

Abstract
Background –
The mesenchymal stem cell (MSC), known to remodel in disease and have an extensive secretome, has recently been isolated from the human heart. However, the effects of normal and diseased cardiac MSCs on myocyte electrophysiology remain unclear. We hypothesize that in disease the inflammatory secretome of cardiac hMSCs remodels and can regulate arrhythmia substrates.
Methods –
Human cardiac MSCs (hMSCs) were isolated from patients with or without heart failure from tissue attached to extracted device leads and from samples taken from explanted/donor hearts. Failing hMSCs or non-failing hMSCs were co-cultured with normal human myocytes (hCM) derived from induced pluripotent stem cells. Using fluorescent indicators, APD, Ca2+ alternans, and spontaneous calcium release (SCR) incidence were determined.
Results –
Failing and non-failing hMSCs from both sources exhibited similar tri-lineage differentiation potential and cell surface marker expression as bone marrow hMSCs. Compared to non-failing hMSCs, failing hMSCs prolonged APD by 24% (p<0.001, n=15), increased Ca2+ alternans by 300% (p<0.001, n=18), and promoted SCR activity (n=14, p <0.013) in hCM. Failing hMSCs exhibited increased secretion of inflammatory cytokines IL-1β (98%, p<0.0001) and IL-6 (460%, p <0.02) compared to non-failing hMSCs. IL-1β or IL-6 in the absence of hMSCs prolonged APD but only IL-6 increased Ca2+ alternans and promoted SCR activity in hCM, replicating the effects of failing hMSCs. In contrast, non-failing hMSCs prevented Ca2+ alternans in hCM during oxidative stress. Finally, non-failing hMSCs exhibited >25 times higher secretion of IGF-1 compared to failing hMSCs. Importantly, IGF-1 supplementation or anti-IL-6 treatment rescued the arrhythmia substrates induced by failing hMSCs.
Conclusions –
We identified device leads as a novel source of cardiac hMSCs. Our findings show that cardiac hMSCs can regulate arrhythmia substrates by remodeling their secretome in disease. Importantly, therapy inhibiting (anti-IL-6) or mimicking (IGF-1) the cardiac hMSC secretome can rescue arrhythmia substrates.
Keywords: electrophysiology, alternans, mesenchymal stem cell, cytokine, COVID-19
Journal Subject Terms: Arrhythmias, Growth Factors/Cytokines, Stem Cells
Graphical Abstract
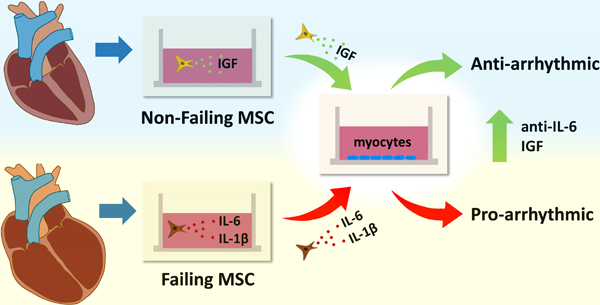
Introduction
The mesenchymal stem cell (MSC) has an extensive secretion profile1, which is consistent with its role as a supportive (i.e. stromal) cell. MSCs can be readily isolated from several sources including bone marrow (BM) and adipose tissue, and they can also originate in the heart by endothelial to mesenchymal transition2. Despite this possibility, there are very few reports of resident MSCs derived from the human heart. Recently, it was shown that MSCs, defined by tri-lineage potential, can be isolated from explanted human hearts and cardiac biopsy samples3–5.
It has been proposed that MSCs are the “sentinel and safe-guards of injury” as demonstrated by their ability to assess injury and then treat damaged tissue6. Consistent with this concept, we have recently shown that BM derived MSCs can suppress cardiac alternans, an important cause of arrhythmia7, in human myocytes by paracrine action8. This result builds upon an earlier study demonstrating that MSCs can enhance SERCA2a function by paracrine activity9, and is in keeping with a subsequent computer simulation showing the benefit of MSC paracrine activity over direct cellular coupling10. However, there are several lines of evidence demonstrating that MSCs remodel in disease11–13 and can secrete inflammatory cytokines1,3,11,12. Among the MSC secretome, IGF-1, IL-1β and IL-6 are suspected to modulate inflammation and have been associated with heart failure14,15. It is unknown if cytokines secreted by cardiac MSCs change in disease. We hypothesize that in disease the inflammatory secretome of cardiac hMSCs remodels and can regulate arrhythmia substrates. An important clinical implication of this study is that targeted anti-inflammatory therapies that are FDA approved for rheumatologic disorders16 and cytokine release syndrome17, and being considered for cardiac disease18,19 or severe COVID-1920, may prove useful as antiarrhythmics.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Isolation of human cardiac MSCs from device leads and human heart samples
Human cardiac MSCs were isolated from device lead tips and explanted/donor hearts. For the device lead tips, human cardiac tissue was carefully separated (pulled or scraped off) from the lead tip under sterile conditions within 1–2 hours after lead extraction (Figure 1). This small amount of tissue was washed with PBS, minced into tiny pieces with scissors, extensively washed with PBS again, and was then digested with 0.2% Collagenase Type 2 (Worthington) for 3 hours in an incubator. Human cardiac tissue from explanted/donor hearts was isolated from the right ventricle RV, in regions without scar, and was maintained in cardioplegia soon after resection. Within 2 hours, it was washed with PBS, minced into tiny pieces, extensively washed with PBS again, and was also digested with 0.2% Collagenase Type 2 for 3 hours in an incubator. The cell isolation and culture protocol are the same after this step for cardiac tissue obtained from leads and explanted/donor hearts. Intermittent vigorous pipetting is done to aid digestion and isolate cells while constantly monitoring under the microscope. Digested tissue with isolated cells was centrifuged, collagenase was removed, and plated onto 35mm plastic dish with media containing F12/DMEM (Gibco), E8, MEM/NEA (Non-essential amino acids), FGF Basic (R&D systems) 20 ng/ml, 20% FBS (Biowest), and Penicillin/Streptomycin. Maintenance media consisted of DMEM Low Glucose (Gibco), MEM/NEA (Non-essential amino acids), FGF Basic (R&D systems) 10 ng/ml, 10% FBS (Biowest), and Penicillin/Streptomycin. This media was replaced every 3–4 days. To enrich for hMSCs, a higher concentration of FBS (20%) was used as others have described previously21, cellular attachment for 2–3 days was utilized, and trypsin selected adherent cells were allowed to proliferate in the isolation medium for 8–10 days. Cells were further proliferated and maintained in the maintenance medium and were typically used between passages 3–5.
