

Abstract
Lung fibrosis and tissue remodeling are features of chronic diseases such as severe asthma, idiopathic pulmonary fibrosis and systemic sclerosis. However, fibrosis-targeted therapies are currently limited. We demonstrate in mouse models of allergen and bleomycin-driven airway inflammation that neutralization of the TNF family cytokine TL1A, through antibody blocking or genetic deletion of its receptor DR3, restricted increases in peribronchial smooth muscle mass and accumulation of lung collagen, primary features of remodeling. TL1A was found as a soluble molecule in the airways, and expressed on the surface of alveolar macrophages, dendritic cells, ILC2, and subpopulations of lung structural cells. DR3 was found on CD4 T cells, ILC2, macrophages, fibroblasts and some epithelial cells. Suggesting in part a direct activity on lung structural cells, administration of recombinant TL1A into the naive mouse airways drove remodeling in the absence of other inflammatory stimuli, innate lymphoid cells and adaptive immunity. Correspondingly, human lung fibroblasts and bronchial epithelial cells were found to express DR3, and responded to TL1A by proliferating and/or producing fibrotic molecules such as collagen and periostin. Reagents that disrupt the interaction of TL1A with DR3 then have the potential to prevent deregulated tissue cell activity in lung diseases that involve fibrosis and remodeling.
Introduction
Severe asthma, idiopathic pulmonary fibrosis (IPF) and systemic sclerosis (SSc; scleroderma with pulmonary fibrosis) are examples of chronic inflammatory disorders characterized by a severe fibrotic and remodeling component of the lungs (1-3). This includes production of extracellular matrix proteins, such as collagen, by lung epithelial cells and fibroblasts, along with proteins such as periostin that can act with collagen to enhance fibrotic activity. Additional consequences are hyperplasia or hypertrophy of smooth muscle cells, and the excessive accumulation of myofibroblasts that express alpha smooth muscle actin and have smooth muscle cell characteristics (1, 4). An increased understanding of the molecules that can drive lung fibrosis and remodeling is essential for elucidating new targets for therapies that may halt or reverse these diseases.
One molecule of interest is the cytokine TL1A, an inducible TNF family protein, that can be made by dendritic cells, macrophages, T cells, and endothelial cells, and that acts through the cell surface receptor DR3 (5). TL1A plays a strong role in orchestrating inflammation in the intestine through activities on T cells and innate lymphoid cells (6-8). Moreover, TL1A has been associated with acute lung inflammation, in that it has been shown to stimulate cytokine production from Th2 cells and innate lymphoid type 2 cells (ILC2) in mouse models with OVA and papain (9-11). Although DR3 has been thought to be primarily expressed on T lymphocytes and innate lymphoid cells (12), some reports have found DR3 on human kidney tubular epithelial cells during renal allograft rejection (13), cells in the mesothelial layer of the peritoneal cavity after bacterial infection (14), and mouse intestinal myofibroblasts (15). Additionally, in the latter study, a reduced number of myofibroblasts was associated with the ability of blocking TL1A to limit fibrosis in the colon driven by DSS or adoptive transfer of CD4 T cells (15). This directly implies that TL1A might be strongly relevant for stimulating tissue cells in other organs where remodeling takes place. In this paper, we show that TL1A is central to fibrosis and remodeling of the lung using mouse models driven by allergen or bleomycin exposure, and that TL1A exhibits remodeling relevant activities in lung fibroblasts and bronchial epithelial cells that express DR3.
Materials and Methods
Mice
Six- to 8-week-old female DR3-deficient mice and WT littermates, derived by Taconic Biosciences (#TF3529), were bred in-house on the C57BL/6 × 129 background. WT C57BL/6 mice were purchased from Jackson Laboratories (#000664). RAG2-deficient mice and RAG2-γc-deficient mice on the BALB/c x 129 background were purchased from Jackson Laboratories (#014593). All experiments complied with the regulations of the La Jolla Institute for Immunology Animal Care Committee.
Experimental lung remodeling protocols
Bleomycin model: Mice were challenged with bleomycin (Sigma, 0.2U/mouse), given intratracheally once as in prior studies (16, 17). Analyses were performed after 7 days. For neutralization, 100 μg of DR3.Fc or control IgG was given i.v. one day prior to injection of bleomycin and every other day until the end of the experiment.
Allergen model: Mice were sensitized intranasally (i.n.) on day 0, 7, and 14 with 200 and 100 μg house dust mite extract protein (HDM; GREER Labs Inc, North Carolina), followed by chronic i.n. challenges of 50 μg of HDM protein administered twice a week for the following 4 weeks as previously described (16). Analyses were performed 24 hours after the last challenge. Mouse DR3.Fc (made in-house by Kyowa Kirin, La Jolla, CA) or isotype control IgG were administered i.p. after the initial sensitization period starting at day 14, and were given every 3 days until the end of the experiment (100 μg/injection/mouse).
Activity of recombinant protein: Mice were given 10 μg of recombinant mouse TL1A (produced in-house by Kyowa Kirin) or PBS intratracheally on days 1 and 2, and sacrificed for analyses one day later on day 3.
Lung analyses
Lung sections were stained as in previous studes (16, 17). For collagen, Masson’s trichrome blue was used and scored with defined areas of interest (AOI) created throughout the entire lung sections, including alveolar and peribronchial regions, to quantify blue color using Image-Pro Plus. Total lung collagen was also measured using a hydroxyproline kit (Sigma, MAK008) or a sircol assay (Biocolor, S1000). For smooth muscle mass, sections were stained with antibody to alpha smooth muscle actin (clone 1A4, ab7817; Abcam, Cambridge, MA, 1:200) and rhodamine Red-X AffiniPure Donkey anti-rabbit (711295152; Jackson Immunoresearch, West Grove, PA, 1:500). Peribronchial smooth muscle mass was evaluated using Image-Pro Plus from multiple random bronchi throughout the lungs. A similar protocol was used with an antibody to periostin (clone ab14041; Abcam) or with H&E. All images, unless indicated, were captured with the Zeiss Scanner 20x objective. Soluble TL1A in BAL fluid was assessed by Western blot using mDR3-Fc and anti-IgG/HRP (109-035-003, Jackson ImmunoResearch).
