

Abstract
Meroterpenoids derived from the polyketide 1,3,6,8-tetrahydroxynaphthalene (THN) are complex natural products produced exclusively by Streptomyces bacteria. These antibacterial compounds include the napyradiomycins, merochlorins, marinones, and furaquinocins and have inspired many attempts at their chemical synthesis. In this review, we highlight the role played by biosynthetic studies in the stimulation of biomimetic and, ultimately, chemoenzymatic total syntheses of these natural products. In particular, the application of genome mining techniques to marine Streptomyces bacteria led to the discovery of unique prenyltransferase and vanadium-dependent haloperoxidase enzymes that can be used as highly selective biocatalysts in fully enzymatic total syntheses, thus overcoming the limitations of purely chemical reagents.
Graphical Abstract
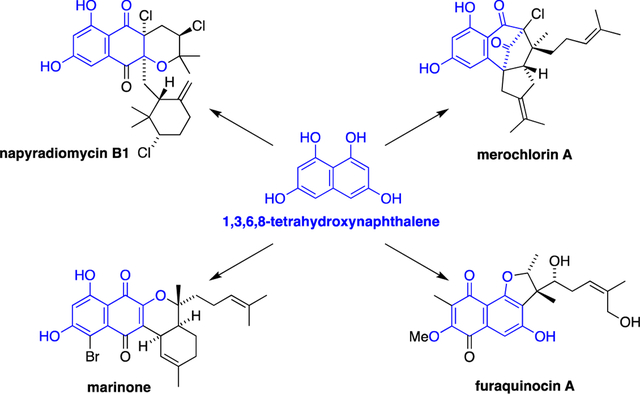
A review of the isolation, 13C-biosynthetic labelling studies, biosynthesis and total synthesis of meroterpenoids is presented.
1. Introduction
Meroterpenoids are natural products of mixed biosynthetic origin that are formed in part from a terpenoid co-substrate.1 These hybrid molecules are broadly produced in nature by bacteria, fungi, plants, algae and animals and combine terpenoid building blocks with polyketides, phenols, alkaloids, and amino acids to achieve amazing chemical diversity.2,3,4,5 Some notable examples include the cofactor ubiquinone and medicines such as vincristine, mycophenolic acid and tetrahydrocannabinol.
During the past four decades, several families of naphthoquinone-based meroterpenoids have been discovered from marine and soil-derived streptomycete bacteria.6 These natural products have been linked, through both biosynthetic speculation and 13C-labelling studies, to 1,3,6,8-tetrahydroxynaphthalene (THN, 1) as the key aromatic polyketide building block (Scheme 1). In bacteria, THN is biosynthesised by the condensation and aromatisation of five malonyl coenzyme A subunits under the control of a THN synthase.7 Bacterial THN synthases possess a simple, type III polyketide synthase homodimeric structure.8 Several THN synthases have been implicated in the biosynthesis of bacterial meroterpenoids, including Mcl179 (merochlorins), NapB110 (napyradiomycins) and Fur111 (furaquinocins). THN undergoes spontaneous aerobic oxidation to give flaviolin (2), which shares a superficial similarity in structure to several bacterial meroterpenoids such as the furaquinocins (e.g. furaquinocin A,12 3) and the napyradiomycins (e.g. napyradiomycin B1,13 4). Indeed, biosynthetic studies have shown that flaviolin is a likely biosynthetic precursor of the furaquinocins.14, However, it has recently been shown that most marine meroterpenoids – such as the merochlorins (e.g. merochlorin A,9 5), marinones (e.g. marinone,15 6) and napyradiomycins – are rather derived directly from THN itself, with oxidation, halogenation and cyclisation steps occurring after the initial prenylation of THN. Various prenyl donors, including prenyl, geranyl and farnesyl pyrophosphate, are initially appended to the nucleophilic C-2 and C-4 positions of THN via electrophilic aromatic substitution reactions catalysed by aromatic prenyltransferase enzymes.16 Subsequently, vanadium-dependent haloperoxidase (VHPO) enzymes catalyse a range of reactions including direct halogenation, oxidative dearomatisation, halocyclisation, and α-hydroxyketone rearrangements, to give complex meroterpenoid molecular architectures such as 4, 5 and 6.17
Scheme 1.
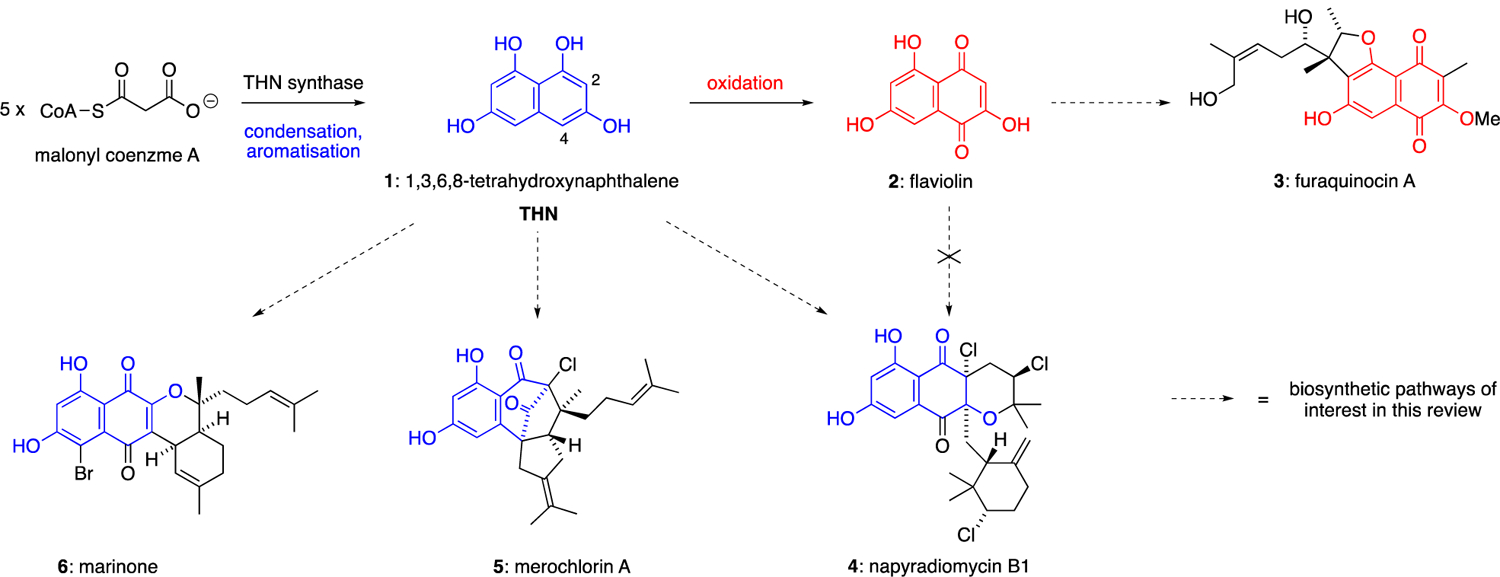
Important families of meroterpenoids biosynthesised from THN (1).
This review has several aims. Firstly, we have distilled over thirty years of isolation work to present all of the known THN-derived meroterpenoids in a logical manner. The modular nature of the biosynthesis of these meroterpenoids presents many opportunities for diversification within each sub-family, so we hope that this section will help the reader to make connections between these unusual natural products. Secondly, we present the early 13C-biosynthetic labelling studies of THN meroterpenoids, which gave valuable information on the origin of the polyketide and terpene building blocks, but not enough to fully elucidate the biosynthetic pathways. As a consequence, the pioneering total syntheses of napyradiomycins by Nakata,18 Tatsuta19 and Snyder20 were uninformed by firm knowledge of their biosynthesis. Thirdly, and in a roughly linear narrative, we describe how the advent of genome analysis has transformed studies on the biosynthesis and total synthesis of complex meroterpenoids. Three case studies on the merochlorins, the napyradiomycins and the marinones are presented in which the discovery of unique prenyltransferase and vanadium-dependent chloroperoxidase (VCPO) enzymes from the respective gene clusters allowed the biosynthetic jigsaw puzzles to be solved piece by piece. This biosynthetic work directly inspired biomimetic total syntheses of THN-derived meroterpenoids by several research groups, in addition to a fully enzymatic total synthesis of napyradiomycins A1 and B1. The use of a unique combination of polyketide synthase, prenyltransferase and VCPO enzymes in one-pot synthetic operations represents the ideal end point in the total synthesis of THN meroterpenoids, although challenges remain in terms of scalability and product diversification in the synthesis of the napyradiomycins. Finally, biosynthetic and synthetic work on the furaquinocins and azamerone are summarised. In both cases, the final key steps of the biosynthetic pathways are unknown, which presents future opportunities for the discovery of new enzymes and biomimetic chemical reactions.
2. Isolation and biological activity
2.1. Napyradiomycins
The napyradiomycins are by far the largest family of THN meroterpenoids, with over fifty natural products reported to date. The simplest member of the family is naphthomevalin (7), which features a naphthoquinone core adorned with prenyl and chlorine substituents at C-2, and geranyl and hydroxyl groups at C-3. All members of the napyradiomycin family can be considered to be functionalised derivatives of naphthomevalin (7) (Figure 1). For example, halocyclisation of the C-2 prenyl substituent gives a large group of tricyclic napyradiomycins with a 6,6,6 ring system, while the C-3 geranyl group can also undergo halocyclisation to form an additional cyclohexane ring. The C-7 position of the left hand aromatic ring can be methylated or not, while the C-5 position can undergo amination and diazotisation to form a series of unusual diazoketone natural products. Intramolecular substitution of the C-2 chlorine substituent with the C-3 tertiary alcohol can form epoxide natural products, or it can be eliminated to introduce further unsaturation. Finally, allylic oxidation of the C-3 geranyl group can occur at various positions, with oxidative macrocyclisation on this substituent also possible. Many combinations of these functionalisations can occur in biosynthesis, which leads to the great diversity of napyradiomycins found in nature.
Figure 1.
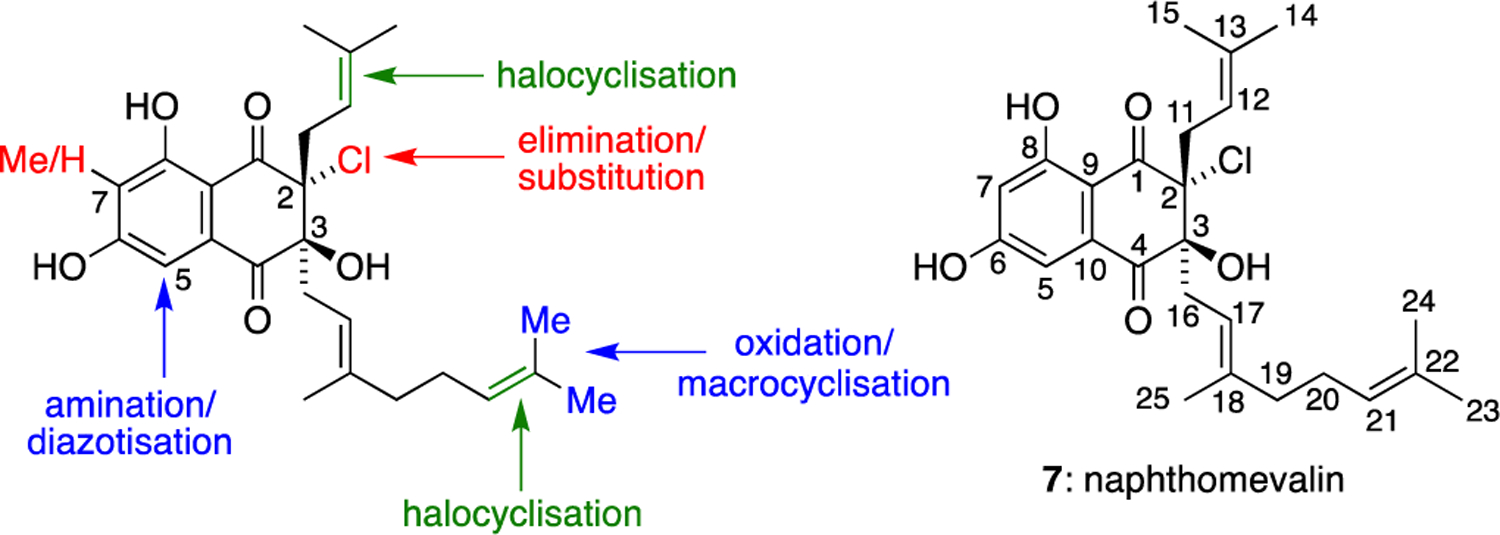
Sites for biosynthetic diversification in the napyradiomycin family of meroterpenoids, and the numbering system used in this article.
The simplest, bicyclic napyradiomycin meroterpenoids are shown in Figure 2. Naphthomevalin (7) itself was isolated from a culture broth of a Streptomyces sp. strain Gö 28, originally found in a soil sample collected at Strathgordon, Australia. This metabolite possesses weak antimicrobial activity against Gram-positive bacteria.21 Treatment of 7 with sodium hydroxide formed the epoxide natural product A80915G (8), which had previously been isolated from Streptomyces aculeolatus A80915, a bacterial strain found in a soil sample collected from Palau, Western Caroline Islands.22 A80915G (8) was found to have good antibacterial activity against aerobic Gram-positive Staphylococcus, Streptococcus and Enterococcus bacterial strains (MIC 2–16 μg/mL) and Gram-positive and Gram-negative anaerobic bacteria. The marine-derived metabolite A80915G-8”-acid (9) produced by Streptomyces sp. MS239 is an oxidised version of 8, and it showed only weak antibacterial activity against Bacillus subtilis ATCC6633 (64 μg/ml).23 SF2415B1 (10), B2 (11), Al (12) and A2 (13) feature methyl substituents at C-7, plus various combinations of C2–C3 epoxide and C-5 diazoketone functional groups. They were isolated from the culture filtrate of S. aculeolatus SF2415.24,25 The producing organism was found in a soil sample collected in Tottori Prefecture, Japan, and these natural products were found to have moderate activities against Gram-positive bacteria.
Figure 2.