Figure 1:

Shown is a myocardial tissue sample obtained from a cardiac device lead extraction procedure as it exists on the tip of the lead after extraction (Panel A) and after removal from the tip (Panel B). In both panels, the arrow points to the tissue sample. Shown at the bottom are cardiac MSCs from device leads from a non-failing heart (Panel C), and cardiac hMSCs from device leads from a failing heart (Panel D) after isolation and plating.
Failing hMSCs were obtained from 6 patients; 2 from leads and 4 from explanted hearts with systolic heart failure (Patient Characteristics Table 1). For non-failing hMSCs, leads from 2 patients without systolic heart failure who underwent lead extraction for lead malfunction and 1 donor heart sample donated for research were used. Patients with a known recent history of infection were excluded. Tissue from the RV of non-failing and failing human heart samples were acquired from the Cleveland Clinic Foundation tissue repository. All protocols were approved by the Cleveland Clinic Foundation Institutional Review Board (IRB no. 2378). Samples were received coded, and no identifying metrics were documented for the study.
Table 1:
Patient Characteristics. ICM – ischemic cardiomyopathy, NICM – non-ischemic cardiomyopathy.
| Group | Age | Sex | EF | Type | Tissue Origin | NYHA Class |
|---|---|---|---|---|---|---|
| Non-Failing | 31 | Male | 60% | NA | RV from Lead | I |
| Non-Failing | 62 | Female | >60% | NA | RV from Lead | I |
| Failing | 70 | Male | 20% | ICM | RV from Lead | III |
| Failing | 59 | Male | 20% | NICM- Sarcoidosis | RV from Lead | II |
| Non-Failing | 65 | Male | >60% | NA | RV from Explant | I |
| Failing | 63 | Female | 21% | NICM-Myocarditis | RV from Explant | II |
| Failing | 49 | Male | 15% | ICM | RV from Explant | III |
| Failing | 64 | Male | 23% | ICM | RV from Explant | IV |
| Failing | 43 | Male | 25% | ICM | RV from Explant | III |
Human myocytes derived from induced pluripotent stem cells
Human cardiac myocytes derived from induced pluripotent stem cells (hCM) were purchased from Cellular Dynamics Inc. Cell pellets in the cryoprecipitate tube were thawed and cultured as monolayers according to the protocol provided by the manufacturer. Cells were plated onto fibronectin coated 25mm coverslips at cell density of 50 × 104 per coverslip and culture media was changed every 2 days until day 14–20 when experiments were performed.
Differentiating human cardiac MSCs to determine tri-lineage potential
After hMSCs are 70–80% confluent, they are cultured in the adipocyte differentiation media, DM-2 (Zenbio), for 1 weeks, and then adipocyte maintenance media, AM-1 (Zenbio), for 2 weeks per the manufacturer instructions. DM-2 contains insulin, dexamethasone, IBMX, and a PPAR-gamma agonist. AM-1 contains insulin and dexamethasone. Adipose vacuoles can be noted within the hMSCs as early as 1 week into differentiation. Adipocytes are confirmed by staining with Oil Red-O per the protocol in the lipid staining kit, ST-R100 (Zenbio). Osteogenesis is induced by exposing hMSCs that are 70–80% confluent to osteogenesis media for 3 weeks with media changes every 3–4 days. Osteogenesis media contains DMEM High Glucose (Sigma), FBS (Sigma), Dexamethasone (Sigma), Penicillin/Streptomycin, Ascorbic acid 2-phosphate (Wako USA), B-glycerophosphate (Calbiochem), and BMP-2. First evidence of calcium mineralization can be noted 2 weeks into the differentiation. Osteogenesis is confirmed by staining with Alizarin Red S (Sigma). Chondrogenesis is induced by performing micromass pellet culture of hMSCs with chondrogenesis media (StemPro) for 3 weeks with media changes every 3 days per protocol outlined in the Chondrogenesis Differentiation Kit by StemPro. Morphological changes with collagen deposition were noted as early as after 1 week. Chondrogenesis was confirmed by staining with Alcian Blue (Sigma). For all cell groups tested (failing hMSCs, non-failing hMSCs, BM hMSCs, dermal fibroblasts), differentiation media changes and image acquisition were all performed on the same day.
ELISA
IGF-1, IL-1β and IL-6 secretion is quantified using Human ELISA kits (R&D Systems) per standard protocol outlined. Supernatant from cell culture (1ml in 24-well plate and 4 days since last media change with fully confluent monolayer of cells confirmed to have similar number of cells in each group) is used for the ELISA measurements. Standard curves with known concentrations are created for each ELISA experiment, and are used to calculate the concentrations of the measured protein from the optical density measurements. Multiple samples within the same group are measured to ensure reliable results.
Culturing bone marrow MSCs
Human BM MSCs were isolated from BM aspirates and purified at the Case Comprehensive Cancer Center Hematopoietic Biorepository and Cellular Therapy Core using protocol previously described22. BM hMSCs are stored in −80C. For use, they were thawed, cultured, proliferated and maintained with the Stromal Cell Maintenance Medium that contains DMEM Low glucose (Gibco), Non-Essential Amino acids (NEA), 10% FBS (Biowest), and Penicillin/Streptomycin. Cells are used between passages 3–5.
