

Graphical Abstract
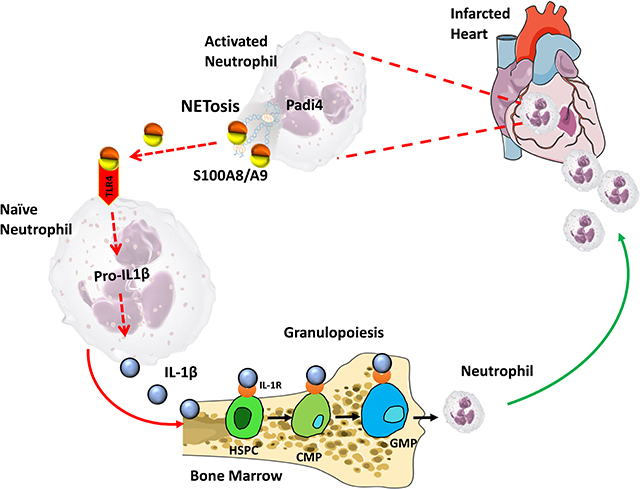
Myocardial infarction (MI) provokes a massive systemic inflammatory response characterized by enhanced infiltration of neutrophils to the ischemic heart via granulopoiesis in the bone marrow (BM)1. In patients with acute coronary syndrome, elevated blood neutrophils and S100A8/A9 is associated with higher incidence of major adverse cardiac events1. We recently discovered that S100A8/A9 is released by infiltrating neutrophils in the infarct to induce granulopoiesis by interacting with TLR4 on naïve neutrophils, priming the NLPR3-inflammasome for release of IL-1β1. However, the earliest events that trigger S100A8/A9 release from the infiltrating-neutrophils remain unclear.
Given the abundance of stimuli that can potentially induce cell death in the infarct (i.e. ROS, ATP, Ca2+, etc.), we hypothesized that S100A8/A9 is released by NETosis, a form of cell-death that involves extrusion of decondensed chromatin along with its entire granular content including S100A8/A9. To test this hypothesis, first, we interrogated RNA-seq data obtained from adult cardiac CD45+ leukocytes 72 hours-post MI (details in Quaife-Ryan et al2). We found robust upregulation of genes encoding NET-associated proteins along with kinases and transcriptional factors (common elements) crucial for NETosis (Figure, A). Because NETosis is initiated by abnormal elevations of either intracellular ROS via NADPH oxidase 2 (i.e. Nox-dependent) or Ca2+ (Nox-independent)3, we examined the expression of the kinases and transcription factors that represent these 2 pathways by sorting neutrophils from the infarct (24 hrs. post-MI). The data revealed a genetic signature for Nox-independent NETosis, dominated by Padi43 (Figure, B), an enzyme that is critical for citrullination of histones and Nox-independent NETosis. Furthermore, we found a robust increase in citrullination of histone moieties (H3Cit+ neutrophils), co-localization of S100A8/A9 with H3Cit, and increased S100A8/A9 levels in the heart as early as 6 hours after MI (Figure, C).
Figure. NETosis is essential for S100A8/A9-induced granulopoiesis.
