

Abstract
The roles distinct B cell subsets play in clonal expansion, isotype switching, and memory B cell differentiation in response to T cell-independent type 2 antigens (TI-2 Ags) has been understudied. Using sorted B cells from VHB1–8 knock-in mice, we evaluated B-1b, marginal zone (MZ), and follicular (FO) B cell responses to the TI-2 Ag, NP-Ficoll. All subsets extensively divided in response to NP-Ficoll. Nonetheless, B-1b cells exhibited significantly increased IgG switching and differentiation into Ab-secreting cells(ASC)—a finding that coincided with increased Ag receptor signaling capacity and Blimp1 expression by B-1b cells. All subsets formed memory cells and expressed markers previously identified for TD memory B cells, including CD80, PDL2, and CD73, although B-1b cells generated the greatest number of memory cells with higher frequencies of IgG- and CD80-expressing cells. Despite memory formation, secondary immunization 4 weeks post-primary immunization did not increase NP-specific IgG. However, boosting occurred in B-1b cell-recipient mice when IgG levels declined. CD80+ memory B-1b cells divided, class switched, and differentiated into ASC in response to Ag in vivo, but this was inhibited in the presence of NP-specific IgG. Furthermore, CD80 blockade significantly increased memory B-1b cell division and differentiation to ASC upon Ag restimulation. Collectively, these findings demonstrate B-1b, MZB, and FOB subsets significantly contribute to the TI-2 Ag-specific memory B cell pool. In particular, we show B-1b cells generate a functional CD80-regulated memory population which can be stimulated to divide and differentiate into ASC upon Ag re-encounter when Ag-specific IgG levels decline.
Introduction
Carbohydrates are displayed on the surface of many types of pathogens, including bacteria, protozoa, helminths, viruses, and fungi (1). Humoral responses to these pathogen-displayed polysaccharides are critical for protection against infection. The regulation of humoral responses to polysaccharides and other T cell independent type 2 antigens (TI-2 Ag) that elicit Ab production in the absence of classical cognate T cell help or other strong innate stimuli is not fully understood. One aspect of B cell responses to TI-2 Ags that remains poorly understood is memory, including the pathways that regulate memory B cell generation and the participation of these cells in recall responses.
The refractoriness of TI-2 immune responses to booster immunization has long been recognized (2–4). Due to this phenomenon, TI-2 Ags (pneumococcal polysaccharide, meningococcal polysaccharide, and haptenated Ficoll, etc.) were originally not thought to generate memory B cells. However, early studies providing evidence for the generation of TI memory suggested Ag-specific Ab (IgM)-mediated inhibition as well as suppressor T cells prevented TI-2 Ag-specific memory cells from responding to secondary challenge (5, 6). A more recent study used adoptive transfer experiments with transgenic (Tg) splenic B cells that express a high affinity NP-specific BCR (VHB1–8) to clearly show classical non-Ab-secreting memory B cells developed following NP-Ficoll immunization (7). Studies using these NP-specific transgenic (Tg) mice suggested an IgM-independent, but Ag-specific IgG-dependent suppression of Ag-specific memory B cells occurs following boosting; however, a subsequent study also suggested deletion of high affinity Ag-specific B cells can occur as a result of TI-2 Ag boosting (8). Indeed, this appears to be the case for some polysaccharide-specific memory B cells that have been generated through TD pathways (9). Although some TI Ags encountered on gram negative bacteria can induce plasmablast-like memory cells with the capacity for expansion and increased Ab production upon pathogen re-exposure (10, 11), this does not readily occur with native polysaccharides. Taken together, these findings suggest the regulation of TI-2 Ag-specific memory formation and reactivation is complex and may depend on several factors, including the nature of the Ag, Ag dose, regulatory cells, and Ab-mediated inhibition.