Surface Marker Expression
The MSCs used in this study were further characterized by their surface marker expression. The protocol for characterizing cells isolated from heart samples published by Monsanto et al.3 was used. Live human BM MSCs and cardiac failing and non-failing hMSCs in passages 3–5 were stained using human anti-CD 117 antibody (R&D Systems), human anti-CD90 antibody (Biolegend), human anti-CD105 antibody (Biolegend), human anti-CD45 (Biolegend), human anti-CD133 (R&D Systems). Stained cells were counted using a benchtop flow cytometer (BD Accuri™ C6, BD Biosciences). Gating and analysis were performed using the BD Accuri software.
Co-culture and cytokine supplementation
Monolayers are formed by hCM plated at 50 ×104 cells per 25mm coverslip coated with fibronectin for each functional experiment. hMSCs (passage 3–5) are plated onto the coverslips with hCM at a density of 15 × 104 cells per 25mm coverslip and maintained in hCM media for 2 days prior to functional measurements. For experiments with cytokine supplementation, concentrations used were comparable to that measured with ELISA. Each factor, IGF-1 (50 ng/ml), VEGF (50 ng/ml), IL-1β (40 ng/ml), IL-6 (20 ng/ml), anti-IL6 monoclonal antibody (anti-hIL-6-IgG, Invitrogen, 100 ng/ml) was maintained in culture for 48hrs prior to and during experimentation. For assessing the PI3K signaling pathway, a PI3K inhibitor, Wortmannin (100 nmol/L) was used.
Assessment of electrophysiological substrates
Intracellular Ca2+ and action potentials were measured from monolayers to assess electrophysiological substrates. Alternans measurements and analysis were performed with Ca2+ staining and imaging using Fluo4 (Sigma), as described previously8. Alternans was measured at a pacing period of 800ms at room temperature. Response to oxidative stress was measured by repeating the same measurement 2 minutes after administration of H2O2 (200 µM). Mean % alternans was determined for each monolayer. Action potentials were measured after staining with the voltage sensitive dye FluoVolt (Sigma) at room temperature as described previously23. Action potential durations (APD50 and APD90) were measured at room temperature when cells were paced at a period of 2000ms. Finally, spontaneous calcium release (SCR) activity was assessed immediately after stopping rapid pacing (periods: 1200ms, 1000ms, 800ms, 600ms, 400ms, then stop pacing) post H2O2 administration. The presence of SCRs was identified by an expert blinded to the groups.
For statistical analysis of continuous variables, unpaired student’s t-test was performed; whereas, for categorical variables (e.g. SCR incidence) Fisher’s exact test was used. Differences among 3 or more groups of continuous variables were compared using 1-way ANOVA followed by Bonferroni test for multiple comparison. Statistical analysis was performed using SPSS version 24.0. A p value < 0.05 was considered statistically significant.
Results
Tri-lineage potential was tested for cells isolated from non-failing and failing hearts as well as from BM hMSCs (positive control) and dermal fibroblasts (negative control). Standard differentiation protocols for osteoblasts, adipocytes and chondrocytes were used. As shown in Figure 2, osteoblasts (Alizarin Red), adipocytes (Oil Red), and chondrocytes (Alcian Blue) were readily differentiated from purified BM hMSCs (positive control, left), hMSCs from a non-failing heart (device lead), and hMSCs from a failing heart (device lead), but not from dermal fibroblasts (negative control). Similar tri-lineage potential was observed for samples from explanted/donor hearts. Furthermore, flow cytometry for BM, non-failing, and failing hMSCs demonstrated the absence of CD45+ cells (leukocytes) and a high percentage of CD90+ and CD105+ cells, which are characteristic of hMSCs (Figure 3). Finally, BM, non-failing and failing hMSCs exhibited similar growth curves (Supplement Figure S1). These results suggest that the composition of cells from failing and non-failing hearts either isolated from cardiac device leads and donor hearts contains a significant and similar amount of hMSCs, with very few leukocytes (CD45+) or endothelial cells.
Figure 2:

Examples of osteoblasts (Alizarin Red, top), adipocytes (Oil Red, middle), and chondroblasts (Alcian Blue, bottom) differentiated from plastic adherent cells isolated from the bone marrow (positive control), a device lead extracted from a patient with a normal EF (non-failing hMSC, middle left) and a failing heart (Failing hMSC, middle right), and human dermal fibroblasts (negative control). After 3 weeks, a similar differentiation potential was observed in all groups, except dermal fibroblasts.
Figure 3:

Surface marker expression by flow cytometry for BM (bone marrow, n=5), non-failing (n=3) and failing hMSCs (n=9). All three MSC types had characteristic MSC surface marker expression (positive for CD 90 and CD 105 and negative for CD45). CD 117 and CD 133 surface expression are shown for further characterization.
The effects of failing and non-failing hMSCs on arrhythmia substrates
We first tested the effects of failing and non-failing hMSCs on electrophysiological substrates measured in hCM monolayers under normal conditions. Shown in Figure 4 are the effects on Ca2+ alternans and APD. The traces (Panel A) show that non-failing hMSCs co-cultured with hCM slightly decreased Ca2+ alternans (left) and APD (right) compared to hCM alone. In contrast, failing hMSCs significantly increased Ca2+ alternans and APD compared to hCM alone. Summary data (Panel B) that includes samples from leads (filled circles) and explanted/donor hearts (empty circles) also show that failing hMSCs significantly increase Ca2+ alternans and APD90 compared to hCM alone; however, non-failing hMSCs had little effect because, possibly, Ca2+ alternans was small to begin with. When analyzed separately, the increase in APD50 with failing hMSCs was greater for explanted/donor hearts compared to leads; however, this difference could not account for the significant difference observed between non-failing and failing hMSCs when the samples were pooled. No other differences were observed between leads and explanted/donor hearts. In separate experiments, we found that hMSCs isolated from BM had no significant effect on Ca2+ alternans, APD50, and APD90 compared to hCM alone (Supplement Figure S2).