Napyradiomycin natural products with uncyclised C-2 prenyl and C-3 geranyl substituents.
Napyradiomycin A1 (14) is perhaps the most well-known member of the napyradiomycin family (Figure 3). Amongst the first of the napyradiomycins to be reported, it was isolated from the culture broth of Streptomyces rubra (formerly Chainia rubra)26 MG802-AF1, which was found in a soil sample collected in Niigata Prefecture, Japan.13 The 6,6,6 ring system of 14 could be formed by chlorocyclisation of the C-2 prenyl side chain of naphthomevalin. Napyradiomycin A1 (14) was found to inhibit the growth of Gram-positive bacteria including multiple drug-resistant strains such as Staphylococcus aureus MS8710 and MS9610.27 Many variants of the napyradiomycin A1 structure have been isolated. For example, 3-dechloro-3-bromo-napyradiomycin-A1 (15) and 4-dehydro-4a-dechloro-napyradiomycin A1 (16) were isolated from the culture broth of a marine-derived actinomycete strain SCSIO 10428.28 In general, it appears that bromination-cyclisation is less common than chlorination-cyclisation in napyradiomycin biosynthesis, so brominated natural products such as 15 are rare. Napyradiomycin analogues 15 and 16 displayed antibacterial activities against three Staphylococcus and Bacillus strains with MIC values ranging from 0.5 to 8 μg/mL, and possessed moderate cytotoxicities against four human cancer cell lines SF-268, MCF-7, NCI-H460, and HepG-2 with IC50 values ranging from 11.5 to 22.8 μM. Napyradiomycin A2, with an oxidised geranyl side chain, was originally isolated from the culture broth of S. rubra MG802-AF1.29 The absolute configuration of the C-21 hydroxyl group was not determined upon initial isolation. However, two stereoisomers were later re-isolated from Streptomyces antimycoticus NT17, which were named napyradiomycin A2a (17) and napyradiomycin A2b (18) and were found to have absolute configurations at C-21 of 16R and 16S, respectively. Both 17 and 18 showed antibacterial activities against clinical bacterial pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) MB5393, and Mycobacterium tuberculosis H37Ra. They also possess moderate cytotoxic activities with IC50 values of 30.4 μM and 28.6 μM against the human liver adenocarcinoma cell line HepG-2.30 Napyradiomycin A3 (19) possesses an enone functional group formed by elimination of the C-2 chlorine substituent. It was isolated from the marine-derived Streptomyces sp. strain CA-271078,31 and was not found to have antibacterial and antifungal properties on the cell lines tested. 18-Hydroxynapyradiomycin A1 (20), 18-oxonapyradiomycin A1 (21), 16-oxonapyradiomycin A2 (22) and 7-demethyl SF2415A3 (23) were all isolated from the culture broth of S. antimycoticus NT17.30 Diazo compound 23 displayed antibacterial activity against S. aureus 209P JC-1 (MIC 2.0 nM/mL), B. subtilis ATCC6633 (1.0 nM/mL), Enterococcus faecalis ATCC19433 (31.6 nM/mL), Enterococcus faecium ATCC19434 (15.8 nM/mL), and Streptococcus pyogenes ATCC12344 (7.8 nM/mL). The antibiotics SF2415A3 (24) and SF2415B3 (25) were isolated from the culture filtrate of S. aculeolatus.24,25 Both 24 and 25 were found to have moderate activities towards Staphylococcus, Enterococcus and Bacillus strains of Gram-positive bacteria, with 24 being more potent. The antibacterial activities of 23 and 24 were found to be almost identical, suggesting that the C-7 methyl group does not affect the biological activity in this series. CNQ525.538 (26) was isolated from Streptomyces sp. CNQ-52532 and found to have moderate cytotoxicity against HCT-116 colon carcinoma cells (IC50 6 μM).
Figure 3.
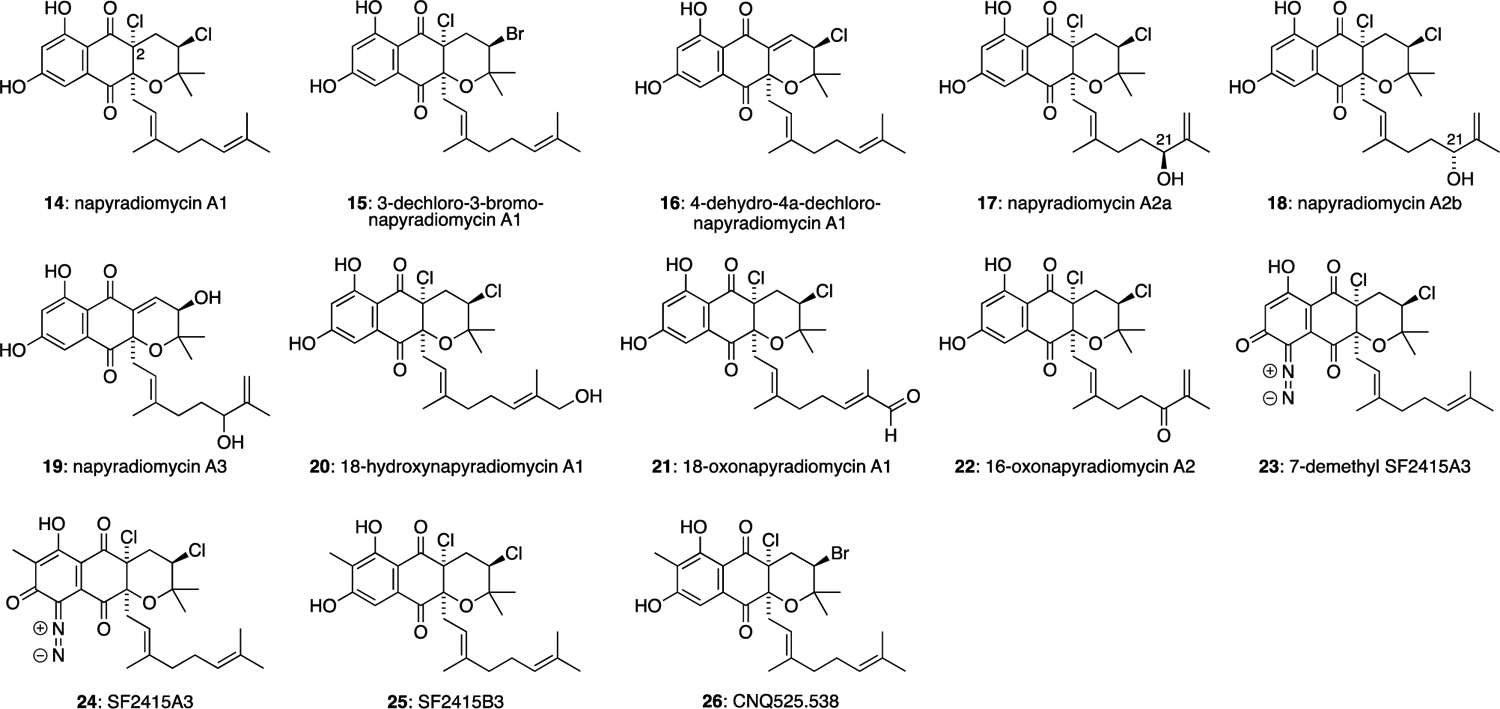
Napyradiomycin natural products with cyclised C-2 prenyl substituents.
The largest sub-family of the napyradiomycins are tetracyclic compounds such as napyradiomycins B1 (4), B2 (27) and B3 (28), which were isolated from culture broth S. rubra MG802-AF1 (Figure 4).13,27 In these natural products, both C-2 prenyl and C-3 geranyl side chains have cyclised to form complex structures with up to three halogenated stereocentres. The halocyclisation of the geranyl side chain is clearly highly stereoselective, as all of these natural products possess the same stereochemical configuration at C-17 and at the newly chlorinated or brominated C-21 position.
Figure 4.
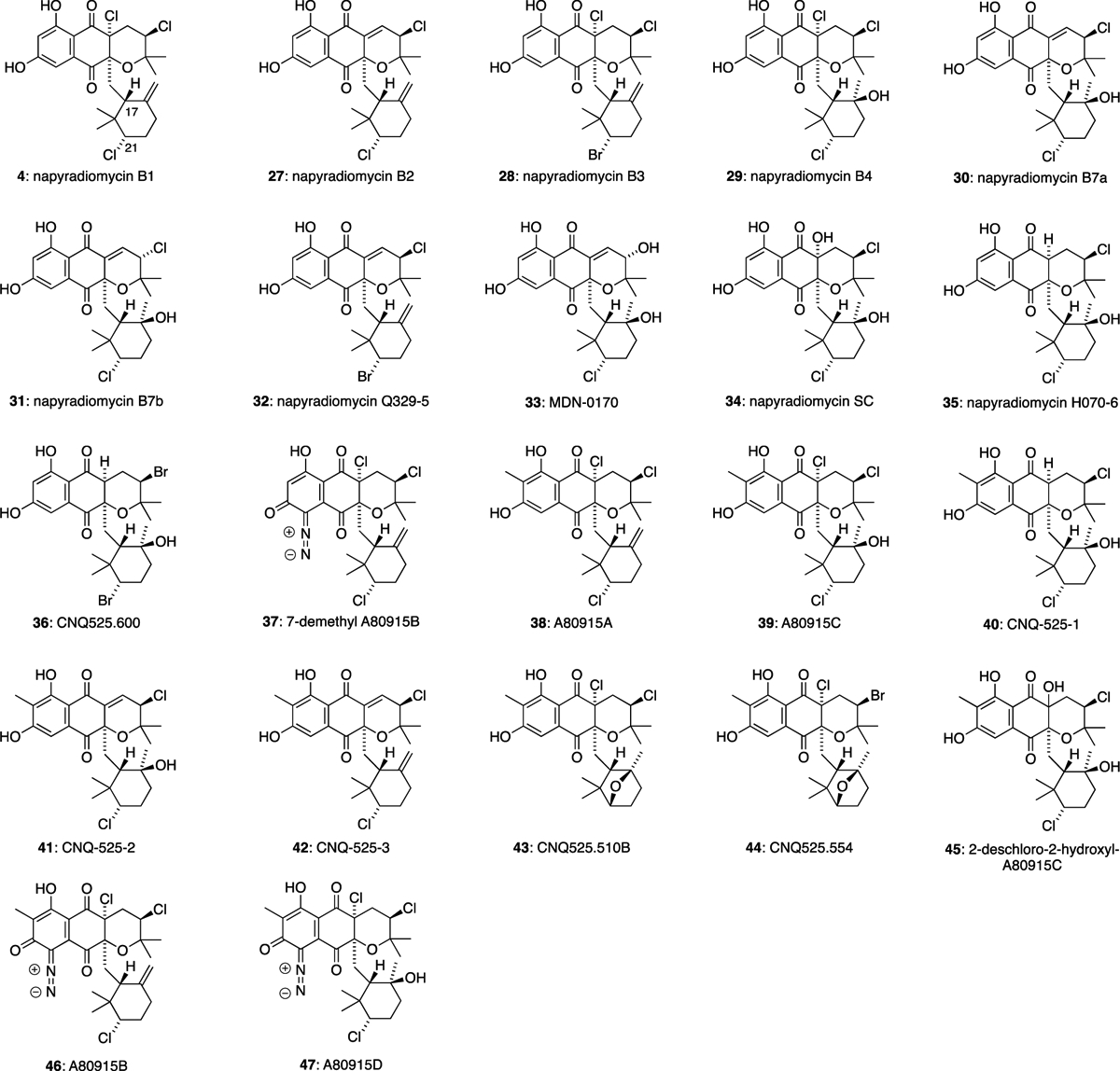
Napyradiomycin natural products with cyclised C-2 prenyl and C-3 geranyl substituents.
Napyradiomycin B2 (27) exhibited good antibacterial activity against MRSA (MIC = 3–6 μg/mL) and moderate activity against M. tuberculosis H37Ra (MIC = 24–48 μg/mL). It also showed moderate cytotoxic activities against HepG-2 (MIC = 27.1 μM).31 Napyradiomycin B4 (29) exhibited moderate activity against MRSA (MIC = 12–24 μg/mL) and M. tuberculosis H37Ra (MIC = 12–24 μg/mL). It also showed moderate cytotoxic activities against HepG-2 (MIC = 41.7 μM).31 Napyradiomycins B7a (30) and B7b (31) are epimers at the C-12 position of their dihydropyran ring, isolated from Streptomyces sp. strain CA-271078.31 Compound 30 showed antibacterial activities against MRSA (MIC = 48 μg/mL), M. tuberculosis H37Ra (MIC = 12–24 μg/mL) and moderate cytotoxic activities against HepG-2 (MIC = 15.6 μM).31 Napyradiomycin Q329–5 (32) was isolated from the culture extracts of marine-derived Streptomyces sp. CNQ-329,33 and it was found to have some cytotoxic activity against the human colon adenocarcinoma cell line (HCT-116) with an MIC = 4.81 μg/mL. MDN-0170 (33) was isolated from Streptomyces sp. CA-271078.34 Unexpectedly, 33 did not show any antibacterial or antifungal properties against MRSA, E. coli, A. fumigatus, and C. albicans. Napyradiomycin SC (34) was isolated from Streptomyces sp. strain CA-271078 and showed antibacterial activity against M. tuberculosis H37Ra (24–48 μg/mL).31 Napyradiomycin H070–6 (35) was isolated from Streptomyces sp. CNH-070, and was found to have moderate cytotoxicity against HCT-116 (IC50 9.42 μM).33 CNQ525.600 (36) was isolated from Streptomyces sp. CNQ-525.32 This compound was found to induce some cytotoxicity against HCT-116 (IC50 49 μM). 7-Demethyl A80915B (37) was isolated from the culture broth of S. antimycoticus NT17.30 Diazo compound 37 showed antibacterial activity against S. aureus 209P JC-1 (MIC 3.7 nM/mL), B. subtilis ATCC6633 (3.7 nM/mL), E. faecalis ATCC19433 (14.8 nM/mL), and E. faecium ATCC19434 (14.8 nM/mL). A80915A (38) and A80915C (39) were isolated from S. aculeolatus A80915.22 A80915A (38) was found to have good antibacterial activity against aerobic Gram-positive bacteria Staphylococcus, Enterococcus and Haemophilus strains (MIC ≤ 1 μg/mL), while 39 was largely inactive. Both 38 and 39 were found to have activity against Gram-positive and Gram-negative anaerobic bacterial strains. CNQ-525–1 (40), CNQ-525–2 (41) and CNQ-525–3 (42) were isolated from Streptomyces sp. CNQ-52535, which was originally found in ocean sediments near La Jolla, California. Metabolites 40 and 42 were found to have the most activity against MRSA, and vancomycin-resistant Enterococcus faecium (VRE), both with MIC values of 1.95 μg/mL. Both 40 and 41 were found to have good cytotoxic activity against HCT-116 with IC50 values of 2.40 and 0.97 μg/mL, respectively. CNQ-525.510B (43) and CNQ-525.554 (44) were isolated from Streptomyces sp. CNQ-525,32 with 43 showing moderate cytotoxicity against HCT-116 (IC50 17 μM). 2-Deschloro-2-hydroxyl-A80915C (45) was isolated from Streptomyces sp. CNQ-525.10 A80915B (46) and A80915D (47) were isolated from S. aculeolatus A80915.22 Diazo compounds 46 and 47 were found to have good antibacterial activity against aerobic Gram-positive bacteria Staphylococcus, Streptococcus, Enterococcus and Haemophilus strains (MIC <1 and <8 μg/mL respectively). Both 46 and 47 also showed good activity against select Gram-positive and Gram-negative anaerobic bacteria.
The C-3 geranyl group of the napyradiomycins can also undergo oxidation and macrocyclisation, usually at the C-7 position of the aromatic ring, to form highly complex natural products such as napyradiomycin C1 (48) (Figure 5). Napyradiomycins C1 (48) and C2 (49) were isolated from a culture broth of S. rubra MG802-AF1,13 and were found to inhibit the growth of Gram-positive bacteria including multiple drug-resistant strains such as S. aureus MS8710 and MS9610. 16-Dechloro-16-hydroxy-napyradiomycin C2 (50) was isolated from the culture broth of S. antimycoticus NT17.30 Napyradiomycins Q329–1 (51), Q329–2 (52) and Q329–3 (53) were isolated from the culture extracts of marine-derived Streptomyces sp. CNQ-329.33 Macrocycle 51 showed good activity against MRSA (16 μg/mL) and strong cytotoxicity against HCT-116 (IC50 = 4.19 μg/mL), whereas 52 and 53 were less active. Napyradiomycin SR (54) was isolated from the culture broth of S. antimycoticus NT17.30 Napyradiomycin Q329–4 (55) was isolated from the culture extracts of marine-derived Streptomyces sp. CNQ-329.33 Both 54 and 55 feature an additional pyran ring formed by cyclisation at the C-6 phenol group. Q329–4 (55) showed moderate cytotoxic activity against HCT-116 (IC50 = 16.1 μg/mL). Napyradiomycin D1 (56) was isolated from marine-derived Streptomyces sp. strain CA-271078,31 and was found to have good antibacterial properties against a clinical isolate of MRSA and M. tuberculosis with MIC values ranging from 12–24 and 24–48 μg/mL, respectively. The structure of 56 possesses a macrocycle formed by cyclisation at the C-8 phenol.
Figure 5.
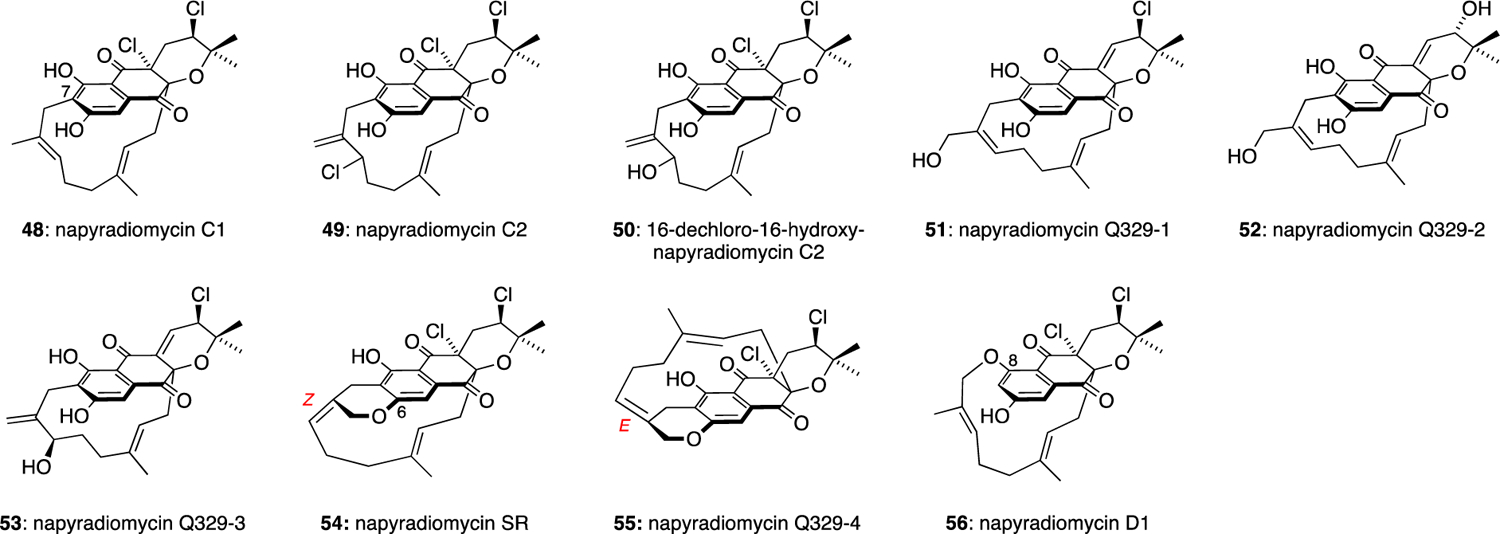
Macrocyclic napyradiomycin natural products.
Azamerone (57), a structurally unique member of the napyradiomycin family, was isolated from Streptomyces sp. CNQ766.36 Azamerone is the first example of a natural product containing a phthalazinone ring system (Figure 6). Its biological activity is relatively modest in comparison to other napyradiomycins, showing only weak in vitro cytotoxicity against murine splenocyte T-cells and macrophages (IC50 40 μM)
Figure 6.
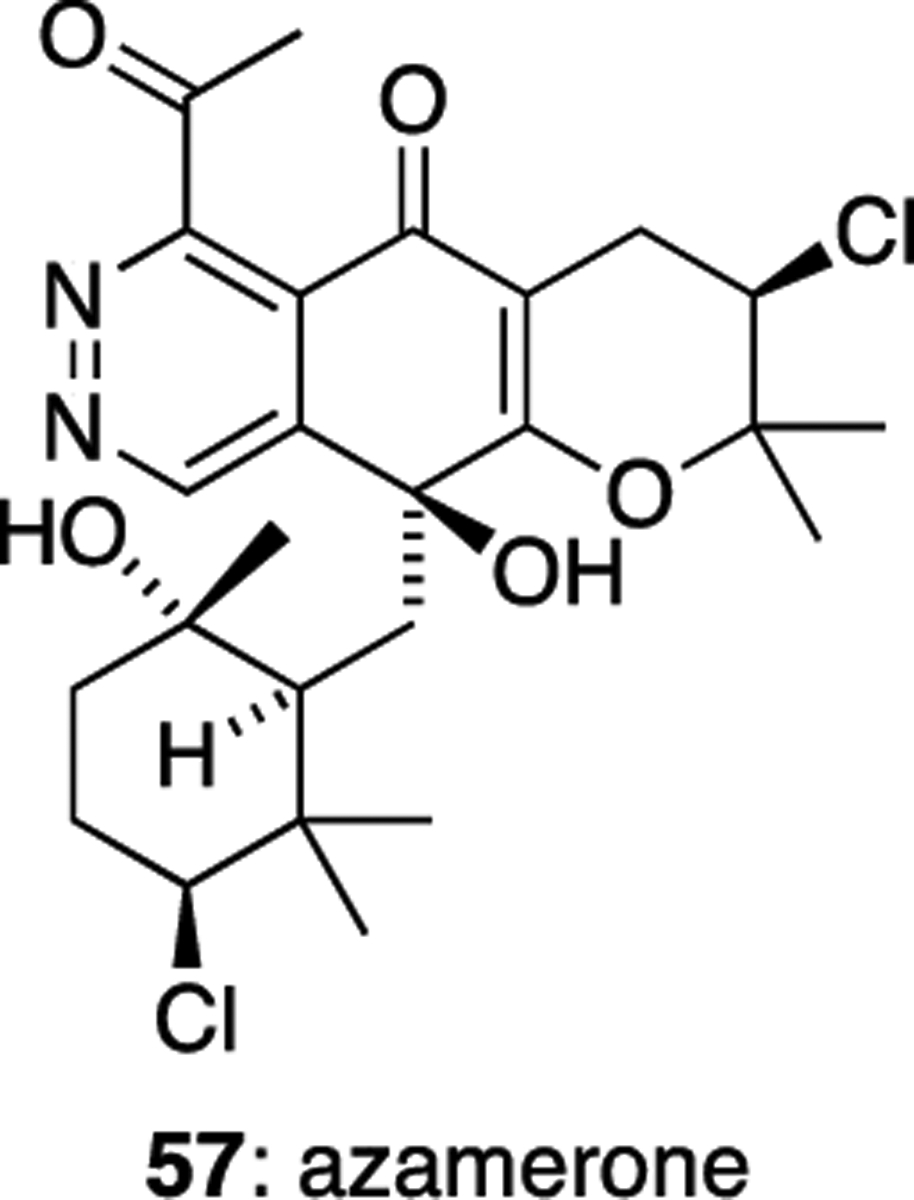
Azamerone (57).
2.2. Merochlorins
Merochlorins A (5), B (58), C (59) and D (60) were isolated in 2012 from Streptomyces sp. strain CNH-189, found in a marine sediment sample collected near Oceanside, California.9 The stereochemically complex, tetracyclic structures of 5 and 58 are particularly unusual (Figure 7). Merochlorin A (5) contains a congested bicyclo[3.2.1]octane core, with four stereocentres and a bridgehead chlorine substituent at C-2, while merochlorin B (58) features three contiguous stereocentres and a chlorinated vinylogous ester. 5 and 58 are also highly active against MRSA strains with MICs in the range of 2−4 μg/mL, whereas merochlorin C (59) showed only weak antibiotic properties. Merochlorins E (61) and F (62) and meroindenon (63) were later isolated in 2019 from the same Streptomyces sp. CNH-189 strain.37 Merochlorins 61 and 62 displayed strong antibacterial activities against B. subtilis, K. rhizophila, and S. aureus, with MIC values of 1−2 μg/mL. Meroindenon (63) was only found to exhibit weak activities against the same Gram-positive bacteria. Merochlorins 59-62 each possess a methyl substituent at C-2, as opposed to the C-2 prenyl substituents of the napyradiomycins.
Figure 7.
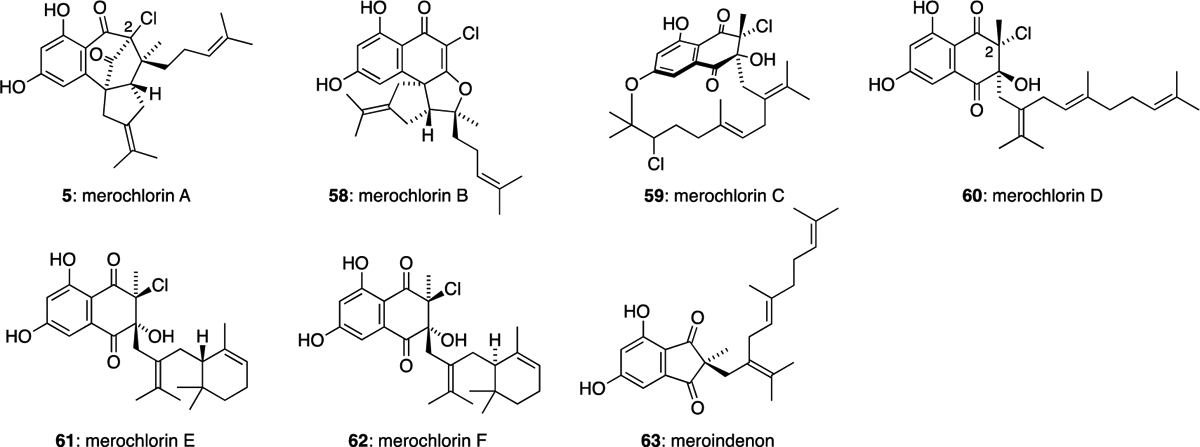
The merochlorin family of meroterpenoids.
Phosphatoquinones A (64) and B (65) also feature a methyl group at C-2 (Figure 8), so they are perhaps biosynthetically related to merochlorins C-F. They were isolated from the cultured broth of Streptomyces sp. TA-0363.38 Both 64 and 65 inhibited the protein tyrosine phosphatase activity prepared from human Ball-1 cells with IC50 of 28 μM and 2.9 μM, respectively.
Figure 8.

Phosphatoquinones A (64) and B (65).
2.3. Naphterpins and marinones
The naphterpins and marinones are closely related meroterpenoid families with a common naphthoquinone ring system and cyclised C-3 geranyl or farnesyl side chains (Figure 9). The terpene derived 6,6 ring systems are all cis-fused in these natural products. The first member of this family to be discovered was naphterpin (66), isolated in 1990 from Streptomyces sp. CL190, which was originally collected from Ishigaki Island, Japan.39 Naphterpin was found to suppress lipid peroxidation in rat homogenate system with an IC50 value of 5.3 μg/mL. Two oxidised members of the naphterpin family, naphterpins B (67) and C (68), were later isolated from the same producing organism Streptomyces sp. CL190.40 Naphterpins 67 and 68 also suppressed lipid peroxidation in rat homogenate system with IC50 values of 6.5 μg/mL and 6.0 μg/mL, respectively. In 2018, naphterpins D (69) and E (70) were isolated from Streptomyces sp. CNQ-509, isolated from the marine sediment collected near La Jolla, California.41 Similar to other members of the naphterpin family, both of these compounds were identified as radical scavengers. Lacking the C-7 methyl substituent of naphterpin, 7-demethylnaphterpin (71) was isolated in 1992 from Streptomyces prunicolor.42 Like naphterpin itself, 71 possesses antioxidative activities and is known to inhibit the lipid peroxidase in rat liver microsomes (IC50 = 9 μg/mL). This molecule was also isolated in 1991 as part of the naphthgeranine family of natural products, so 71 is also sometimes referred to as naphthgeranine A. Several oxidised naphterpin and naphthgeranine natural products have also been isolated, presumably arising from cytochrome P450 mediated C-H oxidations of the terpene-derived ring systems. For example, the naphthgeranines A-E (71–75) were isolated from Streptomyces violaceus Tü 3556,43 a bacterial strain that was originally discovered in a soil sample collected in Nepal. Naphthgeranines A (71) and B (72) showed weak antibacterial and antifungal activities, and moderate cytocidal activity against various tumor cell lines in vitro. In 1995 another member of the family, naphthgeranine F (76), was isolated from the same bacteria S. violaceus Tü 3556.44 This compound was found to have weak antibacterial activity against Gram-positive bacteria (B. subtilis ATCC6051, Bacillus brevis ATCC9999, Streptomyces viridochromogenes Tü 57), with similar MIC values to that of naphthgeranine C (73). Two further analogues of the naphthgeranine family, 12-hydroxy-naphthgeranine A (77) and isonaphthgeranine C (78) were later isolated in 2016 from Streptomyces sp. XZYN-4.45 12-Hydroxy-naphthgeranine A (77) was found to possess moderate activity against Aspergillus niger.
Figure 9.