Flow cytometry
BAL and lung cells were treated with red blood cell lysing buffer (Sigma). Lungs were dissociated using a Lung Dissociation Kit (Miltenyi Biotec) and Gentle MACS (Miltenyi Biotec). Live/Dead cells were stained with Fixable Aqua Dead Cell Staining Kit (Thermo Fisher), and after Fc block with the 2.4G2mAb (eBioscience), cells were stained with the following antibodies: DR3-PE (4C12, BioLegend), TL1A-PerCPef710 (Tandys1a, eBioscience), CD45-V500 (30-F11, BioLegend), SiglecF-BV605 (E50-2440, BD Biosciences), CD11b-APC-Cy7 (M1/70, BD Biosciences), CD11c-PE-Cy7 (HL3, BD Biosciences), Ly6G-Alexa700 (1A8, BioLegend), MHCII-APC (M5/114.15.2, BioLegend), CD4-BV570 (RM4-5, BioLegend), CD8a-PEeFluor610 (53-6.7, eBioscience), B220-BV785 (RA3-6B2, BioLegend), CD127-BV785 (A7R34, BioLegend), ST2-BV605 (DIH9, BioLegend), CD90.2-PECy7 (30-H12, BioLegend), Epcam-PECy7 (G8.8, BioLegend), CD31-BV785 (390, BioLegend). For lineage markers for ILC2 staining: CD3-FITC (145-2C11, eBioscience), CD4-FITC (GK1.5, eBioscience), CD8-FITC (5H-10-1, BioLegend), CD19-FITC (1D3, BioLegend), NK1.1-FITC (PK136, BioLegend), CD11b-FITC (M1/70, BioLegend), CD11c-FITC (HL3, BD Bioscience), Gr1-FITC (RB6-8C5,BioLegend). After fixation and permeabilization using with Foxp3/Transcription Factor Staining Buffer Set (eBioscience), cells were stained intracellularly with Vimentin-APC (280618, R&D). Flow cytometry analysis was performed on a Fortessa (BD Biosciences) and data were analyzed using FlowJo Software (version 10, FlowJo, LLC, Ashland, OR). Live CD45+ lung immune cells were separated into CD4+ T cells (CD3+, CD4+), B cells (CD3-B220+CD11c-), alveolar macrophages (CD11b-CD11c+SiglecF+), DCs (CD11c+MHCII+), and ILC2 (Lin-CD127+CD90.2+ST2+). Live CD45- lung structural cells were separated by staining with Epcam, CD31, CD90.2 and Vimentin.
Stimulation of structural cells
Normal human lung fibroblasts (NHLF, from Lonza, Walkersville, MD) and human bronchial epithelial cells (BEAS-2B cell line, from ATCC), were stimulated with 100 ng/ml of recombinant human TL1A, a dose predetermined to produce maximal response (made in-house by Kyowa Kirin), or varying doses of human recombinant TGF-β (R&D Systems, Minneapolis, MN), for 72h in Epilife media (Life Technologies/Thermo Fisher Scientific, Carlsbad, CA). DR3 was visualized using anti-human DR3 (clone JD3, Biolegend, San Diego, CA). Periostin and collagen protein were visualized with anti-human periostin (clone ABT292 from Millipore) or anti-human collagen (clone MAB4165 from R&D). Proliferation was studied by uptake of 0.5 μCi [3H]-TdR (PerkinElmer, Waltham, MA) added 16 hours before the end of the culture. PCR was performed as previously described (16, 17).
Patient human lung fibroblasts
Fibroblasts were obtained from normal, SSc with pulmonary fibrosis, or IPF patients undergoing lung transplantation at the University of Pittsburgh Medical Center and from donors whose lungs were not used for transplantation as previously described (18). Samples were analyzed for expression of mRNA for DR3 by PCR.
Quantitative real-time PCR
Total RNA was isolated from lungs or in vitro stimulated cells using TRIzol (Invitrogen). RNeasy Fibrous Tissue mini kit (74704; Qiagen Inc, Valencia, CA) was used to further purify RNA from lung samples. Single-strand cDNA was prepared by reverse transcribing 5 μg of total RNA using Transcriptor First Strand cDNA kit (Roche Diagnostics, Indianapolis, IN). Samples were amplified in IQ SYRB Green Supermix (Bio-Rad Laboratories, Hercules, CA) using the primer pairs: collagen, huCollagen, forward 5’-CCT CAA GGG CTC CAA C-3’, reverse, 5’- GGT TTT GTA TTC AAT CAC TGT CTT GC-3’; muCollagen, forward, 5’-GAG CCC TCG CTT CCG TAC TC-3’, reverse, 5’- TGT TCC CTA CTC AGC CGT CTG-3’; huPeriostin: forward, 5’-GAC TCA AGA TGA TTC CCT TT-3’; reverse, 5’-GGT GCA AAG TAA GTG AAG G-3’; muPeriostin, forward, 5’-CCC TCC AGC AAA TTC TGG GCA CCA-3’; reverse, 5’-GAG ACT CAC GTT TTC TTC CCG CAG A-3’; hu alpha smooth muscle actin, forward 5’-CTG GCA TCG TGC TGG ACT CT-3’, reverse, 5’-GATCTCGGCCAGCCAGATC-3’; mu aSMA, forward, 5’-TCT CTA TGC TAA CAA CGT CCT GTCA-3’, reverse, 5’-CCA CCG ATC CAG ACA GAG TAC TT-3’; huDR3, forward, 5’-AAG GCG AAG AAG CAC GAA CGA ATG-3’, reverse, 5’-ACT CCG GCC GAG AAG TTG AGA AAT-3’. All samples were run in triplicate, and the mean values were used for quantification.