Figure 4:

Shown are Ca2+ alternans (ALT) and APD measured in hCM under normal conditions when co-cultured with cardiac hMSCs from non-failing hearts (nonFailing hMSCs) and failing hearts (Failing hMSC). Example traces in Panel A show Ca2+ alternans (left) and APD (right). Summary data are shown in Panel B. For the summary data, filled circles represent samples from device leads and empty circles are from explanted/donor hearts. Levels of significance are shown in the figure.
Spontaneous calcium release (SCR) is typically associated with Ca2+ dysregulation and heart failure. SCR activity following rapid pacing was rarely observed in hCM monolayers when cultured alone or when co-cultured under normal conditions with BM hMSCs, non-failing hMSCs, or failing hMSCs. However, in the presence of H2O2, SCR activity was observed when hCM were cultured with failing hMSCs but not with non-failing hMSCs or hCM alone. Shown in Figure 5 are Ca2+ transients measured in an hCM monolayer co-cultured with failing (top) and non-failing (bottom) hMSCs upon termination of rapid pacing. SCR activity (arrows) alone and preceding full Ca2+ transients after rapid pacing were clearly observed only with failing hMSCs. Under conditions of H2O2 no SCR activity was observed in 14 experiments with non-failing hMSCs; however, 7 out of 19 experiments with failing hMSCs exhibited SCR activity (p <0.02).
Figure 5:

Shown are examples of Ca2+ transient recordings from hCM with H2O2 and co-cultured with either failing (top) or non-failing cardiac hMSCs (bottom) during the termination of rapid pacing. Spontaneous calcium release (SCR) activity (arrow) was observed with failing but not non-failing cardiac hMSCs.
The effects of failing and non-failing hMSCs on arrhythmia substrates under oxidative stress
To test if non-failing hMSCs can suppress heightened Ca2+ alternans, as observed in disease, experiments were repeated under oxidative stress conditions. Shown in Figure 6 are Ca2+ alternans and the effect of failing and non-failing hMSCs on hCM monolayers treated with H2O2. In Panel A, the traces show a heightened level of Ca2+ alternans in hCM with H2O2 in the absence of failing or non-failing hMSCs. When failing hMSCs were co-cultured with hCM treated with H2O2, Ca2+ alternans was not further increased. Interestingly, non-failing hMSCs reduced Ca2+ alternans in hCM treated with H2O2, similar to hCM alone and what we have reported previously with BM hMSCs8. Summary data (Panel B) show results from all leads (filled circles) and all explanted/donor heart samples (empty circles). As in the traces, failing hMSCs did not further increase Ca2+ alternans; however, non-failing hMSCs significantly reduced Ca2+ alternans compared to H2O2+hCM. When analyzed separately, the suppression of Ca2+ alternans by non-failing hMSCs from explanted/donor hearts was greater than that with hMSCs from leads; however, this difference could not account for the significant difference observed between failing and non-failing MSCs when samples were pooled. No other differences were observed between leads and explanted/donor hearts. Finally, there was no statistical difference between non-failing hMSCs and hCM without H2O2 (from Figure 4, shown for reference).
Figure 6:

Shown are Ca2+ alternans (ALT) measured under conditions of oxidative stress (H2O2) in hCM alone or when also co-cultured with cardiac hMSCs from a non-failing heart (nonFailing hMSC) and failing heart (Failing hMSC). All conditions include H2O2 and hCM. Example traces are shown in Panel A and summary data in Panel B. For the summary data, filled circles represent samples from device leads and empty circles are from explanted/donor hearts. For reference, hCM in the absence of H2O2 is shown. There was no significant difference between hCM and non-failing hMSCs. Otherwise, levels of significance are shown in the figure.
Cytokines that may explain the effects of failing and non-failing hMSCs
To better understand the mechanisms of arrhythmia substrates induced by failing hMSCs, cytokines associated with MSCs and heart failure24 were investigated. In preliminary studies, several combinations of such cytokines including TNFα, MCP-1, IL-1β, and IL-6 were tested for their effect on Ca2+ alternans and APD in hCM monolayers. Only IL-6 showed a significant effect on Ca2+ alternans, but both IL-6 and IL-1β accounted for a majority of APD prolongation (Supplement Figure S3). Summarized in Figure 7 (Panel A) are the effects of IL-1β or IL-6 on Ca2+ alternans and APD measured in hCM. Also shown are hCM alone and hCM co-cultured with failing hMSCs for reference. Interestingly, IL-1β tended to decrease Ca2+ alternans compared to hCM alone, whereas IL-6 significantly increased Ca2+ alternans. Also, only IL-6 promoted SCRs (2 of 5 experiments, data not shown). This finding is in line with clinical data showing a greater than 6 times higher mortality risk associated with elevated IL-6 in patients with heart failure, much more than other cytokines25. In contrast, both IL-1β and IL-6 increased APD50 and APD90 compared to hCM alone, similar to the effects of failing hMSCs. In separate experiments, secreted levels of IL-1β (Panel B, left) and IL-6 (Panel B, right) were significantly increased only in failing hMSCs compared to BM hMSCs. Furthermore, levels of secreted IL-1β and IL-6 were very low in hCM and, thus, unlikely to explain both the increase in Ca2+ alternans and APD. These results suggest that IL-1β and IL-6 can explain the increase in APD; however, IL-6 seems to be mainly responsible for Ca2+ alternans and increased SCR activity. Furthermore, even though IL-1β increased APD it did not increase Ca2+ alternans, suggesting that Ca2+ alternans is independent of APD prolongation. To further test the effect of IL-6 on Ca2+ alternans and its potential for targeted therapy, failing hMSC co-cultures were treated with anti-IL-6 monoclonal antibody. This treatment completely rescued Ca2+ alternans induced by failing MSCs compared to hCM alone (p=ns, Figure 7, Panel C). Finally, no SCR activity was observed with anti-IL-6 treatment.
