasing trend in medicinal chemistry of incorporating fluorine substituents in FDA-approved drugs, benzoate analog 6 was obtained in 42% yield (Scheme 1).19
Scheme 1.
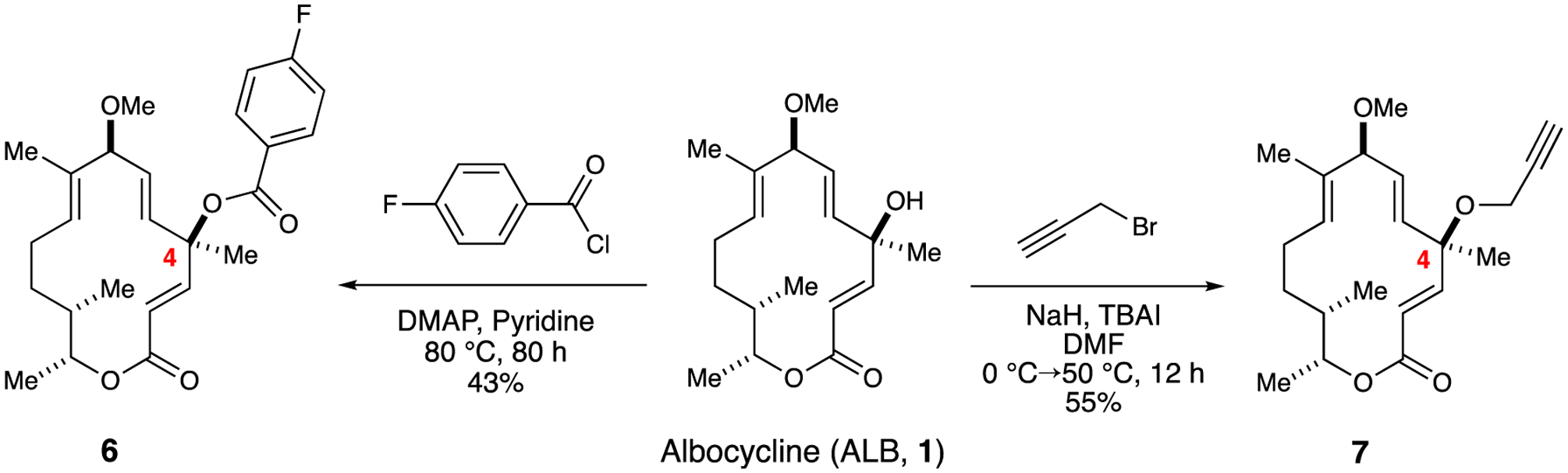
Functionalization of ALB at the C4 carbinol position by acylation to access ester analog 6 or by alkylation to access ether analog 7.
We reasoned that the installation of a propargylic ether at C6 could allow for the rapid generation of a library of ALB congeners by using the venerable copper(I)-catalyzed azide-alkyne cycloaddition (i.e., Click) reaction.20 In addition, an alkyne-functionalized analog of ALB could serve as a chemical probe for use in affinity-based protein profiling (ABPP)21,22 to help identify the target of ALB.10 To this end, we reacted ALB (1) with NaH and propargyl bromide in presence of catalytic tetrabutylammonium iodide (TBAI), which afforded 4-O-propargyl albocycline (7) in 55% yield after column chromatography, in addition 15% of recovered starting material. It is noteworthy that owing to the hindered nature of the tertiary C4 carbinol, the reaction mixture required heating in analogy to the acylation reaction.
Inspection of albocycline’s structure reveals a non-obvious site of functionalization, namely the allylic C16 methyl group. We were inspired by Undheim and co-workers who successfully functionalized the allylic C10 methyl group on the erythromycin framework by employing N-bromosuccinimide (NBS) in the presence of peroxides.23 Accordingly, we attempted to generate a similar allylic bromide derivative on ALB (1) that would allow ready access to a variety of analogs (e.g., amines, ethers, esters, and carbamates) using simple substitution reactions. In the event, we obtained an inseparable 1:1 mixture of allylic bromide regioisomers 8 and 8’ in low yield (Scheme 2). Alternatively, site selective Riley oxidation of 1 at C16 with selenium dioxide (SeO2) proved to be a superior approach allowing access of allylic alcohol 9 in a modest 35%. yield.