Statistical analyses
One-way ANOVA or non-parametric Mann-Whitney U test was used where indicated. A P value < 0.05 was considered statistically significant (*<0.05, **<0.01, ***<0.005, ****<0.001).
Results
TL1A injection into the lungs promotes fibrosis and remodeling
We previously found that one member of the TNF superfamily, LIGHT (TNFSF14), induced fibrosis and tissue remodeling when administered directly to the mouse lungs (16, 17). To assess if another TNF-like protein might also be relevant for lung remodeling, we injected recombinant TL1A (TNFSF15) into the airways of naïve wild-type mice. Two intratracheal injections of rTL1A, given over three days, induced primary remodeling features, namely accumulation of collagen, based on the extent of Masson’s trichrome blue stain, around the bronchioles and in the alveolar regions, and an increase in smooth muscle mass, based on alpha smooth muscle actin staining, around the bronchioles (Fig. 1a). The effect on lung remodeling was generally quicker than has been reported for remodeling in other systems, for example with IL-13 transgenic mice, but is in line with the rapidity with which we found LIGHT could induce a similar lung remodeling response (16, 17), and was likely due to the high dose of the TL1A protein used and that its receptor was readily available on relevant cells to signal for these activities. Because T cells and innate lymphoid cells express DR3, we asked whether these cells were necessary for the remodeling activity of rTL1A, and found that the effect was still evident in RAG2-deficient and RAG2γc-deficient mice that lack one or both of these cell populations (Fig. 1b and c). Showing specificity, rTL1A failed to induce fibrosis and remodeling in mice lacking DR3 (Fig. 1d). Thus, TL1A exerts pro-fibrotic and remodeling activity in the lungs through DR3, independently of T cells and ILC.
Figure 1. TL1A promotes lung fibrosis and remodeling.

Mice were injected i.t. with rTL1A or PBS on two consecutive days and analyzed 24h later. a) WT mice, b) RAG2−/− mice, c) RAG2γc−/− mice, or d) WT vs. DR3−/− mice. Lung sections stained for collagen (Masson’s trichome blue) and alpha smooth muscle actin (red). Dotted lines indicate the bronchus lumen. Sections quantified for collagen deposition (Trichrome score) and smooth muscle mass (aSMA ratio). Data representative of, or means ± s.e.m from, 10 to 20 mice, from 2-3 experiments.
Inhibiting DR3 reduces fibrosis and lung tissue remodeling induced by bleomycin
To determine if the physiological production of TL1A played any role in lung tissue remodeling, we first used intratracheal administration of the antibiotic bleomycin at high dose. We have previously shown that this also rapidly induces upregulation of lung collagen and increases in peribronchial smooth muscle mass over a one week period (17), as opposed to other models where lower doses of bleomycin generally only drive accumulation of lung collagen after 2-3 weeks. In line with our studies of recombinant TL1A, genetic deletion of DR3 significantly attenuated increases in collagen deposition and lung smooth muscle mass (Fig. 2a). Inflammatory lung infiltrates were also affected to an extent in mice lacking DR3 with a trend toward a reduction in specific lung cell populations seen with flow cytometry analyses, albeit not statistically significant (Supplementary Figure 1a). Showing the reduced remodeling response was not due to a developmental defect in the knockout animals or the background of these mice, TL1A blocking with an Fc fusion protein of DR3 (DR3.Fc) in wild-type BL/6 mice replicated the effect and inhibited fibrosis and increases in smooth muscle mass (Fig. 2b). The reduction in collagen deposition observed by scoring the extent of trichrome staining was confirmed with assays of soluble (sircol score) and total collagen (hydroxyproline score) extracted from the whole lungs (Fig. 2c).
Figure 2. Inhibiting DR3 reduces bleomycin-induced lung fibrosis.

a) DR3−/− or WT littermates (C57BL/6 x 129), or b) WT C57BL/6 mice given 100 μg of DR3.Fc or control IgG, were challenged i.t. with bleomycin, and analyzed 7 days later. Lung sections were stained and scored for collagen and alpha smooth muscle actin as in Fig. 1. Data representative of, or means ± s.e.m from, 10 to 20 mice, from 2-3 experiments. c) Trichome score (left) vs. hydroxyproline (middle) and sircol (right) assays for collagen content in the lungs of bleomycin treated WT vs. DR3−/− mice. Data means ± s.e.m from 6 mice. Dotted lines, values in control WT mice not given bleomycin.
TL1A-DR3 neutralization limits allergen-induced lung fibrosis and tissue remodeling
To further implicate TL1A and DR3 as being strong regulators of lung fibrotic activity, we used an allergen-driven model (16) where mice were sensitized and chronically challenged with house dust mite extract (HDM) given intranasally. We have previously shown this protocol also results in fibrosis in the lungs with significant remodeling of the smooth muscle mass around the bronchioles. In mice lacking DR3, a strong decrease in fibrosis was visualized in lung tissue (trichrome blue stain for collagen deposition), along with reduced remodeling (alpha smooth muscle actin staining around the bronchioles) (Fig. 3a). We then tested whether blocking TL1A-DR3 signaling could produce a similar reduction in remodeling features, and specifically if the intervention was performed after the initial Th2 response that characterizes this model had developed (16). Treatment of WT mice with DR3.Fc given starting after day 14 also demonstrated less fibrosis and remodeling in the lungs with a significant decrease in collagen deposition and smooth muscle mass (Fig. 3b). Both CD4+ T cells and eosinophils were significantly reduced in the lungs of HDM-challenged DR3-deficient mice while DR3-Fc-treated mice showed a moderate decrease in the CD4+ T cell and dendritic cell infiltrates but no difference in eosinophilia (Supplementary Figure 1b). These results show that endogenous TL1A-DR3 signaling is central to the development of lung fibrosis and tissue remodeling downstream of two diverse stimulants, bleomycin and house dust mite allergen.