Figure 7:

IL-1β and IL-6 effect on arrhythmia substrates. Panel A shows the effect of IL-1β (n=6) and IL-6 (n=5) on average Ca2+ alternans (Ca2+ ALT), APD50 and APD90 when administered to hCM alone. Levels of significance are compared to hCM where * p < 0.001 (for Ca2+ ALT, n=23), * p = 0.002 (for APD50, n=6), * p < 0.02 (for APD90, n=6). Panel B shows ELISA results for IL-1β (left) and IL-6 (right) in separate populations of bone marrow (BM) hMSCs, non-failing cardiac hMSCs, failing cardiac hMSCs, and hCM alone (n=4). Levels of significance are compared to BM hMSC where * p < 0.001 (for IL-1β), * p = 0.008 (for IL-6). Panel C shows the rescue of Ca2+ alternans induced by failing hMSCs (n=18) with anti-IL-6 (n=18) treatment. Levels of significance are compared to hCM (n=12) where * p < 0.001.
Previously we have shown that the suppression of oxidative stress-induced Ca2+ alternans by BM hMSCs could be attributed to activation of the PI3K-Akt pathway8. To further determine the signaling mechanisms involved, we investigated two important PI3K-Akt activators, IGF-1 and VEGF. Summarized in Figure 8 (Panel A) are the average magnitude of Ca2+ alternans when hCM treated with H2O2 were incubated with IGF-1, VEGF, IGF-1+WORT. Ca2+ alternans in hCM, hCM treated with H2O2 alone, and in hCM co-cultured with non-failing hMSCs are shown for reference. We found that IGF-1 restored Ca2+ alternans to control levels (hCM alone) and non-failing hMSCs (p=ns), but VEFG did not. When WORT (an inhibitor of PI3K-Akt pathway) was incubated along with IGF-1, the suppression of Ca2+ alternans was lost. Shown in Figure 8 (Panel B) are levels of secreted IGF-1 measured in BM hMSCs, non-failing hMSCs, failing hMSCs, and hCM. Levels of secreted IGF-1 was lowest in failing hMSCs compared to all other cell populations, which may explain why they were unable to suppress Ca2+ alternans. Finally, we tested if IGF-1 can also suppress arrhythmia substrates induced by failing hMSCs (Figure 8C). Incubating failing hMSCs with IGF-1 markedly decreased Ca2+ alternans compared to failing hMSCs without IGF-1, and no SCR activity (n=7) was noted in the presence of H2O2 after rapid pacing. Interestingly, IGF-1 also rescued Ca2+ alternans when failing hMSCs were treated with H2O2 (data not shown). Furthermore, incubating failing hMSCs with IGF-1 decreased APD50 and APD90 to control levels (hCM alone, p=ns).
Figure 8:

Panel A shows the effect of IGF-1 (n=6) and VEGF (n=5) on average Ca2+ alternans when incubated with hCM alone. Also shown are the effects of IGF-1 when incubated with Wortmannin (WORT), an inhibitor of PI3K (n=5). All conditions include hCM. Levels of significance are * p < 0.006 compared to hCM alone (n=38). Panel B shows ELISA results for IGF-1 in separate populations of bone marrow (BM) hMSCs (n=4), non-failing hMSCs (n=8), failing hMSCs (n=8), and hCM alone (n=4). Levels of significance are * p < 0.0001 compared to BM hMSC and † p < 0.0001 compared to non-failing hMSC. Panel C shows IGF-1 supplementation rescues the failing phenotype induced by failing hMSCs without oxidative stress. Ca2+ ALT (n=15), APD50 (n=6) and APD90 (n=6) are shown for hCM alone, hCM co-cultured with failing cardiac hMSC (n=21), and with the addition of IGF-1 (n=5). Levels of significance are compared to hCM where * p < 0.001 and † p = 0.05 (for Ca2+ ALT) and † p = 0.08 (non-significant for APD50) and * p = 0.004 (for APD90).
Discussion
We show that hMSCs can be successfully isolated from extracted device lead tips. Furthermore, our main findings show that cardiac hMSCs remodel in disease and can regulate important arrhythmia substrates. More specifically, pro-arrhythmic inflammatory cytokines IL-1β and IL-6 secreted by failing hMSCs enhanced arrhythmia substrates; whereas, secretion of IGF-1 by non-failing hMSCs suppressed arrhythmia substrates. Finally, IGF-1 supplementation and anti-IL-6 rescued the effects of failing hMSCs on hCM. Taken together, these findings suggest that resident cardiac hMSCs can play an important role in human cardiac electrophysiology by remodeling their secretion profiles from an anti-arrhythmic phenotype to a pro-arrhythmic phenotype in disease. Furthermore, these insights suggest novel therapeutic strategies for treating arrhythmias associated with heart failure and, possibly, cytokine syndromes.
Human MSCs isolated from device leads.
Recently, it has been proposed that MSCs are the “sentinel and safe guard for injury” as demonstrated by their ability to assess injury and then treat damaged tissue6. This notion inspired us to develop an innovative approach to obtain cardiac MSCs from extracted cardiac device leads (i.e. pacemaker and defibrillators). Our results suggest that the cells we isolated from cardiac device leads and explanted hearts contains a similar and significant number of MSCs as demonstrated by cell surface marker expression (Figure 3), growth rates (Supplemental Figure S1), and their tri-lineage differentiation potential which is considered the gold standard for verifying MSCs (Figure 2). There are no clear markers that can distinguish between fibroblasts and MSCs; nevertheless, preferential selection in culture and the results mentioned above suggest a similar cellular composition that mostly consists of MSCs. Furthermore, our findings are consistent with a recent report showing that MSCs with multi-lineage potential can be isolated from human cardiac tissue samples3.