Scheme 2.
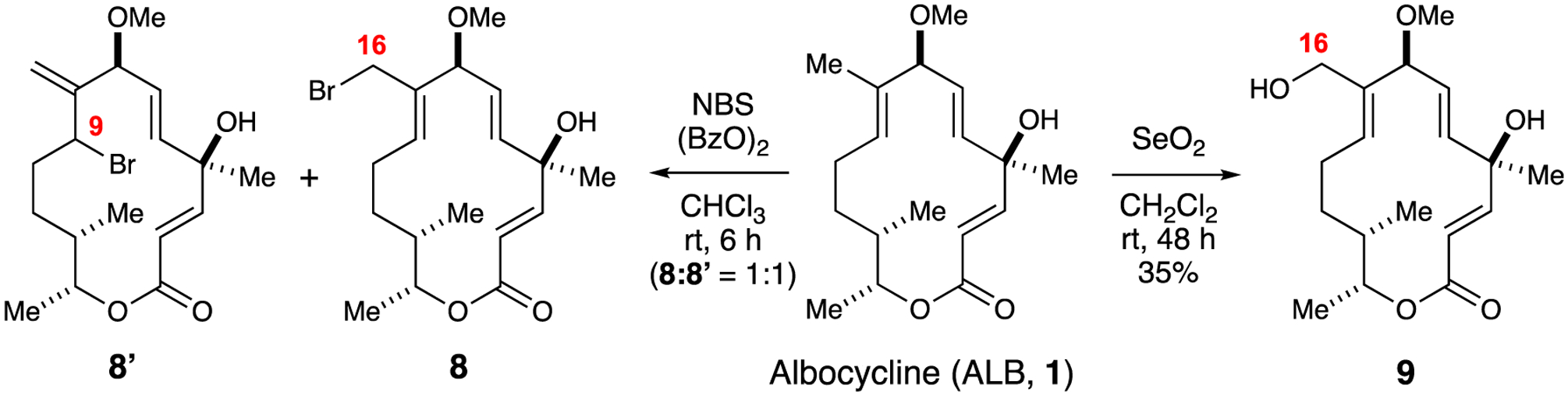
Allylic functionalization at C16 with NBS or SeO2 to generate regioisomeric bromides 8 and 8’ or allylic alcohol 9, respectively.
Establishing a synthetic route to allylic alcohol 9 from ALB allowed ready access to several functional groups such as aldehyde, ether, and azide through simple functional group interconversions. The rationale behind accessing the oxidized derivative of allylic alcohol 9, namely the C16 aldehyde, was the known presence of this moiety in established macrolides as typified by tylosin.24 Accordingly, reaction of allylic alcohol 9 with the Dess-Martin periodinane (DMP) furnished aldehyde 10 in 55% yield (Scheme 3). In order to test the influence of the hydroxyl group at C16 on biological activity enabled by potential hydrogen bonding interactions, we synthesized the 16-methoxyalbocycline (11) using by alkylating the hydroxyl group of 9 with Meerwein’s salt and Proton Sponge in 55% yield.
Scheme 3.