Figure 3. TL1A-DR3 neutralization limits allergen-induced lung tissue remodeling.

a) WT or DR3−/− mice, or b) WT mice administered DR3.Fc or control IgG starting on day 14, were sensitized and challenged i.n. with HDM. After 42 days, lung sections were stained and scored for collagen and alpha smooth muscle actin as in Fig. 1. Data representative of, or means ± s.e.m from, 10 to 20 mice, from 2-3 experiments.
TL1A and DR3 are expressed in immune cells and lung structural cells
TL1A can be expressed on the membrane of cells and also can be cleaved from the membrane and act as a soluble cytokine, similar to TNF (19-21). Our data with recombinant TL1A (Fig. 1) suggested the soluble molecule could be most relevant, and a recent report found elevated levels of soluble TL1A in sputum from severe asthmatics, a population of patients most likely to display lung fibrosis and airway remodeling (22). In line with this, we detected soluble TL1A in the bronchoalveolar fluid of mice with lung remodeling (Fig. 4a). To understand which cells might produce TL1A, we stained both immune lung infiltrates and structural cells for membrane TL1A (Fig. 4b). TL1A was expressed in the naive lung on a subpopulation of epithelial cells (CD31-Epcam+) and on CD31+Epcam- cells, the latter likely a mixture of endothelial cells and pericytes (see Supplementary Fig. 1c, for gating). In mice challenged with bleomycin or HDM, this expression was lost, perhaps reflecting production of the soluble molecule. In addition, membrane TL1A was found after challenge with either bleomycin or HDM on subpopulations of alveolar macrophages, ILC2, and vimentin+CD90- cells that might represent smooth muscle cells. Not surprisingly, based on prior reports, DR3 was found constitutively expressed on CD4 T cells and ILC2 in naive lungs and those receiving bleomycin and HDM (Fig. 4c). DR3 was also expressed on alveolar macrophages in all situations, and on DC after HDM challenge. Most interestingly, DR3 was strongly expressed on vimentin+CD90+ (CD31-Epcam-) cells, a phenotype consistent with fibroblasts. We also observed DR3 on a small population of epithelial cells (Epcam+CD31-), but as our recovery of epithelial cells from the lungs was poor (Supplementary Fig. 1c), this might have been an underestimate of the expression of DR3 on these cells. Lastly, we examined human fibroblasts for expression of DR3 isolated from the lungs of normal individuals, or patients with systemic sclerosis (SSc) with pulmonary fibrosis and patients with idiopathic pulmonary fibrosis (IPF). Significantly, all samples expressed mRNA for DR3 at high levels (Fig. 4d) correlating with the mouse data.
Figure 4. TL1A and DR3 are expressed in immune and structural cells in the lungs.
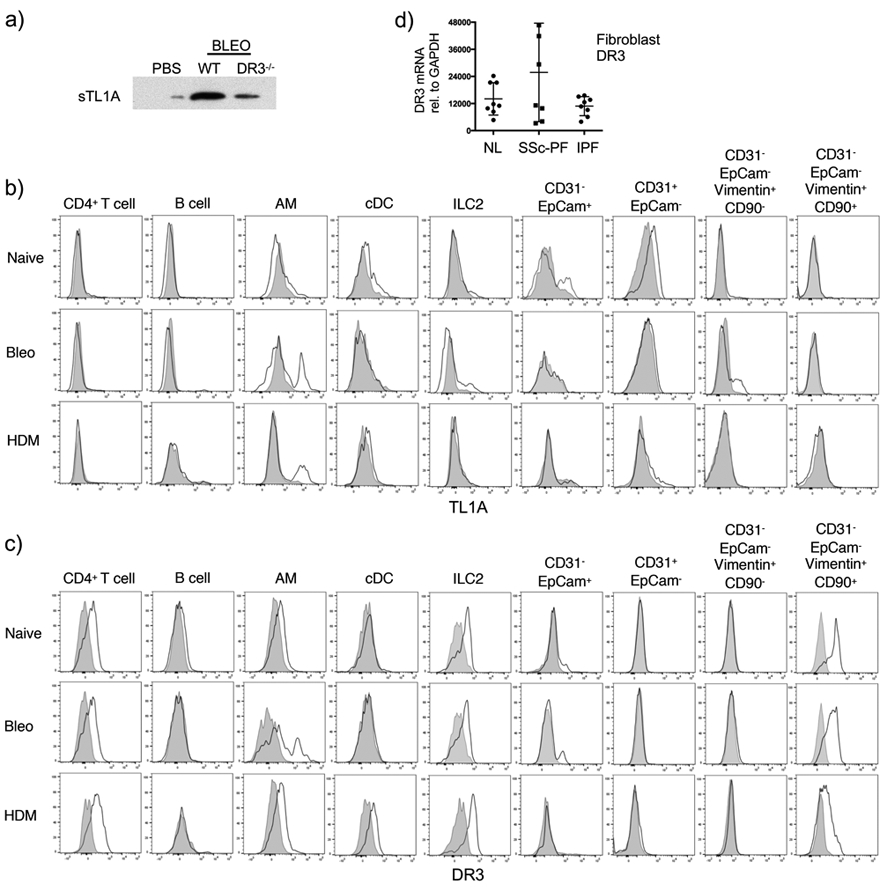
a) Soluble TL1A expression in BAL fluid of wild-type and DR3-deficient mice at day 7 post-bleomycin challenge. b-c) Lung cells from naive, 7-day bleomycin-challenged, or 14-day HDM-challenged wild-type mice were stained for membrane TL1A (b) or DR3 (c) on the indicated populations. d) DR3 mRNA expression in fibroblasts isolated from lung tissue from 8 SSc and 8 IPF patients compared to 7 healthy controls.