Overcoming impaired boosting to TI-2 Ags is a formidable challenge that must be overcome in order to develop efficacious TI-2 Ag-based vaccines that provide protection through establishment of high titers and functional memory B cells which can respond directly to pathogen-derived TI-2 Ags. Protein conjugation to bacterial polysaccharides is one strategy that enables significant boosting in young children (except perhaps with serotype 3-conjugates(4)). In adults, pneumococcal polysaccharide-conjugate vaccines do not boost as well and overall Ab responses have been reported to be similar to those elicited to native pneumococcal polysaccharides (12). Thus, alternative approaches to enhancing these vaccine responses, such as use of B cell activating adjuvants (13), may be needed to improve TI-2 Ag-specific memory B cell formation and responsiveness to secondary Ag encounter.
Work from our lab and others demonstrates a key role for B-1b cells as well as marginal zone (MZ) B cells in contributing to the humoral response to TI-2 Ags, including haptenated Ficoll (10, 14–20). The extent to which these and other B cell subsets contribute to the TI-2 memory B cell pool has not been determined, however it is nonetheless important to consider when developing strategies aimed at improving TI-2 Ag-based vaccines. In the current study, we used the VHB1–8 Tg model system to examine the contribution Ag dose, Ag affinity, and B cell subsets (B-1b, MZ, and FO B cells) play in TI-2 memory B cell generation. Moreover, we examine the phenotypic characteristics of these cells and factors regulating their reactivation.
Materials and Methods
Mice
B1–8hi IgH knock-in (B6.129P2-Ptrpca Ightm1Mnz/J) mice expressing the Igh-V (VHB1–8) TGG->TTG codon 33 mutation (Jackson Laboratory; stock #007594) were on a C57BL/6 background and were bred in-house under SPF conditions. VHB1–8 Tg mice homozygous for CD45.1 and B1–8 genes were used as donors for cell transfer experiments into CD45.2+ C57BL/6 recipient mice (Jackson). Mice were age- and sex-matched for studies. Studies were approved by Wake Forest School of Medicine’s Animal Use Committee.
Immunizations and ELISAs
Mice were immunized with 1 or 25 μg NP40-AECM-Ficoll or TNP65-AECM-Ficoll (Biosearch Technologies) in 200 μl PBS i.p. NP-specific ELISAs were performed as previously described (13). In order to approximate serum concentrations of NP-specific isotypes, we used standards consisting of NP-specific IgMa and IgG mAbs produced in-house from hybridomas generated from NP40-Ficoll-immunized VHB1–8 Tg mice. Allotype-specific IgMa and IgMb biotin-tagged mAbs were obtained from BD Biosciences and used in conjunction with streptavidin-conjugated alkaline phosphatase. For IgMb-specific ELISAs, an NP-specific IgMb standard was not available. In this case, the standard curve was generated using the NP-specific IgMa mAb to approximate NP-specific IgMb concentrations.
B cell phenotyping
Single-cell suspensions (2 × 107/mL) in PBS containing 2% newborn calf serum were incubated with Fc Block (eBioscience) for 15 minutes, followed by staining with fluorochrome-conjugated Abs: CD19, PDL2, CD73 (from BD Biosciences), and CD138 (from eBioscience); B220, CD80, CD11b, CD21/35, CD45.1 (BioLegend), pooled rat anti-mouse-IgG1, -IgG2b, and -IgG3 Abs (Southern Biotech), and NP40-APC for 30 min at room temperature, followed by washing and fixing in 1.5% buffered formaldehyde. Blimp1 staining was performed as previously described (21). NP12-AECM-Ficoll-fluorescein3 (Biosearch) was used in initial staining experiments along with lambda 1 detection mAb (R11–153, BD Biosciences). NP-allophycocyanin (NP40-APC) was used in the majority of experiments and was made by coupling NP-Osu (Biosearch) with allophycocyanin (ProZyme) in N,-N-dimethylformamide. Fluorochrome-labeled isotype controls were used to determine background staining levels of NP-specific B cells. Adoptively transferred memory cells were identified in recipient mice as CD45.1+NP-APC+CD19+FSClow CD138negCFSElow cells. Cells were analyzed using a FACSCantoII or FortessaX20 cytometer (BD Biosciences) with FSC-A/FSC-H doublet exclusion. Data were analyzed using FlowJo analysis software (Tree Star).