The naphterpin family of meroterpenoids.
In 1992, the farnesyl-derived naphthoquinone natural products debromomarinone (79) and marinone (6) were isolated from the culture broth of an unidentified marine actinomycete Streptomyces sp. CNB-632 (Figure 10).15 Both metabolites show significant in vitro antibacterial activity against Gram-positive bacteria. Marinone (6) shows activity against Bacillus subrifis (MIC 1 μg/mL) and debromomarinone (79) is active against S. aureus, S. epidermis, S. pneumoniae, S. pyogenes and S. epidermis (MIC 1–2 μg/mL). In 2000, three more marinone natural products were discovered: isomarinone (80), hydroxydebromomarinone (81) and methoxydebromomarinone (82).46 These natural products were isolated from a taxonomically-novel marine actinomycete strain CNH-099 found in a sediment sample at Batiquitos Lagoon, California. Isomarinone (80) showed moderate in vitro cytotoxicity against the HCT-116 colon carcinoma cell line.
Figure 10.
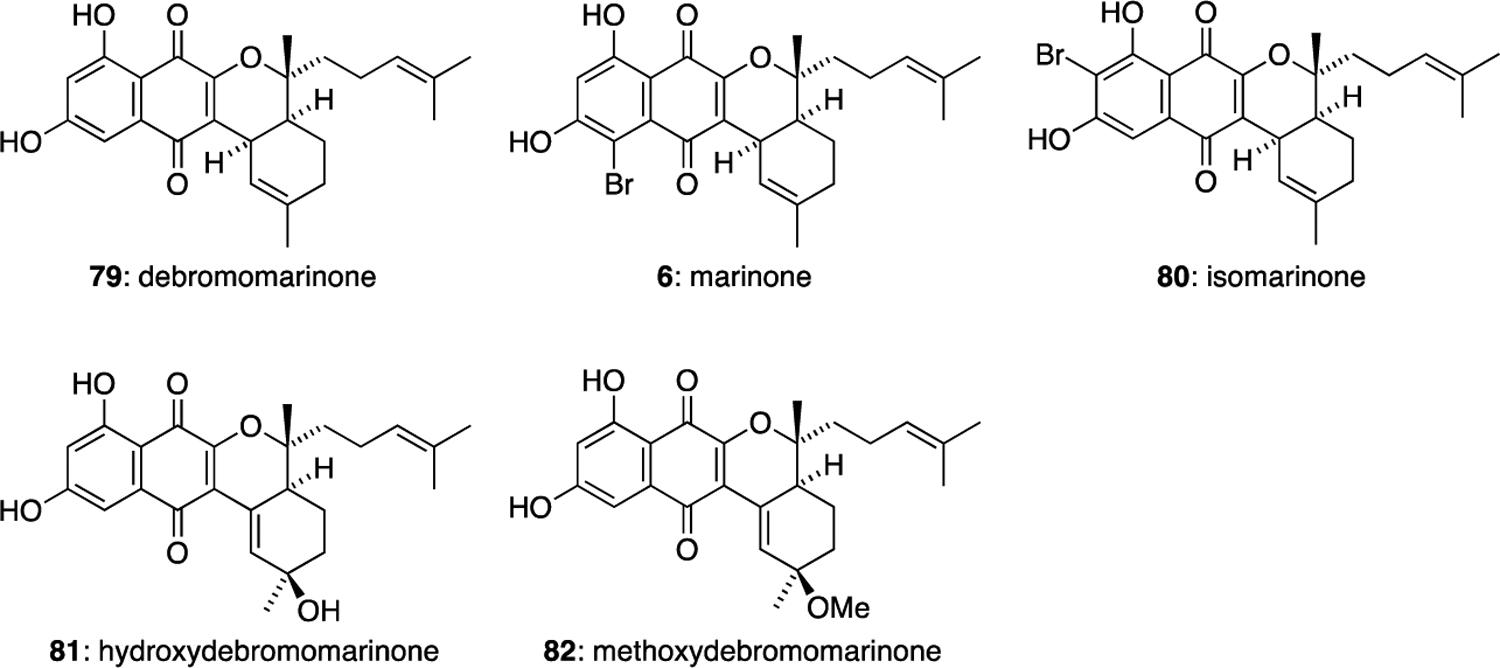
The marinone family of meroterpenoids.
2.4. Furaquinocins, marfuraquinocins and neomarinone
The furaquinocins and the related marfuraquinocins and neomarinone all share a densely functionalised naphthoquinone core fused to a terpene derived dihydrobenzofuran ring system. The furaquinocins A-J differ only in the degree of oxidation of the terpene side chains (Figure 11). Furaquinocins A (3) and B (83) were isolated in 1990 from Streptomyces sp. KO-3988.12 These compounds were not active against Gram-positive or Gram-negative bacteria, fungi or yeast, however 3 and 83 showed cytocidal activities against HeLa S3 cells in vitro at concentrations of 3.1 μg/mL and 1.6 μg/mL, respectively. Furaquinocins C-H (84-89) were later isolated in 1991 from the same Streptomyces sp. KO-3988 strain.47 These meroterpenoids also showed activity against HeLa S3 cells and B1 melonoma cells in vitro. Furaquinocins I (90) and J (91) were isolated in 2011 from Streptomyces reveromyceticus SN-593 strain NRM2.48 PI-220 (92) was isolated from the culture broth of Streptomyces sp. A8059.49 Furaquinocin derivative JBIR-136 (93), with a reduced naphthoquinone ring system, was isolated from Streptomyces sp. 4963H2.50
Figure 11.
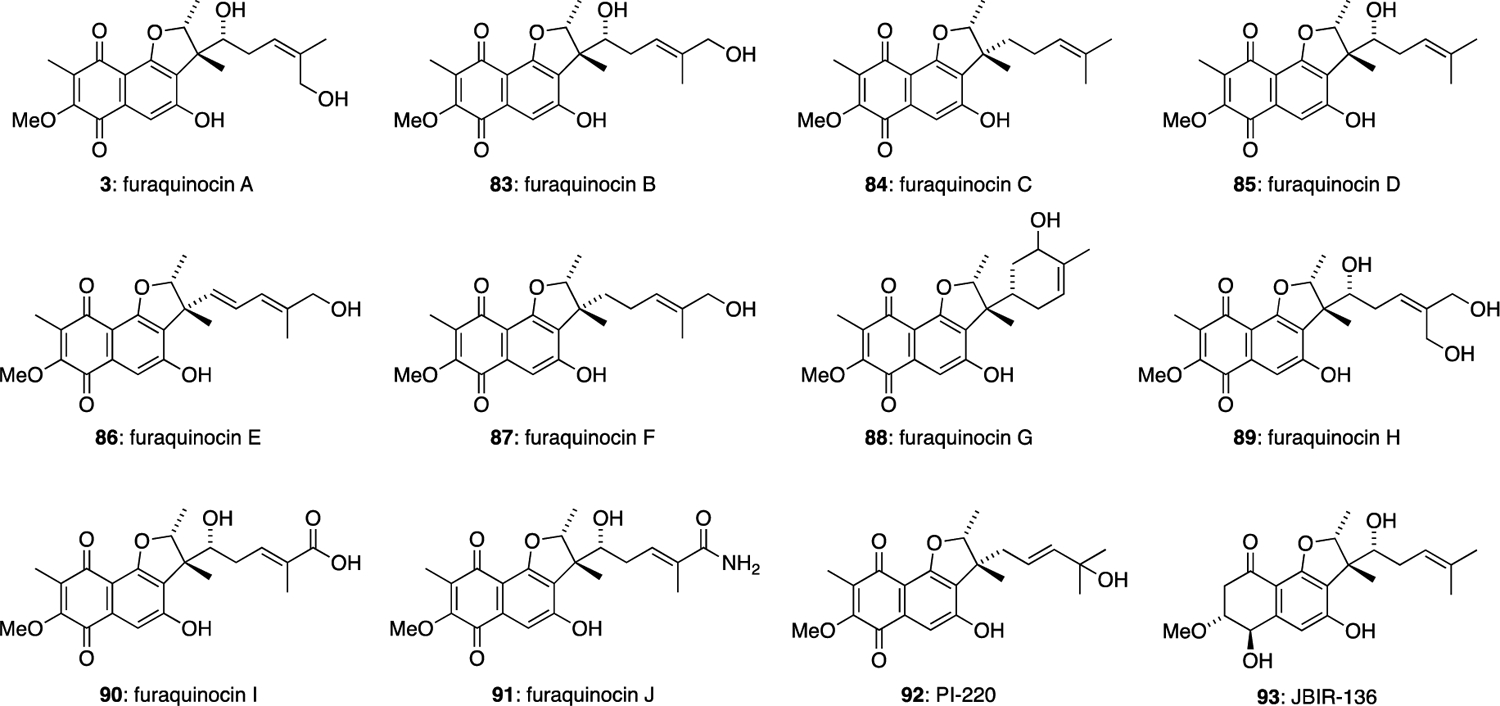
The furaquinocin family of meroterpenoids.
Furanonaphthoquinone I (94) is a regioisomer of furaquinocin C (84), with the benzofuran ring closed at the C-3 phenol (Figure 12). It was isolated from Streptomyces cinnamonensis ATCC 15413.51 The relative configuration of 94 was not determined.
Figure 12.
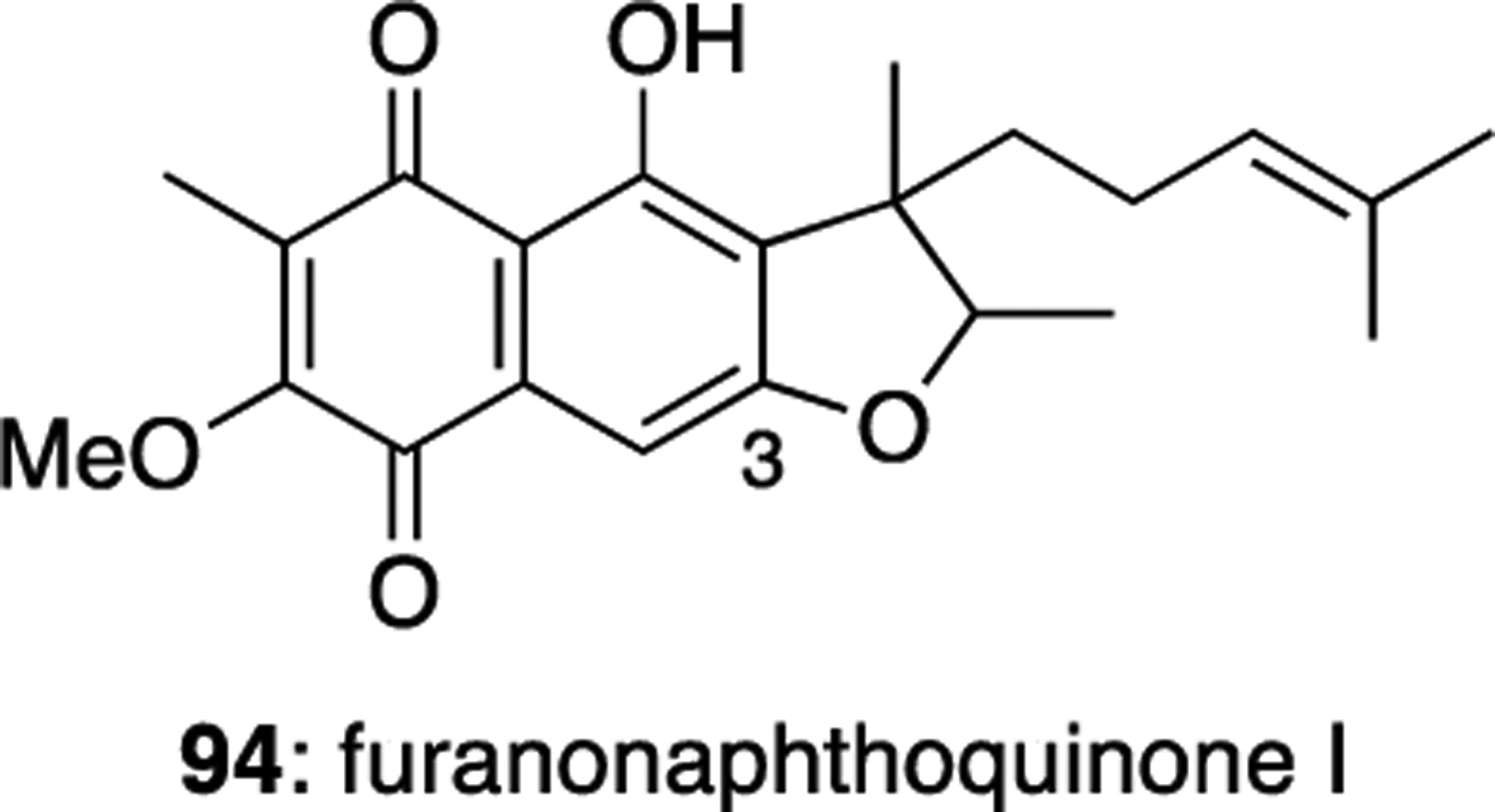
Furanonaphthoquinone I (94).
The marfuraquinocin family (95-98) of meroterpenoids were isolated from the fermentation broth of Streptomyces niveus SCSIO 3406, which originated from a South China Sea sediment sample.52 The marfuraquinocins possess a similar naphthoquinone core to the furaquinocins, but the fused dihydrobenzofuran ring system is derived from a farnesyl, rather than a geranyl, subunit. Further cyclisation of the farnesyl side chain results in a cyclohexane ring in each member of the family. The marfuraquinocins differ only in their configuration at C-13 and their oxidation state at C-11, although the relative configuration at C-16 was also not determined for any of these meroterpenoids (Figure 13). Marfuraquinocins A (95) and C (97) were found to inhibit the cancer cell line NCI-H460, with IC50 values of 3.7 and 4.4 μM, respectively. Marfuraquinocins 95, 96 and 98 exhibited antibacterial activities against S. aureus ATCC 29213 with equivalent MIC values of 8.0 μg/mL. Marfuraquinocins 96 and 98 also showed antibacterial activity against methicillin-resistant Staphylococcus epidermidis (MRSE) shhs-E1 with an MIC value of 8.0 μg/mL.
Figure 13.
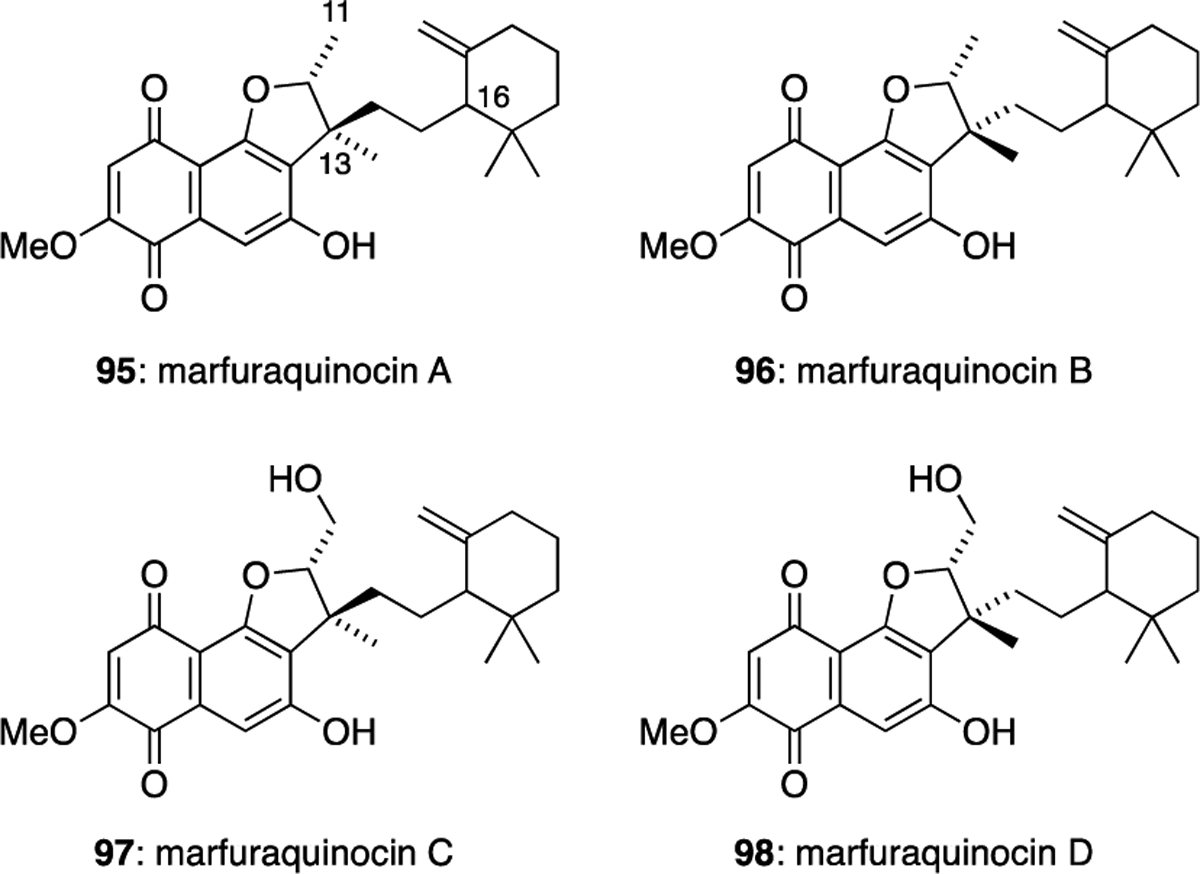
The marfuraquinocin family of meroterpenoids.
Neomarinone (99) was co-isolated with isomarinone from the marine actinomycete strain CNH-099.46 The originally proposed structure of neomarinone was revised to be 99 on the basis of 13C labelling studies, combined with 2D NMR spectroscopy (Figure 14).53 Neomarinone showed moderate in vitro cytotoxicity against HCT-116 with an IC50 value of 10 μM.
Figure 14.
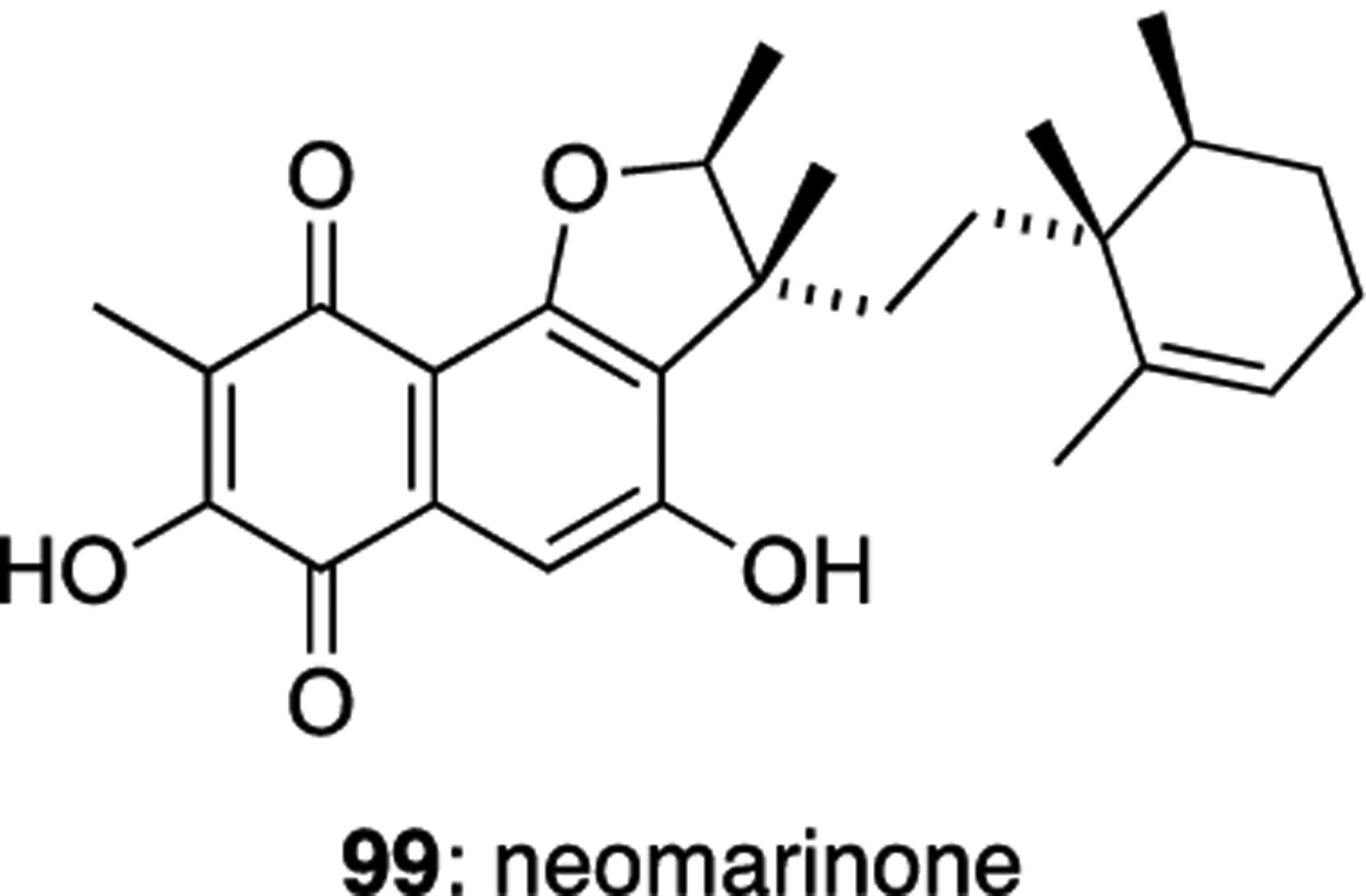
Neomarinone (99).
Arromycin (100) is a meroterpenoid with a slightly different oxidation pattern compared to the furaquinocins and neomarinone (Figure 15), and was produced by a Streptomyces sp. bacterium isolated from an unidentified marine sponge sample collected from Monterey Bay, California.54 This natural product was found to have activity against B. subtilis, E. faecium, L. ivanovii, S. epidermis, S. aureus, and MRSA, with only mild cytotoxic activity against HeLa cells.
Figure 15.
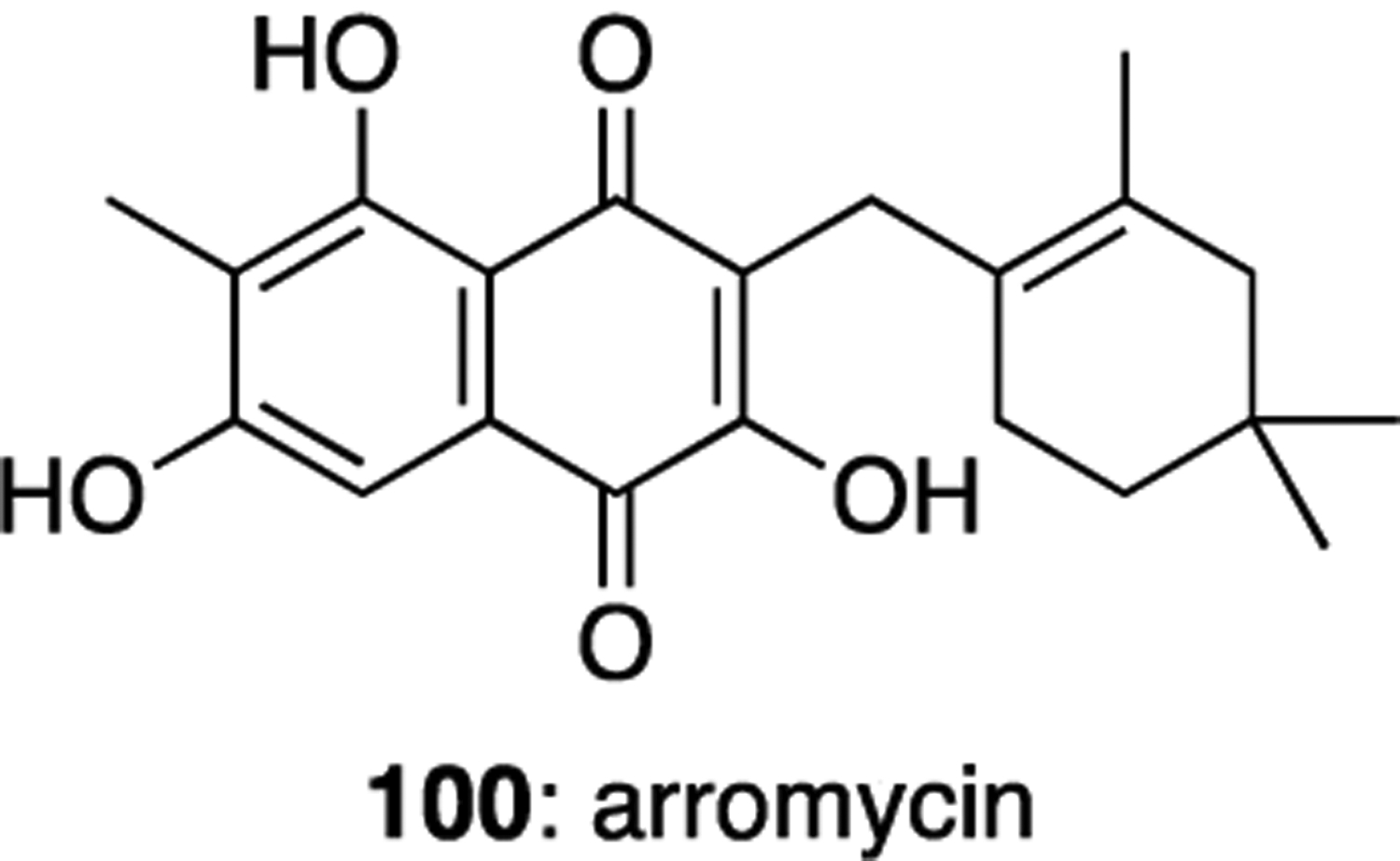
Arromycin (100).
3. Early biosynthetic studies
The early biosynthetic studies of the naphthoquinone-based streptomycete meroterpenoids focused on stable isotope labelling with 13C-labelled acetate to establish their biosynthetic origins. In 1987, Shiomi and colleagues reported preliminary isotope incorporation results with the napyradiomycins only a year after they were first reported.29 Feeding experiments with [2-13C]acetate and [1,2-13C]acetate established that the napyradiomycins are derived from a symmetrical pentaketide, such as 1,3,6,8-tetrahydroxynaphthalene (THN, 1), and two isoprenes, namely the five-carbon dimethylallyl pyrophosphate (DMAPP) and the ten-carbon geranyl pyrophosphate (GPP) (Figure 16). Moreover, the enriched 13C-NMR signals in the napyradiomycin molecules allowed for the authors to reassign multiple carbon assignments in napyradiomycins A1 and C4.
Figure 16.
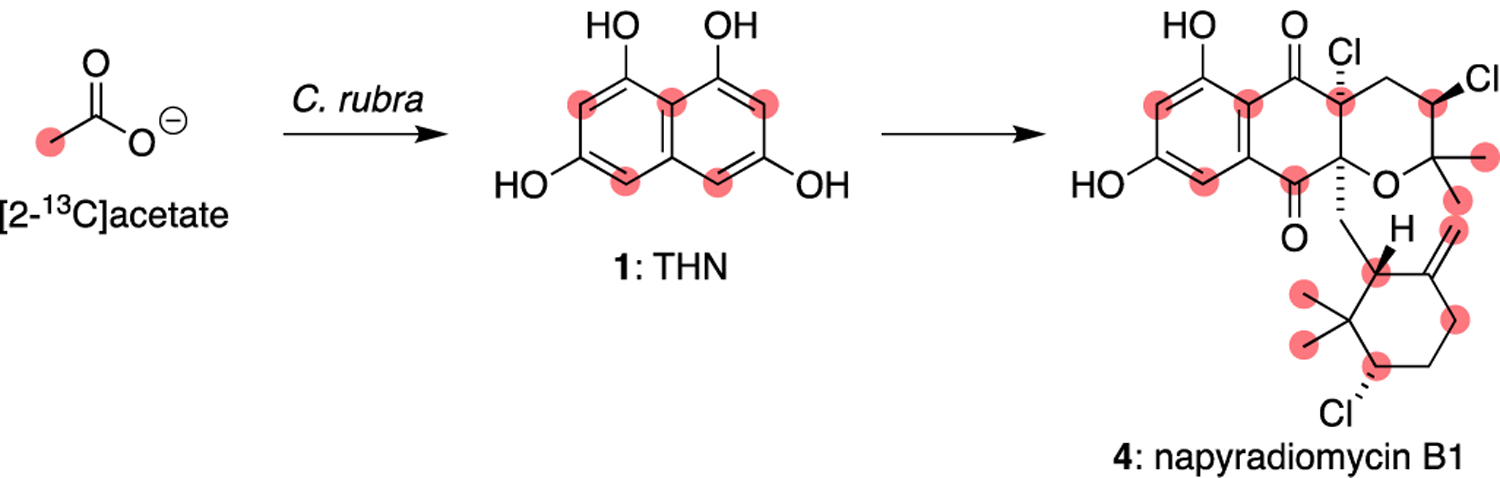
Labelling pattern of napyradiomycin B1 (4) from [2-13C]acetate.29
Soon thereafter, two reports in 1990 by the laboratories of Seto and Omura similarly showed that naphterpin (66)55 and the furaquinocins,56 respectively, most likely also originate from the coupling of THN and GPP precursors (Figures 17 and 18). In all three examples, the isotope labelling patterns established that the isoprene units originate from the mevalonate pathway.
Figure 17.
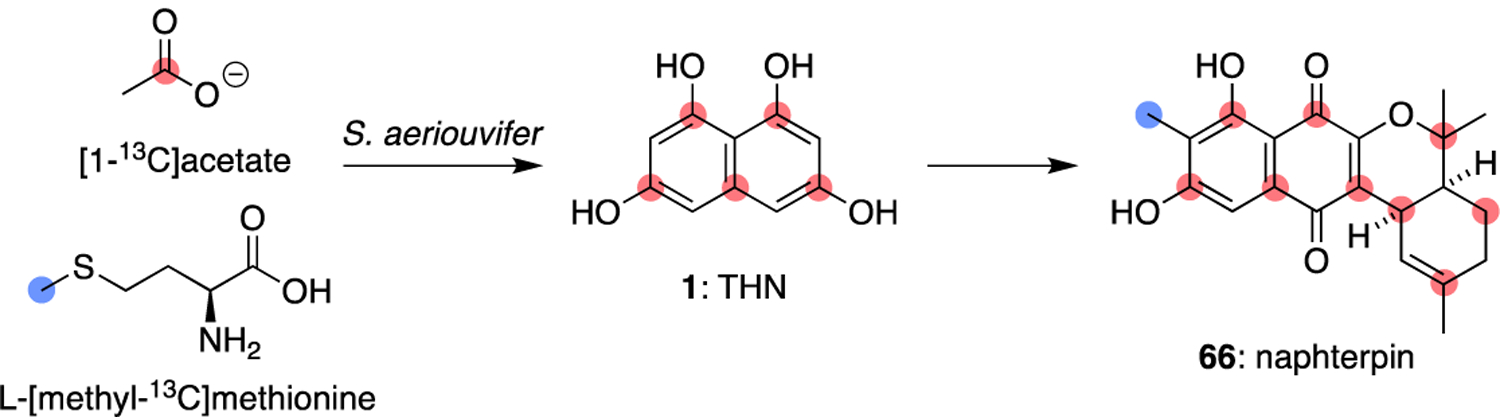
Labelling pattern of naphterpin (66) from [1-13C]acetate and L-[methyl-13C]methionine.55
Figure 18.
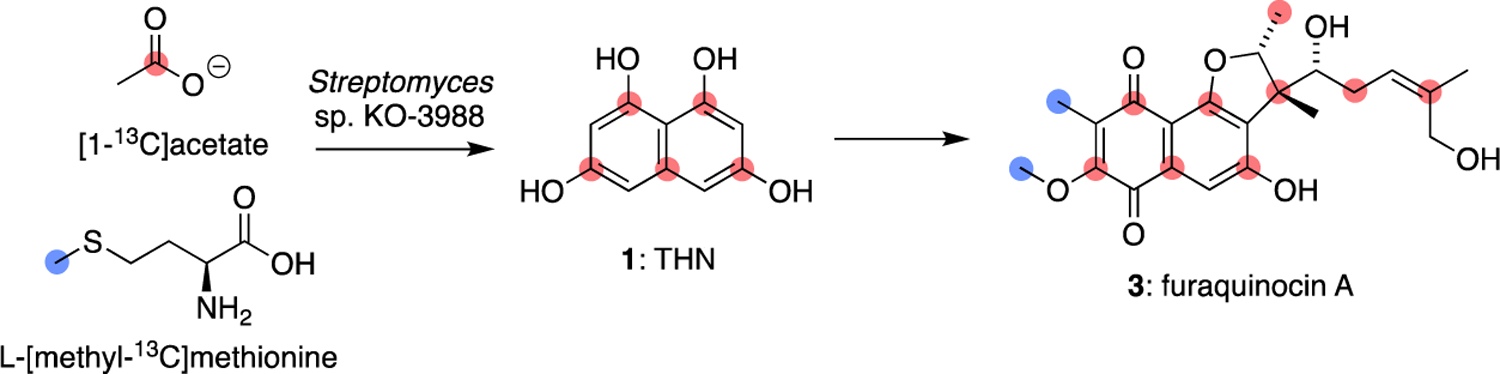
Labelling pattern of furaquinocin A (3) from [1-13C]acetate and L-[methyl-13C]methionine.56
Debromomarinone (79) and neomarinone (99) offered a slight departure regarding the origin of their isoprene units as reported by Moore in 2003.53 While 13C-acetate symmetrically labelled the naphthoquinone moieties indicative of a THN building block, the sesquiterpene-derived carbons of 79 and 99 showed no enrichment (Figure 19). Feeding experiments with 13C-labelled glucose alternatively enriched the terpenoid fragments consistent with a non-mevalonate or methyl D-erythritol-4-phosphate pathway origin of its farnesyl pyrophosphate (FPP) precursor. The 13C-enrichment in the isoprene sidechain of 99 moreover had the added benefit of revealing that the original structure proposed by Fenical46 was incorrect and should be revised as depicted in Figure 19.53
Figure 19.
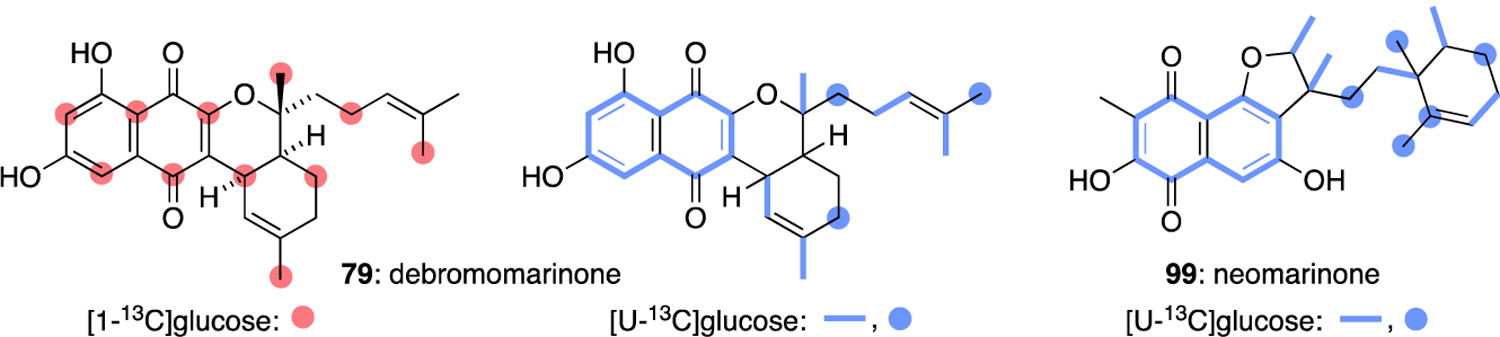
Incorporation of 13C-labelled precursors in debromomarinone (79) and neomarione (99).53
The first connection of clustered biosynthesis genes to chemistry for a naphthoquinone-based meroterpenoid from a Streptomyces bacterium came in 2006 with a pair of papers by the groups of Dairi11 and Heide57 who reported the biosynthetic gene clusters for the similarly structured furaquinocin A (3) and furanonaphthoquinone I (94), respectively. The following year, the Moore group discovered and validated the napyradiomycin biosynthetic gene cluster via heterologous expression studies.10 These early meroterpenoid gene cluster studies confirmed that the common THN fragment is derived from a type III polyketide synthase, namely THN synthase, that had only been discovered in bacteria a few years prior by Horinouchi and colleagues.7 THN synthase is a homodimeric enzyme8 responsible for pigmented melanin production in Streptomyces bacteria in addition to being integral in meroterpenoid biosynthesis. The gene clusters also similarly harboured a complete set of genes encoding isoprenoid biosynthesis via the mevalonate pathway as well as an ABBA prenyltransferase (or two in the case of napyradiomycin) that was first established for naphterpin biosynthesis by Kuzuyama.58 The napyradiomycin biosynthetic gene cluster also featured genes encoding vanadium-dependent haloperoxidases (VHPOs), the first of its kind identified (and later biochemically validated) from a bacterium. These rare prokaryotic halogenating enzymes would later be shown to be synonymous with chlorinated meroterpenoids and become instrumental biocatalysts for the enzymatic construction of these natural products.
4. Early total syntheses of the napyradiomycins
In 1999, Nakata and co-workers reported the first total synthesis of a napyradiomycin natural product, A80915G (8).18 Nakata anticipated that the geranyl and prenyl substituents of 8 could be installed using sequential Stille reactions of a trihalogenated benzene derivative, followed by an intermolecular Diels-Alder reaction to form the desired naphthoquinone core (Scheme 2). The synthesis began with commercially available 2,5-dimethoxy-4-nitroaniline (101) which was converted into the chlorinated intermediate 102 over two steps. Successive bromination and iodination (via intermediate 103), formed the trihalogenated 1,4-dimethoxybenzene derivative 104 in four steps. After extensive optimisation of the reaction conditions, the selective Pd catalysed Stille cross-coupling between geranyl tributyltin 105 and 104 occurred exclusively at the C-I bond to give 106. Prenylation with the analogous prenyl tributyltin 107 under similar Stille reaction conditions was then selective for the C-Br over the C-Cl bond of 106, affording 108 in good yield. Oxidative demethylation with AgNO3 gave the unstable quinone 109, which was immediately subjected to an intermolecular Diels-Alder reaction. Several dienes were assessed, however a successful cycloaddition was only achieved between quinone 109 and Brassard diene 110, with the desired aromatised adduct 111 formed after treatment with aqueous HCl and heating in DMF. Following protection of the free phenol of 111 as a methoxymethyl (MOM) ether to give 112, the epoxide of the target molecule was installed under basic conditions with H2O2 to give 113. Subsequent deprotection of the two phenolic protecting groups with BCl3 completed the total synthesis of (±)-A80915G (8).
Scheme 2.
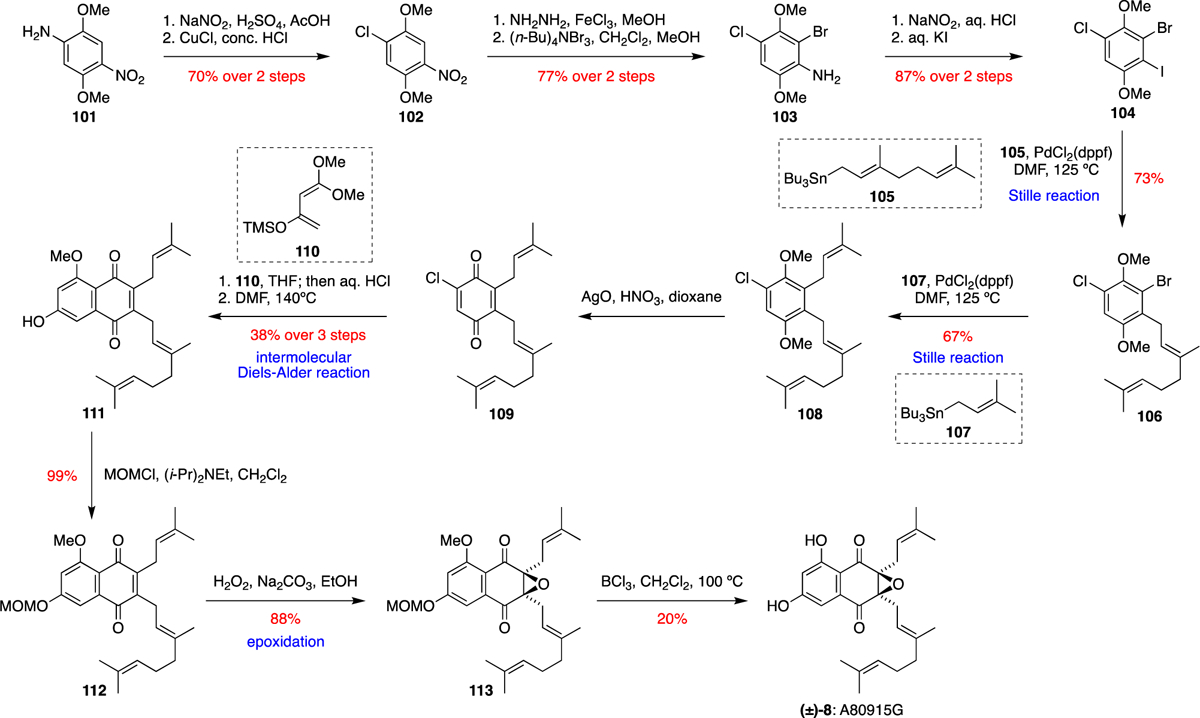
Nakata’s total synthesis of (±)-A80915G (8).18
The first total synthesis of (±)-napyradiomycin A1 (14) was completed by Tatsuta et al. in 2002 via a route that utilised a Hauser annulation, followed by stereo- and regio-selective chlorinations to furnish the natural product’s pyranonaphthoquinone core (Scheme 3).19 The synthesis commenced with the quantitative conversion of commercially available 2,4-dihydroxybenzoic acid (114) to the TBS-protected N,N-diethylamide 115 in three steps. Formylation of 115 was achieved by ortho-lithiation of the amido group followed by a quench with DMF, and addition of PhSO2Na to the resultant aldehyde then gave lactone 116. Methoxymethyl (MOM) protection of the free phenols of 116 then afforded 117. With this intermediate in hand, the key Hauser annulation, which follows a Michael-Dieckmann cascade mechanism, was achieved by deprotonation of 117 with t-BuOLi, followed by addition of the Michael acceptor 118. The initially formed hydroquinone was then oxidised with MnO2 to afford naphthoquinone 119. α-Chlorination of 119 with SOCl2 gave 120, and then installation of the geranyl side chain at C-3 was achieved by a Michael addition of a geranyllithium species, generated in situ by transmetallation of geranyl tributyltin (121). The Michael addition reaction was favoured on the opposite face to the C-12 chlorine substituent, giving intermediate 122 with good diastereoselectivity. Selective deoxygenation of 122 at C-11 proved difficult by direct methods, demanding the use of a four-step protocol. Ketone 122 was first chlorinated with NCS to give intermediate 123, followed by reduction with KHBPh3 to afford alcohol 124. Treatment with 2,2’-dipyridyldisulfide and tributylphosphine afforded α,β-unsaturated ketone 125, followed by selective 1,4 hydride reduction with KHBPh3 to give 126. The final diastereoselective chlorination at C-2 was achieved by treatment of 126 with KHMDS and NCS. Finally, MOM deprotection completed the first total synthesis of (±)-napyradiomycin A1 (14).
Scheme 3.
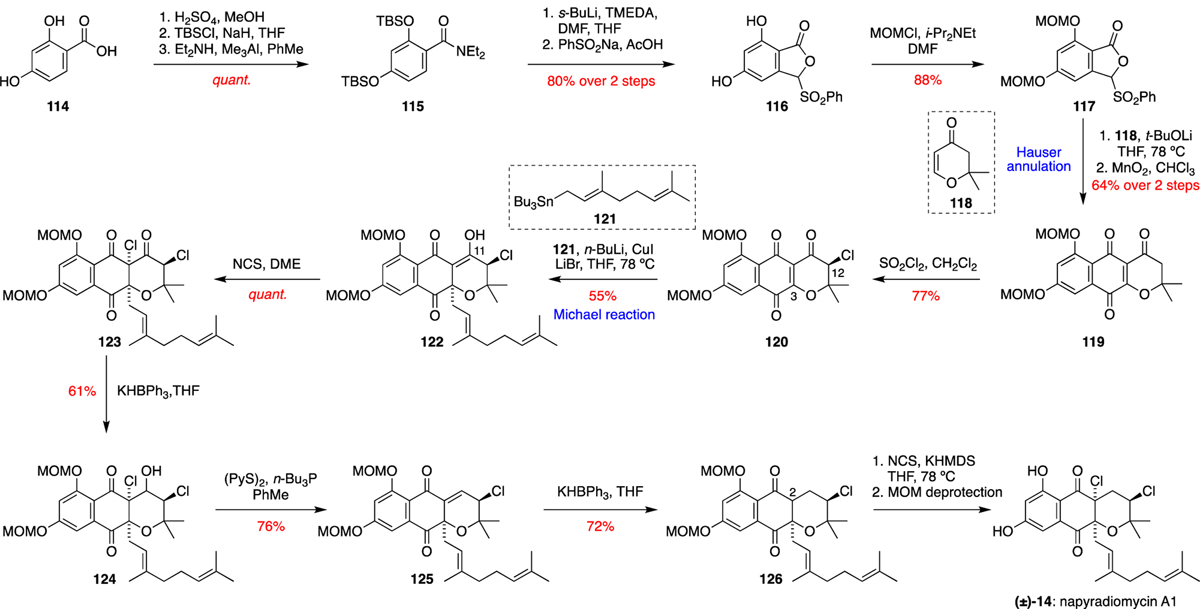
Tatsuta’s total synthesis of (±)-napyradiomycin A1 (14).19
The first enantioselective total synthesis of (–)-napyradiomycin A1 was achieved by Snyder and co-workers in 2009.20 This bio-inspired approach utilised flaviolin (2) as a starting material and incorporated a novel asymmetric chlorination reaction as a key step (Scheme 4). The synthesis of 2 was achieved by an alkali fusion reaction of commercially available chromotropic acid sodium salt (127). The initially formed THN (1) was readily oxidised in air at room temperature to form 2. With the naphthoquinone core of the napyradiomycins already established, treatment of 2 with 3-methylcrotonaldehyde (128) and EDDA resulted in successive Knoevenagel condensation and oxa-6π-electrocyclisation reactions to give a chromene intermediate that was selectively MOM protected at the non-hydrogen bonded C-6 phenol to give 129. Asymmetric alkene chlorination was then accomplished with Cl2, BH3·THF and AcOH in the presence of the chiral ligand (S)-130 to afford the vicinal dichloride 131 in excellent yield and enantioselectivity. Substitution of the C-11 chloride substituent with acetate occurred with retention of stereochemistry to give 132, which was O-methylated and hydrolysed to give 133. The key alkyl substituent at C-3 was then introduced using an acid-catalysed Johnson-Claisen rearrangement employing excess CH3C(OMe)3. The [3,3] sigmatropic rearrangement of the intermediate enol ether 134 afforded methyl ester 135 with the desired relative configuration at C-3. A series of functional group interconversions then converted methyl ester 135 to aldehyde 136 in three steps. Wittig olefination of 136 with the ylide derived from phosphonium salt 137 gave the geranylated intermediate 138. Installation of the C-2 chlorine was then achieved using Tatsuta’s conditions to generate compound 139, and finally a two-step deprotection protocol yielded (–)-napyradiomycin A1 (14).
Scheme 4.