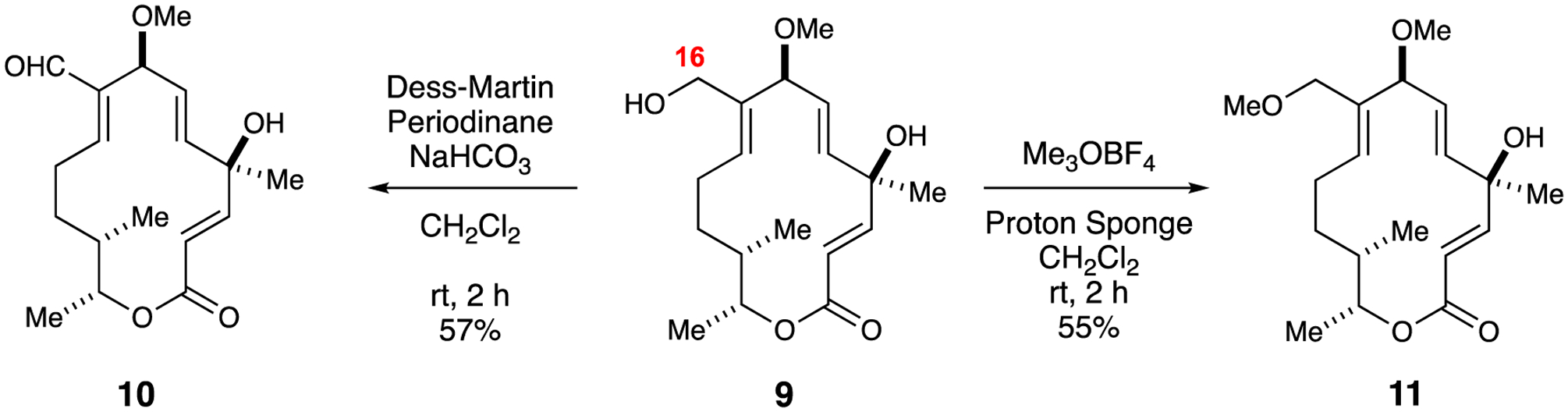
Allylic functionalization at C16 to generate aldehyde 10 and ether 11
The abundance of the azide functionality in medicinal chemistry due to its kinetic stability under physiological conditions, coupled with their unique reactivity patterns make them promising candidates to explore. Initial attempts at activating the allylic alcohol in 9 by preparing sulfonate (e.g., mesylate or tosylate) derivatives for subsequent displacement with NaN3 were unsuccessful. Alternatively, the use of diphenyl phosphoryl azide (DPPA) and DBU allowed the formation of diphenyl phosphate intermediate 12 within the first 20 minutes; however, displacement with azide was observed after stirring an additional 3–4 hours to furnish an inseparable mixture of azide regioisomers 13 and 14, in which the latter is the product of SN2’ displacement and is often observed in allylic substrates according to literature precedent (Scheme 4).25
Scheme 4.
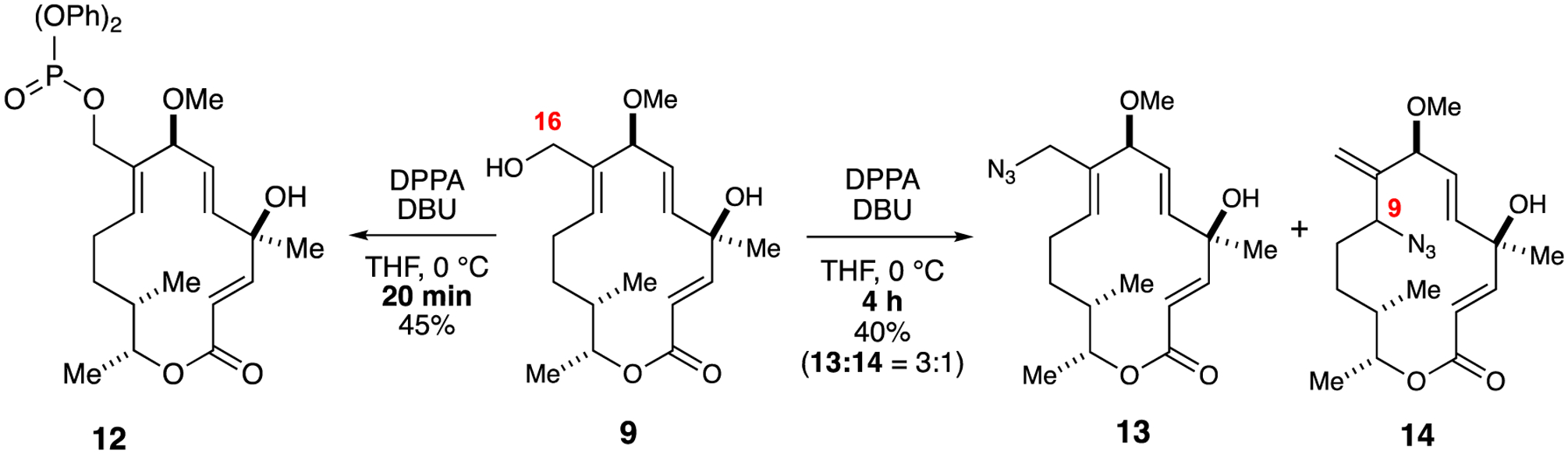
Allylic functionalization at C16 to generate azides 13 and 14 from isolable phosphonate intermediate 12.
Recent reports from Hergenrother and co-workers suggested that compounds exhibiting primary amine functionalities tend to be highly accumulating inside the cell due to their amphiphilic nature, rigidity, and low globularity.26 This in return enhances the activity of the small molecules, in which the rate of compounds crossing porins will be higher than the rate at which they are pumped out. Hence, considering this approach may lead to better efficacy especially against Gram-negative bacteria, as was shown for modified compounds towards E. coli, as a result