Adoptive transfer experiments
For FACS sorting, cells from peritoneal lavage and single cell splenocyte preparations isolated from VHB1–8 Tg mice were subjected to CD43 negative selection (Dynal), followed by incubation with Fc block for 15 min and then staining for peritoneal B-1b cells using fluorochrome-labeled Abs against B220, CD11b, and CD5, and for spleen cells, CD19, CD21/35, CD23, and CD11b. Cells were sorted using a FACSAria with doublet exclusion gating onto B-1b (B220+CD11b+CD5−) and splenic FOB (CD19+CD21/35intCD23+CD11b−) and MZB (CD19+CD21/35hiCD23lowCD11b−) cells. FACS-sorted B cells were CFSE-labeled (CFDA-SE; InvivoGen) and transferred i.v. (5 × 105) into sex-matched wild type CD45.2+ C57BL/6 recipients. In some experiments, purified CD43− splenocytes (>97% CD19+) were sorted into CD23+ and CD23− populations using CD23 magnetic beads (Miltenyi Biotec) whereas CD43-depleted peritoneal cells were further purified using CD11b magnetic beads to obtain B-1b cells (Miltenyi) for transfers. In some memory B-1b cell reactivation experiments, CD19+ B cells were selected using Miltenyi beads. Intraperitoneal transfers of peritoneal CD19+ VHB1–8 Tg memory B-1b cells into WT or mumt recipient mice were performed 3 months post immunization of donors with 25 μg NP40-Ficoll i.p. Recipient mice received 250 μg mouse anti-NP IgG1 (1E9R) generated in house from a subcloned VHB1–8 Tg-derived hybridoma or control mouse IgG (Sigma) i.p. 1 hour prior to immunization with 5 μg NP40-Ficoll i.p.
Ag-induced intracellular calcium flux assay
For NP-Ficoll-induced intracellular calcium flux assays, peritoneal and spleen VHB1–8 Tg cells were labeled with 1 μM Indo-1AM (Invitrogen) according to manufacturer’s instructions, washed, and stained with fluorochrome-labeled mAbs against CD11b, B220, and CD23. Following washing, cells were resuspended in RPMI containing 5% FCS (2×106/ml). Cells were analyzed using a FortessaX20, with baseline readings taken for 30 seconds, followed by addition of 10 μg/ml NP12-AECM-Ficoll-fluorescein3 (Biosearch) and collection of cell events for 5 additional minutes. Data was analyzed using FlowJo analysis software. Baseline intracellular calcium ([Ca2+]i) values were assessed using Indo-1AM violet/Indo-1AM blue ratios for all B cell subsets recorded in the first 30 seconds of acquisition. [Ca2+]i mobilization values for NP-specific B cells following NP-Ficoll-fluorescein addition were specifically assessed by gating on NP-Ficoll-fluorescein-binding B cells within each subset.
In vitro B cell activation assay
Peritoneal cavity cells containing memory B-1b cells derived from VHB1–8 Tg mice were harvested 2–3 months post immunization (i.p.) with 25 μg NP-Ficoll. Cells were CFSE labeled (1μM; Invitrogen) and cultured for 4 days in cRPMI + 10% FCS (1×106/ml) in media alone or with 5 μg/ml NP40-Ficoll, with either 2 μg/ml rat IgG2b or rat anti-mouse CD80 added (LTF-2 and 1G10; InVivoMAb, Bioxcell). On day 4, cells were stained directly in culture wells with fixable Live/Dead dye, NP-APC, and fluorochrome-labeled mAbs to detect CD138, IgM, IgG, and CD45.1 as well as Countbright beads (Thermofisher) for enumeration. Cells were harvested from wells, washed, fixed in 1.5% buffered formaldehyde and analyzed by flow cytometry.
Statistical analyses
Data are shown as means ± SEM. Differences between sample means were assessed using Student’s t-test or one-way ANOVA with Tukey’s post-hoc analysis.