Snyder’s total synthesis of (–)-napyradiomycin A1 (14).20
Although uninformed by firm knowledge of napyradiomycin biosynthesis, these three elegant total syntheses feature several creative synthetic solutions for the napyradiomycin molecular architecture, including successive chemoselective Stille couplings in Nakata’s work, a regioselective Hauser annulation in Tatsuta’s synthesis, and an enantioselective alkene chlorination in Snyder’s synthesis. Furthermore, each of these total syntheses highlights the utility of MOM protecting groups for the C-6 and C-8 phenols, which can be removed under mildly acidic conditions. This protecting group strategy was later used in successful biomimetic syntheses of the napyradiomycins and marinones.
5. Biosynthetic studies and biomimetic total syntheses of the merochlorins
Merochlorins A and B were the first THN-based meroterpenoids to have their biosyntheses fully established. This insight would eventually lead to several biomimetic syntheses as well as a one-pot enzymatic total synthesis. Moreover, the establishment of the merochlorin biosynthetic logic would ultimately help unlock many questions in how meroterpenoids are universally assembled in nature. Fenical and Moore combined forces to report the structures and biosynthesis genes of the merochlorin family of molecules in 2012.9 The 57-kb merochlorin biosynthetic gene cluster mcl was cloned and successfully expressed in the heterologous host strain Streptomyces coelicolor to produce the merochlorins. Together with gene knockout experiments in the native Streptomyces CNH-189 strain on the THN synthase-encoding mcl17, these studies identified the genes responsible for the construction of the merochlorin molecules. That foundational genetic work led to the biochemical characterisation of four enzymes responsible for converting five molecules of malonyl-coenzyme A, DMAPP (140), and GPP (141) to merochlorins A (5) and B (58) (Scheme 5).59 Other than the THN synthase Mcl17, the remaining three enzymes were all novel at the time of their reporting. The branched isoprene unit was shown to be assembled from an unusual head-to-torso condensation of 140 and 141 to give the newly identified isoprene building block isosesquilavandulyl pyrophosphate (142) by the Mcl22 prenyltransferase.60,61 The C-4 alkylation of THN (1) with the branched sesquiterpene substrate 142 was shown to be highly preferred over FPP by the prenyltransferase Mcl23 to give pre-merochlorin (143). Quite extraordinarily, the VHPO enzyme Mcl24 singlehandedly converted 143 to merochlorins A (5) and B (58).
Scheme 5.
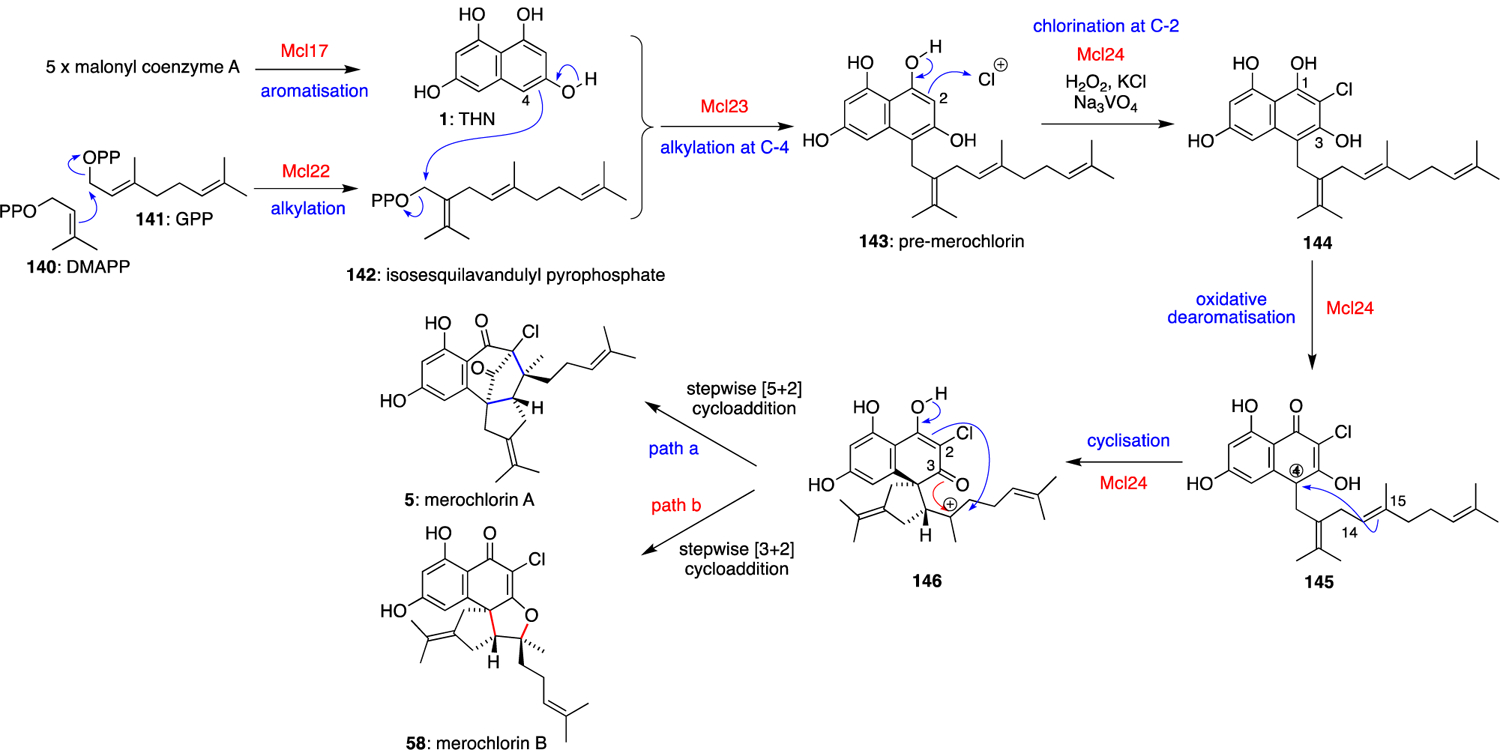
Biosynthesis of merochlorins A (5) and B (58).
Synthetically, this cascade of reactions began with C-2 mono-chlorination of pre-merochlorin (143) to give 144. Subsequent oxidative dearomatisation gave carbocation 145, which after cyclisation to 146, could then undergo stepwise [5+2] or [3+2] cycloaddition to give merochlorin A (5) or B (58), respectively. These enzymes, substrates, and cofactors could also be bundled together into a single reaction vessel to achieve the efficient enzymatic total synthesis of the merochlorins.
Intriguingly, the structural complexity initiated by Mcl24 in the construction of merochlorins A and B showed further chemical diversity depending on the reaction pH.62,63 At all interrogated pHs in the presence of its requisite cosubstrates and cofactors, Mcl24 catalyses the C-2 mono-chlorination and oxidative dearomatisation of pre-merochlorin (143). However, under mildly basic conditions (pH 8.0), the dearomatised intermediate is preferentially hydrated at C-4 to give tertiary alcohol 147 instead of being intercepted by an alkene carbon (Scheme 6). Performing the reaction in the presence of H218O supported the hypothesis that water was the source of this C-4 hydroxyl group instead of co-substrate hydrogen peroxide or atmospheric oxygen. Mcl24 then facilitates a second chlorination at C-2 on this dearomatised substrate 147 to give 148, and a subsequent enzyme-catalysed α-hydroxyketone rearrangement to give 149. This novel in vitro metabolite 149 possesses a di-chlorinated naphthoquinone moiety and C-3 attached prenyl substituent. This 1,2-suprafacial shift is thermodynamically favorable only following di-substitution at the C-2 position in the THN-derived ring, established via both computational and experimental analyses.63 While unexpected, the chemical logic established within this pH-dependent 1,2-alkyl shift rationalised the biosynthesis of the abundant C-3 prenylated metabolites within this natural product family. Initial biosynthetic hypotheses suggested that “promiscuous” prenyltransferases were responsible for this chemically counterintuitive electrophilic prenylation on an electrophilic C-3 carbonyl-derived carbon.58 Instead, this prenylation, dearomatisation and α-hydroxyketone rearrangement sequence constructs the substitution pattern present in many of these THN-derived meroterpenoids.
Scheme 6.

An unexpected α-hydroxyketone rearrangement under basic pH conditions observed in the action of Mcl24 on pre-merochlorin (143).
The chemical logic established within the pH dependence of Mcl24 further rationalised the biosynthesis of merochlorins C (59) and D (60), the two naphthoquinone metabolites isolated from this family. However, instead of Mcl24-catalysed C-2 dihalogenation, the mono-chlorinated, dearomatised, and hydrated product 147 would be methylated at C-2 stereoselectively by a SAM-dependent methyltransferase to give 150 (Scheme 7). Di-substitution at this position enables Mcl24 to facilitate an analogous α-hydroxyketone rearrangement of 150 to generate merochlorin D (60). Genetic evidence has implicated mcl40, a second merochlorin VHPO gene, in the entropically-challenging 15-membered macrocyclic chloro-ether ring formation in the biosynthesis of merochlorin C (59).59 However, any production of 59 by this putative activity has yet to be confirmed using in vitro recombinant enzymology due to protein insolubility and expression issues.
Scheme 7.

Proposed biosynthesis of merochlorins C (59) and D (60).
The first total synthesis of merochlorin A completed in 2013 by Pepper and George closely anticipated its biosynthetic pathway.64 Pivotal to the success of the synthesis was the early introduction of the merochlorin A chlorine substituent at C-2 via the Friedel-Crafts acylation of methyl 3,5-dimethoxyphenylacetate (151) with chloroacetyl chloride to give chloroketone 152 (Scheme 8). Base-induced aromatisation of 152 via a Dieckmann cyclisation gave phenolate anion 153, which was directly alkylated at C-4 with “isosesquilavandulyl bromide” 154 (accessed in three steps from geranyl bromide) to give 155. Unexpectedly, some isomerisation of the C14–C15 alkene occurred during the alkylation under basic conditions. Oxidative dearomatisation of 155 with Pb(OAc)4 in CHCl3 then gave 156, the dimethyl ether of merochlorin A, in 50% yield. Under these conditions, no formation of the merochlorin B ring system was observed, indicating preference for the [5+2] cyclisation over the [3+2] cyclisation pathway with this substrate. Finally, deprotection of 156 was achieved via two successive Krapcho demethylations to give racemic merochlorin A (5).
Scheme 8.