TL1A can act directly on lung fibroblasts
As rTL1A rapidly upregulated collagen and alpha smooth muscle actin expression in the lungs in the absence of DR3-expressing T cells and ILC2 (Fig. 1), and these are products of fibroblasts and/or smooth muscle cells, this suggested that TL1A might partly have functioned via a direct action on lung fibroblasts. This was supported by finding DR3 expression on fibroblasts, but not on putative smooth muscle cells (vimentin+CD90-) in the mouse lung. We tested if this was possible using commercially purchased normal human lung fibroblasts (NHLF) which were also found to have significant expression of surface DR3 (Fig. 5a). The division of fibroblasts can lead to their excessive accumulation in the lung tissue where they produce extracellular matrix proteins. Moreover, fibroblasts expressing alpha smooth muscle actin (myofibroblasts), in addition to mature smooth muscle cells, can contribute to the smooth muscle mass surrounding the bronchioles in patients with asthma and systemic sclerosis (23-25). We then first asked if recombinant human TL1A could promote proliferation of fibroblasts and differentiation into a myofibroblast. rTL1A induced division of NHLF, leading to an approximate doubling of the basal level of proliferation of these cells (Fig. 5b), but it did not induce alpha smooth muscle actin expression in isolation (Fig. 5c). Because TGF-β, a cytokine ubiquitously expressed by many cells in the lungs, is one of the primary factors that can enhance myofibroblast differentiation (26), we tested whether TL1A acted together with TGF-β to promote either division or differentiation of lung fibroblasts. TGF-β alone did not induce proliferation but strongly upregulated alpha smooth muscle actin, as previously reported by many groups in the literature (Fig. 5b, c). However, TGF-β in a dose dependent fashion significantly enhanced the extent of division induced by TL1A (Fig. 5b). Interestingly, TL1A also moderately augmented the induction of alpha smooth muscle actin by TGF-β (Fig. 5c). Lastly, we asked whether TL1A could promote production of extracellular matrix proteins by lung fibroblasts, and found strong induction of collagen, the cardinal feature of lung fibrosis (Fig. 5d). Collectively, these data show that TL1A can directly drive remodeling-relevant activity in lung fibroblasts, and it can work together with TGF-β to enhance the accumulation of myofibroblasts that make collagen, corresponding in part to the requirement of TL1A in contributing to fibrosis and smooth muscle mass observed in the mouse studies.
Figure 5. TL1A promotes hyperplasia and extracellular matrix protein production in lung fibroblasts.

a) Normal human lung fibroblast (NHLF) expression of DR3 (blue) compared to isotype control (red). b) NHLF were cultured with PBS, rTL1A, or rTGF-β alone, or rTL1A and increasing doses of rTGF-β (ng/ml as indicated). Proliferation was assessed after 72hr. Data are means ± s.e.m from 3 replicate cultures. c) NHLF were cultured as in (b) and alpha smooth muscle actin mRNA was assessed after 72hr. Data are from 3 individual replicate cultures. d) NHLF were cultured with PBS or rTL1A, and collagen mRNA and protein expression (red IF staining) analyzed after 72hr (blue, DAPI). Data are from 3 individual replicate cultures. All results representative of 3 experiments.
DR3 activity in lung fibroblasts and epithelial cells promotes periostin expression
Next, we asked whether TL1A could regulate the expression of periostin, a protein that has recently emerged as a potential marker of fibrosis and tissue remodeling associated with steroid-resistant asthma, IPF, and SSc in humans, and that is thought to contribute to lung dysfunction (27-30). Periostin is primarily expressed by fibroblasts and epithelial cells, and has been described to possess a number of activities including acting as a scaffold by binding to ECM proteins like collagen, as well as synergizing with TGF-β to enhance collagen deposition or chemokine production that further contributes to tissue remodeling (27, 31, 32). Significantly, we found that periostin expression was upregulated in human lung fibroblasts after stimulation with TL1A in vitro (Fig. 6a). We additionally tested the human bronchial epithelial cell line BEAS-2B, and found that DR3 was expressed well in these cells. Moreover, they also responded to TL1A by making periostin (Fig. 6b).
Figure 6. TL1A promotes periostin expression in lung fibroblasts and bronchial epithelial cells in vitro.

a) Periostin mRNA and protein expression (green IF staining) in NHLF cultured with PBS or rTL1A. Data from 3 individual replicate cultures. b) DR3 expression (blue) compared to isotype control (red) in human bronchial epithelial cells. Periostin mRNA and protein expression after culture with PBS or rTL1A. Data are from 3 individual replicate cultures.
TL1A regulates periostin production in the lungs
Finally, we then assessed whether TL1A was upstream of periostin production in vivo, to provide additional evidence of the relevance of the prior in vitro findings in lung fibroblasts and bronchial epithelial cells, and as a further feature linking TL1A activity to lung tissue fibrotic activity. The in vivo administration of rTL1A to the airways of naïve mice upregulated periostin protein expression in the bronchial epithelium and in cells in the parenchyma as shown by immunofluorescence (Fig. 7a). Confirming this, an increase in periostin mRNA was also induced in the lungs. Most importantly, when the TL1A-DR3 interaction was inhibited by ligand blocking in the allergen-induced model, we saw an almost complete abrogation of periostin protein expression in both the bronchial epithelium and parenchyma, and correspondingly mRNA expression in the lungs of these mice was reduced (Fig. 7b). These results further the contention that TL1A-DR3 signaling is a major driver of lung fibrosis and tissue remodeling.
Figure 7. TL1A controls lung periostin production in vivo.

Periostin mRNA, and periostin protein immunofluorescent staining (green), in relation to aSMA (red), in a) lungs of WT mice injected i.t. with PBS or rTL1A as in Fig. 1, or (b) lungs of WT mice challenged with HDM and treated with DR3.Fc or IgG as in Fig. 3. Results representative of, or means ± sem from, 3 experiments.