Results
VHB1–8 B cell Ab production and memory formation is influenced by TI-2 Ag dose and affinity
To better understand B cell responses to TI-2 Ags using NP-Ficoll as a model Ag, we utilized VHB1–8 transgenic mice. These mice express a rearranged IgH that bears high specificity for NP when complexed with lambda light chains (22). We first examined the effect of Ag dose and affinity on Ab production and the formation of memory to NP-Ficoll. CD43-depleted CD45.1+ VHB1–8 splenic B cells were transferred into CD45.2+ WT recipient mice. Recipients were immunized with a high (25 μg) or low (1 μg) dose of NP-Ficoll. Alternatively, recipient mice received 25 μg TNP-Ficoll for which VHB1–8 B cells have lower affinity, similar to that which has been described for quasimonoclonal mice with B cells bearing NP specificity (23). As shown in Fig. 1A, immunization with 25 μg NP-Ficoll resulted in rapid NP-specific IgG production which was significantly higher than in mice receiving either 1 μg NP-Ficoll or 25 μg TNP-Ficoll. Consistent with these findings, previous work with quasimonoclonal mice demonstrated 1 μg NP-Ficoll induced plasmablast formation with little AID induction (necessary for IgG switching) in the early response, in contrast to the AID induction seen with a 30 μg dose (24). Of note, WT mice receiving no cells exhibit <5 μg/ml IgG in response to 25 μg NP-Ficoll (13). IgMa levels (derived exclusively from Tg cells) were significantly higher in the high affinity, relative to the low affinity TNP-driven, NP-specific Ab response, regardless of dose (Fig. 1A). Finally, the endogenous IgMb anti-NP response was significantly higher in mice that had been immunized with TNP-Ficoll, suggesting there was less competition exerted from high affinity NP-specific Tg B cells during these responses.
Figure 1. NP-specific Ab production and memory formation by VHB1–8 Tg B cells.

A-C) WT CD45.2+ C57BL/6 recipient mice were given CD43-depleted CFSE-labeled splenic B cells i.v. (4 × 106) and peritoneal B cells i.p. (1.6 × 106) harvested from CD45.1+ VHB1–8 transgenic mice. One day later, mice were immunized with 1 or 25 μg NP40-Ficoll or 25 μg TNP65-Ficoll i.p. A) NP-specific IgMa, IgG, and endogenous IgMb production in recipient mice. B) Frequency and number of VHB1–8 Tg memory B cells (CD45.1+NP-APC+CD19+CD138negCFSElow) in spleen, peritoneal cavity, and bone marrow of recipient mice 21 days post immunization. Right panel indicates frequency of memory B cells which are IgG+ (inclusive of IgG1, IgG2b, and IgG3) among memory cells. C) Representative flow cytometry plots showing CD80 and PDL2 expression by VHB1–8 Tg memory B cells in spleen and peritoneal cavity (left panels) and the frequency of memory B cells that express CD80 (right panel). Asterisks (*) indicate values are significantly different from mice immunized with 25 μg NP-Ficoll (n=4–5 mice/group). Results representative of 2 or more experiments.
We examined the effect of Ag dose and affinity on memory formation and phenotype by CD43− splenic Tg B cells in recipient mice 21 days post immunization. Memory cells were defined as small-sized non-ASC that had divided (CD138negCFSElowFSClow). Division was observed in NP-specific B cells, regardless of dose. NP-specific memory B cell frequencies and numbers were not significantly different between high and low dose NP-Ficoll immunization in spleen, peritoneal cavity and bone marrow, although numbers were slightly lower in mice that had received 1 μg NP-Ficoll (Fig. 1B). In contrast, total memory B cell frequencies and numbers were significantly lower in mice that had received TNP-Ficoll.
Due to the use of the CD45.1 mAb (mouse IgG2a isotype) for identifying transferred Tg B cells in experiments, we were unable to detect IgG2a-switched cells. However, the majority of NP-specific IgG-switched cells present (omitting the CD45.1 mAb) were IgG3+ (~66 ± 2%), followed by IgG2b+ (24 ± 1%), IgG2a (7.5 ± 2%), and IgG1 (2.5 ± 2%). Thus, we detected ~90% of IgG-switched cells using staining for IgG1, 2b, and 3. Although we did not find a difference in the overall total number of memory B cells generated with high or low dose NP-Ficoll, significantly fewer IgG-switched memory B cells were generated with low dose Ag (<10% of memory cells vs >25% of memory cells with high dose), consistent with significantly less IgG production. Furthermore, although significantly fewer memory B cells were generated with low affinity (TNP-Ficoll) Ag, a similar proportion of the memory population had class switched to IgG, as observed with low dose high affinity Ag (Fig. 1B).