George’s biomimetic total synthesis of (±)-merochlorin A (5).64
Shortly after the first total synthesis of merochlorin A, Trauner and co-workers reported the first synthesis of merochlorin B,65 which followed a similar biosynthetically-inspired blueprint (Scheme 9). Friedel-Crafts acylation of 151 using acetyl chloride gave 157, which was aromatised under basic conditions to give the THN derivative 158. Tandem silylation of 158 followed by directed lithiation and chlorination at C-2 gave 159, which was selectively demethylated using BBr3 in the presence of proton sponge to give naphthol 160. Alkylation of 160 at C-4 using alkyl bromide 154 gave 161, the substrate for the key oxidative cyclisation. Although the structure of 161 is very similar to compound 155 used in the George synthesis of merochlorin A, the outcome of oxidative dearomatisation was very different in this case, as treatment with a variant of Koser’s reagent PhI(OH)OTs (generated in situ) gave the protected merochlorin B derivative 162. Clearly, the methyl ether substituent of 161 favours the [3+2] instead of the [5+2] cyclisation pathway by decreasing the nuclophilicity of the C-2 position, and no merochlorin A scaffolds were formed in the oxidative cyclisation. Finally, desilylation of 162 using TBAF completed this concise total synthesis of racemic merochlorin B (58).
Scheme 9.

Trauner’s biomimetic total synthesis of (±)-merochlorin B (58).65
Alongside their elucidation of merochlorin biosynthesis, the Moore group reported a highly divergent and protecting-group-free biomimetic synthesis of merochlorins A and B, plus several analogues (Scheme 10).66 Conjugate addition of in situ generated lithium dimethyl cuprate to ethyl 2-butynoate (163) followed by quenching of the resultant enolate with geranyl bromide (164) gave ester 165, which was reduced with DIBAL-H to give isosesquilavandulol (166). Conversion of 166 into its carbonate 167 allowed its direct coupling to unprotected THN (1) via a Pd catalysed allylation in the presence of BEt3 to give pre-merochlorin (143). Notably, these Pd catalysed coupling conditions did not cause partial isomerisation of the C14–C15 alkene, unlike previous merochlorin total syntheses. Inspired by the multi-functional Mcl24 VCPO enzyme, the Moore group investigated chemical conditions for the chlorination and oxidative cyclisation of 143 to generate merochlorins A and B. After much experimentation, it was found that stoichiometric quantities of N-chlorosuccinimide (NCS) and i-Pr2NH converted 143 into a mixture of merochlorins A and B (5 and 58), alongside deschloro-merochlorin A (168), deschloro-merochlorin B (169), isochloro-merochlorin B (170) and dichloro-merochlorin B (171), which were later confirmed as natural products produced by the merochlorin-producing bacterium Streptomyces sp. CNH-189. This chemical chlorination / oxidative cyclisation cascade is clearly less selective than the Mcl24 catalysed reaction, with optional chlorination of 143 at C-2 and/or C-7 preceding cycloaddition onto the C14–C15 alkene.
Scheme 10.
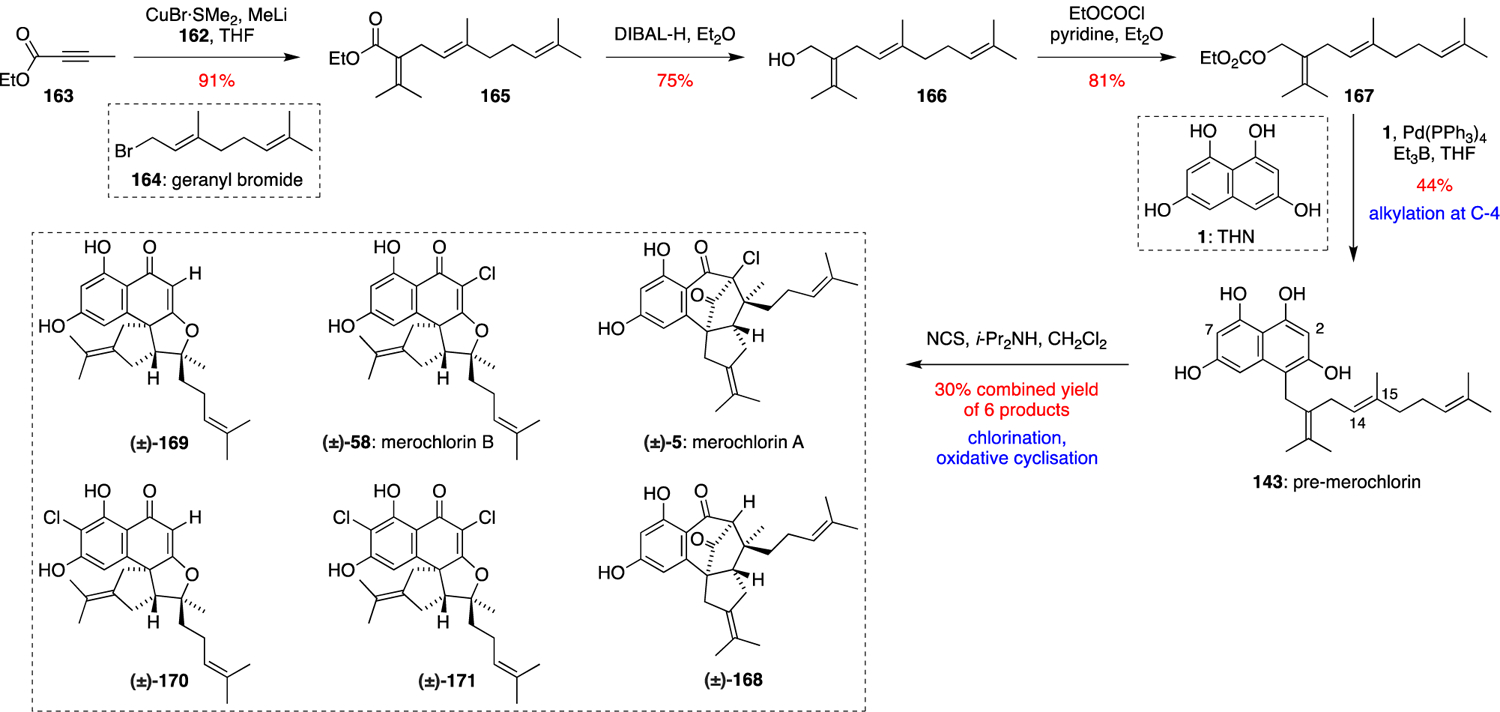
Moore’s biomimetic total synthesis of (±)-merochlorins A (5) and B (58).66
In 2016, Zhang and Tang reported an efficient, divergent total synthesis of merochlorins A and B (Scheme 11).67 Unlike previous syntheses, their approach hinged on oxidative dearomatisation and cyclisation of a functionalised resorcinol derivative, followed by late-stage intermolecular Diels-Alder reactions to complete the total syntheses. Thus, 2-chlororesorcinol (172) was combined with tertiary allylic alcohol 173 via a Friedel-Crafts alkylation to give 174. Oxidative cyclisation of 174 using Pb(OAc)4 then gave a separable mixture of merochlorin A analogue 175 and merochlorin B analogue 176 in a combined yield of 40%. The use of hypervalent iodine oxidants such as PhI(OAc)2 gave slightly inferior results in this divergent reaction. Intermolecular Diels-Alder reaction of tricycle 175 with the Brassard diene 177 gave tetracycle 178. Oxidation of 178 with I2 in MeOH gave di-O-methylated merochlorin A (156), which was deprotected to give racemic merochlorin A (5) using LiCl in DMF according to George’s two-step procedure. Synthesis of merochlorin B (58) was achieved from tricycle 176 via a similar sequence involving intermolecular Diels-Alder reaction with Brassard diene 177, followed by Saegusa oxidation and LiCl-mediated demethylation.
Scheme 11.

Zhang and Tang’s biomimetic total synthesis of (±)-merochlorins A (5) and B (58).67
More recently, the Carreira group achieved the first enantioselective total synthesis of merochlorin A using a non-biomimetic approach (Scheme 12).68 Their synthesis was designed around a key Au(I)-catalysed cascade reaction of an enynyl acetate intermediate 184. This intermediate was prepared via a modified Wittig-Schlosser reaction between the ylide formed from triphenylphosphonium bromide 179 and aldehyde 180. The resultant E-alkenyl iodide 181 was coupled with (R)-182 using a Sonogashira reaction to give 183, which was converted into the key substrate 184 via a series of functional group interconversions. Notably, from this point in the synthesis the sole stereocentre of 184 dictated the formation of (–)-merochlorin A as a single stereoisomer. Au(I)-catalysed cyclisation of 184 using catalyst 185 gave bicyclic ketone 186 via a spectacular cascade featuring successive 1,3-acyloxy migration, Nazarov cyclisation and aldol reaction steps. Triflation and elimination of the secondary alcohol of 186 followed by Tsuji-Wacker oxidation gave diketone 187, which underwent La(III)-mediated Grignard addition of isopropenylmagnesium bromide to the most reactive carbonyl followed by reduction to give tetra-substituted alkene 188. Conjugate addition of the higher order cyanocuprate derived from alkyl iodide 189 to the enone of 188 gave a ketone product as a single diastereomer via addition to the concave face of the bicyclic ring system. Chlorination was achieved by the reaction of “Palau’Chlor” 190 with an intermediate TMS-enol ether, and then hydrolysis of the benzoate ester protecting group gave primary alcohol 191. Oxidation of 191 using PDC in DMF gave a carboxylic acid, which cyclised to give enol lactone 192 on exposure to NaOAc in Ac2O. Reduction of the lactone functionality of 192 with DIBAL-H then gave an intermediate lactol, which broke down to give an enolate-aldehyde species that underwent an intramolecular aldol reaction to form the key bicyclo[3.2.1]octane core of 193. Oxidation of 193 with PCC and desaturation of the resultant ketone using Mukaiyama’s conditions employing phenylsulfinimidoyl chloride 194 then gave enone 175, an intermediate previously synthesised in racemic form in Zhang and Tang’s total synthesis of merochlorin A (Scheme 11). Therefore, a similar end game was then employed by the Carreira group, with an intermolecular Diels-Alder reaction between 175 and Brassard’s diene 177 followed by Saegusa oxidation to give 195, and a final demethylation with LiCl in DMF to give (–)-merochlorin A (5). Although this total synthesis is significantly longer than the earlier, biomimetic approaches, its enantioselectivity allowed the absolute configuration of natural (+)-merochlorin A to be established.
Scheme 12.
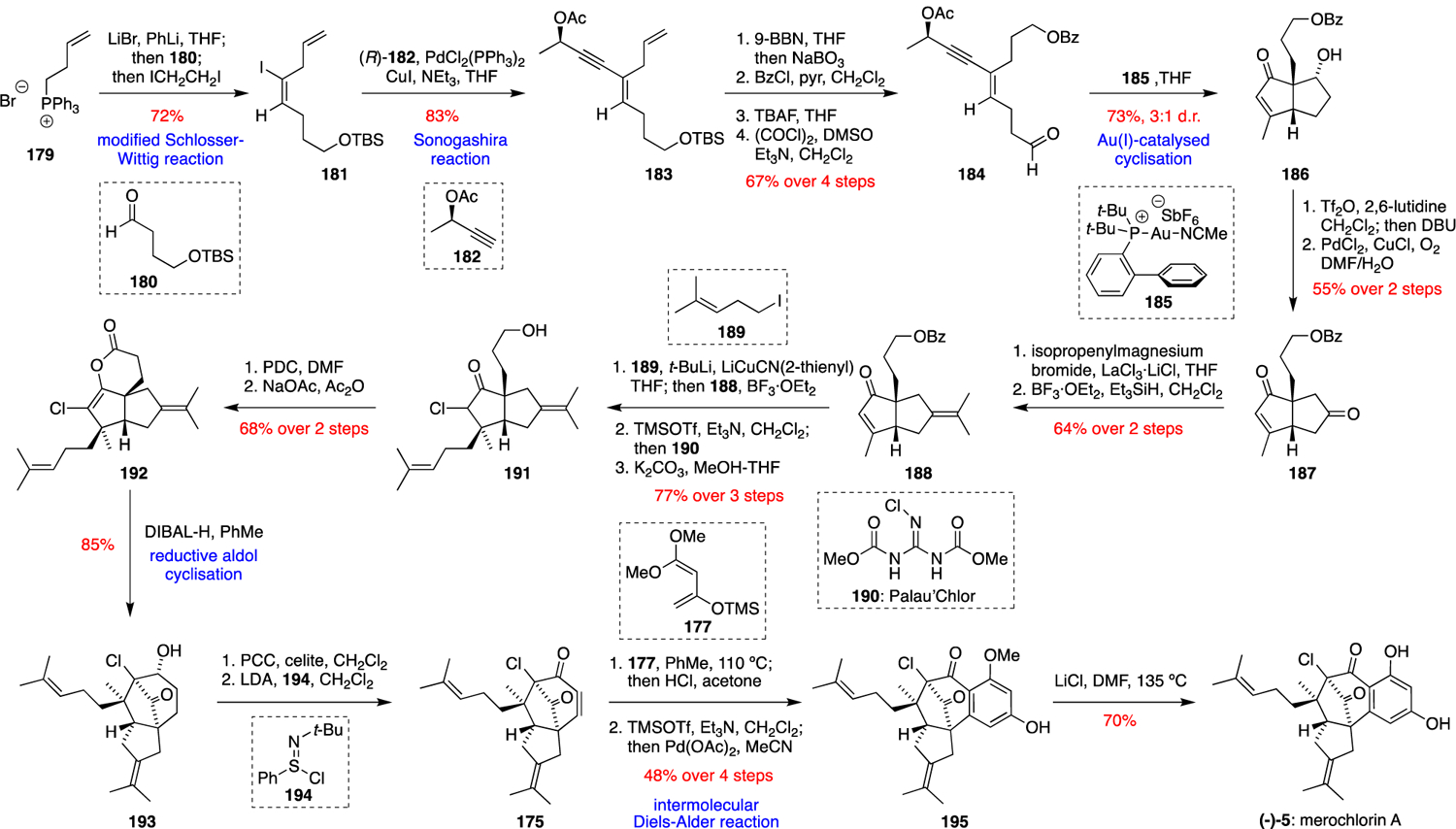
Carreira’s enantioselective total synthesis of (–)-merochlorin A (5).68
6. Biosynthesis and biomimetic total synthesis of the napyradiomycins
The napyradiomycins were the first family of THN-derived meroterpenoids to be definitively linked to their biosynthetic gene clusters via cosmid capture and heterologous expression in a Streptomyces albus host.10 The presence of three VHPO gene homologues in close proximity to the THN synthase suggested their involvement in installing the various chiral chlorinated stereocentres; however this proposal by Moore was novel at the time of initial report, as previously characterised fungal vanadium-dependent chloroperoxidases (VCPOs) failed to exhibit any regio- or enantioselective halogenation activity. While the initial biosynthetic hypothesis has been refined over the past decade, much of the chemical logic established in merochlorin biosynthesis translates to the napyradiomycins (Scheme 13). First, C-4 geranylation of the THN (1) ring with GPP (141) gives 196, which undergoes an analogous C-2 chlorination to give 197 and then dearomatisation and hydration to form 198. A divergent DMAPP (140) C-2 alkylation would enable the required C-2 disubstitution necessary to facilitate the α-hydroxyketone rearrangement of 199 to naphthomevalin (7), the structurally simplest halogenated napyradiomycin family member. Sequential chloronium-mediated cyclisations with either a hydroxyl or alkenyl nucleophile would generate napyradiomycins A1 (14) and B1 (4) respectively, with putative VHPO enzymology establishing the stereocontrol over the majority of these challenging synthetic transformations.
Scheme 13.
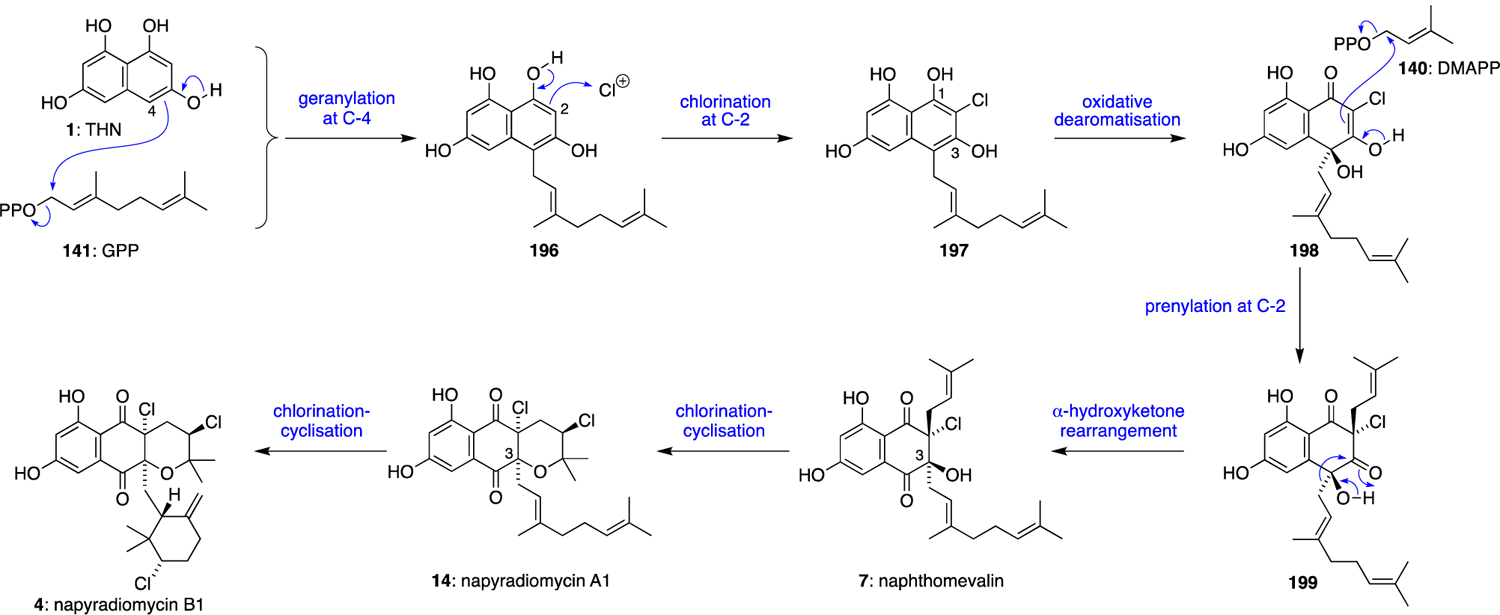
Proposed biosynthesis of the napyradiomycins.
Moore and colleagues reported the first instance of a stereospecific VCPO in natural product biosynthesis in 2011.69 After the isolation of SF2415B1 (10) from microbial cultures, NapH1 facilitated a stereoselective halogenation of the prenyl alkene on this substrate, followed by ring opening of the chloronium intermediate (200) via a 6-endo-tet cyclisation with the adjacent C-3 hydroxyl nucleophile (Scheme 14). The chlorinated tetrahydropyran-containing product matched tricyclic natural product SF2415B3 (25) by all measured parameters, including HPLC retention time, mass spectrometry and NMR spectroscopy. Intriguingly, NapH1 catalysis was not limited to solely using chloride as the halide source; substitution with KBr facilitated an analogous bromonium-induced etherification at a comparatively faster rate due to the ease of oxidising the less electronegative bromide, giving bromo-SF2415B3 (201). However, enzyme inactivation and the competing presence of two structurally uncharacterised bromohydrin products indicated that alternative halides are less optimal co-substrates for efficient VHPO biocatalysis. NapH1 exhibits high substrate specificity, failing to facilitate analogous chloro-cyclisations on structurally similar molecules. However, one equally acceptable substrate is naphthomevalin (7), lacking the C-7 methyl group, which is cyclised to the known natural product napyradiomycin A1 (14).63 This implies that the C-7 methyl substituent is negligibly recognised by NapH1 and could represent a site for future diversification.
Scheme 14.

VHPO-catalysed halogenation-cyclisation of SF2415B1 (10) to give SF2415B3 (25).
Inspired by the proposed biosynthesis of the napyradiomycins outlined in Scheme 13, George and Moore achieved a biomimetic total synthesis of naphthomevalin (7) (Scheme 15).63 In addition to confirming the chemical feasibility of the proposed biosynthetic pathway, the synthesis provided access to potential biosynthetic intermediates that could be used as substrates and synthetic standards for newly discovered enzymes. Methyl 3,5-dimethoxyphenylacetate (151) was converted over four steps into the protected THN derivative 202, which was selectively geranylated at C-4 with geranyl carbonate 203 under Pd catalysis. Oxidative dearomatisation of 204 with Pb(OAc)4 followed by dichlorination at C-2 with NCS gave the geminal dichloride 205. Selective mono-dechlorination of 205 with LDA followed by acetate hydrolysis gave 206, which was diastereoselectively prenylated at C-2 to give 207. MOM deprotection of 207 under mildly acidic conditions gave 199, which underwent a thermal α-hydroxyketone rearrangement on heating in toluene to give racemic naphthomevalin (7). All of the synthetic intermediates in the route were amenable to MOM deprotection, thus giving access to a number of useful enzyme substrates and product standards for further investigation of the biosynthetic pathway.
Scheme 15.
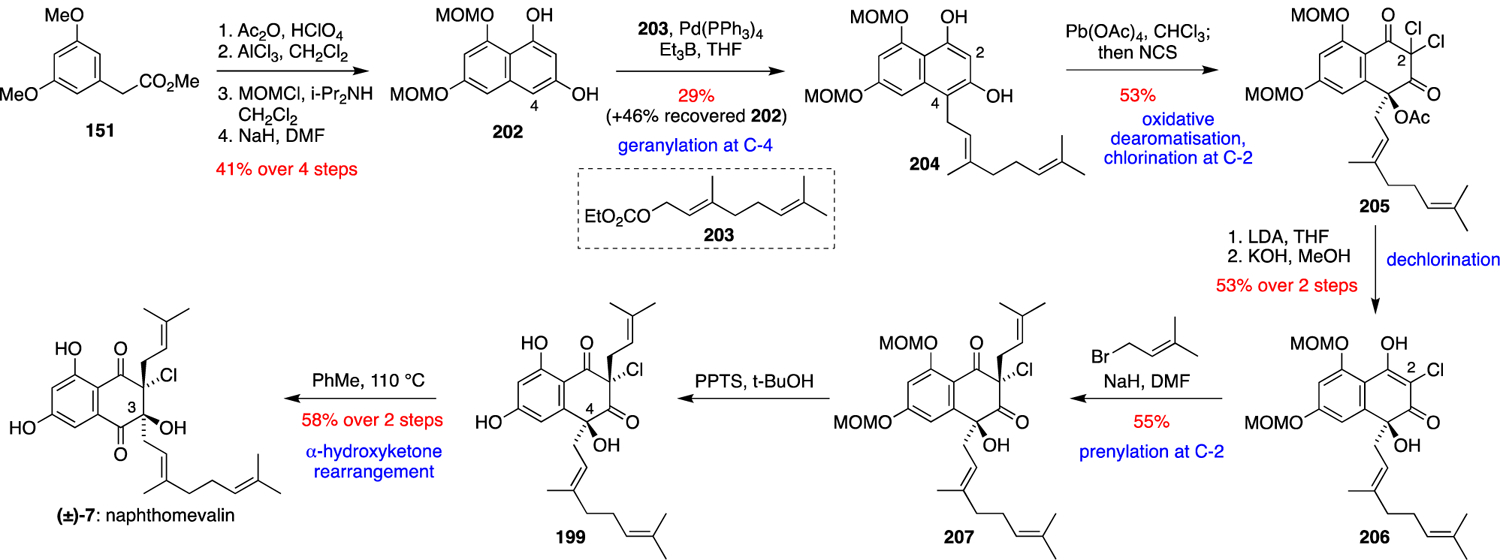
George and Moore’s biomimetic total synthesis of (±)-naphthomevalin (7).63
Mirroring the putative biosynthetic merochlorin C and D pathway, NapT8 catalysed a diastereoselective aromatic prenylation using DMAPP (140) as a prenyl donor at the C-2 chlorinated position of the THN ring of synthetic substrate 198 to give 199 (Scheme 16).63 While catalysis was not completely abolished in the absence of magnesium, it was significantly reduced, highlighting the important but not essential nature of the inorganic cofactor in this ABBA prenyltransferase. C-2 disubstitution enables the thermodynamically favorable α-hydroxyketone rearrangement of the geranyl moiety of 199 to form the naphthoquinone core of naphthomevalin (7). While this reaction does proceed spontaneously in vitro, the addition of VHPO homologue NapH3 dramatically accelerates the migration. Despite having 63–64% sequence similarity to the other two nap VHPO homologs, NapH3 solely catalyses this structural rearrangement, showing no halogenation activity with any napyradiomycin biosynthetic intermediate or model VHPO substrate monochlorodimedone. Intriguingly, when the C-2 prenyl substituent of intermediate 199 is replaced with a chlorine atom, NapH3 fails to efficiently promote a comparable 1,2-shift of the C-4 geranyl moiety, indicating an unknown level of specificity for the C-2 prenyl group.
Scheme 16.
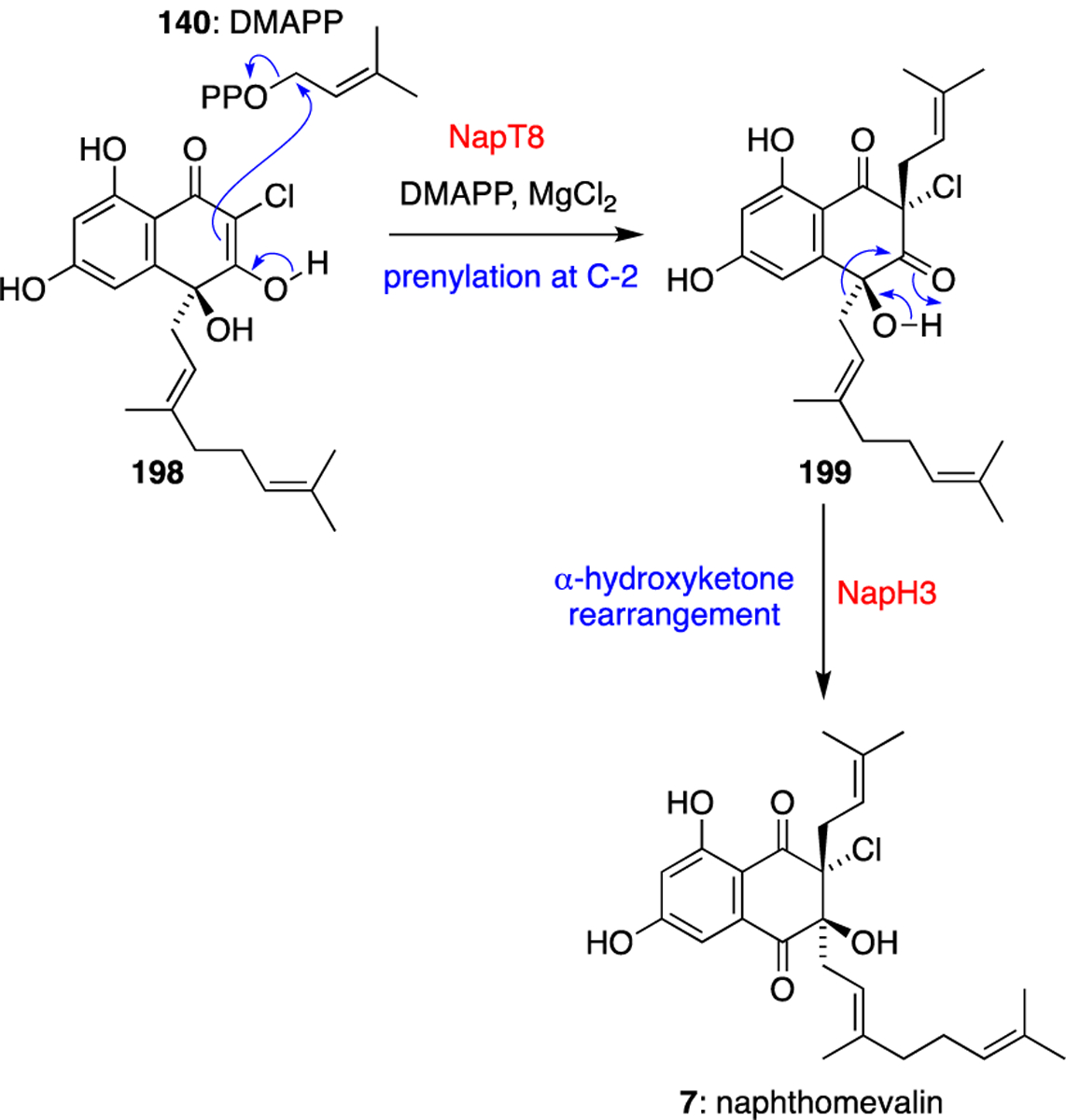
Discovery of C-2 prenylation catalysed by NapT8, and α-hydroxyketone rearrangement catalysed by NapH3.63
The final VHPO in the nap cluster to be identified, NapH4, facilitates a stereoselective chloronium ion formation on the distal alkene of the geranyl moiety of napyradiomycin A1 (14) (Scheme 17).70 In contrast to other established VCPO enzymology, NapH4 uses the proximal alkene to form a chloro-cyclohexane ring while defining two new stereocentres. Cyclisation of the chloronium ion 208 followed by a final deprotonation of tertiary carbocation 209 generates the exocyclic double bond within the 6-membered ring, efficiently producing napyradiomycin B1 (4) in 89% yield when interrogated on the milligram scale. While novel for the bacterial VCPO enzymes, this carbon-carbon bond forming activity parallels a comparable VHPO reaction characterised in red macroalgae discovered in the early 2000s by Carter-Franklin and Butler.71,72 Macroalgal VBPO catalysis stereoselectively brominated and cyclised various terpenoid substrates to form bromocyclohexene-containing natural products. It is intriguing to see how comparable chemistry in the absence of the THN meroterpenoid scaffold has emerged from phylogenetically distinct organisms, suggesting there may be additional chemical diversity encoded within uncharacterized members of this unique enzyme family.
Scheme 17.