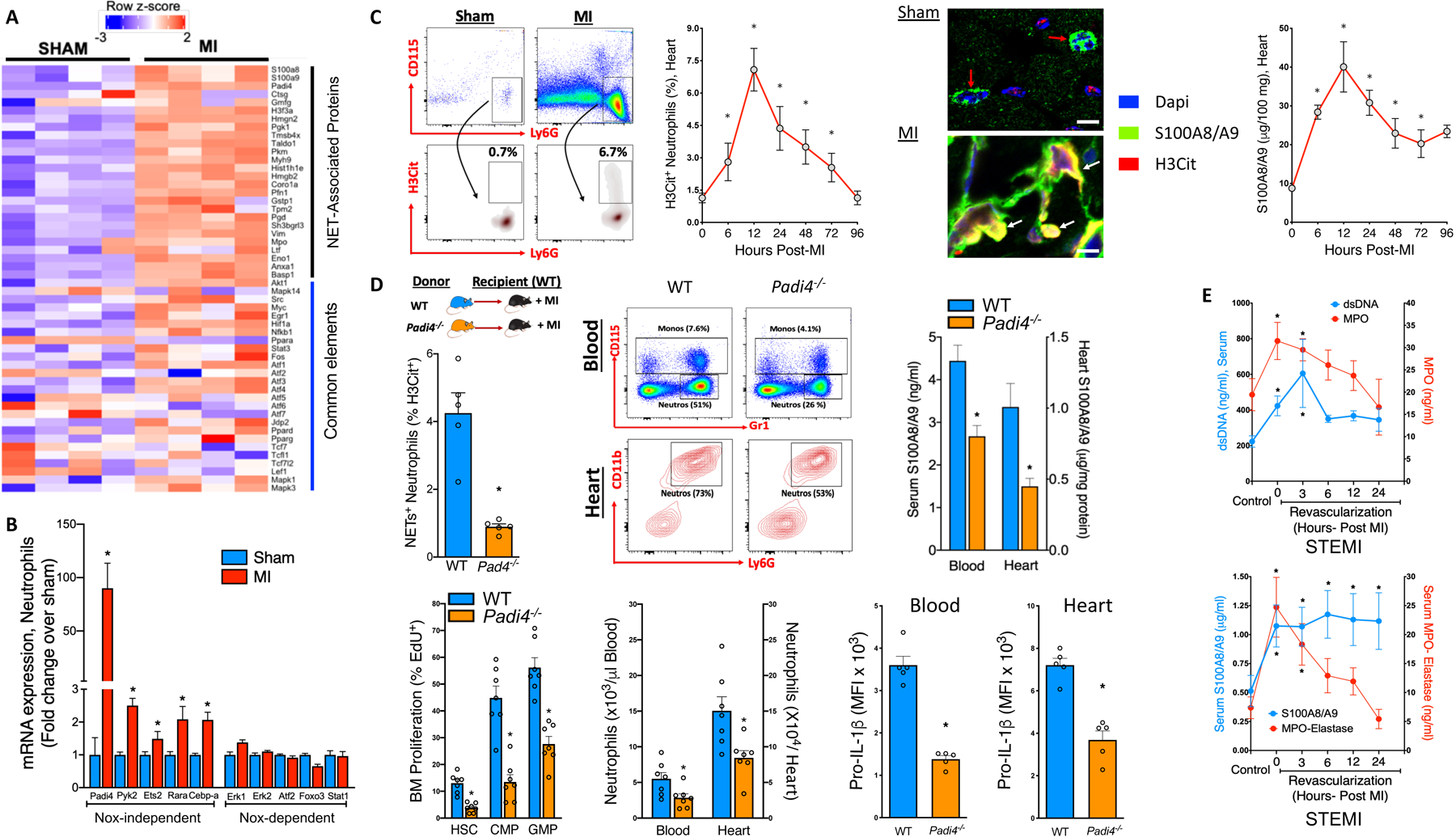
MI was induced in 8–10-week-old male C57BL/6J mice by permanent ligation of the left anterior descending (LAD) coronary artery. A) Cardiac leukocyte heat maps (RNA-seq) displaying log 2-fold change (FDR=0.025) in genes representing NET-associated proteins and elements common to both Nox-dependent and Nox-independent pathways. B) Nox-dependent and Nox-independent NETosis gene expression in cardiac neutrophils (24hr Post-MI). *P<0.05; unpaired student’s t-test (n=4). C) Representative flow cytometry plots (left panel, gated on CD45+ cells) and quantification (right panel) of NETosis in cardiac neutrophils by H3Cit staining. Confocal images (X60) of heart sections showing markers of DNA (Dapi, blue), NETosis (H3Cit, red) and S100A8/A9 (green) expression, red arrows: S100A8/A9 and DNA within the neutrophil, white arrows: externalized NETs with extensive H3Cit staining on decondensed nuclei decorated with S100A8/A9 (colocalized = orange/yellow) scale bars=10μm. Heart S100A8/A9 levels (top right) were quantified by ELISA. P<0.05 c.f. sham control (0); Kruskal-Wallis test and Dunn’s multiple comparison test. D) Experimental overview: BM from male C57BL/6J WT (blue) and Padi4−/− mice (orange) was transplanted to WT recipients and allowed to reconstitute for 6 weeks. MI was induced by LAD ligation and 18hrs later; NETosis, BM hematopoietic stem cells (HSC), common myeloid progenitor (CMP) and granulocyte macrophage progenitor (GMP) cell proliferation (EdU+), blood and heart neutrophils (with representative flow cytometry plots), along with circulating and heart neutrophil pro-IL-1β levels were measured by flow cytometry. Quantification of S100A8/A9 levels in the serum and heart (by ELISA). P<0.05 c.f. WT> WT+ MI; unpaired t-test. E) Quantification of dsDNA (blue line) and myeloperoxidase (MPO, red line), and S100A8/A9 (blue line) and MPO-Elastase (red line) in the serum from control (non-STEMI) and STEMI patients on admission (0) and 3, 6, 12 and 24hrs post-revascularization by PCI. n=15–21/group, *P<0.05 c.f. healthy control group; Mann-Whitney test.
To confirm if Nox-independent NETosis is involved in MI-induced granulopoiesis, we transplanted WT or Padi4−/− BM into WT recipients followed by a MI (Scheme, Figure, D). Padi4, required for Nox-independent NETosis, is expressed almost exclusively in neutrophils. As expected, deletion of Padi4 significantly reduced Nox-independent NETosis. This was associated with suppressed granulopoiesis, fewer neutrophils in the blood and heart and, reduced cardiac and serum S100A8/A9 (Figure, D). Reduced pro-IL-1β expression in cardiac and blood neutrophils from Padi4−/− mice revealed suppressed granulopoiesis1, 4 (Figure D).