Previous work demonstrated high dose (50 μg) heavily haptenated NP190-Ficoll induced memory formation by B cells 15 days post immunization (7). These cells were reported to express lower levels of B220 and CD21/35. This was observed in our study at d21 post immunization using a 25 μg dose of NP40-Ficoll, although we identified variation in CD21/35 and B220 expression levels using the low 1 μg dose, with distinct B220hiCD21/35hi and B220lowCD21low populations present in the CFSEnegCD138neg pool at d21 (data not shown). We assessed the presence of additional memory markers, including CD80 and PDL2, which are present on mouse TD memory B cells (25, 26) as well as on non-human primate TNP-specific splenic B cells months after immunization with TNP-Ficoll (27). As shown in Fig. 1C, a substantial fraction of NP-specific memory B cells expressed CD80 in spleen, peritoneal cavity, and bone marrow, although frequencies were slightly lower in mice immunized with TNP-Ficoll. Expression of CD80 was relatively similar between peritoneal cavity and spleen memory B cells but PDL2 expression was much higher on peritoneal relative to splenic, memory B cells. Collectively, this data demonstrates high affinity, high dose Ag promotes increased IgG production along with increased formation of IgG+ memory B cells relative to lower Ag doses or lower affinity Ag. However, regardless of Ag dose or affinity (and despite decreased overall generation with lower affinity Ag), significant frequencies of Ag-specific CD80+ and PDL2+ memory B cells are generated among the established memory populations.
VHB1–8 B cell subset expansion, switching, and differentiation in response to NP-Ficoll
Previous work by our lab and others has demonstrated a major role for B-1b cells in producing Abs in response to TI-2 Ags, including pneumococcal polysaccharide and haptenated Ficoll (10, 14–20, 27–29). MZB cells also play a role these responses (30), and in some cases, FOB cells have been reported to contribute (31). To further investigate the roles specific subsets play in Ab production and memory formation in response to these types of Ags we again used VHB1–8 Tg mice which have the advantage of eliminating receptor specificity (ie., repertoire) and affinity as a variable among B cell subsets. We found no significant difference in the frequencies of NP-specific B cells among CD19+CD11b+ peritoneal B-1b cells, CD19+CD21/35hiCD1dhi MZ/T2-MZ precursor cells, and the CD19+CD21/35intCD1dint population, largely consisting of FO B cells (Fig. 2A–B). Consistent with this, the frequencies of lambda light chain (λ)1+ B cells for peritoneal B-1b, and splenic FO and MZ B cell subsets were not significantly different from that found for NP-specific B cell frequencies in these subsets (data not shown). However, peritoneal B-1b cells had significantly higher λ1 chain expression (Fig. 2C) and NP binding (Fig. 2D), based on selective gating for these populations. NP-specific peritoneal B-1b (CD11b+) cells from VHB1–8 Tg mice predominantly expressed B220 and CD21/35 at levels similar to peritoneal CD11bneg and FO B cells, although a small fraction (~5–20%) in some animals expressed a B220lowCD21/35lowCD23lowCD5− phenotype (Supplemental Figure 1A), as is observed in the heterogeneous population of CD11b+CD5− (B-1b) cells that we have described in WT and CD19−/− mice (28).
Figure 2. NP-specific B cell subsets in VHB1–8 Tg mice.

A-B) NP-specific B cell frequencies among splenic follicular (FOB), marginal zone (MZB), and peritoneal B-1b cell subsets in VHB1–8 Tg mice. Representative gating strategy is shown in (A) and frequencies of NP12-Fluorescein(Fl)-Ficoll-binding B cells among indicated populations indicated in (B). C-D) Lambda 1 (λ1) light chain expression (C) and NP12-Fl-Ficoll binding levels (D) among VHB1–8 Tg B cell subsets selectively gated for λ1 expression and NP-Ficoll binding, as depicted by the gating within histograms of panels C and A, respectively. Asterisks indicate significant differences between indicated groups (one-way Anova with Tukey’s post-hoc analysis, n=3 mice/group).
