

Abstract
Background
Pulmonary arterial hypertension (PAH) is a fatal disease characterized by profound vascular remodeling in which pulmonary arteries narrow due to medial thickening and occlusion by neointimal lesions, resulting in elevated pulmonary vascular resistance and right heart failure. Therapies targeting the neointima would represent a significant advance in PAH treatment, however our understanding of the cellular events driving neointima formation, and the molecular pathways that control them, remains limited.
Methods
We comprehensively map the stepwise remodeling of pulmonary arteries in a robust, chronic inflammatory mouse model of pulmonary hypertension. This model demonstrates pathologic features of the human disease, including increased right ventricular pressures, medial thickening, neointimal lesion formation, elastin breakdown, increased anastomosis within the bronchial circulation, and perivascular inflammation. Using genetic lineage tracing, clonal analysis, multiplexed in situ hybridization, immunostaining, deep confocal imaging and staged pharmacologic inhibition we define the cell behaviors underlying each stage of vascular remodeling and identify a pathway required for neointima formation.
Results
Neointima arises from smooth muscle cells (SMCs) and not endothelium. Medial SMCs proliferate broadly to thicken the media, after which a small number of SMCs are selected to establish the neointima. These neointimal founder cells subsequently undergoing massive clonal expansion to form occlusive neointimal lesions. The normal pulmonary artery SMC population is heterogeneous and we identify a Notch3-marked minority subset of SMCs as the major neointimal cell of origin. Notch signaling is specifically required for the selection of neointimal founder cells, and Notch inhibition significantly improves pulmonary artery pressure in animals with pulmonary hypertension.
Conclusions
This work describes the first nongenetically driven murine model of PH that generates robust and diffuse occlusive neointimal lesions across the pulmonary vascular bed and does so in a stereotyped timeframe. We uncover distinct cellular and molecular mechanisms underlying medial thickening and neointima formation and highlight novel transcriptional, behavioral and pathogenic heterogeneity within pulmonary artery SMCs. In this model, inflammation is sufficient to generate characteristic vascular pathologies and physiologic measures of human PAH. We hope that identifying the molecular cues regulating each stage of vascular remodeling will open new avenues for therapeutic advancements in the treatment of PAH.
Keywords: Pulmonary hypertension, Neointima, Medial thickening, Notch signaling, Inflammation, Vascular smooth muscle cells, Lineage tracing, Clonal analysis, Cellular heterogeneity, Cellular mechanisms of disease
Introduction
Pulmonary arterial hypertension (PAH) is a fatal disease characterized by progressive narrowing of the pulmonary vasculature for which there are limited therapies and no cure1. PAH arises as a consequence of many different etiologies including heritable mutations, congenital heart disease, systemic inflammatory conditions, viral infection, and drug and toxin exposure2. Despite this heterogeneity, all forms of PAH demonstrate profound remodeling of the pulmonary arteries, in which the smooth muscle layer of the vessel wall (the ‘media’) thickens and abnormal ‘neointima’ cells accumulate beneath the endothelial layer, eventually occluding the vessel lumen, increasing pulmonary vascular resistance and leading to right heart failure1. These shared histologic features suggest that diverse upstream triggers converge on a core arterial remodeling program common to all forms of PAH. Current therapies target pulmonary vasoconstriction with limited long-term success as the underlying vasculopathy remains unchecked3, 4. Approaches effectively targeting the core mechanisms of vascular remodeling, including neointima formation and expansion, would represent a breakthrough in the treatment of PAH.
Key challenges in understanding the cellular pathophysiology of PAH include determining which cells are involved in medial thickening and neointima formation, delineating the steps by which remodeling occurs, and uncovering the molecular signals which control each of those steps. Early events in remodeling have been difficult to study as patients often present with advanced disease5. In PAH, there is thought to be dysregulated signaling between endothelial and smooth muscle cells, resulting in increased vascular tone and abnormal rates of cellular proliferation and apoptosis6. The cellular origin of the neointima is currently a subject of active debate in the field of pulmonary vascular biology with both smooth muscle and endothelial cells proposed to differentiate into neointima and generate occlusive lesions7–11. Many molecules and processes have been implicated in vascular remodeling in PAH12, but their specific roles have been difficult to investigate as chronic hypoxia exposure, the dominant murine model, does not reproduce the complex vasculopathy seen in human PAH, including formation of occlusive neointimal lesions13.
Here we establish an inflammation-driven murine model of pulmonary hypertension that reproduces key features of human PAH pathology, including medial thickening and robust neointimal lesion formation. Using lineage tracing and clonal labeling we demonstrate that both the neointima and the expanded media are derived from vascular smooth muscle cells (VSMCs) of the artery wall rather than the endothelium, informing a current controversy in the field. We define distinct cell behaviors for medial thickening and neointima formation and identify a Notch3-marked minority subset of VSMCs as the neointimal cell of origin in multiple mouse models of PH, highlighting how heterogeneity within the normal VSMC population is exploited to give rise to disease pathology. Finally, we identify a requirement for Notch signaling specifically in the selection of the neointimal founder cells that, when blocked, leads to significant improvement in pulmonary artery pressure in affected animals.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
HDM mouse model of pulmonary hypertension
10 week old BALB/c female mice were obtained from Charles River (strain 028). Lyophilized house dust mite extract (Stallergenes Greer, XPB70D3A25) was diluted to a concentration of 20μg Derp1 protein per 50μl with sterile PBS and 50μl of the resulting solution was delivered intranasally to animals briefly anesthetized by isoflurane inhalation. Animals were dosed five days per week for the indicated periods. Control animals received an equal volume of sterile PBS delivered in the same manner and following the same dosing schedule as those receiving dust mite solution. All animal experiments were approved by the Stanford University Institutional Animal Care and Use Committee.
Statistical analysis
Statistical tests used for each figure are detailed in figure legends. All statistical analysis and plotting were done in the statistical language R (www.R-project.org) using RStudio (RStudio Inc).
Population-level lineage tracing
Lineage tracing was carried out using either Acta2-CreER44 combined with a TdTomato Cre reporter45 (Jax strain #007914), Gja5-CreER46 combined with the mTmG Cre reporter47 (Jax strain #007676), Myh11-CreER49 (Jax strain #019079) combined with the multicolor Rainbow Cre reporter48, or Notch3-CreER58 combined with the TdTomato Cre reporter45. In all cases, 3mg of tamoxifen (Sigma, T5648; 10mg/ml stock dissolved in corn oil) was administered by oral gavage two weeks prior to the first exposure to house dust mite solution. Baseline labeling in all cases was assessed three days after tamoxifen administration. Both male and female animals were used from each of these genotypes (with the exception of Myh11-CreER which is found on the Y chromosome), at a variety of ages, and all showed pulmonary artery remodeling following similar kinetics to those seen when HDM exposure was initiated in 10 week old BALB/c females.
Notch inhibition with DBZ
DBZ stock was made by dissolving 10mg DBZ (Tocris, 4489) in 72μl DMSO (Sigma, D8418), and was stored protected from light at 4 degrees C for up to 2 months and was injected at 10μl/g of body weight every other day for two weeks. Vehicle solution was prepared and delivered identically, with 20μl DMSO substituted for the DBZ stock. Because intestinal goblet cell metaplasia is a known outcome of systemic Notch inhibition59, 71 animals were weighed daily and removed from the experiment if they lost more than 15% of their starting weight.
Full detailed methods including murine physiologic measurements, tissue handling, patient characteristics for human tissue, immunohistochemistry and antibody staining, clonal analysis, and quantitative multiplexed fluorescent in situ hybridization are available in the Expanded Methods.
Results
Pulmonary hypertension in mice following chronic inflammation
In recent years a central role for perivascular inflammation and pro-inflammatory signals within the adventitia in the pathogenesis of PAH has been increasingly appreciated14–24. We utilized a mouse model of chronic pulmonary inflammation25 and found that it effectively induces pulmonary hypertension and vascular pathologies similar to human PAH. In this model, mice are exposed to the common aeroallergen house dust mite (HDM) daily by intranasal administration for 2 to 8 weeks. Following chronic HDM administration, mice developed elevated right ventricular pulse pressure (RVP), a proxy measurement for pulmonary arterial pressure, and also exhibit a trend toward hypertrophy of the right ventricle, an adaptation to increased pulmonary resistance (Figure 1a–c). Though HDM is delivered to the airways, the inflammatory response rapidly centers and organizes around veins and arteries26 (Figure 1d & e; Figure I in the Supplement). The elevated RVP seen in this model is similar to that seen following hypoxia exposure. When mice were jointly exposed to HDM and hypoxia, RVP elevation was severe (Figure 1a), and RV hypertrophy was further increased (Figure 1b), indicating inflammation and hypoxia can act in combination to produce severe disease. All subsequent analysis was performed on animals housed in normoxia.
Figure 1: Chronic inflammation causes pulmonary hypertension in mice.