Functional effects of human MSCs isolated from failing and non-failing hearts
In the absence of oxidative stress, failing hMSCs significantly increased Ca2+ alternans, APD90, and APD50, which are well established heart failure phenotypes. These results suggest that failing hMSCs may contribute to the electrophysiological phenotype observed in heart failure patients. It is difficult to remark on the extent of this contribution because remodeling of ion channels26,27 and Ca2+ regulation28 in myocytes are already known to contribute to a similar electrophysiological phenotype associated with heart failure. Unlike failing hMSCs, non-failing hMSCs did not increase Ca2+ alternans, APD90, and APD50, suggesting that in a normal heart hMSCs are not proarrhythmic. It has been previously shown that non-myocytes, such as fibroblasts, isolated from both infarcted and non-infarcted rat hearts have the same prolonging effect on APD when plated at a high density and APD shortening effect when plated at a lower density29. These results, which are inconsistent with ours, provide functional evidence that the hMSCs we isolated have properties unlike fibroblasts. In that study, Vasquez et al.29 preferentially selected for fibroblasts by using cells that attach within 30 minutes. Our isolation protocol allowed cells to attach for 2–3 days (see Methods), which is similar to other protocols used to isolate MSCs3,5. Furthermore, all our tissue samples were taken from areas without scar.
Importantly, under conditions of oxidative stress, non-failing hMSCs decreased Ca2+ alternans, which is consistent with our previous findings using BM hMSCs8. Furthermore, IGF-1 that is secreted at high levels in BM hMSCs and non-failing hMSCs but not in failing hMSCs, rescued the increase in Ca2+ alternans and APD caused by failing hMSCs (Figure 8C) even in the presence of oxidative stress. These results suggest that cardiac hMSCs, like BM hMSCs, can suppress arrhythmia substrates. We have previously shown using conditioned media and transwell inserts that the suppression of Ca2+ alternans persists8, and in the current study we observed no significant effect of BM hMSCs (Supplement Figure S2) or non-failing hMSCs on APD (Figure 4). These results strongly suggest that at the cell densities we tested, the effects reported are largely paracrine. These results may explain how BM MSCs can reduce arrhythmia in patients with acute MI30 and promote repair31 by paracrine action.
The significant differences between failing and non-failing MSCs (including their secretome) that we report suggest the intriguing possibility that in disease, cardiac hMSCs remodel and can induce arrhythmia substrates that are often observed in patients with heart failure (e.g. cardiac alternans, prolonged repolarization, and spontaneous calcium release). Previous studies have shown that non-myocytes in the heart can remodel in disease32. For example, plastic adherent stromal cardiac cells have been shown to change their secretion profile in disease and aging to a pro-inflammatory phenotype expressing high levels of cytokines, including TNFα, IL-1β and IL-633 that are associated with heart failure14,15,25. Similarly, it was recently shown that among MSCs isolated from failing human hearts, IL-1β is increased and IGF-2 is decreased compared to other stem cell populations isolated from the same human hearts3, which is consistent with our findings.
Molecular signals responsible for electrophysiological action of cardiac MSCs
We found that IL-1β and IL-6 are increased in failing hMSCs, which may explain enhanced arrhythmia substrates. While both IL-1β and IL-6 prolonged APD, only IL-6 increased Ca2+ alternans. De Jesus et al.34 found in a mouse model of MI with severe inflammation induced with LPS, that non-selective IL-1 inhibition (both IL-1α and IL-1β) with anakinra decreased Ca2+ alternans, which is inconsistent with our result. One possible explanation for this is that the mouse relies more on SERCA2a function to remove cytoplasmic Ca2+ compared to humans35, which might make Ca2+ alternans more sensitive to changes in IL-1. Another possible explanation is that IL-1β is a known potent activator of IL-6 in vivo and antagonists targeting IL-1β have been shown to also decrease IL-636. Importantly, our results show that IL-6 inhibition rescued the failing Ca2+ phenotype induced by the diseased hMSCs. IL-6 is abundant in cardiac cells37,38 and is known to remodel cardiac ion channels and cause Ca2+ dysregulation39. Thus, within the MSC inflammatory secretome, IL-6 may be an important mechanism of arrhythmia substrates, which raises the intriguing possibility of novel therapeutic strategies targeting non-myocytes and their inflammatory secretome.
We also found that IGF-1 is increased in non-failing hMSCs, which may explain the suppression of arrhythmia substrates. IGF-1 levels were much higher (Figure 8B) in BM hMSCs and non-failing hMSCs compared to failing hMSCs, which were unable to suppress Ca2+ alternans. IGF-1 supplementation rescued the failing phenotype induced by failing hMSCs on both normal myocytes (Figure 8C) and myocytes under oxidative stress (Figure 8A). Finally, Wortmannin, a PI3K inhibitor, blocked the effect of IGF-1 supplementation (Figure 8A). These results are consistent with our previous study showing that BM hMSCs suppress Ca2+ alternans by activation of the PI3K/Akt pathway8. While not directly tested in the present study, there might be a link between IGF-1 and IL-6 as reported previously, where IGF-1 activation was found to decrease IL-6 secretion40. Future work in this area might better elucidate the signaling pathways between IGF-1 and IL-6 in cardiac tissue.
Clinical Implications
We show that extracted device leads are a novel source of human cardiac MSCs. This represents a unique and untapped resource, because thousands of device leads are extracted across the U.S. each year that are currently destined to be discarded.
There is now renewed and growing interest in understanding the action of specific inflammatory components and targeted anti-inflammatory therapy14,18,34. Herein, we show that specific inflammatory cytokines in heart failure contribute differentially to arrhythmia mechanisms (IL-1β vs IL-6), and that IL-6 may be partly responsible for Ca2+ mediated arrhythmia substrates seen in heart failure, which may explain the higher mortality risk noted clinically in patients with elevated IL-614,25. Additionally, our results suggest that elevated cytokines41, especially IL-642 may explain arrhythmia in patients with severe COVID-19 and concerns using hydroxychloroquine, which can further prolong QT in such patients43. Novel therapies that inhibit (e.g. IL-6) or mimic (e.g. IGF-1 pathway activation) specific cytokines may be effective at treating arrhythmias associated with heart failure or cytokine storm such as COVID-1920. Furthermore, our results warrant studying the antiarrhythmic efficacy of targeted anti-inflammatory drugs (tocilizumab, sarilumab, canakinumab or anakinra) currently used in rheumatology, gastroenterology, oncology, and in cardiology for atherosclerosis and pericarditis.