VCPO-catalysed chlorination-cyclisation of napyradiomycin A1 (14) to give napyradiomycin B1 (4).70
As shown in Scheme 18, napyradiomycin biosynthesis is initiated by the C-4 geranylation of THN (1) via ABBA prenyltransferase NapT9 in the presence of Mg2+.70 This enzyme reaction is highly specific for the 10-carbon GPP (141) as a prenyl donor, failing to prenylate THN in the presence of shorter 5-carbon pyrophosphates (DMAPP or IPP - isopentenyl pyrophosphate) or in the absence of its metal ion cofactor. The highly oxygen-sensitive 4-geranyl THN product 196 is then monochlorinated at C-2 and dearomatised and hydrated at C-4 by NapH1 to give 198. In contrast to Mcl24 catalysis with its branched sesquiterpene substrate, the geranylated napyradiomycin biosynthetic intermediate is not further C-2 halogenated by NapH1 even after the addition of superstoichiometric quantities of hydrogen peroxide. This phenomenon is further observed with marH1 in marinone/naphterpin biosynthesis described in section 8, and generates additional interest in understanding the biophysical factors that dictate VHPO catalysis in actinomycetes.
Scheme 18.

Discovery of the C-4 geranylation of THN catalysed by NapT9, and C-2 chlorination and oxidative dearomatisation catalysed by NapH1.70
7. Enzymatic total synthesis of the napyradiomycins
Complete elucidation of the napyradiomycin biosynthetic pathway from THN and organic pyrophosphate precursors highlighted that only two enzyme families were necessary to facilitate their construction: Mg2+-dependent aromatic ABBA prenyltransferases (NapT8, NapT9), and VHPOs (NapH1, NapH3, NapH4).70 These two enzyme types had orthogonal redox and metal cofactor requirements, and as each of the Nap proteins exhibited high in vitro substrate specificity, Moore hypothesised that a controlled and sequential addition of recombinant biosynthetic enzymes could biocatalytically generate useful quantities of napyradiomycins from basic building blocks (Scheme 19). One-pot reaction conditions were built around optimal buffers for the most synthetically challenging NapH4 reaction to minimise the accumulation of late stage biosynthetic intermediates. Navigating the oxygen-sensitive NapT9 prenylation reaction with the oxidative switch required for NapH1 VHPO catalysis proved to be the greatest limitation to yield; however following the optimisation of these first two steps, the addition of DMAPP, hydrogen peroxide and all remaining Nap enzymes at once facilitated the production of napyradiomycin intermediates that mirrored the biosynthetic pathway. The addition of all five Nap enzymes alongside their organic substrates, hydrogen peroxide cosubstrates, and inorganic metal salts enabled the production of 4.6 mg (18% yield) of napyradiomycin B1 (4) after a 24-hour reaction time. Omitting the final addition of NapH4 instead produced napyradiomycin A1 (14) (5.4 mg, 22%) within the same timescale. The chemoenzymatic application of 0.2–1 mol % recombinant napyradiomycin enzymes constructed all five stereocentres of 4 in a protecting group-free manner without the need to isolate individual enzymatic products, highlighting the utility of biocatalysis in complex natural product synthesis.
Scheme 19.
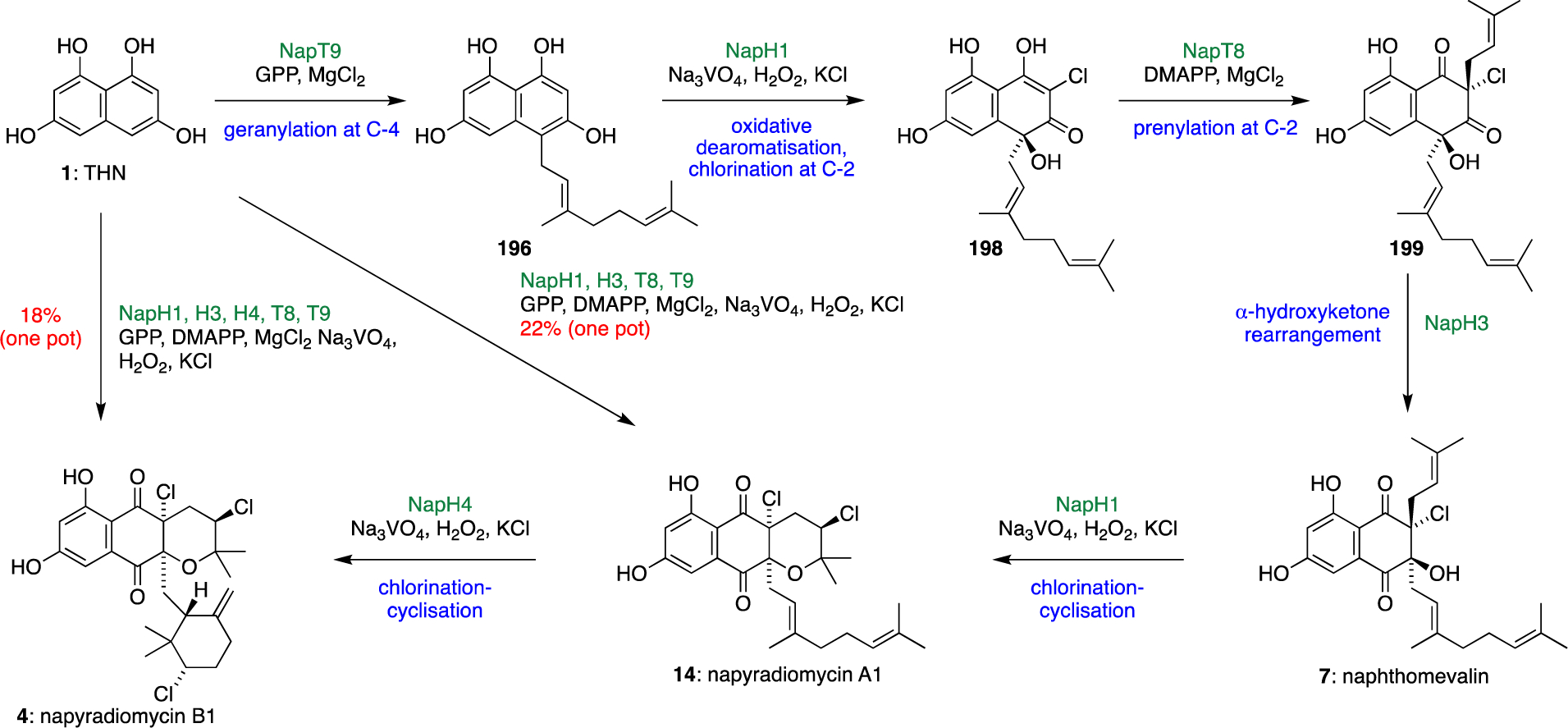
Fully elucidated biosynthetic pathway from THN to napyradiomycin B1 (4) and enzymatic, one-pot total syntheses of napyradiomycins A1 (14) and B1 (4).70
8. Biosynthesis and biomimetic total synthesis of the marinones
Early isotope labelling studies revealed that both the naphterpin and marinone families of natural products are derived from THN, despite their unusual oxidation pattern that is markedly different from that of THN. Additionally, the naphterpins and marinones contain terpene substituents at the C-3 position, despite THN being nucleophilic only at C-2 and C-4. From these observations, it was predicted that the naphterpins and marinones might be formed via a similar α-hydroxyketone rearrangement as that observed in the merochlorin and napyradiomycin biosyntheses. The key difference between the naphterpin and marinone biosynthetic pathways is the length of the terpene sidechain, with the naphterpins incorporating a geranyl subunit while the marinones assimilate a farnesyl group. With this in mind, the biosynthesis of these families of natural products was proposed from THN (1), as outlined for marinone in Scheme 20. Firstly, 1 undergoes alkylation with farnesyl pyrophosphate (210) at C-4 to give intermediate 211. This reaction is putatively catalysed in vivo by the aromatic prenyltranferase CnqP3 or CnqP4.73 Next, 211 would undergo VCPO-catalysed oxidative dearomatisation and dichlorination at C-2 to give geminal dichloride 212, which is then subjected to a VCPO-catalysed α-hydroxyketone rearrangement to shift the farnesyl substituent from C-4 to C-3, giving 213. The sequence of chlorination, dearomatisation and 1,2-shift is analogous to the reaction catalysed by the VCPO Mcl24 in merochlorin biosynthesis (Scheme 6). At this point, mildly basic conditions would convert 213 into the α-chloroepoxide 214, which could then undergo a reductive dehalogenation to give hydroxynaphthoquinone 215. Oxidation and alkene isomerisation of 215 would then set up the final intramolecular hetero-Diels-Alder reaction of 216 to construct the tetracyclic scaffold of debromomarinone (79). Finally, late-stage vanadium-dependent bromoperoxidase (VBPO) catalysed bromination at C-5 or C-7 would give marinone (6) or isomarinone (80), respectively. Despite the absence of chlorine in these natural products, it was envisaged that cryptic chlorination would play a key role in selectively oxidising the THN ring and promoting the predicted α-hydroxyketone rearrangement. Initial investigation of the putative marinone genome from Streptomyces sp. CNQ-509 supported this hypothesis through the clustering of VCPO and aromatic prenyltransferase homologues with a THN synthase.
Scheme 20.
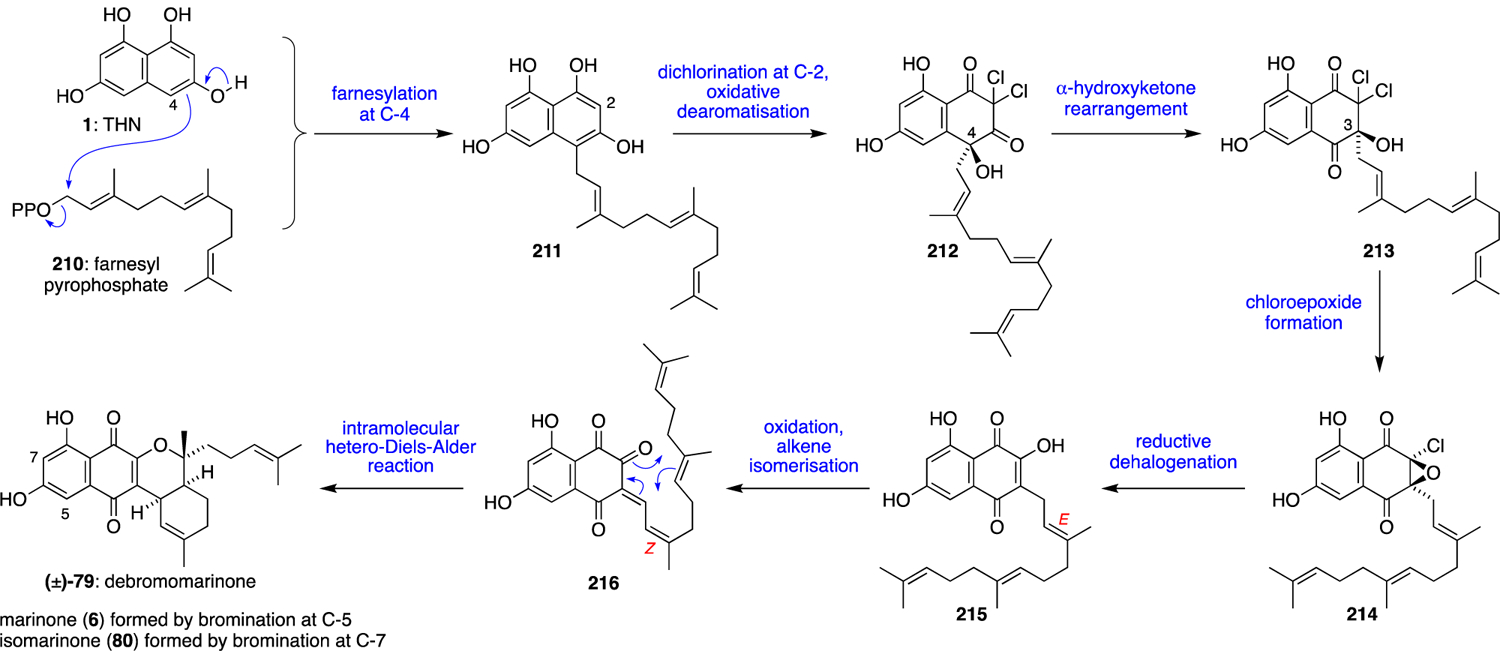
Proposed biosynthesis of the marinones.
Naphterpin (66), isolated from Streptomyces sp. CL190, differs from marinone (6) only by the absence of one prenyl group, as it is derived from a cyclised geranyl rather than a farnesyl sidechain.39 In 2005, Kuzuyama and co-workers described three genes from the naphterpin biosynthetic gene cluster (BGC), including a THN synthase and an aromatic prenyltransferase.58 The naphterpin prenyltransferase Orf2 (renamed NphB74) was found to have relaxed substrate specificity toward various hydroxyl-containing aromatic acceptors; however, THN (1) was never evaluated as a substrate. This prenyltransferase was confirmed to be crucial in naphterpin biosynthesis, as a mutant Streptomyces sp. CL190 strain containing a disruption in the Orf2 gene abolished naphterpin production.
Recently, George and Moore completed the first total syntheses of debromomarinone (79) and 7-demethyl naphterpin (71) using a biomimetic approach (the synthesis of debromomarinone is presented in Scheme 21).75 The successful strategy followed the same sequence of key reactions as the proposed biosynthesis, starting from the protected THN derivative 202, which was previously used in biomimetic studies towards the synthesis of the napyradiomycins (Scheme 15). Consequently, the design of the synthesis allowed access to many anticipated biosynthetic intermediates for use in later in vitro studies. The synthesis began with farnesylation at C-4 of the di-MOM protected THN 202 with farnesyl carbonate 217 via a Pd catalysed Tsuji-Trost reaction to give 218. This intermediate was then oxidatively dearomatised with Pb(OAc)4 and dichlorinated with NCS at C-2 to give 219 in a one-pot process. Direct acetate hydrolysis of 219 was impossible due to competing fragmentation via a haloform-type reaction. This was resolved using a 3-step sequence of LDA-mediated dechlorination, acetate hydrolysis, and re-chlorination at C-2 of chloroenol 220 with NCS to give geminal dichloride 221. Heating 221 in toluene thermally induced the desired α-hydroxyketone rearrangement, shifting the farnesyl side chain from the C-4 to the C-3 position to give 222 in 54% yield. Treatment of 222 with Cs2CO3 formed the α-chloroepoxide 223, which was reduced with Zn in MeOH to give 224. Oxidation of hydroxynaphthoquinone 224 with TEMPO/PhI(OAc)2 gave tricycle 225 through an oxa-6π-electrocyclisation. Heating 225 in EtOH with SnCl4 resulted in a cascade of retro-oxa-6π-electrocyclisation, alkene isomerisation, and a final intramolecular hetero-Diels-Alder reaction to afford (±)-debromomarinone (79) in low yield.
Scheme 21.