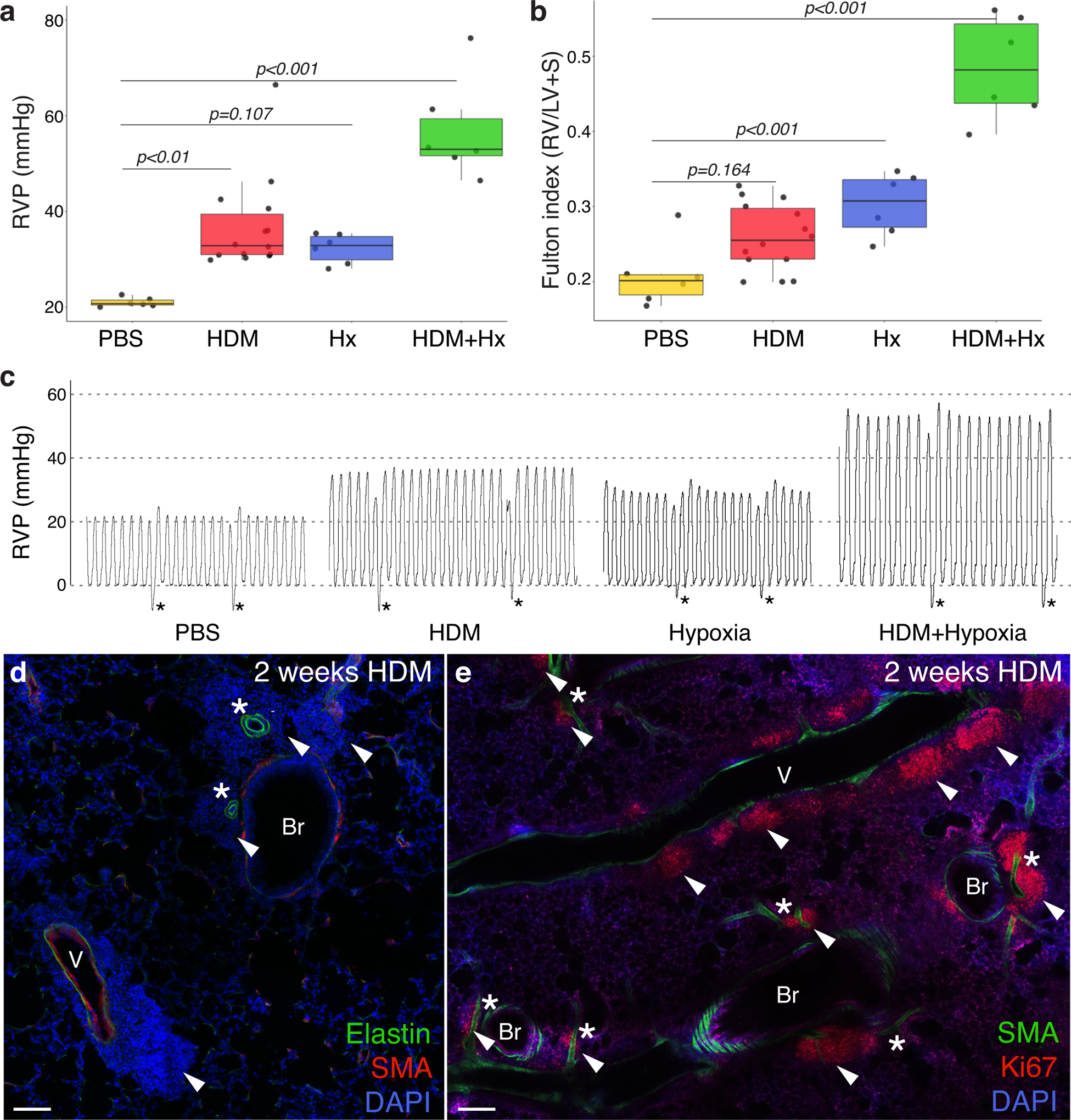
a, Right ventricular pulse pressure (RVP) elevation in mice exposed to 6 weeks of house dust mite (HDM) extract in PBS. Control animals were treated with intranasal PBS for the same duration. RVP was significantly elevated in HDM-exposed animals when compared to control (PBS). Hx, hypoxia. b, Right ventricular hypertrophy, expressed as the ratio of the mass of the right ventricle to that of the left ventricle plus septum (Fulton Index), was observed in all three exposure groups, though HDM-exposed animals fall short of significance. Comparisons in a & b were assessed by one-way ANOVA, showing a significant difference in RVP [F(3, 28)=19.95, p=4.05×10−7] and Fulton index [F(3,28)=38.67 p=4.32×10−10] across groups. Tukey’s HSD post-hoc test was carried out to derive the reported between-group comparison p-values. Normality checks and Levene’s test for homogeneity of variance were carried out and the assumptions were met. Dots, individual animal measurements. (a & b, PBS n=6 mice, HDM n=14 mice, Hx n=6 mice, HDM+Hx n=6 mice) c, Representative traces from a pressure catheter inserted into the right ventricle of anesthetized animals from each treatment group. Difference between pressure minimum and maximum averaged for three peaks to calculate RVP value for each animal. Peaks immediately before and after a breath (asterisk) were excluded when calculating RVP. d, Regions of DAPI-positive inflammatory cells (arrowheads) are prominent around pulmonary arteries (asterisks) and veins (v) following HDM exposure in immunostained lung sections. Br, bronchi. Representative confocal images shown from >20 mice examined. e, Clusters of actively proliferating inflammatory cells (arrowheads) marked by nuclear Ki67 staining organize a sustained local inflammatory response throughout HDM exposure and are found along arteries, veins and bronchi. Clusters around arteries and veins marked with arrowheads. Representative images shown, n=4 mice. Scale bar d & e, 100μm.
Medial thickening and neointimal lesion formation follow a reproducible stepwise time course
Sustained HDM exposure leads to a stereotyped pattern of progressive artery remodeling that follows a reproducible time course, with neointimal lesions present in nearly all pulmonary artery branches by 8 weeks (Figure 2a, b & d). Within the first two weeks of HDM exposure, mice develop significant medial hypertrophy, in which the media triples in thickness from 2.9 μm to 8.6 μm (p<0.001; Figure 2c). The media remains significantly thickened through the subsequent six weeks of remodeling. During weeks 2 through 4 the first neointimal lesions are established in which isolated clusters of smooth muscle α-actin expressing cells appear between the internal elastic lamina and the endothelium (Figure 2a, b & d). These neointimal cells are morphologically different from media VSMCs. While media VSMCs are short (~50μm), crescent-shaped cells and oriented orthogonally to the vessel axis, neointima cells are long (~150μm), thin and oriented parallel to the vessel axis (Figure 2b), similar to the longitudinally-arrayed neointima cells seen in patients with PAH27 (Figure 2e–g). Between weeks 4 and 8, there is robust expansion of the neointima (4–6 weeks and 6–8 weeks, each p<0.001; Figure 2d) with 97% of arteries containing neointima after 8 weeks (Figure 2d). Previously reported models of pulmonary vascular remodeling – weekly pulsed HDM administration28, and endothelial injury by the VEGFR-inhibitor SU5416 combined with hypoxia exposure (Su/Hx)29 – were quantitated in similar fashion and while significant medial thickening was observed in both models, neointimal lesions were statistically smaller and fewer when compared to chronic HDM exposure (Figure II in the Supplement).
Figure 2: Chronic inflammation results in reproducible progressive artery remodeling in mice, including neointima formation and expansion, that closely mimics human disease.
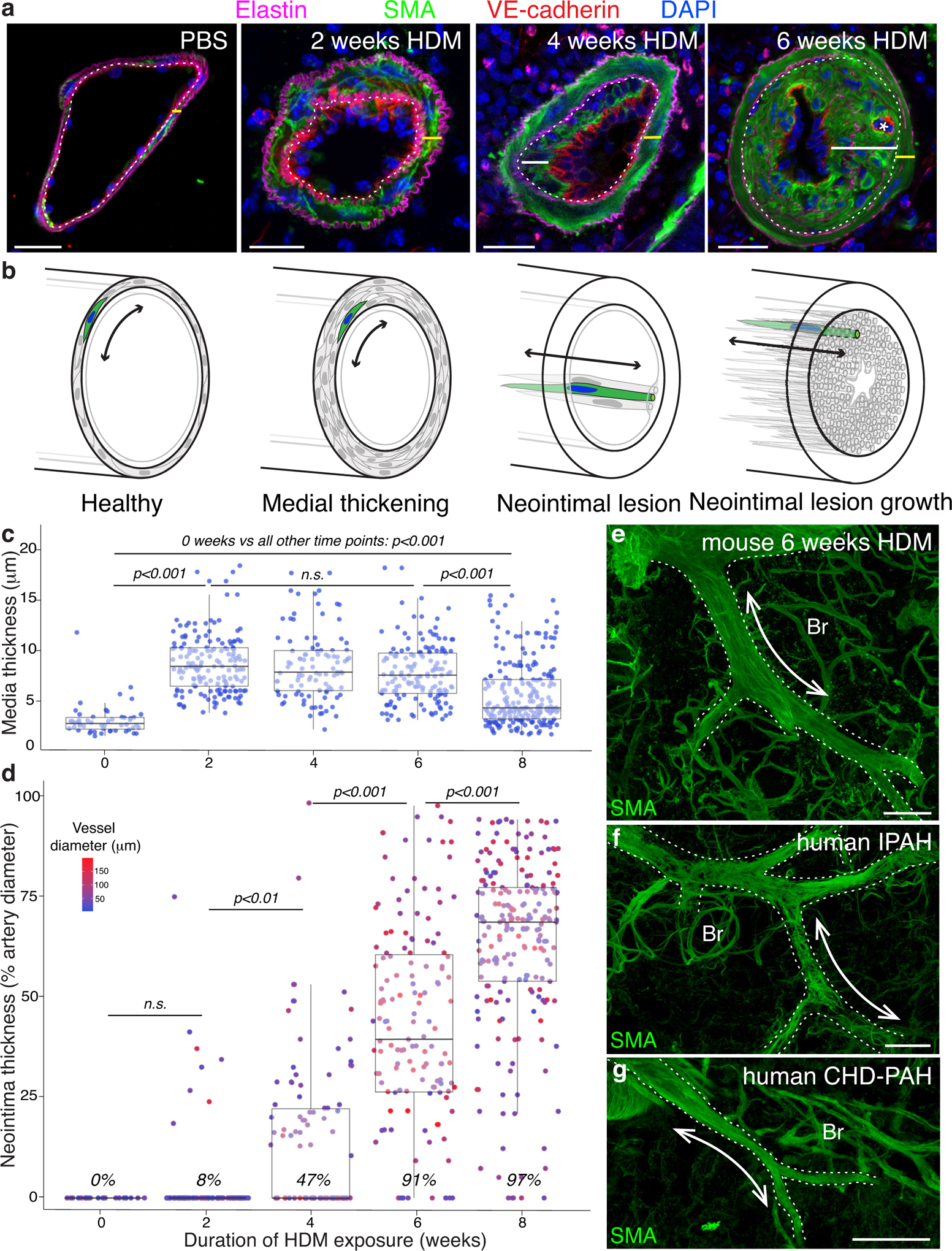
a, Immunostained sections of pulmonary arteries demonstrate progressive remodeling following HDM exposure. The medial layer (yellow bar) located between the internal (dotted line) and external elastic laminae (elastin, magenta) comprising smooth muscle α-actin (SMA, green) expressing cells expands, with an increase in thickness and cell number (2 weeks HDM). SMA positive neointimal cells (white bar) appear between the internal elastic lamina and the endothelium (VE-cadherin, red, 4 weeks HDM) and grow over the next two weeks to fully occlude the vascular lumen (6 weeks HDM). Occasional neointimal “recanalization” is observed as small secondary endothelial tubes (asterisk) grow through the lesion (6 weeks HDM). DAPI, blue. Representative images shown from >25 animals evaluated for each timepoint. Scale bar, 20μm. b, Schematics depicting circumferential orientation of smooth muscle cells between elastin layers (black lines) in the media of both control arteries (“Healthy”) and those with thickened media (“Medial thickening”), and longitudinal orientation of neointima cells between internal elastin layer and endothelial cell layer (grey line; “Neointimal lesion” and “Neointimal lesion growth”). Double-headed arrows depict orientation of cells. c & d, Pulmonary artery remodeling following HDM exposure follows a reproducible time course, with medial thickening (c) occurring solely between weeks 1 and 2. Neointima (d) first appears between weeks 2 and 4 and expands thereafter until by 8 weeks neointima occupies 75% of artery diameter, and 97% of arteries contain neointimal lesions. During the period when neointima first appears (weeks 2–4), no significant difference in vessel diameter between arteries with neointima and those without was observed (p=0.92). Due to homogeneity of variance assumptions not being met for standard ANOVA analysis, a one-way Welch’s ANOVA was performed showing a significant difference in medial thickness [F(4,707)=51.08, p <0.001] and neointima thickness [F(4,707)=204.28, p<0.001] between groups. A Games-Howell nonparametric post-hoc test was used for between group comparisons, with p-values reported as above for both c & d. Dots, individual vessel measurements. Number of animals evaluated for quantification at each timepoint: 0wk n=3, 2wk n=3, 4wk n=3, 6wk n=5, 8wk n=2. Dot color in d indicates vessel diameter. Percent of scored vessels with neointimal lesions in italics. e, Longitudinally oriented SMA-positive neointimal cells run along the length of the artery in mouse neointimal lesions following 6 weeks of HDM exposure, shown in a confocal z-stack projection of an immunostained vibratome section. Representative image from >20 mice evaluated. Scale bar, 100μm. f & g, Human PAH neointimal lesions (representative images from n=2 patients with idiopathic PAH, f, or congenital heart disease-PAH, g) shown in confocal z-stack projections of immunostained vibratome sections, are also composed of long thin SMA-positive cells aligned parallel to the longitudinal axis of the arteries. e-g, Dotted lines outline arteries; Br, bronchus; Scale bar f & g, 200μm.