of overcoming their outer membrane by favoring diffusion and prolonged accumulation inside the bacteria similar to what is typically encountered with Gram-positives.26 Accordingly, we were motivated to incorporate amine functionality on the framework of ALB in order to test if there is an increased spectrum of activity. The presence of the α,β-unsaturated enone motif in ALB between C2 and C3 is a potential site for incorporating primary or secondary amines by conjugate (Michael) addition. Moreover, previous work conducted by Colby and co-workers showed that aza-Michael addition of amines to α-methylene-γ-lactones resulted in enhancing solubility, biological activity, and pharmacokinetic profile of drugs.27 Consequently, initial efforts focused on reacting ALB with secondary amines such as pyrrolidine. However, ALB was recovered and no desired adduct was detected. Alternatively, thiols are potentially another class of nucleophiles that can undergo thia-Michael addition with unsaturated enones.28 Smith and co-workers previously reported the 1,4-addition of thiophenol to the α,β-unsaturated lactone of ALB during the course of a semi-synthetic campaign toward of melanogenesis inhibitors.29 Accordingly, we were inspired to incorporate a fragment bearing both amine and thiol functionalities to ensure the incorporation of amine analog within the ALB scaffold. To this end, 2-mercaptoethylamine was reacted with 1 in CH2Cl2 at rt for 4–6 h, which afforded β-amino-sulfide adduct 15 as a 2:1 ratio of diastereomers at C3 (LCMS) in 40% yield (Scheme 5). Both diastereomers were readily subsequently separated by reverse-phase column chromatography.
Scheme 5.