We performed adoptive transfers of FACS sort-purified CFSE-labeled B-1b, MZ, and FO B cells from VHB1–8 Tg mice to assess differences in their ability to divide, class switch, and differentiate into ASC. NP-specific cells from each subset exhibited complete CFSE loss 6 days post immunization, indicative of extensive division (Fig. 3A). Consistent with this, we did not detect significant differences in the frequencies of NP-specific B-1b, MZ B, or FOB cells in recipient spleens 6 days following immunization (Fig. 3B). However, NP-specific B-1b cells were significantly increased in the peritoneal cavity relative to FOB and MZB. NP-specific B cell frequencies were very low (<0.1%) in inguinal lymph nodes and bone marrow for all subsets relative to spleen and peritoneal cavity at this time point (Fig. 3B).
Figure 3. NP-specific B cell expansion, isotype switching, and differentiation by distinct VHB1–8 Tg B cell subsets 6 days post NP-Ficoll immunization.

A-E) B-1b (B220+CD11b+CD5−), FOB (CD19+CD21intCD23+CD11b−) and MZB (CD19+CD21/35hiCD23lowCD11b−) from CD45.1+VHB1–8 Tg mice were FACS-sorted, CFSE labeled and transferred i.v. (5 × 105) into WT recipients. Recipients were immunized with 25 μg NP40-Ficoll on d1. On day 7, tissues were harvested, with CD45.1+ NP-specific CD19+ B cells analyzed by flow cytometry. A) CFSE levels in Tg NP-specific and non-specific splenic B cells recovered from recipient mice. B) CD45.1+ NP-specific B cell frequencies among leukocytes on d7 in recipient spleen, peritoneal cavity (PerC), inguinal lymph node (LN), and bone marrow. C-D) FSC levels for CD45.1+ NP-specific B cells in recipients (C) and CD21/35 expression on donor CD45.1+ NP-specific splenic B cells (D) in immune and naïve recipients. Shaded histograms indicate expression in endogenous (recipient) B cells. E) Frequencies of IgG+, CD138+, and IgG+CD138+ cells among CD45.1+ NP-specific cell populations in spleen, PerC, LN, and bone marrow. Asterisks indicate significant differences between indicated groups (p<0.05, n=3–5/group). Results representative of two or more experiments.
Spleen B-1b and MZ B cells had significantly increased cell size (ie., higher FSC levels) on d6 relative to FOB cells (Fig. 3C), suggesting they may be more activated or undergoing increased ASC differentiation. CD11b expression was still present on adoptively transferred NP-specific B-1b cells in the spleen and peritoneal cavity at this time point. CD11b expression was low to negative on i.v.-adoptively transferred splenic FOB and MZB cells. However, recipients of MZB cells harbored NP-specific B cells expressing CD11b in the peritoneal cavity (not shown), although they were at significantly lower frequencies than in B-1b cell recipients (Fig. 3B). This may be due to the presence of B-1b cells within these sorted splenic populations, as adoptive transfers of normal spleen B cells lead to reconstitution of a population of CD11b+ B-1-like cells (16). The lack of CD11b expression by resting splenic B-1b cells along with other defining markers makes identification of these cells difficult. Interestingly, one week post transfer, naïve CD45.1+ B-1b cells transferred i.v. expressed a CD21/35int/hi phenotype in the spleen, whereas MZB remained CD21/35hi and FOB remained CD21int, suggesting that recirculating B-1b cells may have additional phenotypic alterations upon entry into the spleen, including upregulation of CD21/35 to levels typically found on MZB cells(Fig. 3D). Nonetheless, for all subsets, CD21/35 expression was decreased on NP-specific splenic and peritoneal B cells following immunization (Fig. 3D).