Chronic HDM exposure also leads to a number of other pathologies seen in human PAH1 including proliferating nodes of inflammatory cells around veins and arteries with immune cell infiltration of neointimal lesions5, 30 (Figure 1d & e; Figure 3a), elastin breakdown and remodeling resulting in gaps in the internal elastic lamina31 (Figure 3b & c), and progressive development of intimal lesions (“arterialization”32–34) in the pulmonary veins (Figure 3d–g). While these animals do not develop plexiform lesions, we observed endothelial re-canalization through obstructive neointimal lesions (asterisk, 6wk HDM, Figure 2a) as well as robust expansion of the bronchial circulation35, 36 with increased anastomosis with the pulmonary circulation37 (Figure 3h–j). Like neointimal lesion formation, these features of human PAH are all absent in the most widely used mouse model of PH, chronic hypoxia exposure13. Unlike other mouse models which produce neointimal lesions38–40, chronic HDM exposure has the significant advantages of being robust, with rapid onset of disease, and non-genetic, permitting the use of the full range of mouse genetic tools to investigate the mechanisms of lesion formation.
Figure 3: PH mice demonstrate numerous pathologic features of human IPAH.
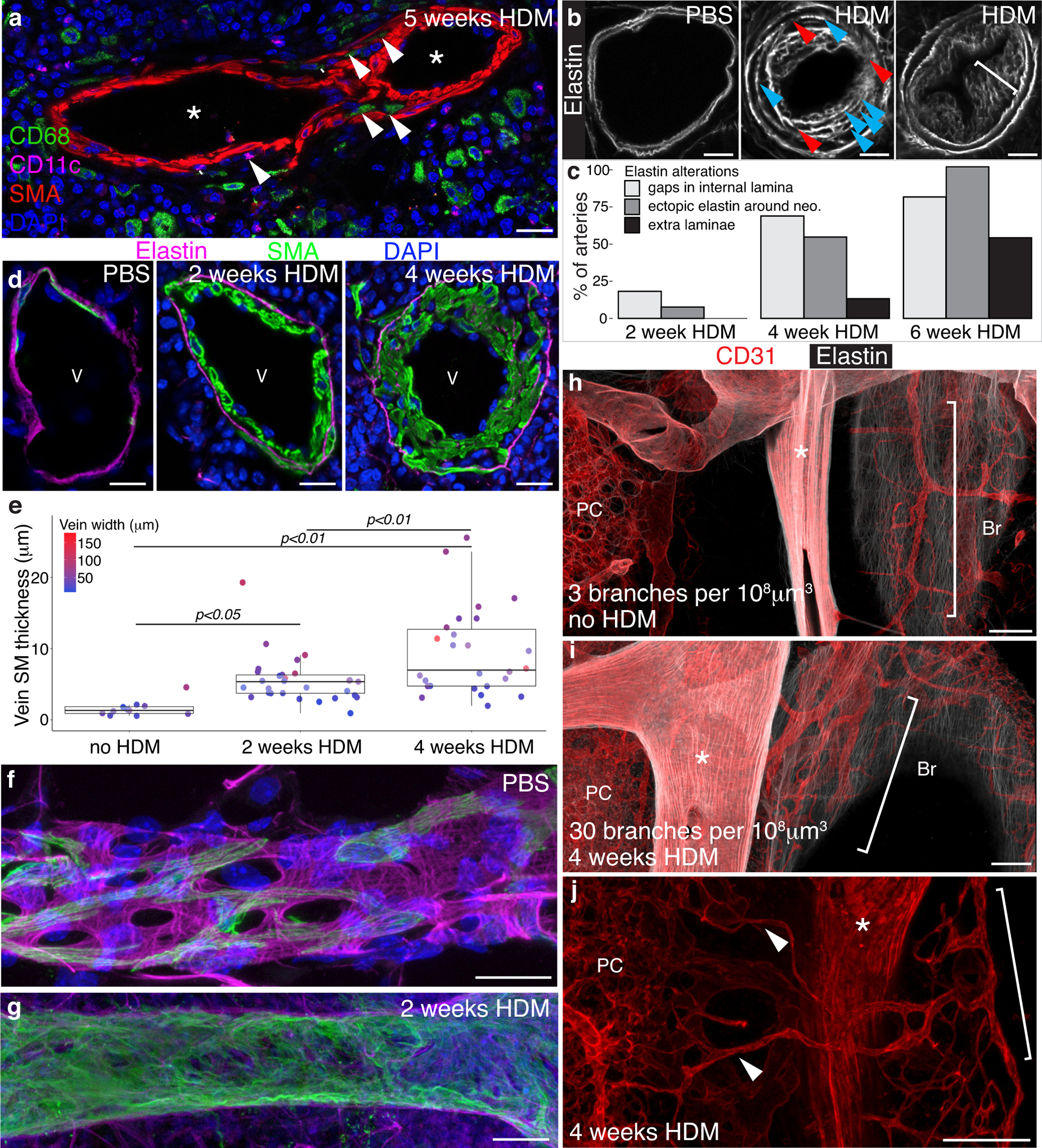
a, Following 2 weeks of HDM exposure, inflammatory cells including CD68+ macrophages and CD11c+ dendritic cells surround arteries and are found within the walls of remodeling arteries (arrowheads). Representative image shown, 5 mice evaluated. b, Lesions from HDM-exposed mice show irregularities of the elastin layers including degradation of the internal elastin layer, with numerous apparent thinnings or gaps (red arrowheads), formation of supernumerary laminae (blue arrowheads), a phenomenon termed “onion skinning” in human PAH, and ectopic elastin deposition surrounding neointimal cells throughout the lesions (white bracket). Representative images shown from >50 mice evaluated. c, Temporal progression of the elastin changes illustrated in b (n=4 mice). Following HDM exposure veins develop neointima, similar to the “arterialization” of veins reported in PAH. Control veins (d & f) have a single elastin layer (magenta) with a sparse covering of smooth muscle cells (SMA, green) on both the external and lumenal sides. After two weeks of HDM exposure, the inner face of the elastin layer is lined with long thin SMA-positive cells roughly aligned with the long axis of the vein, as observed in neointima formation in arteries. As with the artery neointima, vein neointima expands significantly over the following weeks (d & e). Dots in e, individual vein measurements from n=4 mice. Dot color indicates vessel width. Due to homogeneity of variance assumptions not being met for standard ANOVA analysis, a one-way Welch’s ANOVA was performed showing a significant difference in vein smooth muscle layer thickness [F(2,63)=12.75, p<0.001] between groups. A Games-Howell nonparametric post-hoc test was used for between-group comparisons. f, Confocal z-stack projection of a control vein demonstrating a porous elastin layer (magenta) with sparse smooth muscle cells (SMA, green) wrapping healthy vein. g, Confocal z-stack projection of a vein from a mouse exposed to HDM daily for 2 weeks demonstrates near complete muscularization of the vein by smooth muscle cells (SMA, green). b, d, f & g Representative images shown from >20 mice evaluated. Scale bars in a, b, d, f & g, 20μm. h-j, In mice the bronchial circulation (brackets), which brings oxygenated blood to the tissues of the lung, wraps large airways (Br) and veins, and is found between arteries (asterisks) and airways. h, In healthy animals, connections between the bronchial and pulmonary circulations (PC) are rare. i & j, In HDM exposed animals, similar to human PAH, the bronchial vessels become more extensively branched (30 branches per 108 μm3 in HDM vs. 3 branches per 108 μm3 in PBS controls) and anastomosis (arrowheads, j) with pulmonary capillaries is observed. Images representative of 6 PBS control mice and 5 HDM treated mice shown. Scale bars h-j, 100μm.
Neointima is generated by smooth muscle cells and not endothelium
The reproducible time course of vascular remodeling (Figure 2) makes it possible to order events and dissect the cellular and molecular mechanisms responsible for the pathologies. Both neointimal cells and the expanded SMCs in arteries of PAH patients41 (Figure 2f & g), HDM-exposed mice (Figure 2a & e), as well as other mouse models of pulmonary artery remodeling (Figure IIb & c) express smooth muscle proteins, and recent studies in mice have shown that thickened media arises from pre-existing SMCs28, 42. However, detection of endothelial markers within the neointima and pronounced endothelial dysfunction in PAH19, 43 has led to the proposal that neointima arises by endothelial to mesenchymal transition7–11, 22. To identify the cellular origin of the neointima, we used the Cre-loxP system to heritably mark and follow cell types of interest during the remodeling of the pulmonary arteries following HDM exposure (Figure IIIa in the Supplement). To distinguish between smooth muscle cells and endothelium as potential sources for neointima, we marked SMCs using Acta2-CreER44 combined with the R26TdTomato Cre reporter45 and marked artery endothelial cells using Gja5-CreER46 combined with the R26mTmG Cre reporter47. Acta2-CreER marks smooth muscle cells of the airways, arteries and veins, but not pericytes. Tamoxifen was administered to healthy mice to heritably label the cell type of interest, either smooth muscle or endothelial cells, after which mice were rested for two weeks and then exposed to daily intranasal HDM for 8 weeks to assure robust neointima formation. Only Acta2-lineage traced smooth muscle cells contributed to either the media or the neointima (Figure 4a & c), with no contribution from Gja5-lineage labeled endothelial cells to either (Figure 4b & c). The vast majority of both medial and neointimal cells were Acta2-lineage labeled, though unlabeled patches of neointima were observed, likely reflecting labeling inefficiency (Figure IVa–c in the Supplement). Acta2 lineage-labeled cells also generate neointima in pulmonary veins (Figure IVd & e in the Supplement). These results confirm that preexisting functional smooth muscle cells proliferate to expand the medial layer28, 42. Direct measurement of proliferation using daily EdU administration during the first two weeks of HDM exposure further supports that VSMCs of the media proliferate to thicken the artery wall (Figure 4d & e; Figure V in the Supplement). Most importantly, these results demonstrate that media VSMCs generate cells that breach the internal elastic lamina to form the neointima, and do not support a model in which endothelial to mesenchymal transition generates the neointima.
Figure 4: Smooth muscle not endothelium generates expanded media and neointima following HDM exposure.