Discussion
Lung fibrosis and tissue remodeling are common features of many chronic inflammatory disorders such as asthma, IPF, and SSc, but knowledge of molecules that promote these responses is still limited. In this study, we reveal a new role for TL1A-DR3 signaling in contributing to lung fibrosis and remodeling, potentially independent of its activity in enhancing adaptive immunity that has been reported previously in the literature. We elucidate new functional activities of TL1A in lung structural cells, demonstrating significant effects on proliferation, collagen production, or periostin expression, in fibroblasts or myofibroblasts and bronchial epithelial cells, all phenotypes that were strongly reduced when DR3 signaling was abrogated in mouse models of lung fibrosis. Our data highlight an unappreciated role of TL1A/DR3 signaling as a central driver of lung fibrotic activity downstream of several inflammatory insults, and suggest that TL1A and DR3 could be targets for therapeutics aimed at reducing fibrosis and tissue remodeling in humans with severe lung disease.
TL1A could potentially contribute to lung tissue deregulation in both indirect and direct manners. It has previously been linked with induction of acute lung inflammation from experiments showing reduced eosinophilia and production of the Th2 cytokines IL-4, IL-5, IL-13, and IL-9 in the lungs of TL1A-deficient mice in models driven by ovalbumin and papain. DR3 was found to be expressed by Th2 cells and type 2 innate lymphoid cells, and was shown to either enhance the expansion of these cells or their ability to secrete Th2 cytokines (9-11). By contributing to a Th2-dominant environment, TL1A could therefore be instrumental in generating the initial response that creates an environment favorable to fibrosis and tissue remodeling in, for example, asthmatics that progress from moderate to severe. Our results now add to this literature, but highlight a potential downstream role of TL1A independent of the Th2/ILC2 response, whereby TL1A may provide inflammatory signals to lung structural cells that directly rather than indirectly drive the fibrotic and remodeling response. Indeed, we found that DR3 was expressed in mouse and human lung fibroblasts and bronchial epithelial cells, and TL1A induced several remodeling-related activities in these cells, including their ability to proliferate and/or upregulate collagen and periostin. Importantly, these effects were observed in vitro in the absence of other Th2 factors, and further substantiating this new activity, in vivo injection of rTL1A into the lungs of mice demonstrated that a tissue remodeling phenotype could be induced in the absence of Th2 cells and ILC2. This leads to the contention that if TL1A is expressed and available in the lungs, and DR3 is present on both adaptive immune cells and tissue structural cells, it will be a major contributor to the tissue remodeling and fibrosis that is seen in chronic lung disease. We primarily focused on fibroblasts, and in the mouse lung they constitutively expressed DR3. This was further supported by mRNA data from human lung fibroblasts from normal donors as well as those with SSc and IPF, and by surface expression seen in normal human lung fibroblasts from a commercial source. However, we found a small subset of epithelial cells that were DR3 positive in the mouse, and the human bronchial epithelial cell line, BEAS-2B, also expressed DR3, suggesting that lung epithelial cells could also be a major target of TL1A activity in addition to fibroblasts. It is possible the mouse data were an underestimate of the true expression of DR3 on epithelial cells, as we recovered only small numbers of these cells, or that given 6 or 7 distinct bronchial epithelial subsets have recently been described, DR3 expression might be restricted to only a subpopulation of these cells. Future studies are then needed to further understand the extent of DR3 expression on epithelial cells in the lung and the range of potential activities of TL1A on these cells. Additional questions remain about whether extrinsic factors can positively or negatively modulate the availability of DR3 on the various structural cell types.
The exact source of TL1A that might be crucial to remodeling in the lung is not known, but prior publications have observed TL1A to be made by macrophages, dendritic cells, T cells, and endothelial cells (33-36). Our mouse staining data for membrane TL1A further support multiple possible sources in the lungs, including alveolar macrophages, DC, ILC2, epithelial cells and endothelial cells or pericytes (CD31+Epcam- cells). Another intriguing idea is that TL1A could be a product of the structural cells that are central to tissue remodeling, and it is of interest in future studies to determine if normal lung cells or those from patients with severe lung disease can produce TL1A. This could imply that an autocrine feed-back loop might exist whereby fibroblasts or epithelial cells autonomously drive their own deregulated activity during the later stages of chronic disease that may be independent of adaptive immunity, a notion previously discussed as potentially being central to several diseases, including rheumatoid arthritis as well as SSc and IPF. We observed surface TL1A on a subset of epithelial cells in the naive mouse lung, and on a subset of vimentin+CD90- cells in bleomycin-challenged mice that might have been smooth muscle, although none on fibroblasts, but given that TL1A can be cleaved from the membrane, this data might also be an underestimation of whether lung structural cells could be a primary source of TL1A. In fact, there is some support for this in the literature in that synovial fibroblasts and chondrocytes, and intestinal subepithelial myofibroblasts, were found to be capable of making TL1A after stimulation with TNF or IL-1 (37-39).
Our studies suggest that either TL1A or DR3 are potential targets for therapeutics aimed at limiting the development or persistence of tissue remodeling and fibrosis in severe lung disease, but raise additional questions that will need to be addressed before translational work can be pursued. It is becoming increasingly apparent that patients with severe asthma, SSc and IPF are heterogenous in terms of the extent and type of lung inflammatory response that underlies their clinical symptoms. Understanding whether DR3 or TL1A are differentially expressed in the lung tissue of subsets of these patients will be important, including whether the expression of DR3 in individual patients varies between lymphoid cell types and structural cells of the lung. Lastly, as TL1A can be made as a soluble molecule (19-21), a further question relates to whether soluble TL1A is found in biological fluids of these patients (serum, bronchoalveolar lavage, sputum) and if this can be used as a biomarker to indicate progression of disease or simply patients amenable to therapeutic interventions targeting TL1A and DR3. In this regard, a recent study found soluble TL1A was more strongly expressed in the sputum of a subset of severe eosinophilic asthmatics compared to mild asthmatics after challenge with allergen (22), suggesting this could inform future treatments.