By 6 days post immunization, roughly half of NP-reactive B-1b cells and MZB cells had class switched to IgG (IgG1, 2b, or 3) in the spleen (Fig. 3E). This was significantly increased over that observed for IgG-switched FOB cells (36%). A similar trend was observed in the peritoneal cavity and lymph node (Fig. 3E). Lower frequencies of IgG-switched cells were found among B cells localized in bone marrow. Strikingly, the frequency of NP-specific B-1b cells that had undergone differentiation to Ab-secreting plasmablast cells (i.e., CD138+) in the spleen on d6 (~50%) was significantly increased over that observed for MZB cells (20%) and FOB cells (15%) (Fig. 3E). NP-specific B-1b cells yielded significantly increased frequencies of NP-specific IgG+ plasmablasts (~30%) relative to MZB and FOB recipients in the spleen. In the bone marrow, the frequency of B-1b cells that had undergone plasmablast differentiation was also significantly higher (2-fold) than that for MZB and FOB (Fig. 3E). We detected NP-reactive B cells in the blood of B-1b cell recipients on d6 (0.2 ± 0.05% of total blood cell population), supporting the capacity of TI-2 Ag-activated B-1b cells to recirculate. The frequency of NP-specific blood B-1b cells that were IgG+ (29 ± 9%) and IgG+CD138+ (1 ± 0.2%) was similar to that found in the bone marrow, raising the possibility these cells were in the process of seeding the bone marrow. Minimal plasmablast frequencies were found for B cells on day 6 in the lymph node (<1%) and peritoneal cavity (<10%), consistent with the major role for the spleen in fostering TI-2 Ab responses (32, 33). Thus, despite finding similar frequencies of NP-specific splenic B cells derived from B-1b, MZB and FOB Tg B cells 6 days post immunization, B-1b and MZB cells exhibited increased class switching over FOB cells, and B-1b cells exhibited significantly increased differentiation to plasmablasts relative to MZB and FOB cells.
Contribution of VHB1–8 B cell subsets to humoral responses
We assessed NP-specific Ab responses in WT recipient mice following adoptive transfers of FACS sort-purified B-1b, MZ, and FO B cells from VHB1–8 Tg mice to determine if Ab responses paralleled differences in cellular responses. As shown in Fig. 4, B-1b cells elicited a rapid IgM and IgG response to NP-Ficoll that was significantly higher than that observed with FOB and MZB cells. This is consistent with the increased isotype switching and plasmablast cell differentiation observed with donor B-1b cells relative to MZB and FOB cells at d6 (Fig. 3E). IgMa (derived solely from Tg cells) was 3 to 6-fold higher in B-1b cell recipients than other recipients 7 days post immunization and total IgM (derived from both endogenous B cells and Tg cells) showed a similar pattern, indicating the majority of IgM and IgG produced was derived from Tg cells (not shown). Importantly, MZ B cells produced significantly more IgG than FOB 2–3 weeks post immunization along with a delayed, but significantly increased and more sustained IgMa response relative to FOB recipients (Fig. 4A). Boosting of mice with NP40-Ficoll on d30 did not result in increased IgMa or IgG in any of the recipients indicating mechanisms of suppression (such as IgG (7), which remained at substantial levels at this time point) prevent boosting by all subsets. Thus, VHB1–8 Tg peritoneal B-1b cells mount a rapid and transiently heightened IgG and IgM response to NP-Ficoll relative to MZB and FOB cells. All transferred populations contributed to sustained IgG production which likely elicits suppression during secondary responses as previously shown for NP-specific IgG1 (7).
Figure 4. Heightened NP-specific Ab production, Ag-induced [Ca2+]i increases, and Blimp1 expression by VHB1–8 Tg B-1b cells.