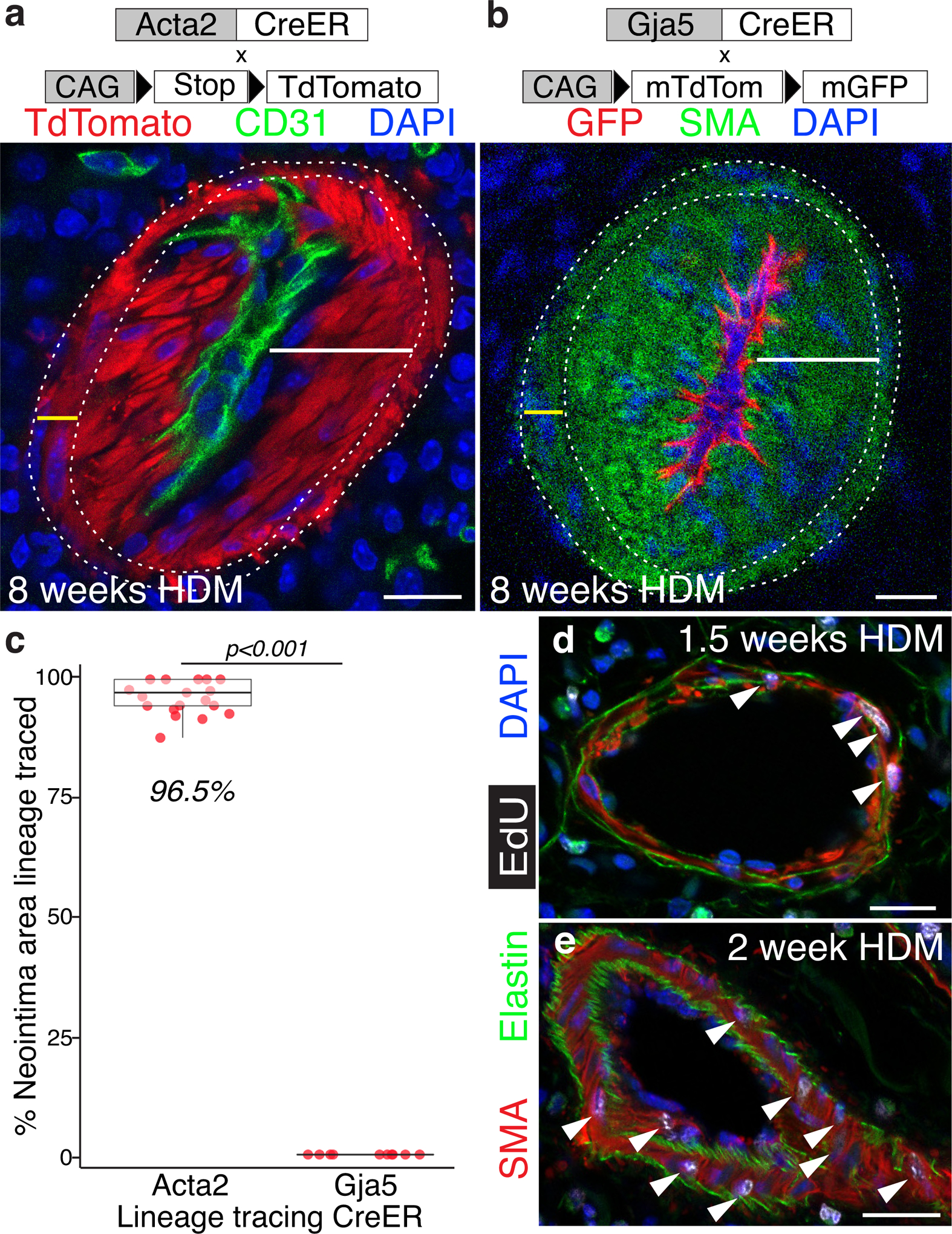
a, Following 8 weeks HDM exposure both the expanded media (yellow bar, between dotted lines) and neointima (white bar) are labeled by genetic lineage trace of pre-existing Acta2-CreER-expressing SMCs (marked by heritable expression of tdTomato, red) demonstrating that smooth muscle cells are the cell of origin for the expanded media and neointima. b, Genetic lineage tracing demonstrates that Gja5-CreER-marked pre-existing endothelial cells (which express GFP following Cre-mediated recombination, red) contribute to neither the media nor the neointima (SMA, green), but rather remain in the endothelium. a & b Position of elastic laminae is indicated by dotted lines. n=5 Acta2-CreER mice and n=2 Gja5-CreER mice with >1000 vessel cross-sections reviewed per genotype, distributed across all lobes of the lung. c, Percent neointima lineage labeled with Acta2-CreER or Gja5-CreER following 8 weeks of HDM exposure. Dots, individual vessel measurements. Median value indicated in italics. A Welch two-sample t-test found a significant difference between Acta2- and Gja5-CreER groups [t(20)=123.86, p< 2.2×10−16, d=38.2]. Quantification completed on 2 mice/genotype. d & e, Daily EdU administration during the first two weeks of HDM exposure shows cell division among VSMCs in media (arrowheads) demonstrating that the media thickens through proliferation. EdU incorporation into nuclei of proliferating cells shown in white. Note higher number of EdU marked cells at 2 weeks. n=2 mice. See Figure V in the Supplement for PBS controls and additional time points. Scale bar, 20μm.
Single cell analysis reveals distinct cell behaviors at each stage of remodeling
The above results demonstrate that smooth muscle cells contribute to both medial hypertrophy and neointima formation. To determine whether all VSMCs contribute equally to both processes, or whether a VSMC subpopulation is active at either step, we employed a multicolor labeling strategy (Figure IIIb in the Supplement) in which the VSMCs of healthy animals were marked with a randomly distributed mix of one of three potential fluorescent proteins using the Rainbow multicolor Cre reporter48 crossed with Myh11-CreER, a smooth muscle specific CreER49 (Figure 5a, d & e). If many or all cells contribute to medial expansion or neointima formation, with limited cell migration, the random distribution of the three colors should be maintained. In contrast, if only a small subset of SMCs contributes, then patches of single color should emerge in the remodeled tissue, consistent with rare progenitors dividing while other cells remain quiescent (Figure VI in the Supplement). Following three weeks of HDM exposure, the cells of the thickened media retain a random distribution of colors (Figure 5b, d & e), consistent with many or all of the VSMCs dividing a small number of times to drive expansion. At this stage, the first neointima cells were already visible as loose clusters of 3–10 cells oriented parallel to the long axis of the artery in the subendothelial space (Figure 5b’). Each group was composed of cells of multiple colors interspersed, with most clusters containing all three colors (Figure 5e), indicating that neointima is founded by multiple cells within a local area that independently cross the internal elastin layer. After robust neointimal expansion (6 weeks HDM), however, large patches of coherent single color cells were observed in neointimal lesions with 3–5 patches per vessel cross-section (Figure 5c–e and Figure VII in the Supplement), consistent with a small number of neointimal founder cells undergoing a striking clonal expansion to generate bundles of 30–60 longitudinal cells. This clonal expansion suggests neointimal lesions do not grow through continued recruitment of new cells from the smooth muscle, but rather there is a bottleneck selection event in which neointima emerges from a restricted pool of founder cells. We hypothesized a potential source of neointimal founder cells could be a VSMC subset present within the healthy artery wall.
Figure 5: Distinct cell behaviors underlie medial expansion and neointimal lesion formation.
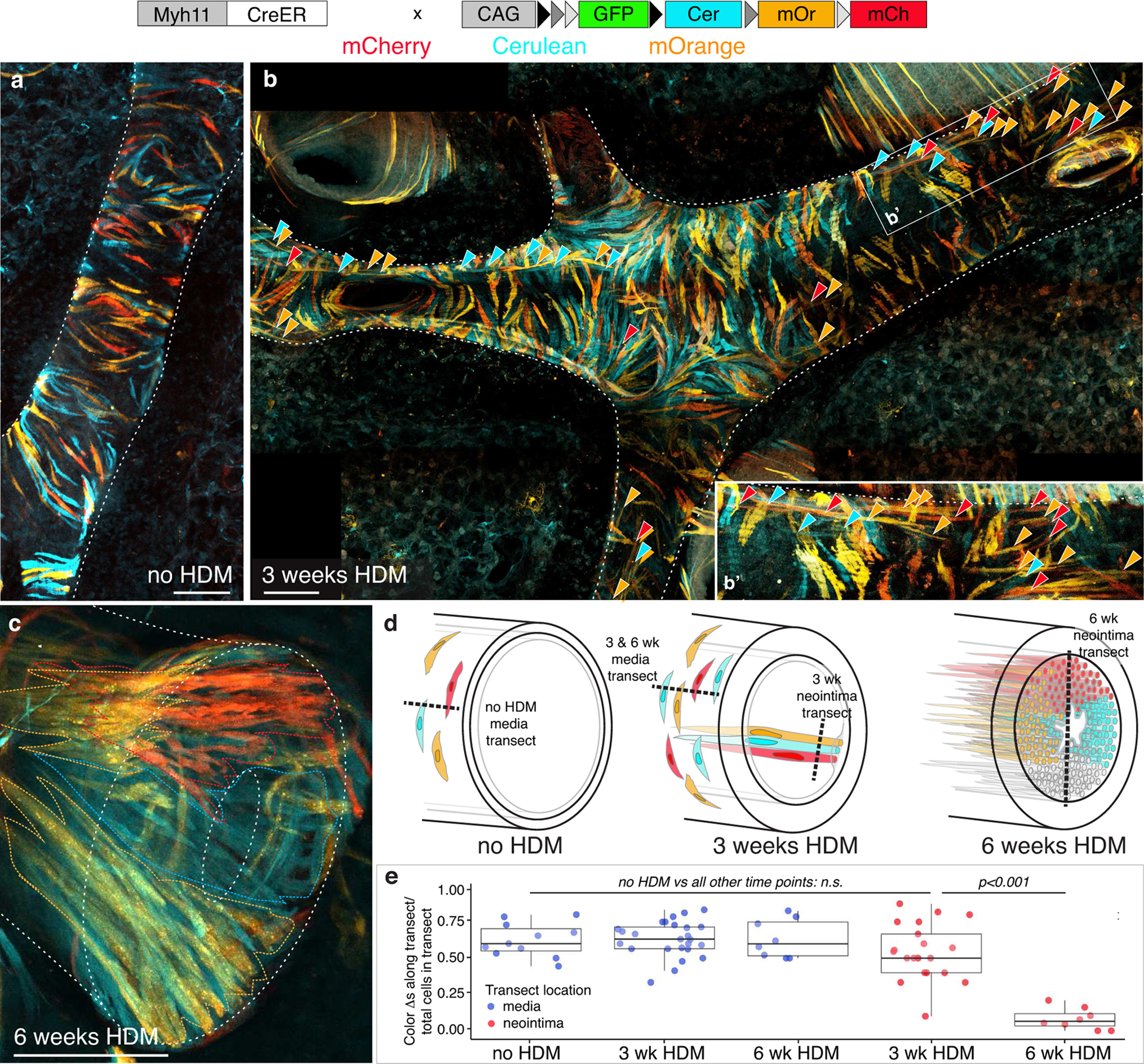
a, In healthy mice, a single dose of tamoxifen to Myh11-CreER; Rainbow animals resulted in randomly distributed labeling of SMCs within the media such that an individual labeled cell will heritably express either Cerulean, mOrange, or mCherry. Medial SMCs are oriented orthogonal to axis of artery. 2 animals examined. Scale bar, 50μm. b, Following 3 weeks HDM the distribution of colors remains random in the thickened media, suggesting many or all VSMCs divide a small number of times to expand the media. >100 arteries from 2 animals examined. Early neointima cells (arrowheads, highlighted in inset, b’), oriented parallel to axis of artery, are visible beneath media cells. Early neointima clusters are composed of a small number of cells of different colors. >200 clusters scored from 2 animals. Scale bar, 100μm. c, In contrast to the random distribution of colors in the media, single color bundles of neointima cells (oriented parallel to the axis of the artery) are seen after expansion of neointimal lesions, indicating that lesion growth occurs by local proliferation of a small number of neointima founder cells. >100 arteries from 2 animals examined. Colored dotted lines indicate boundaries of single-color bundles of neointima cells. Scale bar, 50μm. a-c, Representative projections of confocal z-stack of cleared vibratome sections. mCherry, red; mOrange, orange; Cerulean, cyan. White dotted lines outline arteries and lumens for cross-sectional images. d, Schematics illustrating cell dynamics among VSMCs and their progeny during artery remodeling. Random color distribution of labeled cells prior to HDM exposure (“no HDM”) is maintained in medial layer following medial thickening (“3 weeks HDM”) suggesting many or all medial cells proliferate to expand the media. Small clusters of longitudinally oriented neointimal cells are seen following 3 weeks of HDM exposure with subsequent clonal expansion of a small number of neointimal founder cells following 6 weeks of HDM as demonstrated by large single-color bundles in the neointima. e, The shift from a random distribution of cell labels in media (blue dots) and early neointima (red dots, 3 wk HDM) to bundles of single color neointima after 6 weeks HDM exposure (red dots, 6 wk HDM) is represented by counting the number of color changes between neighboring cells along transect lines (schematized as dashed lines in d) through either media or neointima. n= 2 mice per timepoint. Dots, individual transect measurements. Comparison assessed by one-way ANOVA which showed a significant difference in color change/transect [F(4,68)=25.58, p=5.95×10−13] across groups. Tukey’s HSD post-hoc tests were carried out to derive the reported between group comparison p-values. Normality checks and Levene’s test for homogeneity of variance were carried out and the assumptions were met. See Figure VI in the Supplement for further information on experimental design.