George and Moore’s biomimetic total synthesis of (±)-debromomarinone (79).75
This biomimetic total synthesis of debromomarinone (79) is lengthy for a target molecule of only moderate complexity, but it gave access to several proposed biosynthetic intermediates, as each compound in the sequence could be MOM deprotected under mild acidic conditions. Utilising this synthetic route, the proposed biosynthetic intermediates shown in Scheme 22 were synthesised as standards for biosynthetic studies. To investigate each biosynthetic step, three VCPO homologues marH1, marH2 and marH3 were individually cloned from the putative Streptomyces sp. CNQ-509 marinone biosynthetic gene cluster. After expression and purification, two of the recombinant proteins (MarH1, MarH3) exhibited halogenation activity through a monochlorodimedone (MCD) assay and were further interrogated using synthetic substrates. MarH2 was not found to have any halogenating activity and was later discovered to lack the key vanadate-coordinating histidine residue in comparison to other VCPO enzymes. In vitro assays with pre-marinone (211) and the recombinant MarH enzymes were conducted and characterised using reversed-phase HPLC to monitor the formation of biosynthetic intermediates. In the presence of sodium vanadate and two stoichiometric equivalents of hydrogen peroxide, MarH1 was shown to catalyse the oxidative dearomatisation of 211 at C-4 to give 226 and monochlorination at C-2, yielding 227. Subsequently, in vitro incubation of MarH3 with 227 catalysed the second chlorination event at C-2, followed by the α-hydroxyketone rearrangement to give 213. This sequence of reactions could also be achieved in a one-pot reaction of 211 with MarH1, MarH3, sodium vanadate and stoichiometric hydrogen peroxide.
Scheme 22.
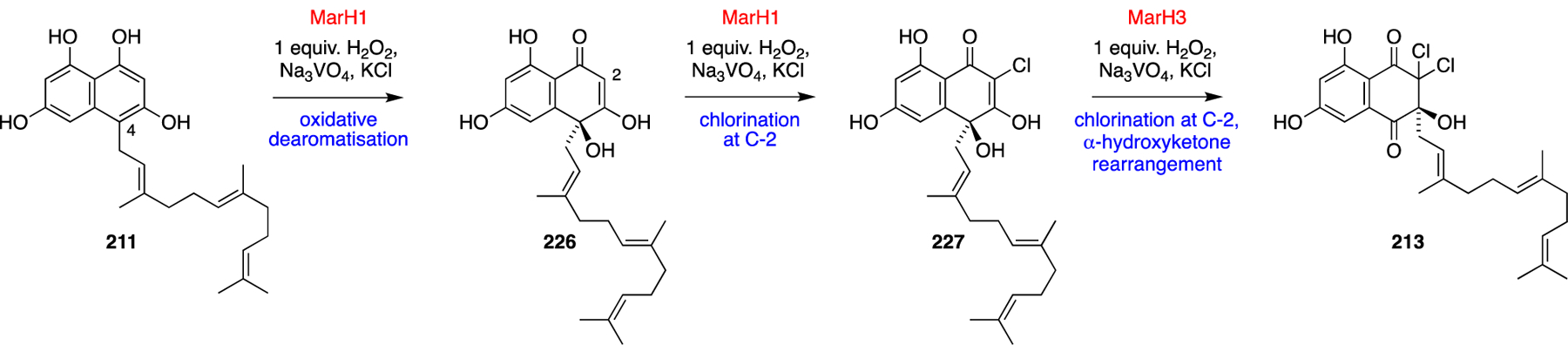
Discovery of MarH1 and MarH3 VCPOs.75
Following the earlier biomimetic synthetic work, a more concise and divergent total synthesis of the naphterpin and marinone families of natural products was devised (Scheme 23).76 Without the constraints imposed by the THN building block used in the previous biomimetic synthesis, it was envisioned that the naphthoquinone ring system could be installed later in the synthesis through an intermolecular Diels-Alder reaction. This allowed for a concise, modular strategy that rapidly built the molecular complexity of these natural products. The total synthesis began with commercially available 2,5-dimethoxyphenol (228), which was coupled to propargylic carbonate 229 in the presence of DBU and copper (II) chloride. Propargyl ether 230 then underwent a thermal rearrangement to give chromene 231. Oxidation of 231 with PhI(O2CCF3)2 gave quinone 232, which participated in a regioselective intermolecular Diels-Alder reaction with Brassard diene 233 at room temperature to give naphthoquinone 234, after the aerobic oxidation of a presumed hydroquinone intermediate. With 234 in hand, a similar endgame was employed to that used in the biomimetic synthesis. Heating 234 with MgBr2 in MeCN induced the desired retro-6π-electrocyclisation and alkene isomerisation to give the reactive enone 216, which then underwent an intramolecular hetero-Diels-Alder reaction to form debromomarinone (79). Cleavage of the labile C-8 methyl ether was also observed under these reaction conditions. Finally, bromination with NBS occurred exclusively at C-7 to furnish isomarinone (80). Total syntheses of 7-demethylnaphterpin (71), naphterpin (66), and naphterpins B (67) and C (68) were also achieved using this Diels-Alder-based strategy.
Scheme 23.
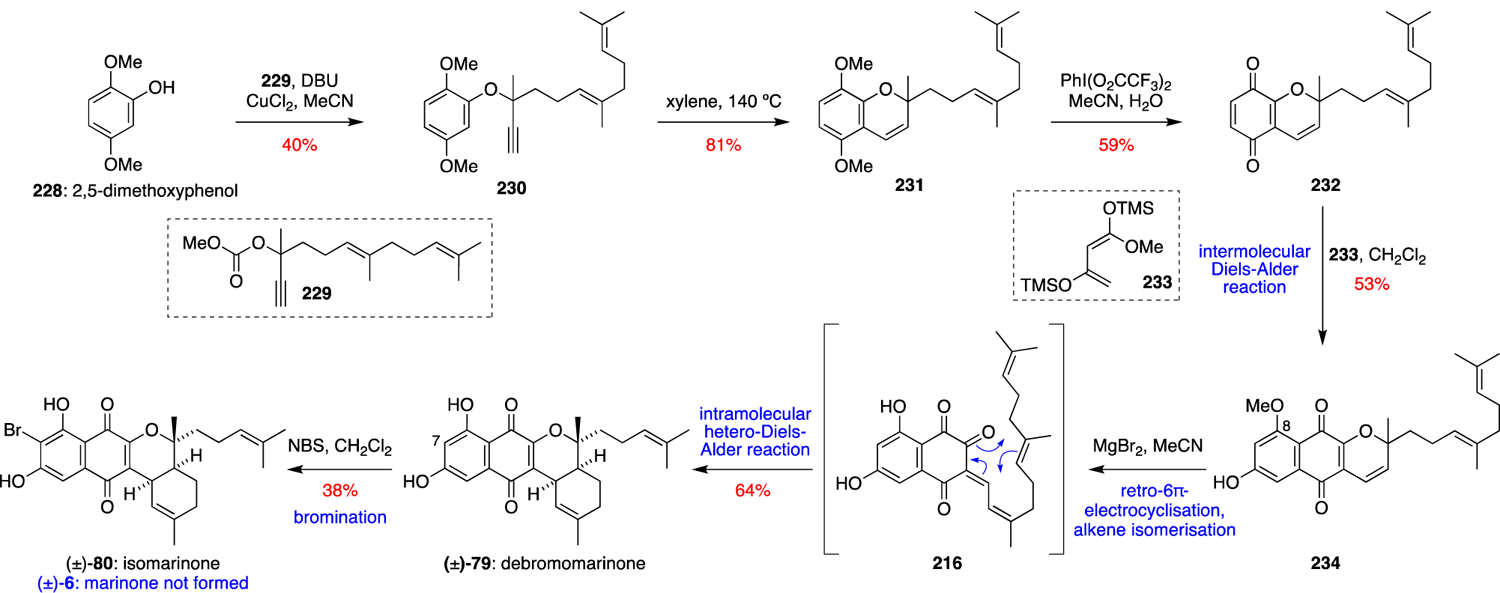
George’s total synthesis of (±)-isomarinone (80).76
9. Biosynthesis and total synthesis of the furaquinocins and neomarinone
Early isotope labelling studies showed that the furaquinocins were similarly constructed around a symmetric THN precursor, and structurally elaborated with mevalonate-derived terpenes and methyl and methoxy substituents derived from the two C1 units of L-methionine.56 The Dairi group later reported the furaquinocin BGC in 2006 via heterologous expression of the 25-kb fur cluster in Streptomyces lividans TK23 to produce furaquinocin D (85).11 The biosynthesis has largely been proposed based on genetic manipulation and comparison to known enzymes in other meroterpenoid biosynthetic pathways. Other than the putative THN synthase (fur1), key genes were identified such as C-methyltransferase fur4, O-methyltransferase fur6, prenyltransferase fur7, and cytochrome P450 enzyme fur8. With this knowledge, the predicted biosynthetic pathway is as outlined in Scheme 24. In contrast to the other described meroterpenoid biosyntheses wherein THN prenylation precedes further oxidation dearomatisation or halogenation reactions, in furaquinocin biosynthesis, monooxygenase Fur2 oxidises half of the naphthalene ring system to flaviolin (2), which is then C- and O- methylated to generate 2-methoxy-3-methylflaviolin (235). Further investigation by Kuzuyama and co-workers into prenyltransferase Fur7 via heterologous protein expression confirmed 235 to be the preferred in vitro substrate when incubated with GPP and independent of divalent cations.14 Intriguingly, two compounds, Fur-P1 (236) and Fur-P2 (237), were produced in a 10:1 ratio, indicating that Fur7 has the ability to simultaneously reverse prenylate at the 6-carbon and forward prenylate the 7-oxygen respectively. Although it is plausible that Fur-P2 was the primary enzymatic product that is subsequently converted to Fur-P1 via a Claisen rearrangement, attempts to catalyse this [3,3]-sigmatropic rearrangement in the presence of Fur7 were unsuccessful. Through in vivo studies, Fur-P1 (236) was found to undergo conversion to furaquinocin C (84), unambiguously confirming that 236 is a late stage biosynthetic intermediate. Cyclisation of substrate 236 would generate furaquinocin C (84), which could be further oxidised by cytochrome P450 fur8 on the homoprenyl side chain to access other members of the furaquinocin family. In the case of Dairi’s study, heterologous expression of the BGC and HPLC analysis showed the predominant formation of furaquinocin D (85) with only trace amounts of what was thought to be di-hydroxylated product furaquinocin A (3). Furanonaphthoquinone I (94), the regioisomer of furaquinocin C (84) is likely synthesised via an analogous strategy diverging via the unidentified cyclase that constructs the furan ring providing a different regiospecificity. Additional biochemical validation of fnq26, the prenyltransferase implicated in the biosynthesis of 94 generated a mixture of forward and reverse C-3 geranylated compounds when incubated with flaviolin (2).77 However, 2-methoxy-3-methylflaviolin (235) was never investigated as a potential substrate in this study. Both neomarinone (99) and marfuraquinocins (95-98) biosynthesis likely follow a comparable biosynthetic scheme utilising FPP instead.53 The longer terpenoid sidechain is then likely cyclised and structurally rearranged to produce various cyclohexene-containing meroterpenoids through a series of unidentified enzymes.
Scheme 24.
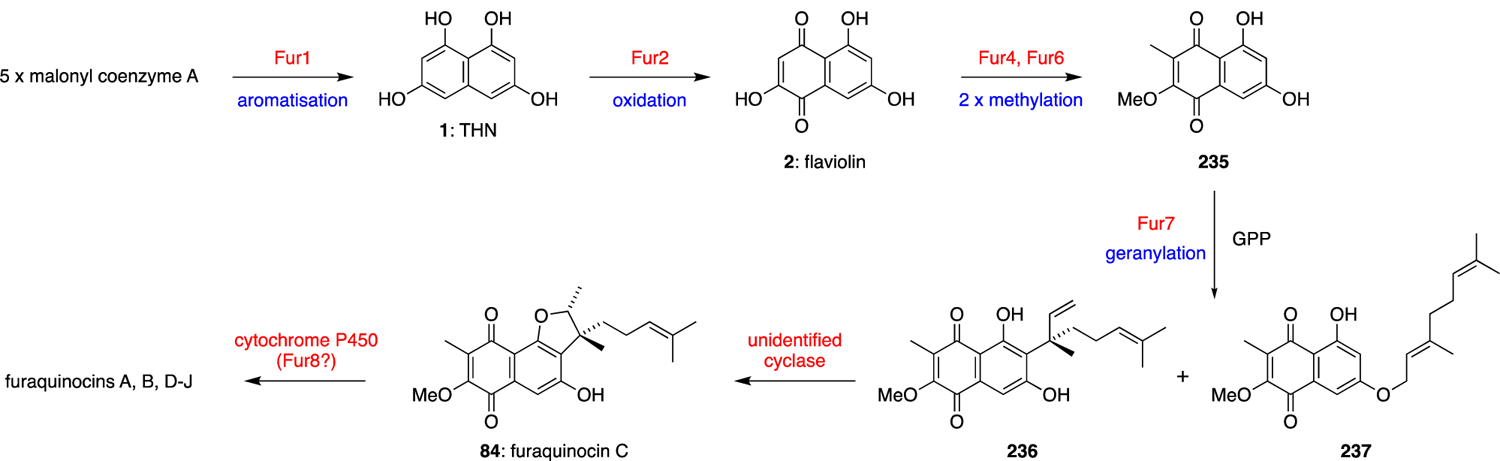
Proposed biosynthesis of the furaquinocins.
In Dairi’s initial heterologous expression of the furaquinocin BCG, another gene, fur3, was also reported. This gene possessed significant similarity to a PLP-dependent aminotransferase, despite the absence of an amination step in the furaquinocin biosynthesis. As a result, this gene was disregarded until 2012, when Kuzuyama reported that deletion of the fur3 gene abolished heterologous production of furaquinocins.78 This interesting result suggested that the aminotransferase enzyme was responsible for converting THN (1) or flaviolin (2) into a key biosynthetic intermediate. Heterologous expression of a plasmid containing fur1, fur2 and fur3 resulted in the accumulation of 8-amino-flaviolin (238) as outlined in Scheme 25. The absence of an aniline functionality in any furaquinocin product insinuated that 8-amino-flaviolin might be a shunt product unrelated to the furaquinocin biosynthesis. Surprisingly, the addition of compound 238 to a culture in which fur3 had been deleted, restored heterologous production of furaquinocin D (85). These cumulative results suggest that fur3 is essential for furaquinocin biosynthesis, and that 238 is an early stage biosynthetic intermediate. However, this revised biosynthetic proposal would necessitate a deamination step, for which the required genes have yet to be discovered.
Scheme 25.

Proposed biosynthesis of 8-amino-flaviolin (238).
The first total synthesis of (–)-furaquinocin C (84) was reported by Smith et al. in 1995.79 Their concise strategy involved two stereoselective organocuprate Michael additions to assemble the dihydrofuran ring system, followed by an intermolecular Diels-Alder reaction (Scheme 26). The synthesis began with (R)-(+)-angelicalactone (239), which was synthesised in five steps from D-ribonolactone. Conjugate addition of the cuprate derived from iodide 240 and subsequent trapping of the resultant enolate with acetyl chloride in the presence of HMPA furnished lactone 241 as a single diastereomer. Treatment of 241 with NaH and PhSeOCl gave the crude unsaturated lactone 242, which was subjected to a Michael reaction with Me2CuLi without purification to give 243. Lactone 243 was then subjected to two consecutive silylations via silyl ether 244 to give bis(silyl ether) 245, followed by a regioslective intermolecular Diels-Alder reaction with bromoquinone dienophile 246 to afford (–)-furaquinocin C (84).
Scheme 26.
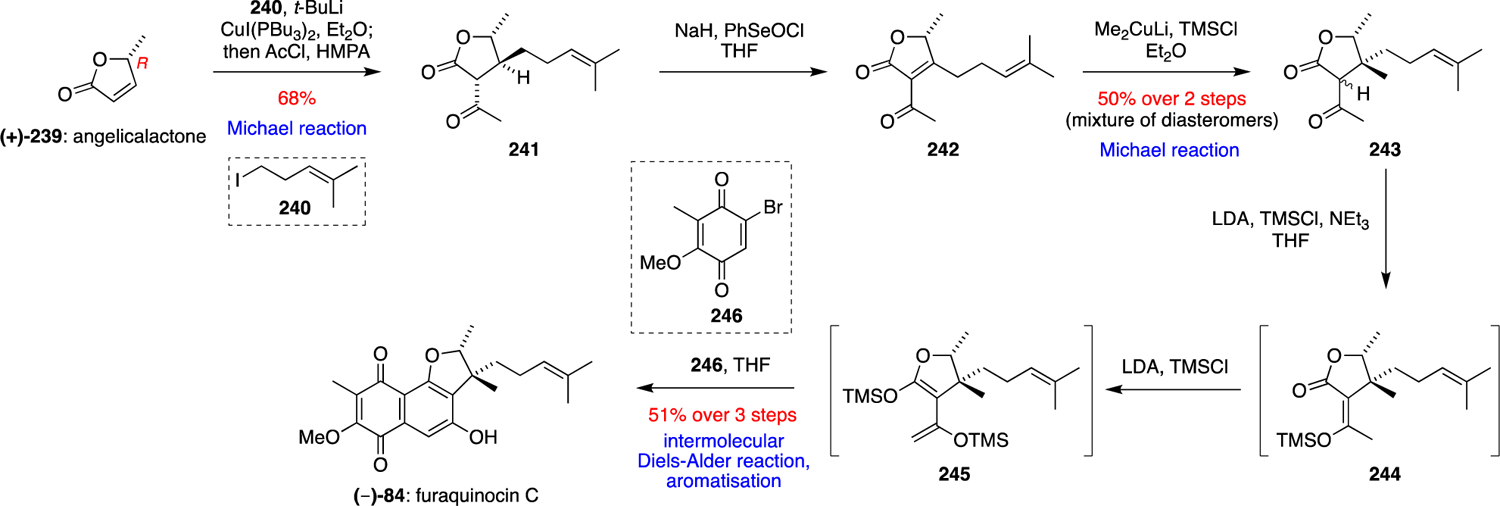
Smith’s total synthesis of (–)-furaquinocin C (84).79
In the same issue of the Journal of the American Chemical Society that Smith’s total synthesis of furaquinocin C was published, Suzuki and co-workers published their synthesis of furaquinocin D (85).80 Their convergent strategy also allowed the later synthesis of furaquinocins A (3), B (83) and H (89) to be completed, and the synthesis of 89 is presented in Scheme 27.81 The synthesis began with 4-(benzyloxy)-2-methoxy-3-methylphenol, which was converted to the intermediate aryl iodide 247 in five steps. Aryl iodide 247 was then subjected to a Sonogashira cross-coupling reaction with alkyne 248 to give the disubstituted alkyne 249. Treatment of 249 with PdCl2 gave dihydrofuran 250 as a single product in high yield via an oxypalladation-protodemetalation cascade. Saponfication of the methyl ester of 250 with NaOH in MeOH gave a sodium carboxylate, which was subsequently treated with acetyl chloride and 4-(dimethylamino)pyridine to give mixed anhydride intermediate 251. Dieckmann condensation of 251 involving intramolecular nucleophilic addition of the cyclic enol ether to the anhydride followed by aromatisation and acetylation then gave the furanonaphthalene structure of 252 in an elegant cascade reaction. The intermediate furanonaphthalene 252 was then converted to aldehyde 253 in three simple steps. With this aldehyde 253 in hand, the next challenge was to introduce the oxidised terpene sidechain, while controlling the stereochemistry of the C-10 hydroxyl group. Firstly, methylene transfer to aldehyde 253 was achieved through a Corey-Chaykovsky reaction using (S)-254 to give epoxide 255 with good diastereoselectivity. Tin-lithium exchange of vinylstannane 256 then gave a vinyllithium species, which reacted with epoxide 255 to give the desired alcohol 257 in good yield. Acid-catalysed MEM and acetyl deprotection followed by silylation of the liberated alcohols was achieved over two steps to give 258. Benzyl deprotection of 258 was accomplished selectively under hydrogen transfer conditions with no saturation of the sidechain, and subsequent oxidation with DDQ gave quinone 259. Finally, silyl deprotection with TBAF afforded (–)-furaquinocin H (89). Ring-opening of the key epoxide intermediate 255 with differentially oxidised vinylstannane nucleophiles allowed access to furaquinocins A, B and D in a divergent manner.
Scheme 27.
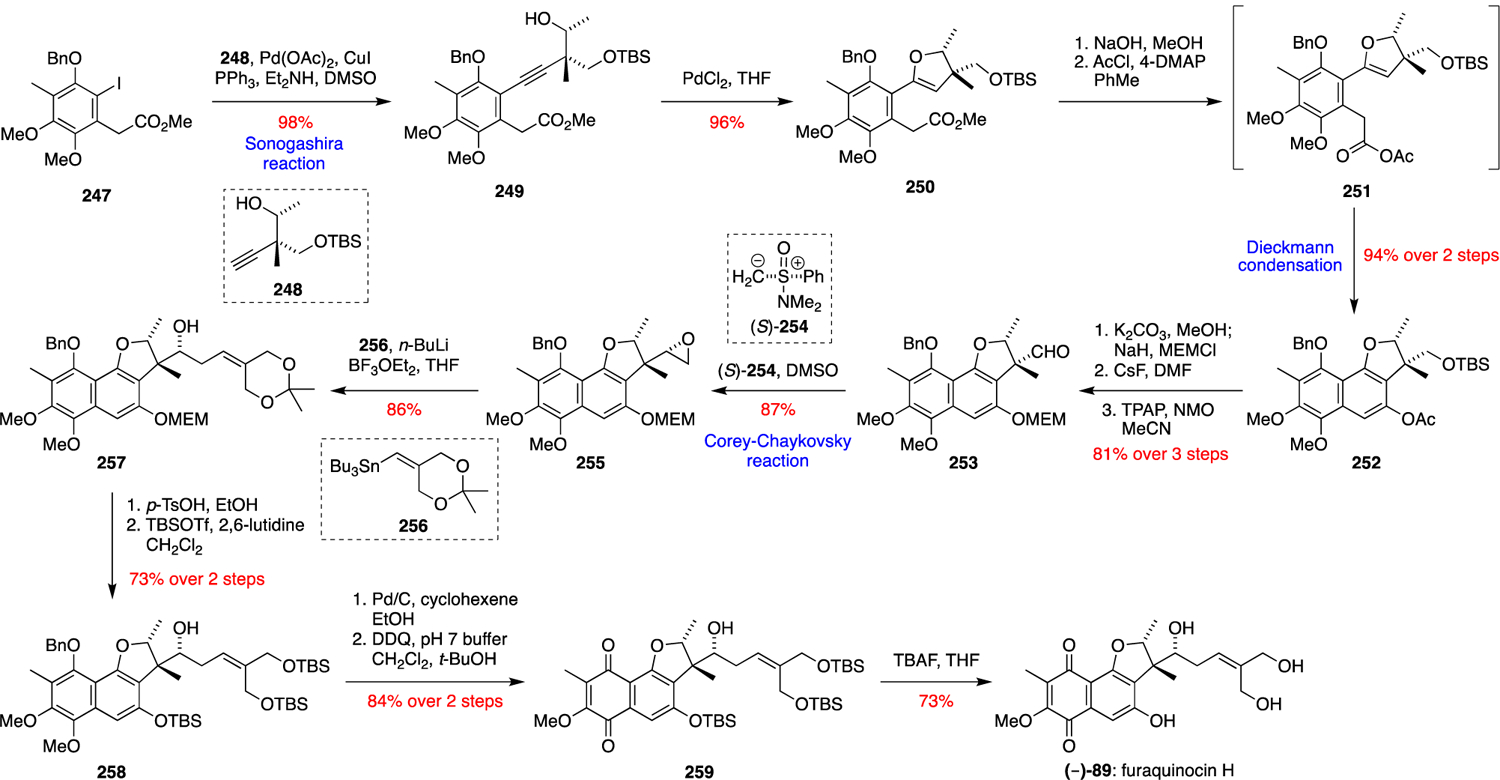
Suzuki’s total synthesis of (–)-furaquinocin H (89).81
In 2002, Trost and co-workers published the first total synthesis of furaquinocin E.82 The following year, Trost went on to complete the total synthesis of (–)-furaquinocins A, B and E (Scheme 28).83 The divergent total synthesis began with asymmetric allylic dialkylation of 2-iodoresorcinol (260) with allylic carbonate (261). In the presence of Pd2(dba)3 and (R,R)-ligand 262, the desired product 263 was achieved in excellent yield and diastereoselectivity. Reductive Heck cyclisation of 263 gave a cyclised product with the second allyl moiety cleaved under reaction conditions that are standard, except for the use of the hindered base pentamethylpiperidine, PMP. Acetate protection of the free phenol then gave intermediate 264. The dimethylbenzofuran intermediate 264 was then converted to the (E,E)-diene 265 in five steps, including a Horner-Wadsworth-Emmons reaction of an intermediate aldehyde to install the side chain. Reduction of the methyl ester of 265 followed by silyl protection gave 266 in good yield. Lithium-halogen exchange of bromide 266 was achieved prior to the addition of the squaric acid imine 267, which after addition was hydrolysed under mild acidic conditions to give 268. Thermal rearrangement of 268 followed by oxidation in air gave the desired naphthoquinone. Finally, desilylation of the protected hydroxyl groups afforded the natural product (–)-furaquinocin E (86). Trost’s strategy also gave access to (–)-furaquinocins A (3) and B (83). These syntheses utilised the same intermediate nitrile compound 264, which then underwent a sequence of reduction, allylation and cross metathesis steps to install the desired side chains, followed by the same squaric acid approach to establish the naphthoquinone core.
Scheme 28.
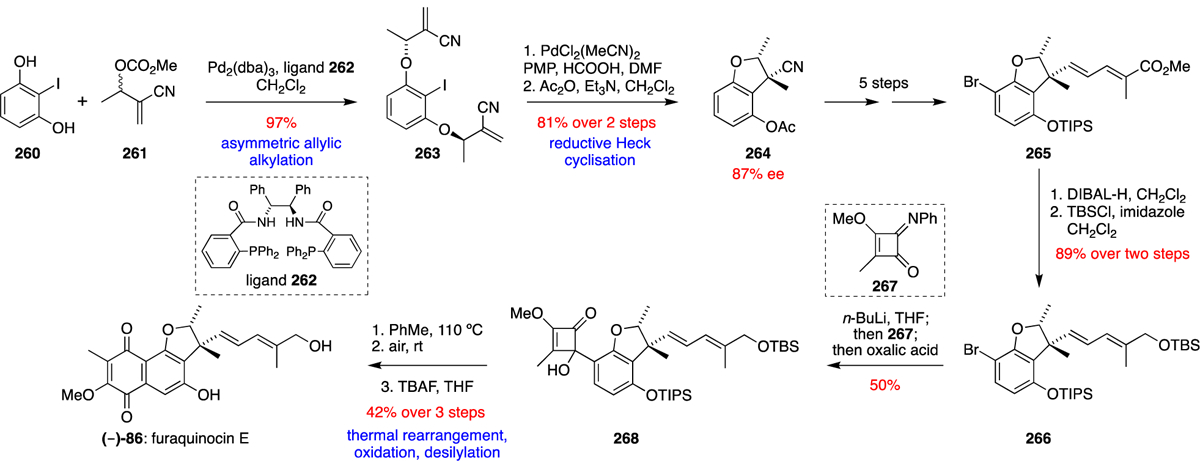
Trost’s total synthesis of (–)-furaquinocin E (86).83
In 2009, Pérez Sestelo et al. completed the first total synthesis of (+)-neomarinone (99) via a convergent, intermolecular Diels-Alder-based strategy that builds on Smith’s earlier synthesis of furaquinocin C.84 The neomarinone synthesis began with commercially available methyl (R)-lactate, which was TBS-protected and selectively reduced with DIBAL-H to give aldehyde 269 (Scheme 29). A TiCl4-catalysed Knoevenagel condensation of 269 with ethyl acetoacetate (270) then gave the α,β-unsaturated 1,3-dicarbonyl compound 271. Michael addition of lithium dimethylcuprate to 271 then gave 272, which underwent spontaneous lactonisation upon TBAF-mediated desilylation to give 273. Treatment of lactone 273 with NaH and PhSeCl gave the intermediate selenide, which then underwent oxidation with H2O2 and selenoxide elimination to form the desired enone 274. A second Michael reaction was achieved on enone 274 using the organocuprate formed from iodide 275, which was synthesised in nine steps from (R)-methylcyclohexanone. The resultant 1,3-dicarbonyl compound 276 was then reacted with LDA and TMSCl to form the electron-rich diene 277, which participated in a regioselective Diels-Alder reaction with bromoquinone 246 to give, after aromatisation, naphthoquinone 278. Treatment with aqueous HClO4 cleaved the methyl ether to afford (+)-neomarinone (99).
Scheme 29.

Sarandeses and Pérez Sestelo’s total synthesis of (+)-neomarinone (99).84
In 2016, Xie and co-workers developed an enantioselective synthesis to form both enantiomers of PI-220 utilising a ruthenium-catalysed asymmetric hydrogenation to establish the initial stereochemistry (Scheme 30).85 The synthesis began with racemic ketone 279, which underwent asymmetric hydrogenation with chiral spiro ruthenium catalyst (Sa,R,R)-280 under 50 atm of hydrogen. The alcohol product 281 was obtained in 97% ee with high anti-selectivity (anti/syn = 95:5). Iodination was then achieved using I2 in the presence of n-BuLi, followed by Swern oxidation to give (R)-282 in 88% ee. After recrystallisation, the ee value increased to 99% (80% yield). The intermediate ketone 282 then underwent a Horner-Wadsworth-Emmons (HWE) reaction with phosphonate 283 to afford the α,β-unsaturated ester 284 as a 1:1 mixture of E and Z isomers. The ester 284 was then cyclised via an intramolecular reductive Heck reaction to establish the desired dihydrobenzofuran core 285. Demethylation of the protected phenol followed by silyl protection and bromination of the aromatic ring then gave aryl bromide 286. Reduction of the ester of 286, followed by Swern oxidation of the resulting primary alcohol furnished aldehyde 287. Wittig reaction of 287 with 283 in the presence of methylmagnesium bromide yielded α,β-unsaturated ester 288. Addition of methylmagnesium bromide to the newly formed ester gave a tertiary alcohol, which was subsequently silyl protected using 1-(trimethylsilyl)-1H-imidazole (TMSIm) to afford 289. With this intermediate in hand, the same squaric acid methodology employed in Trost’s total synthesis of (–)-furaquinocins A, B and E was used to install the desired naphthoquinone core. Lithium-halogen exchange of bromide 289 with n-BuLi generated the desired organolithium species, which was then treated with imine 267. Hydrolysis of the resulting imine under acidic conditions followed by thermal rearrangement and oxidation in air gave naphthoquinone 290. Finally, desilylation of the protected hydroxyl groups furnished (+)-PI-220 (92).
Scheme 30.