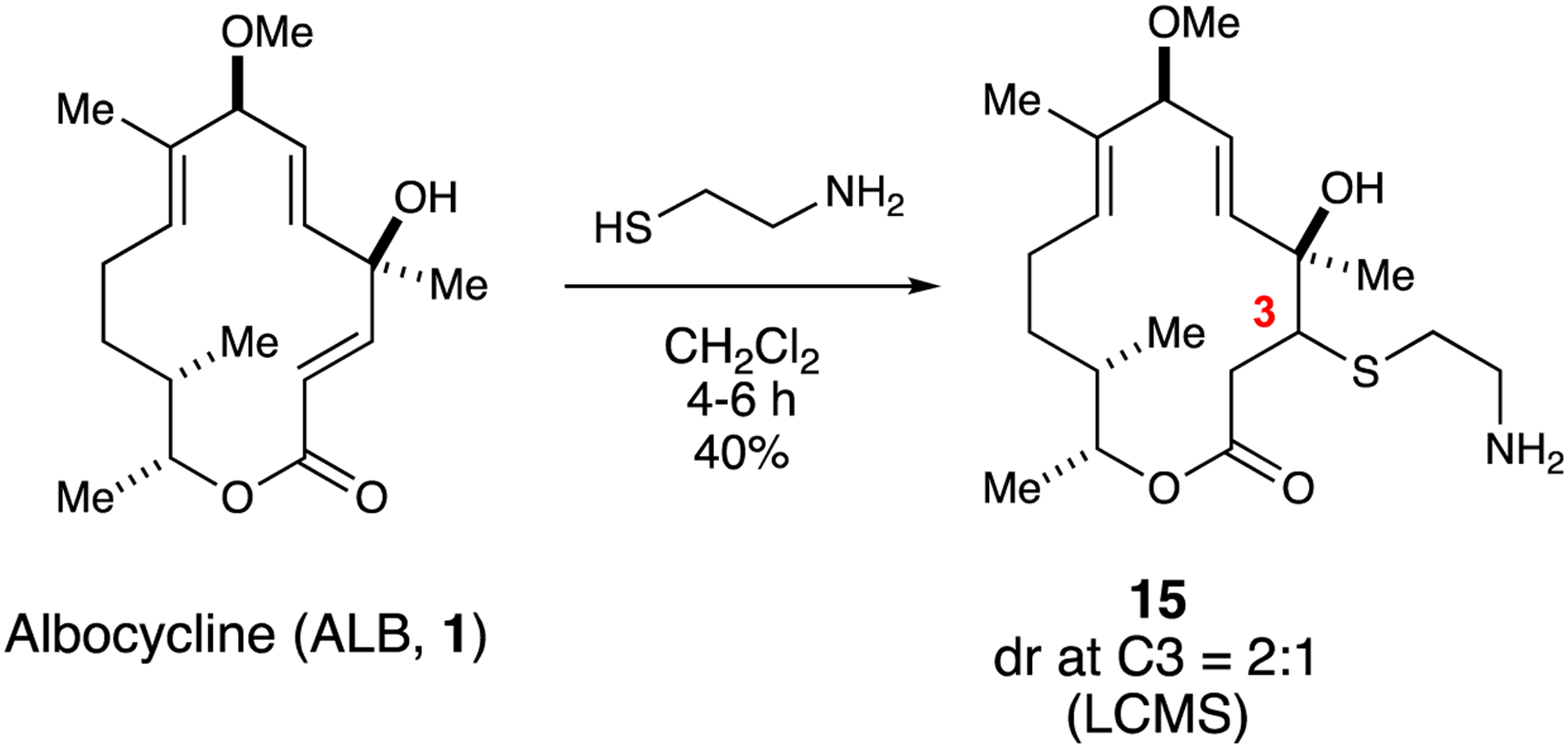
Synthesis of P-amino-sulfide adduct 15 (dr = 2:1) of ALB.
To evaluate the potency of ALB and its semi-synthetic analogs, MIC assays were employed against S. aureus (UAMS-1) and (USA300) strains. The results of those assays are shown in Table 1. Against resistant S. aureus strains, only two analogs 5 and 6 displayed promising biological activity compared to ALB. The O-desmethyl congener of ALB, cineromycin B (5), was 4–8 fold less potent than ALB against (UAMS-1) and two-fold less potent than ALB against USA-300. The fact that C7 modification didn’t destroy bioactivity suggest this site should be further explored. Notably, the p-fluorobenzoyl C4 ester of albocycline 6 was nearly equipotent with ALB against MSSA strain (UAMS-1) and 2-fold more potent than ALB against the MRSA strain (USA-300). While analogs 9 and 10 showed moderate potency, activity was completely destroyed for analogs 7, 11, 12, and 15. On the other hand, testing the biological activity of major and the minor diastereomers of β-amino-sulfide adduct 15 against various Gram-negative strains (i.e., A. baumannii, E. coli, and P. aeruginosa) using kanamycin as positive control revealed poor biological activity, suggesting that inability of 15 to targeting Gram-negative pathogens (see supporting information).