In summary, we reveal new activities of TL1A in structural cells of the lungs that are highly relevant for lung remodeling that is seen in severe diseases such as asthma and SSc, as well as IPF. Using mouse models, we demonstrate that TL1A-DR3 activity is directly or indirectly involved in promoting several cardinal features of these diseases. Given that TL1A has previously been shown to be a regulator of Th2 cells and ILC2 associated with acute lung inflammatory responses, our results further extend the biology of this molecule and suggest it is likely to be a central mediator of both acute and chronic lung inflammation and a realistic target for therapy of lung fibrosis and tissue dysregulation.
Supplementary Material
Key Points.
Disrupting TL1A-DR3 interactions abrogates mouse lung fibrosis and airway remodeling.
Airway fibroblasts and epithelial cells express DR3 and respond to TL1A.
Neutralizing TL1A might be a therapy for airway remodeling in asthma, SSc, and IPF.
Acknowledgements:
We acknowledge LJI Facility cores, DLAC, Microscopy and Flow cytometry cores.
This work was supported by funds from Kyowa Kirin Pharmaceutical Research, Inc. to M.C. and NIH grant AI070535 to M.C. and D.B.
References
- 1.Lekkerkerker AN, Aarbiou J, van Es T, and Janssen RA. 2012. Cellular players in lung fibrosis. Curr Pharm Des 18: 4093–4102. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez IE, and Eickelberg O. 2012. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 380: 680–688. [DOI] [PubMed] [Google Scholar]
- 3.Ho YY, Lagares D, Tager AM, and Kapoor M. 2014. Fibrosis--a lethal component of systemic sclerosis. Nat Rev Rheumatol 10: 390–402. [DOI] [PubMed] [Google Scholar]
- 4.Guida G, and Riccio AM. 2019. Immune induction of airway remodeling. Semin Immunol 46: 101346. [DOI] [PubMed] [Google Scholar]
- 5.Chinnaiyan AM, O’Rourke K, Yu GL, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, and Dixit VM. 1996. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 274: 990–992. [DOI] [PubMed] [Google Scholar]
- 6.Shih DQ, Michelsen KS, Barrett RJ, Biener-Ramanujan E, Gonsky R, Zhang X, and Targan SR. 2011. Insights into TL1A and IBD pathogenesis. Advances in experimental medicine and biology 691: 279–288. [DOI] [PubMed] [Google Scholar]
- 7.Bamias G, Jia LG, and Cominelli F. 2013. The tumor necrosis factor-like cytokine 1A/death receptor 3 cytokine system in intestinal inflammation. Current opinion in gastroenterology 29: 597–602. [DOI] [PubMed] [Google Scholar]
- 8.Siakavellas SI, and Bamias G. 2015. Tumor Necrosis Factor-like Cytokine TL1A and Its Receptors DR3 and DcR3: Important New Factors in Mucosal Homeostasis and Inflammation. Inflamm Bowel Dis 21: 2441–2452. [DOI] [PubMed] [Google Scholar]
- 9.Meylan F, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D, Bundoc V, Hodges M, Shevach EM, Keane-Myers A, Wang EC, and Siegel RM. 2008. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 29: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, Sciume G, Richard AC, Hayes ET, Gomez-Rodriguez J, Chen X, Paul WE, Wynn TA, McKenzie AN, and Siegel RM. 2014. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol 7: 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richard AC, Tan C, Hawley ET, Gomez-Rodriguez J, Goswami R, Yang XP, Cruz AC, Penumetcha P, Hayes ET, Pelletier M, Gabay O, Walsh M, Ferdinand JR, Keane-Myers A, Choi Y, O’Shea JJ, Al-Shamkhani A, Kaplan MH, Gery I, Siegel RM, and Meylan F. 2015. The TNF-Family Ligand TL1A and Its Receptor DR3 Promote T Cell-Mediated Allergic Immunopathology by Enhancing Differentiation and Pathogenicity of IL-9-Producing T Cells. J Immunol 194: 3567–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richard AC, Ferdinand JR, Meylan F, Hayes ET, Gabay O, and Siegel RM. 2015. The TNF-family cytokine TL1A: from lymphocyte costimulator to disease co-conspirator. J Leukoc Biol 98: 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Lamki RS, Wang J, Tolkovsky AM, Bradley JA, Griffin JL, Thiru S, Wang EC, Bolton E, Min W, Moore P, Pober JS, and Bradley JR. 2008. TL1A both promotes and protects from renal inflammation and injury. J Am Soc Nephrol 19: 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perks WV, Singh RK, Jones GW, Twohig JP, Williams AS, Humphreys IR, Taylor PR, Jones SA, and Wang ECY. 2016. Death Receptor 3 Promotes Chemokine-Directed Leukocyte Recruitment in Acute Resolving Inflammation and Is Essential for Pathological Development of Mesothelial Fibrosis in Chronic Disease. Am J Pathol 186: 2813–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, Wallace KL, Chen J, Pothoulakis C, Koon HW, and Targan SR. 2014. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol 7: 1492–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, Zuraw BL, Ware CF, Broide DH, and Croft M. 2011. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nature Medicine 17: 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, and Croft M. 2015. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol 136: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, and Feghali-Bostwick CA. 2011. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum 63: 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cassatella MA, Pereira-da-Silva G, Tinazzi I, Facchetti F, Scapini P, Calzetti F, Tamassia N, Wei P, Nardelli B, Roschke V, Vecchi A, Mantovani A, Bambara LM, Edwards SW, and Carletto A. 2007. Soluble TNF-like cytokine (TL1A) production by immune complexes stimulated monocytes in rheumatoid arthritis. J Immunol 178: 7325–7333. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, and Zhang L. 2005. Identification of naturally secreted soluble form of TL1A, a TNF-like cytokine. J Immunol Methods 298: 1–8. [DOI] [PubMed] [Google Scholar]