To validate these findings in humans, we measured various markers of NETosis and found a marked increase in serum double-stranded DNA (dsDNA), myeloperoxidase (MPO), elastase, which paralleled S100A8/A9 levels in STEMI patients from the time of admission through post-revascularization by percutaneous coronary intervention (PCI) (Figure E). Importantly, neutrophil counts post-MI (<24hrs) in these patients was associated with greater S100A8/A9 levels and incidence of MACE1.
Currently, no approved anti-inflammatory therapy exists to mitigate MI-induced leukocytosis despite overwhelming evidence demonstrating that neutrophils instigate inflammation and promote heart failure. Furthermore, inhibition of NETs in experimental-MI have been shown to be cardioprotective5. We propose that interventions to reduce NETosis and/or S100A8/A9/IL-1β release1, 4, particularly during the acute inflammatory phase could represent a departure from the status quo in managing post-MI inflammation and the subsequent heart failure.
Funding:
This work was supported by NIH funding to PRN (HL122505, HL137799) and AAL (HL124266 and HL138488). A.J.M is supported by the National Health and Medical Research Council (NHMRC) project grants (APP1106154 and APP1142938) and a CSL Centenary Award.
Footnotes
Disclosures: None
Detailed analytical methods and reagents will be made available to other investigators on a reasonable request.
References
- 1.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, et al. Neutrophil-derived s100a8/a9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;141:1080–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136:1123–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan MA, Palaniyar N. Transcriptional firing helps to drive netosis. Sci Rep. 2017;7:41749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, et al. Targeting interleukin-1beta reduces leukocyte production after acute myocardial infarction. Circulation. 2015;132:1880–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y, Wagner DD. Vwf-mediated leukocyte recruitment with chromatin decondensation by pad4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]