Notch3 marks a subset of VSMCs that are the cell of origin for the neointima in multiple models of neointima formation
Pulmonary artery VSMCs are derived from the lung mesenchyme50, 51. Heterogeneity of VSMCs, including those of the pulmonary arteries, has been observed in a number of contexts52–54. Prior studies have demonstrated groups of VSMCs with increased contribution to distal muscularization following chronic hypoxia exposure42. We therefore searched for markers of subsets of VSMCs that might prospectively identify a neointima cell of origin within the VSMCs of healthy arteries. Notch3 is expressed in VSMCs55 and is required for their maturation during development56. NOTCH3/Notch3 protein expression is elevated in human PAH and in hypoxia-exposed mice, and is required for a rise in RVP in mice following hypoxia exposure57. Using quantitative multiplexed fluorescent in situ hybridization (QM-FISH), we found that NOTCH3/Notch3 transcripts localize to neointimal lesions in CHD-PAH patient tissue, chronic HDM exposure, and a PH model driven by endothelial loss of PHD239 (Figure VIII in the Supplement). Together, these findings make Notch3 an appealing candidate for further investigation. QM-FISH revealed three populations of Acta2+ artery SMCs in healthy mice (Figure 6a–a”): those expressing high levels of Notch3 (“Notch3hi” cells), those where Notch3 expression was undetectable or low (“Notch3lo” cells), and intermediate cells (“Notch3mid” cells). All pulmonary artery cross sections examined contained each of these types. We examined the contribution of Notch3-expressing cells to the neointima using the Notch3-CreER mouse line58, in which CreER is expressed from the endogenous Notch3 locus, combined with the R26TdTomato Cre reporter45 (Figure IIIa in the Supplement). Notch3-CreER labels a minority (~15%) of artery SMCs following a single dose of tamoxifen in healthy animals (Figure 6b & b’), roughly consistent with the proportion of Notch3hi VSMCs as measured by in situ hybridization. QM-FISH shows that a strong majority of these TdTomato-labeled cells in healthy arteries are Notch3hi (Figure IXc & d in the Supplement). Tripling the tamoxifen dose increased labeling only slightly, to 18% of artery SMCs (Figure IXa & b in the Supplement), suggesting that a single 3mg tamoxifen dose effectively labels the Notch3hi population. After 6 weeks of HDM exposure we saw a striking enrichment of Notch3-CreER lineage labeled cells in the neointima, where a median 71% of neointima cells are labeled (Figure 6c). Notch3-CreER lineage labeled neointima (Figure 6d) is also found in the Su/Hx mouse model of PH that forms rare neointimal lesions (Figure II in the Supplement). Notch3-CreER lineage labeling of neointima was not complete, therefore we cannot exclude the possibility that other populations within the SMCs also contribute to the neointima. These results demonstrate that all VSMCs do not have equal potential to contribute to neointima, but rather that Notch3-marked VSMCs are the dominant neointimal cell of origin.
Figure 6: Notch3-expressing subset of vascular smooth muscle cells are the cell of origin for neointima.
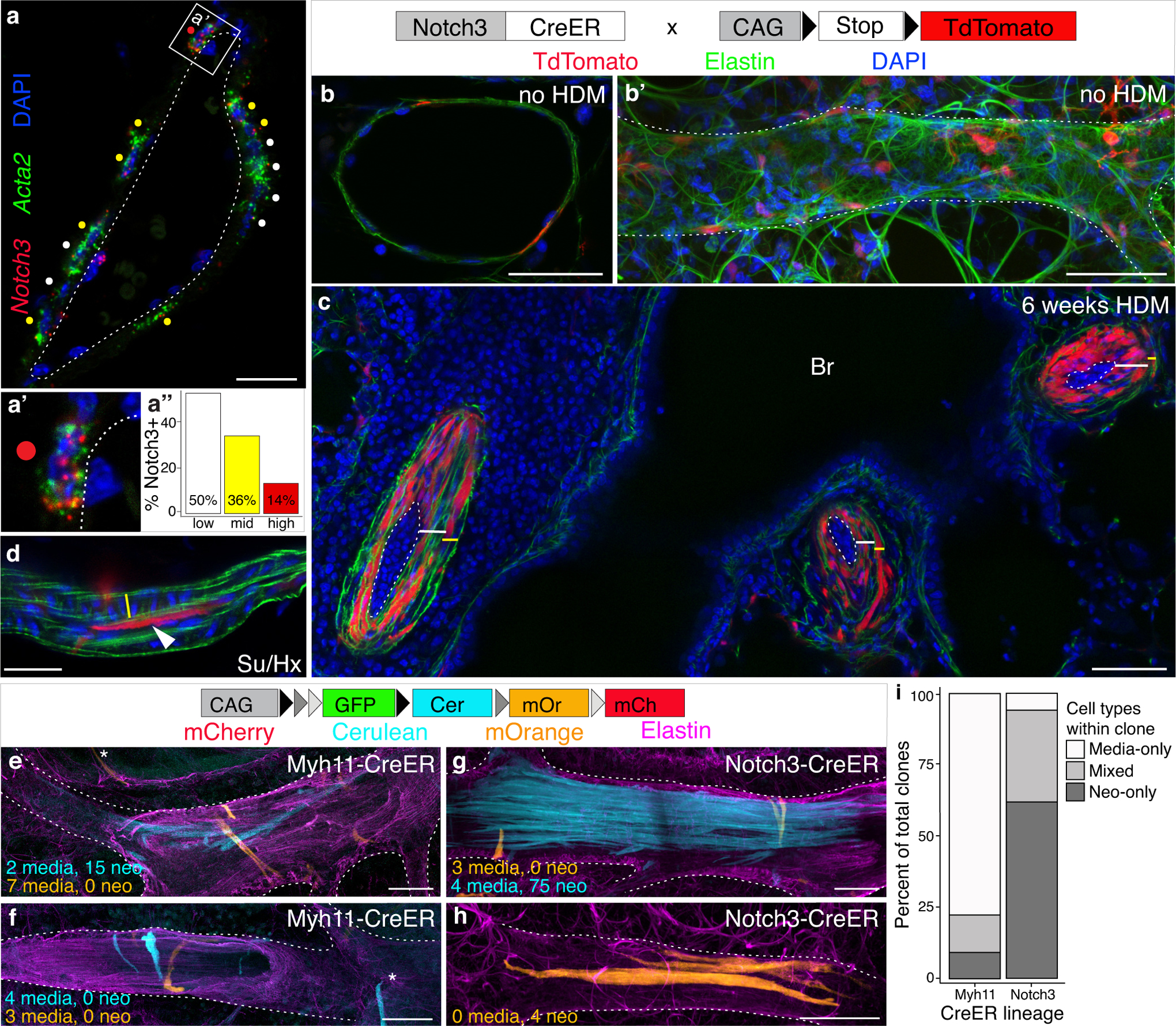
a, Quantitative multiplexed fluorescent in situ hybridization (QM-FISH) for Acta2 (green) and Notch3 (red) mRNA reveals a Notch3-expressing VSMC sub-population around healthy pulmonary arteries. a’, Example of an Acta2+ cells with high Notch3 expression. Scale bar, 20μm. a”, Notch3 levels within Acta2+ cells around arteries quantitated for 192 cells from 1 mouse. None/low (white), 0–2 Notch3 punctae per Acta2+ cell; Mid (yellow), 3–7 punctae; High (red), 8 or more punctae. Dotted line marks boundary between media and endothelium. b, Notch3-CreER labeling (red) marks approximately 15% of SMCs in healthy arteries, seen both in cross-section and along the artery length, b’, in an immunostained vibratome section. >100 arteries from 5 animals examined. Dotted line outlines artery in b’. c, Following 6 weeks HDM exposure the majority of neointima cells (white bars; 41–88%; median 71%) are labeled by lineage trace of pre-existing Notch3-CreER-expressing VSMCs (marked by heritable expression of tdTomato, red) demonstrating that Notch3-expressing cells are the cell of origin for the neointima (1560 cells scored from 2 animals). Dotted lines mark boundary between endothelium and neointima; yellow bars mark thickened media. b & c, Scale bar, 50μm. d, Notch3-lineage cells (red, marked with arrowhead) also generate neointima (arrowhead) in mice treated with SU5416 and exposed to four weeks hypoxia (Su/Hx), an established model of pulmonary artery remodeling; n=2 mice. Elastin, green; DAPI, blue. Scale bar, 20μm. e-h, Clones were generated in healthy animals using Notch3-CreER (n=2 mice) or Myh11-CreER (n=3 mice) combined with the Rainbow multicolor Cre reporter. e, f & i, The majority of Myh11-CreER clones in remodeled arteries were composed solely of media cells, with only 22 of 115 clones (19%) containing neointima (i). g-i, In HDM-exposed Notch3-lineage labeled mice, 121 of 134 (90%) clones contained neointima, demonstrating that neointimal progenitors are strongly enriched in the Notch3-marked population in healthy arteries. White dotted lines outline arteries. Scale bar e-h, 50μm.