- 21.Konsta M, Bamias G, Tektonidou MG, Christopoulos P, Iliopoulos A, and Sfikakis PP. 2013. Increased levels of soluble TNF-like cytokine 1A in ankylosing spondylitis. Rheumatology 52: 448–451. [DOI] [PubMed] [Google Scholar]
- 22.Machida K, Aw M, Salter BM, Ju X, Mukherjee M, Gauvreau GM, O’Byrne PM, Nair P, and Sehmi R. 2020. Role of TL1A/DR3 Axis in the Activation of ILC2s in Eosinophilic Asthmatics. Am J Respir Crit Care Med. [DOI] [PubMed] [Google Scholar]
- 23.Gizycki MJ, Adelroth E, Rogers AV, O’Byrne PM, and Jeffery PK. 1997. Myofibroblast involvement in the allergen-induced late response in mild atopic asthma. Am J Respir Cell Mol Biol 16: 664–673. [DOI] [PubMed] [Google Scholar]
- 24.Brewster CE, Howarth PH, Djukanovic R, Wilson J, Holgate ST, and Roche WR. 1990. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am J Respir Cell Mol Biol 3: 507–511. [DOI] [PubMed] [Google Scholar]
- 25.Larsen K, Malmstrom J, Wildt M, Dahlqvist C, Hansson L, Marko-Varga G, Bjermer L, Scheja A, and Westergren-Thorsson G. 2006. Functional and phenotypical comparison of myofibroblasts derived from biopsies and bronchoalveolar lavage in mild asthma and scleroderma. Respir Res 7: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, and Thomas PE. 2003. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278: 12384–12389. [DOI] [PubMed] [Google Scholar]
- 27.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, and Izuhara K. 2006. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 118: 98–104. [DOI] [PubMed] [Google Scholar]
- 28.Kanemitsu Y, Matsumoto H, Izuhara K, Tohda Y, Kita H, Horiguchi T, Kuwabara K, Tomii K, Otsuka K, Fujimura M, Ohkura N, Tomita K, Yokoyama A, Ohnishi H, Nakano Y, Oguma T, Hozawa S, Nagasaki T, Ito I, Inoue H, Tajiri T, Iwata T, Izuhara Y, Ono J, Ohta S, Tamari M, Hirota T, Yokoyama T, Niimi A, and Mishima M. 2013. Increased periostin associates with greater airflow limitation in patients receiving inhaled corticosteroids. The Journal of allergy and clinical immunology 132: 305–312 e303. [DOI] [PubMed] [Google Scholar]
- 29.Parulekar AD, Atik MA, and Hanania NA. 2014. Periostin, a novel biomarker of TH2-driven asthma. Curr Opin Pulm Med 20: 60–65. [DOI] [PubMed] [Google Scholar]
- 30.Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, Fry CD, White ES, Sisson TH, Tayob N, Carnemolla B, Orecchia P, Flaherty KR, Hershenson MB, Murray S, Martinez FJ, Moore BB, and Investigators C. 2012. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 303: L1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, Muller SJ, and Fahy JV. 2010. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proceedings of the National Academy of Sciences of the United States of America 107: 14170–14175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchida M, Shiraishi H, Ohta S, Arima K, Taniguchi K, Suzuki S, Okamoto M, Ahlfeld SK, Ohshima K, Kato S, Toda S, Sagara H, Aizawa H, Hoshino T, Conway SJ, Hayashi S, and Izuhara K. 2012. Periostin, a matricellular protein, plays a role in the induction of chemokines in pulmonary fibrosis. Am J Respir Cell Mol Biol 46: 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, Cho YH, Ullrich S, Kanakaraj P, Carrell J, Boyd E, Olsen HS, Hu G, Pukac L, Liu D, Ni J, Kim S, Gentz R, Feng P, Moore PA, Ruben SM, and Wei P. 2002. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 16: 479–492. [DOI] [PubMed] [Google Scholar]
- 34.Bamias G, Martin C 3rd, Marini M, Hoang S, Mishina M, Ross WG, Sachedina MA, Friel CM, Mize J, Bickston SJ, Pizarro TT, Wei P, and Cominelli F. 2003. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol 171: 4868–4874. [DOI] [PubMed] [Google Scholar]
- 35.Prehn JL, Mehdizadeh S, Landers CJ, Luo X, Cha SC, Wei P, and Targan SR. 2004. Potential role for TL1A, the new TNF-family member and potent costimulator of IFN-gamma, in mucosal inflammation. Clin Immunol 112: 66–77. [DOI] [PubMed] [Google Scholar]
- 36.Bamias G, Mishina M, Nyce M, Ross WG, Kollias G, Rivera-Nieves J, Pizarro TT, and Cominelli F. 2006. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci U S A 103: 8441–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Wang X, Fahmi H, Wojcik S, Fikes J, Yu Y, Wu J, and Luo H. 2009. Role of TL1A in the pathogenesis of rheumatoid arthritis. J Immunol 183: 5350–5357. [DOI] [PubMed] [Google Scholar]
- 38.Tubak V, Hatarvolgyi E, Krenacs L, Korpos E, Kusz E, Duda E, Monostori E, and Rauch T. 2009. Expression of immunoregulatory tumor necrosis factor-like molecule TL1A in chicken chondrocyte differentiation. Can J Vet Res 73: 34–38. [PMC free article] [PubMed] [Google Scholar]
- 39.Bamias G, Filidou E, Goukos D, Valatas V, Arvanitidis K, Panagopoulou M, Kouklakis G, Daikos GL, Ladas SD, and Kolios G. 2017. Crohn’s disease-associated mucosal factors regulate the expression of TNF-like cytokine 1A and its receptors in primary subepithelial intestinal myofibroblasts and intestinal epithelial cells. Transl Res 180: 118–130 e112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.