Table 1.
MIC (μg/mL) values for ALB (1) and its corresponding analogs against S. aureus (UAMS-1) and (USA-300).
| Compounds | MIC (μg/mL) S. aureus | |
|---|---|---|
| UAMS-1 (MSSA) | USA300 (MRSA) | |
| ALB (1) | 2 | 2 |
| Cineromycin (5) | 8–16 | 4 |
| 6 | 1–4 | 1 |
| 7 | >256 | ND |
| 9 | 32 | ND |
| 10 | 32 | ND |
| 11 | 128 | ND |
| 12 | >256 | >256 |
| 15 (major) | 128 | 256 |
 Equipotent with ALB (1)
Equipotent with ALB (1)
 More potent than ALB (1)
More potent than ALB (1)
ND = Not Determined
In conclusion, the natural product macrolactone albocycline (ALB, 1) has been cultured from S. maizeus and synthetically modified to access analogs at the C2–3 enone, C4 tertiary alcohol, and C16 allylic methyl group. The O-desmethyl congener of 1, cineromycin B (5) was isolated during the culturing of S. maizeus. All compounds were subjected to MIC evaluation against Gram-positive MSSA and MRSA. While most modifications resulted in loss of activity, cineromycin B (5) and the C4 benzoate derivative 6 were the most promising with the latter analog being two-fold more potent than ALB against MRSA. Future work aimed at determining the target of albocycline will assist in guiding structure-activity relationships in order to discover novel analogs that address the need for drugs that treat resistant pathogens including MRSA.
Supplementary Material
Acknowledgements
The NIH (GM126221) and Temple University supported this research. Support for the NMR facility at Temple University by a CURE grant from the Pennsylvania Department of Health is gratefully acknowledged. We thank Dr. David P. Labeda (National Center for Agricultural Utilization Research, United States Department of Agriculture) for kindly providing S. maizeus. We thank Prof. Ann Valentine for optimizing the protocol for culturing S. maizeus, as well as Drs. Robert Stanley and Carol Manhart for kindly providing autoclave facilities. We thank Dr. Bettina Buttaro for assistance with MIC evaluation against Gram-negative pathogens. Finally, we thank Dr. Charles DeBrosse, Director of the Temple University NMR Facility in the Department of Chemistry, for kind assistance with NMR experiments.
Abbreviations
- MRSA
methicillin-resistant Staphylococcus aureus
- VISA
vancomycin-intermediate Staphylococcus aureus
- VRSA
vancomycin-resistant Staphylococcus aureus
- SAR
structure-activity relationship
- MIC
Minimum Inhibitory Concentration
- NPs
natural products
- ALB
albocycline
- ABPP
affinity-based protein profiling
- TBAI
tetrabutylammonium iodide
- NBS
N-bromosuccinimide
- DPPA
diphenyl phosphoryl azide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
General methods, isolation protocol, minimum inhibitory concentration (MIC) method, synthetic methods for the synthesis and characterization of 6, 7, 9–12, and 15 Spectral data (1H and 13C NMR) for 6, 7, 9–12, and 15, and 2D-NMR for ALB (1) and analog 9.