Clonal analysis reveals cell dynamics of lesion formation
To quantitatively assess the enrichment for neointima founder cells within the Notch3 population versus the broader media VSMC population, we generated clones in healthy animals using Notch3-CreER58 and Myh11-CreER49 combined with the Rainbow multicolor Cre reporter48 (Figure 6e–h). To assess the potential of individual cells and measure heterogeneity within each of the above populations, labeling frequency was strictly limited, such that rare single cells and their descendants could be clearly distinguished. Tamoxifen was administered at low doses to ensure labeled cells were rare and well separated at the start of the experiment (Figure IIIc in the Supplement). Following neointimal lesion formation, the percentage of clones containing neointima originating from Myh11-labeled general media VSMCs was 19% (Figure 6e, f & i). This demonstrates variability among media cells and suggests that only a minority of media cells have the ability to make neointima. The observed 19% of media cells that contribute to neointima is roughly consistent with the 14% of Notch3hi transcribing cells observed in healthy arteries (Figure 6a) and the 15–18% of healthy artery VSMCs genetically marked by Notch3-CreER (Figure 6b; Figure IX in the Supplement). If Notch3 expression does define the minority subpopulation in the media that generates neointima, then clones made with Notch3-CreER should contain neointima at an overwhelming rate. In fact, the percentage of neointima-containing clones originating in Notch3-marked VSMCs was 90% (Figure 6g–i), demonstrating that Notch3-marked VSMCs are the dominant neointimal progenitors.
Analysis of the arrangement and distribution of cells within artery clones lets us deduce principles governing cell behavior in remodeling pulmonary arteries. Clones of neointimal cells observed in both genotypes remain closely grouped, with cells nearly always maintaining contact with at least one fellow clone member (Figure 6e, g & h). Neointimal clones are most often seen arranged in bundles, with cells grouped side by side and clones are generally less than 2 cell lengths long (Figure 6g & h), suggesting that the dominant mode of cell division expands lesions radially rather than longitudinally, consistent with the focal occlusions seen in PAH1. End to end cell movement within expanding lesions, however, is not absent. Neointimal cells can be longitudinally staggered (Figure 6e), allowing single clones to extend along the length of the vessel and span artery branch points while still maintaining contact with fellow clone members. Unlike neointima, clones that remain in the media are generally not coherent, nearly always containing cells that have lost contact with fellow clone members. Clonally-related media cells can be separated by hundreds of microns (>250μm separate mOrange clone members in Figure 6g), suggesting that cell movement in the media is substantial. Closely spaced media cell pairs, presumably reflecting recent cell divisions, are found in both end to end (mOrange clone, Figure 6f) and side by side (mOrange clones, Figure 6e & g) configurations, suggesting multiple modes of division and cell movement are possible among media cells. These results demonstrate that media and neointima cells behave differently and suggest that proliferation in each compartment may be independently regulated and targetable.
Notch signaling is required for neointima formation
To determine whether Notch signaling plays a role in medial thickening, neointima establishment and/or growth we again took advantage of the stereotyped time course of vascular remodeling following HDM exposure. HDM-exposed animals were treated with either the γ-secretase inhibitor Dibenzazepine59 (DBZ), which blocks Notch signaling by inhibiting ligand-dependent cleavage of Notch receptors, or vehicle during three two week windows: the periods during which media expands (weeks 0–2 of HDM exposure), during which neointima is established (weeks 2–4), and during which neointima grows to occlude the lumen (weeks 4–6). No difference in medial thickening was detected between DBZ and vehicle-treated animals during weeks 0–2 of HDM exposure (Figure 7a, d & g), demonstrating that Notch pathway activity is not required for the general SMC proliferation that underlies medial expansion (Figure 4d & e). In contrast, animals treated with DBZ during weeks 2–4, when the neointima is established, show little or no neointima formation (Figure 7b, e & g). This result indicates that the mechanism by which smooth muscle cells translocate and switch their fate to become pathogenic neointima cells is a Notch-dependent process. During the subsequent period of neointima expansion (weeks 4–6 of HDM exposure), however, Notch-blockade again had no effect (Figure 7c, f & g), as during media thickening, suggesting that the requirement for Notch activity is confined to the short period when neointima is established, after which neointimal expansion is driven by other mechanisms. These results also support the conclusion that the transition from VSMC to neointima occurs during a defined window and that lesion expansion does not occur through continued recruitment and fate-switching of medial cells. This functional requirement for Notch signaling suggests that Notch may be a potential therapeutic target for preventing neointima formation and thereby treating early lesions in PAH. Indeed, we found that pharmacological Notch inhibition during neointima establishment (Figure 7h) significantly improves hemodynamics in these animals.
Figure 7: Neointimal founder cell selection is Notch-dependent.
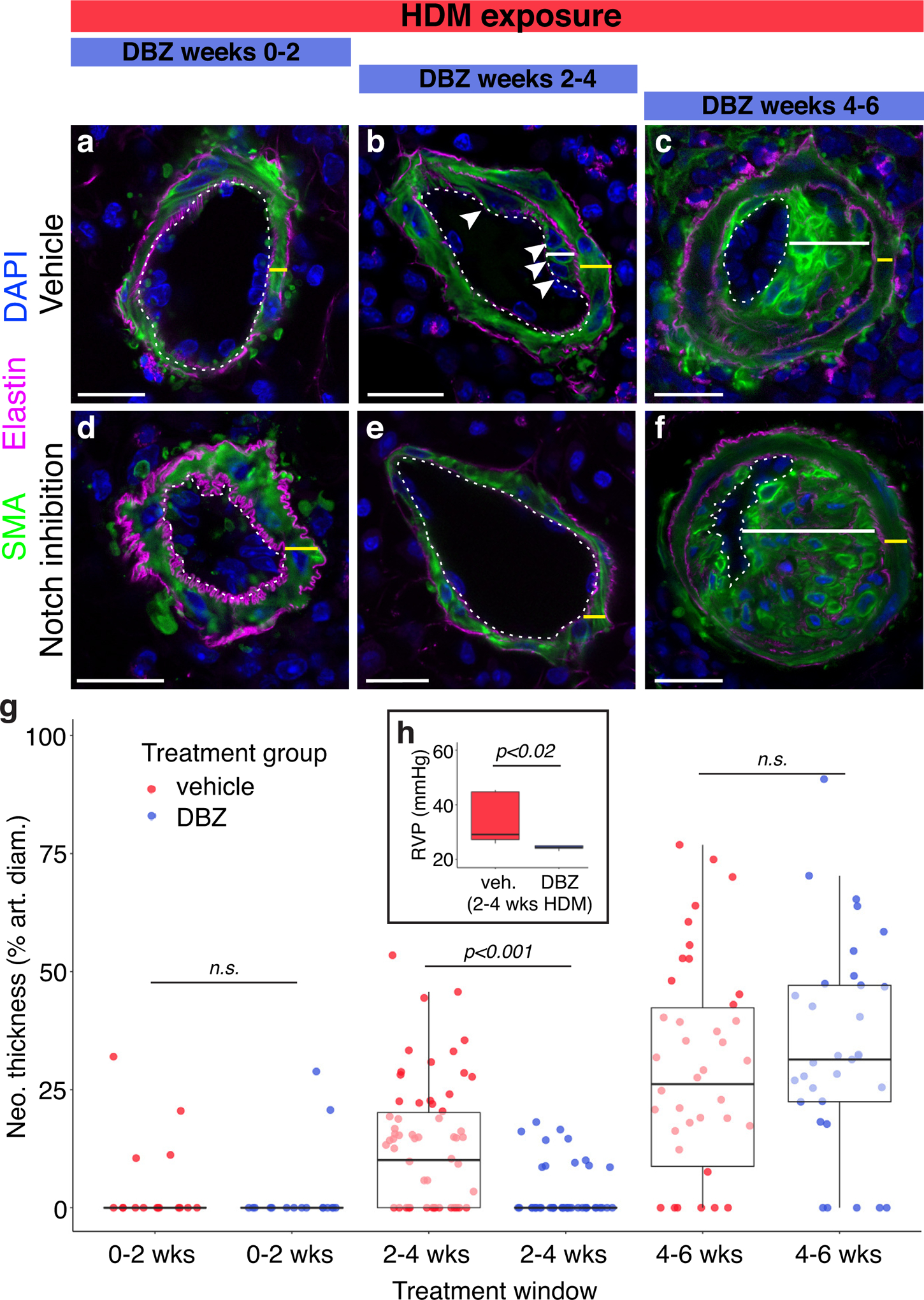
To test the role of Notch signaling in each stage of vascular remodeling, mice were given either vehicle (a-c) or the Notch inhibitor DBZ (d-f) for staggered two-week windows (blue bars, top) during chronic HDM exposure. Blocking Notch signaling prevents formation of the first neointima cells (arrowheads, b & e, DBZ or vehicle given during weeks 2–4 of HDM exposure; harvest at 4 weeks). DBZ treatment had no effect on the medial thickening (a & d, DBZ or vehicle during weeks 0–2 of HDM exposure; harvest at 2 weeks) or expansion of already established neointima (c & f, DBZ during weeks 4–6 of HDM exposure, harvest at 6 weeks), indicating that Notch signaling is required only for neointima founder cell selection. Representative confocal images shown. SMA, green; elastin, magenta; DAPI, blue; media, yellow bars; neointima, white bars. Dotted lines mark the boundary between endothelium and media/neointima. Scale bar, 20μm. Establishment of neointima (g, weeks 2–4) is significantly reduced following Notch inhibition (blue dots) versus vehicle (red dots), but neointimal growth (weeks 4–6) is unchanged. Between group differences were assessed by two-way ANOVA showing a statistically significant interaction between the effects of Treatment and Treatment Window on neointima thickness [F(2,276)=9.182 p<0.001]. Tukey’s HSD post hoc tests were carried out to derive the reported between-group comparison p-values. Normality checks were carried out and assumptions were met. Dots, individual vessel measurements. h, Notch-blockade with DBZ treatment (blue box) during weeks 2–4 of HDM exposure significantly reduces RVP compared to vehicle-treated animals (red box). 6 animals per treatment group. Significance assessed with Mann-Whitney Wilcoxon rank sum test.
Discussion
Here we present a model of pulmonary hypertension in mice which reproduces many key features of the human disease, including progressive vascular remodeling and the formation of complex neointimal lesions. We define a process of temporally and spatially distinct steps in the development of pulmonary vascular pathology, providing a framework to identify the genes and cell-cell signaling regulating each step. Using genetic lineage tracing, we demonstrate that neointima arises from vascular smooth muscle cells without contribution from the endothelium. Single cell labeling reveals that smooth muscle cells employ distinct cell behaviors at each stage of remodeling and that a small molecularly-defined subpopulation seeds the neointima and undergoes subsequent clonal proliferation to make occlusive lesions without continued recruitment from the media (Figure 8).
Figure 8: Staged events in pulmonary artery remodeling and development of PH in this model.