Xie’s total synthesis of (+)-PI-220 (92).85
The same synthetic strategy was also employed by Xie to form (–)-PI-220 utilising the opposite (Sa,S,S) chiral spiro ruthenium catalyst, which was found to match the absolute configuration of natural PI-220.
10. Biosynthesis and total synthesis of azamerone
The unique phthalazinone ring system of azamerone (57) presents an interesting biosynthetic conundrum, although its isolation from Streptomyces sp. CNQ-766 alongside the diazoketo-functionalised napyradiomycins such as SF2415A3 (24) and A80915D (47) suggests a common biogenetic pathway. Indeed, feeding experiments on Streptomyces sp. CNQ-766 with [1,2-13C2]acetate showed that the bicyclic phthalazinone unit of 57 derives from a symmetrical THN building block, in common with other members of the napyradiomycin family.86 Furthermore, the C-8 methyl group was the only carbon atom of 57 not enriched by [1,2-13C2]acetate, but the C-8 methyl was enriched by L-[methyl-13C]methionine (Scheme 31). The origin of the diazo group of 47, and the pyridazine of 57 was investigated using labelling studies with [15N]nitrite, which showed selective incorporation of 15N at the distal diazo nitrogen atom of 47, and at N-2 of 57. This implies that the napyradiomycin diazo group is installed in a stepwise manner via nucleophilic attack of an aminonaphthoquinone 291 on nitrous acid, and the results also strongly suggest that 24 and 47 are the biosynthetic precursors of 57. Finally, to confirm the biosynthetic link between azamerone and the napyradiomycins, labelled [2-15N, 9-13C]-SF2415A3 (24) was prepared biosynthetically, and fed back to Streptomyces sp. CNQ-766 culture. HPLC-MS and NMR analysis of the resultant azamerone (57) clearly showed 13C enrichment at C-8 and 15N enrichment at N-2, thus proving that the pyridazine ring of 57 is derived from the diazo group of 24. Presently, the mechanism for the conversion of 47 into 57 is unclear, although it could involve oxidative cleavage of the diazonaphthoquinone ring system of A80915D (47), followed by a 1,2-shift of the monoterpene unit from C-4 to C-3. The pyridazine ring system could then be formed by cyclisation of the intermediate diketone 292, followed by aromatisation via dehydration and decarboxylation steps to give 57.
Scheme 31.

Key labelling studies in azamerone (57) biosynthesis.
The first total synthesis of azamerone was reported in 2019 by Burns and co-workers, who employed an enantioselective, convergent approach that unified boronic hemiester and quinone diazide coupling partners (Scheme 32).87 The first enantioenriched building block, the boronic hemiester, was prepared from geranyl acetate (293) in four steps. Racemic chlorocyclisation of 293 using a two-step strategy previously developed by Snyder gave chlorocycle 294. Resolution of (±)-294 by chiral derivatisation with (S)-α-methoxyphenylacetic acid gave diastereomeric esters that were separable by flash chromatography. After ester hydrolysis, chlorocycle 294 was obtained in 90% ee. Dehydration of 294 then gave exocyclic alkene 295, which was hydroborated to give the stable boronic hemiester 296. Quinone diazide building block was synthesised from prenylquinone 297 using an enantioselective choroetherification catalysed by a chiral titanium complex derived from ligand 298. The resultant chloropyran 299 was formed in 40% yield and 84% ee. Although the stereoselectivity and efficiency of this chloroetherification reaction is modest, it is to date the closest synthetic equivalent of the NapH4-catalysed chloronium-initiated prenyl cyclisation involved in the biosynthesis of most napyradiomycin natural products. The Burns group methodology therefore has good potential for future applications in the total synthesis of more common members of the napyradiomycin family. The ortho-quinone of 299 was then isomerised to a more stable para-quinone under acidic conditions, and further chlorinated to give 300. Conversion of 300 into azide 301 facilitated its coupling to boronic hemiester 296 under Pd catalysed conditions to give phenol 302 via construction of the key C5–C1’ bond. Dechlorination, TBS-protection and oxidative dearomatisation of 302 then gave para-quinol 303, which underwent a spectacular [4+2]-cycloaddition/oxidation cascade with tetrazine 304, catalysed by the bisboron complex 305 to give 306. Silyl deprotection under acidic conditions converted 306 into (–)-azamerone (57).
Scheme 32.
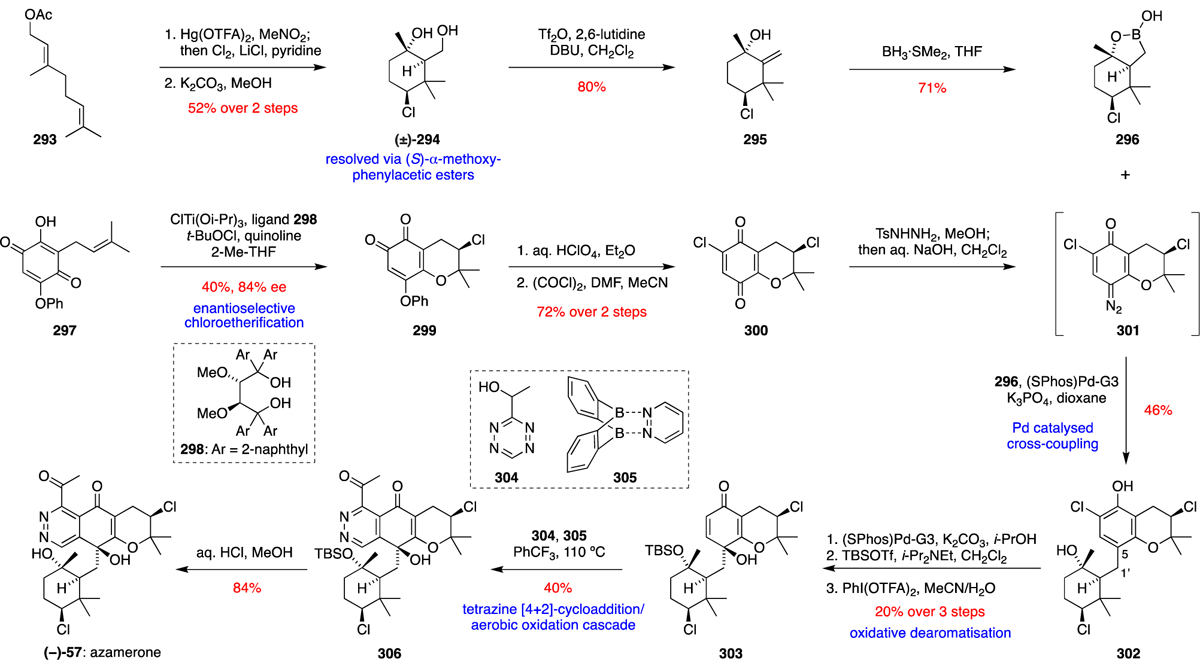
Burns’s total synthesis of (–)-azamerone (57).87
The growing interest in the unique structure of azamerone was further indicated by a recent synthetic approach from Gademann and co-workers (Scheme 33).88 In this study, the known hydroquinone 307 was converted into dialdehyde 308 via a tandem Mannich reaction with piperidine and aqueous formaldehyde, followed by CrO3-mediated oxidation. Condensation of 308 with hydrazine formed the pyradazine ring of 309, which underwent aerobic oxidation to give quinone 310. The cyclohexane unit of azamerone was synthesised starting from methyl acetoacetate (311), which was converted to epoxide 312 in 5 steps. BF3-induced cyclisation of 312 gave ester 313 as a single stereoisomer. Silylation, epoxidation and reduction of 313 resulted in primary alcohol 314, which could potentially be coupled with quinone 310 in a future total synthesis of azamerone (57).
Scheme 33.

Gademann’s synthetic approach to azamerone (57).88
11. Conclusions
Since the isolation of the first napyradiomycins in the mid-1980s, THN meroterpenoids have inspired many creative total syntheses, alongside the development of new synthetic methods of potential broad utility. However, the biosynthesis of these natural products was initially unclear. Compounds such as napyradiomycin B1, with its three chlorinated stereocentres and unusual prenylation pattern, and azamerone, with its unique phthalazinone ring system, must have appeared to isolation chemists as “Molecules from Mars” (as Baldwin memorably termed the cryptic manzamine natural products89). In particular, the presence of multiple halogenated stereocentres in the napyradiomycins implied the existence of highly stereoselective vanadium-dependent haloperoxidase (VHPO) enzymes, which had previously been characterised as unselective, promiscuous biocatalysts in marine biochemistry. Gradually, a unified biosynthetic hypothesis for the origin of THN meroterpenoids was developed, based on early labelling studies combined with a more powerful genomic approach that unveiled many new polyketide synthase, prenyltransferase, and stereoselective VHPO enzymes. The resultant biosynthetic logic was successfully applied in biomimetic total syntheses of several meroterpenoid families, including the merochlorins, napyradiomycins and marinones. Some of these biomimetic syntheses also provided substrates and synthetic standards that subsequently aided the elucidation of the function of novel enzymes, thus creating an interdependent cycle of discovery between total synthesis and biosynthesis. The current state-of-the-art in marine meroterpenoid synthesis is the fully enzymatic total synthesis of napyradiomycin B1, in which a combination of prenyltransferase and VHPO enzymes were used as biocatalysts in a one-pot operation. To date, napyradiomycin B1 has eluded total synthesis by purely chemical means, which indicates the difficulty of the synthesis of halogenated stereocentres.
Several exciting avenues for research on the biosynthesis and synthesis of known THN-derived meroterpenoids remain. For example, the final steps in the biosynthesis of the marinones and naphterpins remain to be deciphered, potentially involving a stereoselective hetero-Diels-Alder reaction. The concluding steps in the biosynthesis of azamerone are equally enticing, with a unique biosynthetic cascade involving diazotisation and oxidative dearomatisation steps to be interrogated. Further study of the structure and function of the napyradiomycin and merochlorin vanadium-dependent haloperoxidase enzymes could stimulate the discovery of new catalysts for stereoselective halogenation. Alternatively, the directed evolution of these enzymes could give rise to less specific biocatalysts for the scalable, selective halogenation of a broad range of organic substrates. Finally, there are many more natural product families derived from the oxidative dearomatisation and halogenation of THN that are awaiting their discovery from marine bacteria. The isolation and characterisation of these molecules will inspire further synergistic cycles of biosynthetic studies and total synthesis.
13. Acknowledgements
This work was supported by an Australian Research Council Future Fellowship (FT170100437) awarded to J.H.G., a grant from the US National Institutes of Health (AI047818) to B.S.M., and faculty research start-up funds provided by the University of California, Santa Cruz to S.M.K.M.
Footnotes
Conflicts of interest
There are no conflicts to declare.
14. References
- 1.Geris R and Simpson TJ, Nat. Prod. Rep, 2009, 26, 1063–1094. [DOI] [PubMed] [Google Scholar]
- 2.Matsuda Y and Abe I, Nat. Prod. Rep, 2016, 33, 26–53. [DOI] [PubMed] [Google Scholar]
- 3.Li S-M, Nat. Prod. Rep, 2010, 27, 57–78. [DOI] [PubMed] [Google Scholar]
- 4.Gallagher KA, Fenical W and Jensen PR, Curr. Opin. Biotechnol, 2010, 21, 794–80. [DOI] [PubMed] [Google Scholar]
- 5.Appendino G, Chianese G and Taglialatela-Scafati O, Curr. Med. Chem, 18, 2011, 1085–1099. [DOI] [PubMed] [Google Scholar]
- 6.Moore BS, Synlett, 2018, 29, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funa N, Ohnishi Y, Fujii I, Shibuya M, Ebizuka Y and Horinouchi S, Nature, 1999, 400, 897–899. [DOI] [PubMed] [Google Scholar]
- 8.Austin MB, Izumikawa M, Bowman ME, Udwary DW, Ferrer JL, Moore BS and Noel JP, J. Biol. Chem, 2004, 279, 45162–45174. [DOI] [PubMed] [Google Scholar]
- 9.Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W and Moore BS, J. Am. Chem. Soc, 2012, 134, 11988–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC and Bradley S Moore, J. Biol. Chem, 2007, 282, 16362–16368. [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki T, Hayashi Y, Kuzuyama T, Furihata K, Itoh N, Seto H and Dairi T, 2006, J. Bacteriol, 188, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komiyama K, Funayama S, Anraku Y, Ishibashi M, Takahashi Y and Omura S, J. Antibiot, 1990, 43, 247–252. [DOI] [PubMed] [Google Scholar]
- 13.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Isshiki K, Takeuchi T, Umezawa H and Iitaka Y, J. Antibiot, 1986, 39, 494–501. [DOI] [PubMed] [Google Scholar]
- 14.Kumano T, Tomita T, Nishiyama M and Kuzuyama T, J. Biol. Chem, 2010, 285, 39663–39671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pathirana C, Jensen PR and Fenical W, Tetrahedron Lett, 1992, 33, 7663–7666. [Google Scholar]
- 16.Tello M, Kuzuyama T, Heide L, Noel JP and Richard SB, Cell. Mol. Life Sci, 2008, 65, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winter JM and Moore BS, J. Biol. Chem, 2009, 284, 18577–18581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takemura S, Hirayama A, Tokunaga J, Kawamura F, Inagaki K, Hashimoto K and Nakata M, Tetrahedron Lett, 1999, 40, 7501–7505. [Google Scholar]
- 19.Tatsuta K, Tanaka Y, Kojima M and Ikegami H, Chem. Lett, 2002, 31, 14–15. [Google Scholar]
- 20.Snyder SA, Tang Z-Y and Gupta R, J. Am. Chem. Soc, 2009, 131, 5744–5745. [DOI] [PubMed] [Google Scholar]
- 21.Henkel T and Zeeck A, J. Antibiot, 1991, 44, 665–669. [DOI] [PubMed] [Google Scholar]
- 22.Fukuda DS, Mynderse JS, Baker PJ, Berry DM and Boeck LD, J. Antiobiot, 1990, 43, 623–633. [DOI] [PubMed] [Google Scholar]
- 23.Motohashi K, Irie K, Toda T, Matsuo Y, Kasai H, Sue M, Furihata K and Seto H, J. Antibiot, 2008, 61, 75–80. [DOI] [PubMed] [Google Scholar]
- 24.Shomura T, Gomi S, Ito M, Yoshida J, Tanaka E, Amano S, Watabe H, Ohuchi S, Itoh J, Sezaki M, Takebe H and Uotani K, J. Antibiot, 1987, 40, 732–739. [DOI] [PubMed] [Google Scholar]
- 25.Gomi S, Ohuchi S, Sasaki T, Itoh J and Sezaki M, J. Antibiot, 1987, 40, 740–749. [DOI] [PubMed] [Google Scholar]
- 26.Goodfellow M, Williams ST and Alderson G, System. Appl. Microbiol, 1986, 8, 55–60. [Google Scholar]
- 27.Shiomi K, Iinuma H, Hamada M, Naganawa H, Manabe M, Matsuki C, Takeuchi T and Umezawa H, J. Antibiot, 1986, 39, 487–493. [DOI] [PubMed] [Google Scholar]
- 28.Wu Z, Li S, Li J, Chen Y, Saurav K, Zhang Q, Zhang H, Zhang W, Zhang W, Zhang S and Zhang C, Mar. Drugs, 2013, 11, 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Takeuchi T and Umezawa H, J. Antibiot, 1987, 40, 1213–1219. [DOI] [PubMed] [Google Scholar]
- 30.Motohashi K, Sue M, Furihata K, Ito S and Seto H, J. Nat. Prod, 2008, 71, 595–601. [DOI] [PubMed] [Google Scholar]
- 31.Carretero-Molina D, Ortiz-López FJ, Martín J, Oves-Costales D, Díaz C, de la Cruz M, Cautain B, Vicente F, Genilloud O and Reyes F, Mar. Drugs, 2020, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farnaes L, Coufal NG, Kauffman CA, Rheingold AL, DiPasquale AG, Jensen PR and Fenical W, J. Nat. Prod, 2014, 77, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng Y-B, Jensen PR and Fenical W, Eur. J. Org. Chem, 2013, 18, 3751–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacret R, Pérez-Victoria I, Oves-Costales D, de la Cruz M, Domingo E, Martín J, Díaz C, Vicente F, Genilloud O and Reyes F, Mar. Drugs, 2016, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soria-Mercado IE, Prieto-Davo A, Jensen PR, and Fenical W, J. Nat. Prod, 2005, 68, 904–910. [DOI] [PubMed] [Google Scholar]
- 36.Cho JY, Kwon HC, Williams PG, Jensen PR and Fenical W, Org. Lett, 2006, 8, 2471–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryu M-J, Hwang S, Kim S, Yang I, Oh D-C, Nam S-J and Fenical W, Org. Lett, 2019, 21, 5779–5783. [DOI] [PubMed] [Google Scholar]
- 38.Kagamizono T, Hamaguchi T, Ando T, Sugawara K, Adachi T and Osada H, J. Antibiot, 1999, 52, 75–80. [DOI] [PubMed] [Google Scholar]
- 39.Shin-ya K, Imai S, Furihata K, Hayakawa Y, Kato Y, Vanduyne GD, Clardy J and Seto H, J. Antibiot, 1990, 43, 444–447. [DOI] [PubMed] [Google Scholar]
- 40.Takagi H, Motohashi K, Miyamoto T, Shin-ya K, Furihata K and Seto H, J. Antibiot, 2005, 58, 275–278. [DOI] [PubMed] [Google Scholar]
- 41.Park J-S and Kwon HC, Mar. Drugs, 2018, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin-ya K, Shimazu A, Hayakawa Y and Seto H, J. Antibiot, 1992, 45, 124–125. [DOI] [PubMed] [Google Scholar]
- 43.Wessels P, Gohrt A and Zeeck A, J. Antibiot, 1991, 44, 1013–1018. [DOI] [PubMed] [Google Scholar]
- 44.Volkmann C, Hartjen U, Zeeck A and Fiedler H-P, J. Antibiot, 1995, 48, 522–524. [DOI] [PubMed] [Google Scholar]
- 45.Lu C, Yang C and Xu Z, Rec. Nat. Prod, 2016, 10, 437–440. [Google Scholar]
- 46.Hardt H, Jensen PR and Fenical W, Tetrahedron Lett, 2000, 41, 2073–2076. [Google Scholar]
- 47.Ishibashi M, Funayama S, Anraku Y, Komiyama K and Omura S, J. Antibiot, 1991, 44, 390–395. [DOI] [PubMed] [Google Scholar]
- 48.Panthee S, Takahashi S, Takagi H, Nogawa T, Oowada E, Uramoto M and Osada H, J. Antibiot, 2011, 64, 509–513. [DOI] [PubMed] [Google Scholar]
- 49.Kagamizono T, Kawashima A, Kishimura Y, Yamagishi M, Tsuchida Y, Kondo H and Hanada K, Biosci. Biotechnol. Biochem, 1993, 57, 766–769. [Google Scholar]
- 50.Kawahara T, Nagai A, Takagi M and Shin-ya K, J. Antibiot, 2012, 65, 579–581. [DOI] [PubMed] [Google Scholar]
- 51.Sedmera P, Pospíšil S and J Novák J Nat. Prod, 1991, 54, 870–872. [Google Scholar]
- 52.Song Y, Huang H, Chen Y, Ding J, Zhang Y, Sun A, Zhang W and Ju J, J. Nat. Prod, 2013, 76, 2263–2268. [DOI] [PubMed] [Google Scholar]
- 53.Kalaitzis JA, Hamano Y, Nilsen G and Moore BS, Org. Lett, 2003, 5, 4449–4452. [DOI] [PubMed] [Google Scholar]
- 54.Wong WR, Oliver AG and Linington RG, Chem. Biol, 2012, 19, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin-ya K, Furihata K, Hayakawa Y and Seto H, Tetrahedron Lett, 1990, 42, 6025–6026. [Google Scholar]
- 56.Funayama S, Ishibashi M, Komiyama K and Omura S, J. Org. Chem, 1990, 55, 1133–1135. [Google Scholar]
- 57.Haagen Y, Glück K, Fay K, Kammerer B, Gust B, and Heide L, Chembiochem, 2006, 7, 2016–2027. [DOI] [PubMed] [Google Scholar]
- 58.Kuzuyama T, Noel JP and Richard SB, Nature, 2005, 435, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS and Moore BS, Angew. Chem. Int. Ed, 2014, 53, 11019–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teufel R, Methods Enzymol, 2018, 604, 425.-. [DOI] [PubMed] [Google Scholar]
- 61.Gao J, Ko T-P, Chen L, Malwal SR, Zhang J, Hu X, Qu F, Liu W, Huang J-W, Cheng Y-S, Chen C-C, Yang Y, Zhang Y, Oldfield E, and Guo R-T, Angew. Chem. Int. Ed, 2018, 57, 683–687. [DOI] [PubMed] [Google Scholar]
- 62.McKinnie SMK, Miles ZD and Moore BS, Methods Enzymol, 2018, 604, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miles ZD, Diethelm S, Pepper HP, Huang DM, George JH and Moore BS, Nat. Chem, 2017, 9, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pepper HP and George JH, Angew. Chem. Int. Ed, 2013, 52, 12170–12173. [DOI] [PubMed] [Google Scholar]
- 65.Meier R, Strych S and Trauner D, Org. Lett, 2014, 16, 2634–2637. [DOI] [PubMed] [Google Scholar]
- 66.Diethelm S, Teufel R, Kaysser L and Moore BS, Angew. Chem. Int. Ed, 2014, 53, 11023–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang H, Liu X, Li Q, Li L, Zhang J-R and Tang Y, Org. Biomol. Chem, 2016, 14, 198–205. [DOI] [PubMed] [Google Scholar]
- 68.Brandstätter M, Freis M, Huwyler N and Carreira EM, Angew. Chem. Int. Ed, 2019, 58, 2490–2494. [DOI] [PubMed] [Google Scholar]
- 69.Bernhardt P, Okino T, Winter JM, Miyanaga A and Moore BS, J. Am. Chem. Soc, 2011, 133, 4268–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKinnie SMK, Miles ZD, Jordan PA, Awakawa T, Pepper HP, Murray LAM, George JH and Moore BS, J. Am. Chem. Soc, 2018, 140, 17840–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carter-Franklin JN, Parrish JD, Tschirret-Guth RA, Little RD and Butler A, J. Am. Chem. Soc, 2003, 125, 3688–3689. [DOI] [PubMed] [Google Scholar]
- 72.Carter-Franklin JN and Butler A, J. Am. Chem. Soc, 2004, 126, 15060–15066. [DOI] [PubMed] [Google Scholar]
- 73.Leipoldt F, Zeyhle P, Kulik A, Kalinowski J, Heide L and Kaysser L, PLoS One, 2015, 10, e0143237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuzuyama T, J. Antibiot, 2017, 70, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murray LAM, McKinnie SMK, Pepper HP, Erni R, Miles ZD, Cruickshank MC, López-Pérez B, Moore BS and George JH, Angew. Chem. Int. Ed, 2018, 57, 11009–11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Murray LAM, Fallon T, Sumby CJ and George JH, Org. Lett, 2019, 21, 8312–8315. [DOI] [PubMed] [Google Scholar]
- 77.Haagen Y, Unsöld I, Westrich L, Gust B, Richard SB, Noel JP and Heide L, FEBS Lett, 2007, 581, 2889–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Isogai S, Nishiyama M and Kuzuyama T, Bioorg. Med. Chem. Lett, 2012, 22, 5823–5836. [DOI] [PubMed] [Google Scholar]
- 79.Smith AB, Sestelo JP and Dormer PG, J. Am. Chem. Soc, 1995, 177, 10755–10756. [Google Scholar]
- 80.Saito T, Morimoto M, Akiyama C, Matsumoto T and Suzuki JK. Am. Chem. Soc, 1995, 117, 10757–10758. [Google Scholar]
- 81.Saito T, Suzuki T, Morimoto M, Akiyama C, Ochiai T, Takeuchi K, Matsumoto T and Suzuki K, J. Am. Chem. Soc, 1998, 120, 11633–11644. [Google Scholar]
- 82.Trost BM, Thiel OR and Tsui H-C, J. Am. Chem. Soc, 2002, 124, 11616–11617. [DOI] [PubMed] [Google Scholar]
- 83.Trost BM, Thiel OR and Tsui H-C, J. Am. Chem. Soc, 2003, 125, 13155–13164. [DOI] [PubMed] [Google Scholar]
- 84.Pena-López M, Martiónez MM, Sarandeses LA and Sestelo JP, Chem. Eur. J, 2009, 15, 910–916. [DOI] [PubMed] [Google Scholar]
- 85.Pu L-Y, Chen J-Q, Li M-L, Li Y, Xie J-H and Zhou Q-L, Adv. Synth. Catal, 2016, 358, 1229–1240. [Google Scholar]
- 86.Winter JM, Jansma AL, Handel TM and Moore BS, Angew. Chem. Int. Ed, 2009, 48, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Landry ML, McKenna GM and Burns NZ, J. Am. Chem. Soc, 2019, 141, 2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schnell SD, Linden A and Gademann K, Org. Lett, 2019, 21, 1144–1147. [DOI] [PubMed] [Google Scholar]
- 89.Moses JE and Adlington RM, Chem. Commun, 2005, 5945–5952. [DOI] [PubMed] [Google Scholar]