Limitations
Due to the lack of clear specific markers, it can be difficult to distinguish between MSCs and fibroblasts. However, our isolation procedure, cell surface marker expression, growth rate, and tri-lineage differentiation potential suggest a similar composition for failing and non-failing hearts. Experiments were performed in an unsealed chamber open to room air, which may not mirror the ischemic oxygen-deplete environment where cells would need to function in disease. While we do show that levels of certain cytokines (IL-1β, IL-6, and IGF-1) change in failing vs. non-failing hMSCs and they can regulate arrhythmia substrates, we cannot rule out the possibility that other cytokines are playing a role. Additionally, though hCM used in our study provide the advantage of experimentation in human myocytes, they are considered immature. Finally, to focus on the effects of failing and non-failing hMSCs we did not use failing myocytes. We chose this method to be consistent with our previously published work using BM hMSCs8, and because the formation of myocyte monolayers co-cultured with hMSCs (2 days) is problematic with adult, diseased myocytes.
Supplementary Material
What Is Known?
Atrial and ventricular arrhythmias are frequently observed with inflammatory myocardial disease including heart failure.
Mesenchymal stem cells, which have an extensive inflammatory-mediating secretome, exist in the heart.
What the Study Adds?
Extracted device leads are a novel source of human cardiac mesenchymal stem cells.
Human cardiac mesenchymal stem cells remodel their secretion profiles from an anti-arrhythmic phenotype (IGF-1) to a pro-arrhythmic phenotype in heart failure (IL-1β and especially IL-6).
IGF-1 supplementation or anti-IL-6 can rescue the pro-arrhythmic phenotype induced by failing human cardiac mesenchymal stem cells in myocytes.
Acknowledgments:
The authors thank Life Banc of Northeastern Ohio for providing access to non-failing heart tissue from unmatched organ donors after family consent.
Sources of Funding: This work was supported in part by grants from NIH HL123012, NIH HL149369 (KRL), NIH HL139006 (JDF), and AHA 17CPOST33650005 (SKV), and generous support from Curtis and Sara Moll (JAM).
Nonstandard Abbreviations and Acronyms
- MSC
mesenchymal stem cell
- hMSC
human mesenchymal stem cell
- BM
bone marrow
- hCM
Human cardiac myocytes derived from induced pluripotent stem cells
- SCR
Spontaneous calcium release
- WORT
Wortmannin
- IGF-1
Insulin-like growth factor 1
- IL-6
Interleukin 6
- IL-1β
Interleukin 1β
- VEGF
Vascular endothelial growth factor
- APD90
Action potential duration at 90% repolarization
- APD50
Action potential duration at 50% repolarization
- H2O2
Hydrogen peroxide
- TNFα
Tumor necrosis factor
- MCP-1
monocyte chemoattractant protein 1
- LPS
Lipopolysaccharides
- SERCA2a
sarcoplasmic reticulum Ca2+-ATPase
Footnotes
Disclosures: None
References:
- 1.Wagner W, Roderburg C, Wein F, Diehlmann A, Frankhauser M, Schubert R, Eckstein V, Ho AD. Molecular and secretory profiles of human mesenchymal stromal cells and their abilities to maintain primitive hematopoietic progenitors. Stem Cells. 2007;25:2638–2647. [DOI] [PubMed] [Google Scholar]
- 2.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monsanto MM, White KS, Kim T, Wang BJ, Fisher K, Ilves K, Khalafalla FG, Casillas A, Broughton K, Mohsin S, et al. Concurrent isolation of three distinct cardiac stem cell populations from a single human heart biopsy. Circ Res. 2017;121:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramani B, Subbannagounder S, Palanivel S, Ramanathanpullai C, Sivalingam S, Yakub A, SadanandaRao M, Seenichamy A, Pandurangan AK, Tan JJ, et al. Generation and characterization of human cardiac resident and non-resident mesenchymal stem cell. Cytotechnology. 2016;68:2061–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garikipati VNS, Singh SP, Mohanram Y, Gupta AK, Kapoor D, Nityanand S. Isolation and characterization of mesenchymal stem cells from human fetus heart. PLoS One. 2018;13:e0192244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caplan AI. Mscs: The sentinel and safe-guards of injury. J Cell Physiol. 2016;231:1413–1416. [DOI] [PubMed] [Google Scholar]
- 7.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking t-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. [DOI] [PubMed] [Google Scholar]
- 8.Sattayaprasert P, Nassal DM, Wan X, Deschenes I, Laurita KR. Mesenchymal stem cells suppress cardiac alternans by activation of pi3k mediated nitroso-redox pathway. J Mol Cell Cardiol. 2016;98:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeSantiago J, Bare DJ, Semenov I, Minshall RD, Geenen DL, Wolska BM, Banach K. Excitation-contraction coupling in ventricular myocytes is enhanced by paracrine signaling from mesenchymal stem cells. J Mol Cell Cardiol. 2012;52:1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayourian J, Cashman TJ, Ceholski DK, Johnson BV, Sachs D, Kaji DA, Sahoo S, Hare JM, Hajjar RJ, Sobie EA, et al. Experimental and computational insight into human mesenchymal stem cell paracrine signaling and heterocellular coupling effects on cardiac contractility and arrhythmogenicity. Circ Res. 2017;121:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Wei H, Deng L, Cong X, Chen X. Expression and secretion of interleukin-1beta, tumour necrosis factor-alpha and interleukin-10 by hypoxia- and serum-deprivation-stimulated mesenchymal stem cells. FEBS J. 2010;277:3688–3698. [DOI] [PubMed] [Google Scholar]
- 12.Cieslik KA, Trial J, Entman ML. Mesenchymal stem cell-derived inflammatory fibroblasts promote monocyte transition into myeloid fibroblasts via an il-6-dependent mechanism in the aging mouse heart. FASEB J. 2015;29:3160–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naftali-Shani N, Levin-Kotler LP, Palevski D, Amit U, Kain D, Landa N, Hochhauser E, Leor J. Left ventricular dysfunction switches mesenchymal stromal cells toward an inflammatory phenotype and impairs their reparative properties via toll-like receptor-4. Circulation. 2017;135:2271–2287. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Chen J, Huang D, Ke J, Wu W. A meta-analysis of proinflammatory cytokines in chronic heart failure. Heart Asia. 2014;6:130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: Mediators and markers. Cardiology. 2012;122:23–35. [DOI] [PubMed] [Google Scholar]