References:
- (1).Newman DJ; Cragg GM J. Nat. Prod 2012, 75, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Newman DJ; Cragg GM Future Med. Chem 2009, 1, 1415. [DOI] [PubMed] [Google Scholar]
- (3).Solomon SL; Oliver KB Am. Fam. Physician 2014, 89, 938. [PubMed] [Google Scholar]
- (4).Koyama N; Yotsumoto M; Onaka H; Tomoda H J. Antibiot 2013, 66, 303. [DOI] [PubMed] [Google Scholar]
- (5).Hassoun A; Linden PK; Friedman B Crit. Care 2017, 21, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zucchi TD; Almeida LG; Moraes LAB; Consoli FL Ind. Crops. Prod 2014, 52, 264. [Google Scholar]
- (7).Bolton MD; Thomma BP; Nelson BD Mol. Plant Pathol 2006, 7, 1. [DOI] [PubMed] [Google Scholar]
- (8).Reusser F J Bacteriol 1969, 100, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Miyairi N; Takashima M; Shimizu K; Sakai H J Antibiot (Tokyo) 1966, 19, 56. [PubMed] [Google Scholar]
- (10).Liang H; Zhou GF; Ge YH; D’Ambrosio EA; Eidem TM; Blanchard C; Shehatou C; Chatare VK; Dunman PM; Valentine AM; Voelz VA; Grimes CL; Andrade RB Bioorg. Med. Chem 2018, 26, 3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tanner D; Somfai P Tetrahedron 1987, 43, 4395. [Google Scholar]
- (12).Chatare VKA, R. B Angew. Chem. Int. Ed 2017, 56, 5909. [DOI] [PubMed] [Google Scholar]
- (13).Bergy ME; Hoeksema H; Johnson LRE; Kinch DG; Google Patents: 1972.
- (14).Wright PM; Seiple IB; Myers AG Angew. Chem. Int. Ed 2014, 53, 8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Slechta L; Cialdella J; Hoeksema H J. Antibiot 1978, 31, 319. [DOI] [PubMed] [Google Scholar]
- (16).Terekhova LP; Galatenko OA; Kulyaeva VV; Malkina ND; Boikova YV; Katrukha GS; Shashkov AS; Gerbst AG; Nifantiev NE Russ. Chem. Bull 2007, 56, 815. [Google Scholar]
- (17).Johnson TC; Chin MR; Han TX; Shen JP; Rana T; Siegel D J. Am. Chem. Soc 2016, 138, 6068. [DOI] [PubMed] [Google Scholar]
- (18).Thomas RC; Chidester CG J. Antibiot 1982, 35, 1658. [DOI] [PubMed] [Google Scholar]
- (19).Shah P; Westwell AD J Enzyme Inhib Med Chem 2007, 22, 527. [DOI] [PubMed] [Google Scholar]
- (20).Kolb HC; Sharpless KB Drug Discov. Today 2003, 8, 1128. [DOI] [PubMed] [Google Scholar]
- (21).Wright MH; Sieber SA Nat. Prod. Rep 2016, 33, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gersch M; Kreuzer J; Sieber SA Nat. Prod. Rep 2012, 29, 659. [DOI] [PubMed] [Google Scholar]
- (23).Gunnes S; Undheim K Bioorg. Med. Chem 2007, 15, 119. [DOI] [PubMed] [Google Scholar]
- (24).Suchodolski JS; Dowd SE; Westermarck E; Steiner JM; Wolcott RD; Spillmann T; Harmoinen JA BMC Microbiol 2009, 9, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Torikai K; Suga H J. Am. Chem. Soc 2014, 136, 17359. [DOI] [PubMed] [Google Scholar]
- (26).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Nature 2017, 545, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Woods JR; Mo H; Bieberich AA; Alavanja T; Colby DA J. Med. Chem 2011, 54, 7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Woods JR; Mo H; Bieberich AA; Alavanja T; Colby DA MedChemComm 2013, 4, 27. [Google Scholar]
- (29).Sunazuka T; Obata R; Zhuorong L; Takamatsu S; Komiyama K; Omura S; Smith AB Tetrahedron Lett 1994, 35, 2635. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.