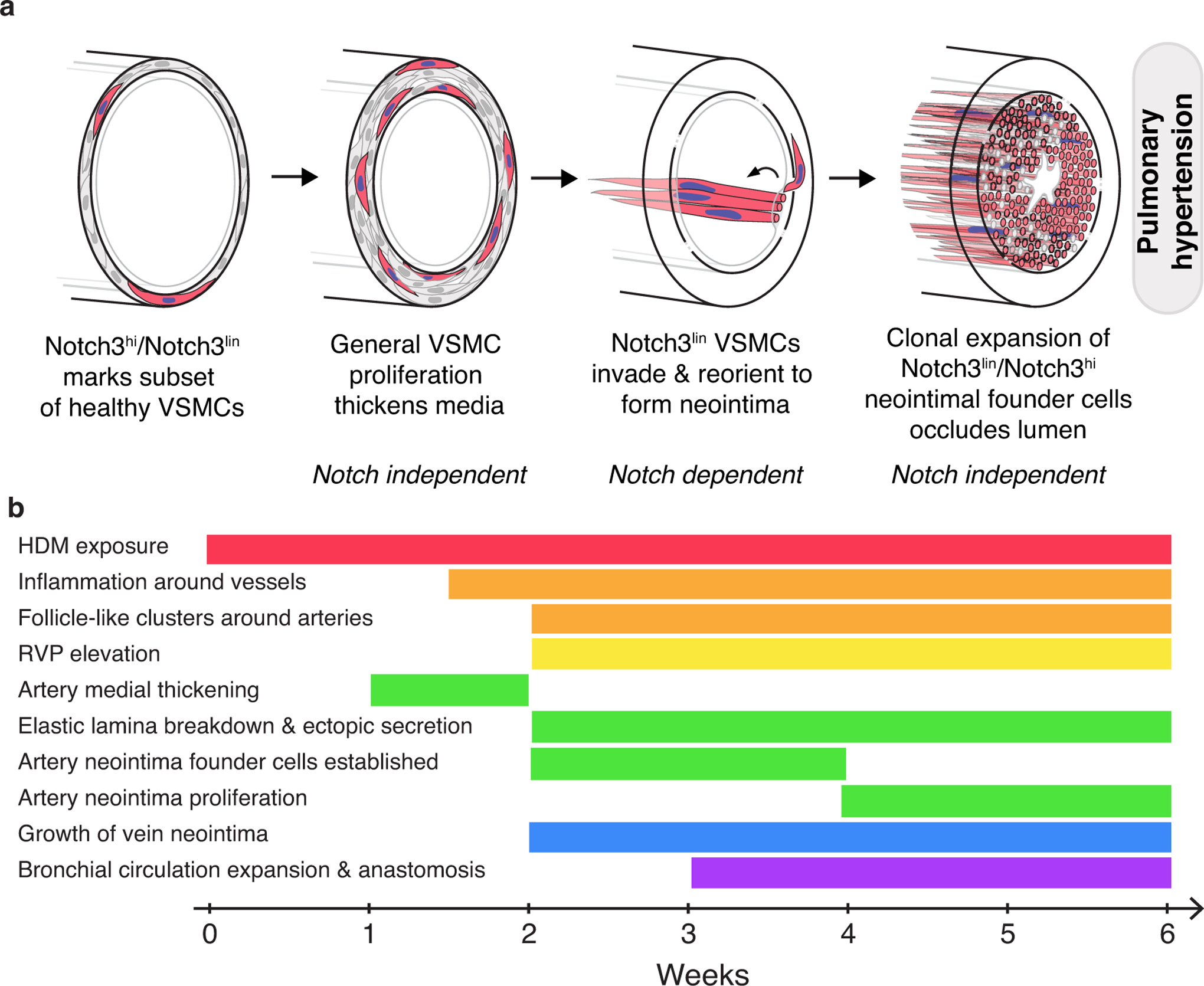
a, The vascular smooth muscle cells in the media of healthy arteries are molecularly heterogeneous. In the schematic, the minority population distinguished by high levels of Notch3 transcription (“Notch3hi”) and labeling by Notch3-CreER (“Notch3lin”) is colored red. Following HDM exposure and accompanied by perivascular inflammation, general proliferation of most or all VSMCs leads to medial thickening, possibly through direct reception of inflammatory cytokines by VSMCs. A subsequent breakdown of the internal elastic lamina may increase access to endothelial signals by VSMCs. A small number of smooth muscle cells, most Notch3hi/Notch3lin, traverse the internal elastic lamina to found the neointima, possibly in response to endothelium-derived Notch ligands. Once established, neointima founder cells clonally expand and eventually occlude the vessel lumen. Notch signaling is specifically required for neointimal establishment; the signals (and their sources) that serve as the proliferative cues driving medial and neointimal expansion are currently unknown. Inflammatory cues may act at any or all steps. b, The major events in vascular remodeling following HDM exposure occur in a defined sequence and with reproducible timing. Key changes to the lung vasculature in HDM-exposed animals, arranged (from top to bottom) in the order in which they appear. Timeline indicates weeks of HDM exposure in BALB/c animals. Events are grouped by category: inflammatory changes, orange; hemodynamic changes, yellow; pulmonary artery changes, green; pulmonary vein changes, blue; bronchial circulation changes, purple.
We show that high levels of Notch3 expression mark a minority subset of healthy VSMCs as measured by both in situ hybridization and genetic labeling with Notch3-CreER. Notch3 has been implicated in maintaining an undifferentiated VSMC state, and contact-mediated Notch signaling between endothelium and VSMCs is important for vascular patterning both during development and postnatally56, 60. In the inflammation-driven HDM model, a Notch3-marked subset acts as the major cell of origin for neointima. In the Su/Hx model, driven by endothelial damage and hypoxia, Notch3 lineage cells also generate neointima. As the neointima is established, Notch3-marked medial cells migrate toward the endothelium through a compromised elastin layer31, a process that fails to occur in the setting of Notch inhibition. This suggests that contact between Notch3-expressing VSMCs and endothelium may be needed for smooth muscle cell conversion to neointimal cells, and that endothelium may be the source of Notch ligands required for neointimal establishment. In contrast, proliferation of VSMCs within the media that precedes neointimal establishment, as well as proliferation of neointimal cells following establishment, are independent of Notch signaling. It remains unclear whether Notch signaling in VSMCs is sufficient to induce neointima formation in the absence of inflammation. We hope to identify molecules that control medial thickening and neointimal proliferation, and ultimately induce regression of established pulmonary vascular lesions by employing similar approaches to those used to identify the role of Notch signaling in neointimal establishment.
PAH is a consequence of many different etiologies that lead to a shared pattern of vascular pathologies1, 2, 61. The HDM model reproduces core features of vascular pathology shared between divergent forms of PAH, offering a tractable system for studying the mechanisms controlling each stage of the process. Here, inflammation is sufficient to trigger this complex series of events. The vascular changes seen in this model are likely driven by a combination of direct reception of inflammatory cytokines by VSMCs, coupled with inflammatory stimulation of neighboring cells including endothelium and adventitial fibroblasts resulting in abnormal signaling to VSMCs62. Endothelial dysfunction has long been appreciated in PAH, leading to dysregulated production of signaling molecules12. Elastin breakdown may further increase the accessibility of those signals to SMCs, promoting increased proliferation and migration.
Our work highlights the potential for uncovering mechanisms of disease by studying cell populations and behaviors with single cell resolution. Large scale single cell profiling efforts have uncovered previously unappreciated diversity within healthy cell types63–67. Here we have uncovered a functional role for a molecularly-defined subset of VSMCs in generating neointimal lesions in PAH, opening the possibility that this subpopulation may play a similar progenitor role in other diseases marked by intimal proliferation68–70. More generally, we propose that distinct subsets of cells in healthy tissues respond differently to disease stimuli. Identification of these subsets will allow us to define the specific molecular characteristics that enable their preferential contribution to pathology, offering new avenues for therapy development.
Supplementary Material
Clinical Perspective: What Is New?
In mice, chronic inflammation is sufficient to develop PH with vascular changes that closely mimic human disease including diffuse, occlusive neointimal lesions.
Neointima arises from a minority subset of vascular smooth muscle cells marked by high levels of Notch3 expression and not from endothelial cells, revealing novel heterogeneity within the healthy artery media and arguing against endothelial to mesenchymal transition in PAH.
The transformation of a Notch3-marked smooth muscle cell to a neointima cell occurs through a Notch-dependent process, highlighting a druggable window during which lesion formation can be prevented.
What Are the Clinical Implications?
Chronic house dust mite exposure can be used as a model for pre-clinical PAH drug trials given its stereotyped and temporally defined generation of complex vascular lesions, allowing specific signaling axes to be manipulated during precise stages of vasculopathy via pharmacologic or inducible genetic manipulations.
The Notch3+ vascular smooth muscle cell subset may serve as target for PAH therapies given its increased potential for pathologic transformation.
Acknowledgements:
M.E.K. conceived, performed and interpreted experiments, wrote the manuscript and secured funding. L.C.S. conceived, performed and interpreted experiments and wrote the manuscript. A.A.F., M.B., M.M., F.Z., W.Z., D.H., X.T., R.J.M. and E.S. performed experiments. A.A. performed statistical analysis and acquired patient samples. L.M. provided reagents. K.N. secured funding and provided feedback. R.J.M and E.S. provided expertise and feedback. The authors wish to thank J. Feinstein, D. Cornfield, and M. Rabinovitch for valuable insights and discussion, and Y. Ouadah and A. Gillich for comments on the manuscript. We thank the Pulmonary Hypertension Breakthrough Initiative for their assistance in providing patient samples and Stanford Medicine’s Animal Histology Service and HistoTech (Hayward, CA) for paraffin sample preparation.
Funding Sources:
This work was supported by Stanford Maternal & Child Health Research Institute Ernest & Amelia Gallo Endowed Postdoctoral Fellowship to L.C.S.; a Human Biology Research Exploration fellowship to A.A.F.; NIH 5T32HL129970-03 to L.C.S. and A.A; a Max-Kade Foundation postdoctoral fellowship to M.B.; National Heart Lung and Blood Institute R01 HL128734, Department of Defense PR161256, SPARK Translational Research Program in the Stanford University School of Medicine, and a Pulmonary Hypertension Association career development award to E.S.; American Heart Association 16SDG30030006, Stanford Spectrum-Child Health Research Institute seed grants, and Bravo Family endowed faculty scholarship to M.E.K.; and Vera Moulton Wall Center for Pulmonary Vascular Disease research grants to R.J.M., E.S. and M.E.K.
Non-standard Abbreviations and Acronyms:
- PAH
pulmonary arterial hypertension
- HDM
house dust mite extract
- RVP
right ventricular pulse pressure
- SMC/VSMC
smooth muscle cell/vascular smooth muscle cell
- QM-FISH
quantitative multiplexed in situ hybridization
- DBZ
Dibenzazepine (γ-secretase inhibitor; blocks Notch receptor signaling)
Footnotes
Publisher's Disclaimer: Disclaimer: The manuscript and its contents are confidential, intended for journal review purposes only, and not to be further disclosed.
Disclosures:
E.S. is listed as inventor on patent applications “Use of FK506 for the treatment of Pulmonary Arterial Hypertension” (Serial No 61/481317) and “Enzastaurin and Fragile Histidine Trial (FHIT) Increasing Agents for the Treatment of Pulmonary Hypertension” (PCT/US2018/033533). All other authors declare no competing interests.