- 16.Scott LJ. Tocilizumab: A review in rheumatoid arthritis. Drugs. 2017;77:1865–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, Przepiorka D, Farrell AT, Pazdur R. Fda approval summary: Tocilizumab for treatment of chimeric antigen receptor t cell-induced severe or life-threatening cytokine release syndrome. Oncologist. 2018;23:943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 19.Imazio M, Lazaros G, Brucato A, Gaita F. Recurrent pericarditis: New and emerging therapeutic options. Nature reviews. Cardiology. 2016;13:99–105. [DOI] [PubMed] [Google Scholar]
- 20.Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab treatment in covid-19: A single center experience. J Med Virol. 2020;92:814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooke G, Rossetti T, Ilic N, Murray P, Hancock S, Pelekanos R, Atkinson K. Points to consider in designing mesenchymal stem cell-based clinical trials. Transfus Med Hemother. 2008;35:279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love Z, Wang F, Dennis J, Awadallah A, Salem N, Lin Y, Weisenberger A, Majewski S, Gerson S, Lee Z. Imaging of mesenchymal stem cell transplant by bioluminescence and pet. J Nucl Med. 2007;48:2011–2020. [DOI] [PubMed] [Google Scholar]
- 23.McPheeters MT, Wang YT, Werdich AA, Jenkins MW, Laurita KR. An infrared optical pacing system for screening cardiac electrophysiology in human cardiomyocytes. PLoS One. 2017;12:e0183761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman MHT, Groot HE, Leach IM, Karper JC, van der Harst P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc Med. 2018;28:369–379. [DOI] [PubMed] [Google Scholar]
- 25.Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: An overview. Heart. 2004;90:464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beuckelmann DJ, Näbauer M, Erdmann E. Alterations of k+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ.Res. 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 27.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. [DOI] [PubMed] [Google Scholar]
- 28.Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted sarcoplasmic reticulum ca2+ atpase 2a gene delivery to restore electrical stability in the failing heart. Circulation. 2012;126:2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Afzal MR, Samanta A, Shah ZI, Jeevanantham V, Abdel-Latif A, Zuba-Surma EK, Dawn B. Adult bone marrow cell therapy for ischemic heart disease: Evidence and insights from randomized controlled trials. Circ Res. 2015;117:558–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MJ, Chou CC, Frantzen M, Miyamoto T, Kar S, Lee S, Shah PK, Martin BJ, Lill M, Forrester JS, et al. Intravenous mesenchymal stem cell therapy early after reperfused acute myocardial infarction improves left ventricular function and alters electrophysiologic properties. Int J Cardiol. 2006;111:231–239. [DOI] [PubMed] [Google Scholar]
- 32.Cheng K, Malliaras K, Smith RR, Shen D, Sun B, Blusztajn A, Xie Y, Ibrahim A, Aminzadeh MA, Liu W, et al. Human cardiosphere-derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. JACC Heart Fail. 2014;2:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savi M, Bocchi L, Sala R, Frati C, Lagrasta C, Madeddu D, Falco A, Pollino S, Bresciani L, Miragoli M, et al. Parenchymal and stromal cells contribute to pro-inflammatory myocardial environment at early stages of diabetes: Protective role of resveratrol. Nutrients. 2016;8:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Jesus NM, Wang L, Lai J, Rigor RR, Francis Stuart SD, Bers DM, Lindsey ML, Ripplinger CM. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm. 2017;14:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J Physiol (Lond). 1994;476:279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridker PM. From c-reactive protein to interleukin-6 to interleukin-1: Moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aromolaran AS, Srivastava U, Ali A, Chahine M, Lazaro D, El-Sherif N, Capecchi PL, Laghi-Pasini F, Lazzerini PE, Boutjdir M. Interleukin-6 inhibition of herg underlies risk for acquired long qt in cardiac and systemic inflammation. PloS one. 2018;13:e0208321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayfield AE, Kanda P, Nantsios A, Parent S, Mount S, Dixit S, Ye B, Seymour R, Stewart DJ, Davis DR. Interleukin-6 mediates post-infarct repair by cardiac explant-derived stem cells. Theranostics. 2017;7:4850–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ali A, Boutjdir M, Aromolaran AS. Cardiolipotoxicity, inflammation, and arrhythmias: Role for interleukin-6 molecular mechanisms. Front Physiol. 2018;9:1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sukhanov S, Higashi Y, Shai SY, Vaughn C, Mohler J, Li Y, Song YH, Titterington J, Delafontaine P. Igf-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in apoe-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:2684–2690. [DOI] [PubMed] [Google Scholar]
- 41.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. Covid-19: Consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai Q, Huang D, Ou P, Yu H, Zhu Z, Xia Z, Su Y, Ma Z, Zhang Y, Li Z, et al. Covid-19 in a designated infectious diseases hospital outside hubei province, china. Allergy. 2020. [DOI] [PubMed]
- 43.Sapp JL, Alqarawi W, MacIntyre CJ, Tadros R, Steinberg C, Roberts JD, Laksman Z, Healey JS, Krahn AD. Guidance on minimizing risk of drug-induced ventricular arrhythmia during treatment of covid-19: A statement from the canadian heart rhythm society. Can J Cardiol. 2020;36:948–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.