References:
- 1.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53:1801887. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG and Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2018;53:1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spiekerkoetter E, Kawut SM and de Jesus Perez VA. New and emerging therapies for pulmonary arterial hypertension. Annu Rev Med. 2019; 70:45–59. doi: 10.1146/annurevmed-041717-085955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau EMT, Giannoulatou E, Celermajer DS and Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14:603–614. doi: 10.1038/nrcardio.2017.84. [DOI] [PubMed] [Google Scholar]
- 5.Tuder RM, Marecki JC, Richter A, Fijalkowska I and Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranchoux B, Harvey LD, Ayon RJ, Babicheva A, Bonnet S, Chan SY, Yuan JX and Perez VJ. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series). Pulm Circ. 2018;8:2045893217752912. doi: 10.1177/2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG and Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129:692–703. doi: 10.1161/CIRCULATIONAHA.113.003734. [DOI] [PubMed] [Google Scholar]
- 8.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L and Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target Slug. Circulation. 2016;133:1783–1794. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 10.Kwapiszewska G, Crnkovic S and Stenmark KR. A twist on pulmonary vascular remodeling: endothelial to mesenchymal transition? Am J Respir Cell Mol Biol. 2018;58:140–141. doi: 10.1165/rcmb.2017-0314ED. [DOI] [PubMed] [Google Scholar]
- 11.Frid MG, Kale VA and Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 12.Lilly B We have contact: endothelial cell-smooth muscle cell interactions. Physiology (Bethesda). 2014;29:234–241. doi: 10.1152/physiol.00047.2013. [DOI] [PubMed] [Google Scholar]
- 13.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, et al. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. Am J Physiol Lung Cell Mol Physiol. 2012;302:L977–991. doi: 10.1152/ajplung.00362.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar R and Graham B. How does inflammation contribute to pulmonary hypertension? Eur Respir J. 2018; 51:1702403. doi: 10.1183/13993003.02403-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham BB, Chabon J, Gebreab L, Poole J, Debella E, Davis L, Tanaka T, Sanders L, Dropcho N, Bandeira A, et al. Transforming growth factor-beta signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation. 2013;128:1354–1364. doi: 10.1161/CIRCULATIONAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar R, Mickael C, Kassa B, Sanders L, Koyanagi D, Hernandez-Saavedra D, Freeman S, Morales-Cano D, Cogolludo A, McKee AS, et al. Th2 CD4(+) T cells are necessary and sufficient for schistosoma-pulmonary hypertension. J Am Heart Assoc. 2019;8:e013111. doi: 10.1161/JAHA.119.013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mickael CS and Graham BB. The role of type 2 inflammation in schistosoma-induced pulmonary hypertension. Front Immunol. 2019;10:27. doi: 10.3389/fimmu.2019.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar R, Mickael C, Chabon J, Gebreab L, Rutebemberwa A, Garcia AR, Koyanagi DE, Sanders L, Gandjeva A, Kearns MT, et al. The causal role of IL-4 and IL-13 in Schistosoma mansoni pulmonary hypertension. Am J Respir Crit Care Med. 2015;192:998–1008. doi: 10.1164/rccm.201410-1820OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian W, Jiang X, Sung YK, Shuffle E, Wu TH, Kao PN, Tu AB, Dorfmuller P, Cao A, Wang L, et al. Phenotypically silent bone morphogenetic protein receptor 2 mutations predispose rats to inflammation-induced pulmonary arterial hypertension by enhancing the risk for neointimal transformation. Circulation. 2019;140:1409–1425. doi: 10.1161/CIRCULATIONAHA.119.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol. 2014;193:597–609. doi: 10.4049/jimmunol.1303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinovitch M, Guignabert C, Humbert M and Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenmark KR, Frid M and Perros F. Endothelial-to-mesenchymal transition: an evolving paradigm and a promising therapeutic target in PAH. Circulation. 2016;133:1734–1737. doi: 10.1161/CIRCULATIONAHA.116.022479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, et al. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mushaben EM, Hershey GK, Pauciulo MW, Nichols WC and Le Cras TD. Chronic allergic inflammation causes vascular remodeling and pulmonary hypertension in BMPR2 hypomorph and wild-type mice. PLoS One. 2012;7:e32468. doi: 10.1371/journal.pone.0032468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD and Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med. 2004;169:378–385. doi: 10.1164/rccm.200308-1094OC. [DOI] [PubMed] [Google Scholar]
- 26.Rydell-Tormanen K, Johnson JR, Fattouh R, Jordana M and Erjefalt JS. Induction of vascular remodeling in the lung by chronic house dust mite exposure. Am J Respir Cell Mol Biol. 2008;39:61–67. doi: 10.1165/rcmb.2007-0441OC. [DOI] [PubMed] [Google Scholar]
- 27.Wagenvoort CA and Wagenvoort N. Pathology of pulmonary hypertension. New York; 1977. [Google Scholar]
- 28.Crnkovic S, Marsh LM, El Agha E, Voswinckel R, Ghanim B, Klepetko W, Stacher-Priehse E, Olschewski H, Bloch W, Bellusci S, et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J Pathol. 2018;244:485–498. doi: 10.1002/path.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bueno-Beti C, Hadri L, Hajjar RJ and Sassi Y. The Sugen 5416/hypoxia mouse model of pulmonary arterial hypertension. Methods Mol Biol. 2018;1816:243–252. doi: 10.1007/978-1-4939-8597-5_19. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, Minami M, Inagaki T, Miyagawa S, Sawa Y, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2015;112:E2677–2686. doi: 10.1073/pnas.1424774112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, Olley PM and Cutz E. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. 1986;55:632–653. [PubMed] [Google Scholar]
- 32.Leopold JA. Pulmonary venous remodeling in pulmonary hypertension: the veins take center stage. Circulation. 2018;137:1811–1813. doi: 10.1161/CIRCULATIONAHA.118.033013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fayyaz AU, Edwards WD, Maleszewski JJ, Konik EA, DuBrock HM, Borlaug BA, Frantz RP, Jenkins SM and Redfield MM. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. 2018;137:1796–1810. doi: 10.1161/CIRCULATIONAHA.117.031608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorfmuller P, Humbert M, Perros F, Sanchez O, Simonneau G, Muller KM and Capron F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893–902. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 35.Ghigna MR, Guignabert C, Montani D, Girerd B, Jais X, Savale L, Herve P, Thomas de Montpreville V, Mercier O, Sitbon O, et al. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur Respir J. 2016;48:1668–1681. doi: 10.1183/13993003.00464-2016. [DOI] [PubMed] [Google Scholar]
- 36.Galambos C, Sims-Lucas S, Abman SH and Cool CD. Intrapulmonary bronchopulmonary anastomoses and plexiform lesions in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2016;193:574–576. doi: 10.1164/rccm.201507-1508LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westöö CN, Peruzzi N, Lovric G, van der Have O, Jeremiasen I, Mokso R, de Jesus Perez V, Brunnström H, Bech M, Galambos C, et al. Distinct types of plexiform lesions in pulmonary arterial hypertension identified by synchrotron-based phase contrast micro-CT [abstract]. Circulation. 2019;140. [Google Scholar]
- 38.Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E and Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Z, Li M, Wharton J, Zhu MM and Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2alpha. Circulation. 2016;133:2447–2458. doi: 10.1161/CIRCULATIONAHA.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korman B, Bell R, White RJ, Garcia-Hernandez M, Wu E, Slattery P, Huertas N, Duemmel S, Nuzzo M, Rahimi H, et al. TNF-α drives progressive obliterative pulmonary vascular disease and represents a novel model of connective-tissue disease associated pulmonary arterial hypertension (CTD-PAH) [abstract]. Arthritis Rheumatol. 2019;71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4613. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheikh AQ, Misra A, Rosas IO, Adams RH and Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med. 2015;7:308ra159. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huertas A, Guignabert C, Barbera JA, Bartsch P, Bhattacharya J, Bhattacharya S, Bonsignore MR, Dewachter L, Dinh-Xuan AT, Dorfmuller P, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur Respir J. 2018;51:1700745. doi: 10.1183/13993003.00745-2017. [DOI] [PubMed] [Google Scholar]
- 44.Wendling O, Bornert JM, Chambon P and Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. doi: 10.1002/dvg.20448. [DOI] [PubMed] [Google Scholar]
- 45.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beyer S, Kelly RG and Miquerol L. Inducible Cx40-Cre expression in the cardiac conduction system and arterial endothelial cells. Genesis. 2011;49:83–91. doi: 10.1002/dvg.20687. [DOI] [PubMed] [Google Scholar]
- 47.Muzumdar MD, Tasic B, Miyamichi K, Li L and Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 48.Rinkevich Y, Lindau P, Ueno H, Longaker MT and Weissman IL. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature. 2011;476:409–413. doi: 10.1038/nature10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 50.Kumar ME, Bogard PE, Espinoza FH, Menke DB, Kingsley DM and Krasnow MA. Defining a mesenchymal progenitor niche at single-cell resolution. Science. 2014;346:1258810. doi: 10.1126/science.1258810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greif DM, Kumar M, Lighthouse JK, Hum J, An A, Ding L, Red-Horse K, Espinoza FH, Olson L, Offermanns S, et al. Radial construction of an arterial wall. Dev Cell. 2012;23:482–493. doi: 10.1016/j.devcel.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stenmark KR, Frid MG, Graham BB and Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res. 2018;114:551–564. doi: 10.1093/cvr/cvy004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majesky MW. Vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2016;36:e82–86. doi: 10.1161/ATVBAHA.116.308261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennett MR, Sinha S and Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML and Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108:161–164. doi: 10.1016/s0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 56.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW and Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289–1297. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fre S, Hannezo E, Sale S, Huyghe M, Lafkas D, Kissel H, Louvi A, Greve J, Louvard D and Artavanis-Tsakonas S. Notch lineages and activity in intestinal stem cells determined by a new set of knock-in mice. PLoS One. 2011;6:e25785. doi: 10.1371/journal.pone.0025785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 60.Mack JJ, Mosqueiro TS, Archer BJ, Jones WM, Sunshine H, Faas GC, Briot A, Aragon RL, Su T, Romay MC, et al. NOTCH1 is a mechanosensor in adult arteries. Nat Commun. 2017;8:1620. doi: 10.1038/s41467-017-01741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heath D and Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18:533–547. [DOI] [PubMed] [Google Scholar]
- 62.Huertas A, Tu L, Humbert M and Guignabert C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: more than a spectator. Cardiovasc Res. 2020;116:885–893. doi: 10.1093/cvr/cvz308. [DOI] [PubMed] [Google Scholar]
- 63.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. Mapping the mouse cell atlas by microwell-seq. Cell. 2018; 172:1091–1107.e17. doi: 10.1016/j.cell.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480. doi: 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- 67.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv. 2020:742320. doi: 10.1101/742320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR and Jorgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016;119:1313–1323. doi: 10.1161/CIRCRESAHA.116.309799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacobsen K, Lund MB, Shim J, Gunnersen S, Fuchtbauer EM, Kjolby M, Carramolino L and Bentzon JF. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight. 2017;2:e95890. doi: 10.1172/jci.insight.95890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, Sheikh AQ, Ding L, Qin L, Fernandez-Hernando C, et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun. 2018;9:2073. doi: 10.1038/s41467-018-04447-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B and Ciaccio PJ. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 72.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. doi: 10.1172/JCI65592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shen Z, Lu Z, Chhatbar PY, O’Herron P and Kara P. An artery-specific fluorescent dye for studying neurovascular coupling. Nat Methods. 2012;9:273–276. doi: 10.1038/nmeth.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Metzger RJ, Klein OD, Martin GR and Krasnow MA. The branching programme of mouse lung development. Nature. 2008;453:745–750. doi: 10.1038/nature07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Susaki EA, Tainaka K, Perrin D, Yukinaga H, Kuno A and Ueda HR. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat Protoc. 2015;10:1709–1727. doi: 10.1038/nprot.2015.085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.