A) B-1b, FOB, and MZB cells from CD45.1+ VHB1–8 Tg mice were FACS-sorted, CFSE labeled and transferred i.v. (5 × 105) into WT recipients. Mice were immunized on d1 and d30 post transfer with 25 μg NP-Ficoll. NP-specific serum IgMa and IgG concentrations were measured by ELISA. Asterisks (*) indicate significant differences compared to FOB group, hash marks (#) indicate significant differences compared to MZB group (p<0.05, n=4–5/group). Results representative of 2 independent experiments. B) NP12-Ficoll-induced [Ca2+]i responses in peritoneal B-1b (CD11b+B220+) and splenic CD23+ and CD23− B220+ B cells from VHB1–8 Tg mice. For baseline readings (first 30 seconds) ([Ca2+]i) values were assessed for Indo-1AM violet/Indo-1AM blue ratios of bulk B cell subsets (ie., both non-Ag- and Ag-specific). Following addition of NP12-Ficoll-fluorescein, NP-binding cells could be visualized and this population (Ag-specific) was gated within each subset to assess changes in ([Ca2+]i) values for NP-Ficoll-fluorescein binding B cells beyond the 30 second time point. Results representative of those obtained from 2–3 mice. C) Blimp1 expression by naïve NP-specific VHB1–8 Tg peritoneal B-1b, splenic FOB and MZB cells. Left panels indicate representative Blimp1 staining (solid line) and control staining with rat IgG PE substituted for Blimp1-PE mAb in intracellular stain for each NP-APC-binding subset. Right graph indicates mean MFI Blimp1 values after subtraction from isotype control values for each individual subset (n=3 mice/group). Results obtained from 2 separate experiments.
The increased levels of differentiation to ASC and high level of NP-specific Ab produced by B-1b cells relative to FOB and MZB cells expressing the same high affinity receptor suggested there may be unique features of B-1b cells that enable them to produce high levels of Abs early in the response to TI-2 Ags. We hypothesized the higher level of binding due to increased receptor expression (Fig. 2C–D), may enable enhanced BCR signaling by these cells. In support of this, we found [Ca2+]i levels were substantially higher in NP-specific B-1b cells following NP-Ficoll stimulation relative to NP-specific splenic CD23+ and CD23− B cells (Fig. 4B). A previous study also indicated resting B220lowCD5+/− peritoneal B1 cells from normal mice express low levels of Blimp1 (34). Although the naïve NP-specific B-1b cells (CD11b+) from VHB1–8 Tg mice used for this assay were CD21+CD23+, with comparable B220 and CD86 expression to MZB and FOB and only slightly greater cell size (FSC) (Supplemental Fig. 1B), they expressed low, but detectable levels of Blimp1 (Fig. 4C). In contrast, Blimp1 expression levels in NP-specific FOB and MZ B cells were barely above background control staining. Taken together, this data indicates naïve NP-specific VHB1–8 Tg B-1b cells are distinct from NP-specific VHB1–8 Tg FOB and MZB cells in that they have increased Ag binding capacity, more robust increases in intracellular [Ca2+]i levels in response to Ag, and express Blimp1 in the resting state. These factors may contribute to their significantly enhanced Ab responses to NP-Ficoll relative to FOB and MZB cells.
Contribution of VHB1–8 Tg B cell subsets to B cell memory
To better understand the potential for boosting TI-2 Ab responses, we examined memory formation by adoptively transferred B cell subsets 21 days post immunization. CD45.1+FSCloCFSElo-negCD138neg B cells were considered memory cells. As shown in Fig. 5A, B-1b cells had significantly higher numbers of memory cells in spleen and peritoneal cavity relative to FOB and MZB cells, whereas memory cell numbers were higher for FOB and MZB cells in inguinal lymph nodes. B-1b cells also had memory B cells circulating in the blood (data not shown). The frequency of class-switched IgG+ memory was similar between B-1b and MZB cells in spleen and bone marrow but was greater for B-1b cells in PerC and iLN relative to other subsets (Fig. 5B). Although low numbers of memory cells were recovered from inguinal LN, FOB and MZB yielded higher numbers in LN (2 to 3-fold) relative to B-1b cells. Considering the total cellularity of lymph nodes throughout the body, these differences raise the possibility that FOB and MZB memory cells make a considerable contribution to the memory pool but are differentially distributed relative to B-1b cell memory cells.
Figure 5. Generation of memory by distinct VHB1–8 Tg B cell subsets in response to NP-Ficoll.
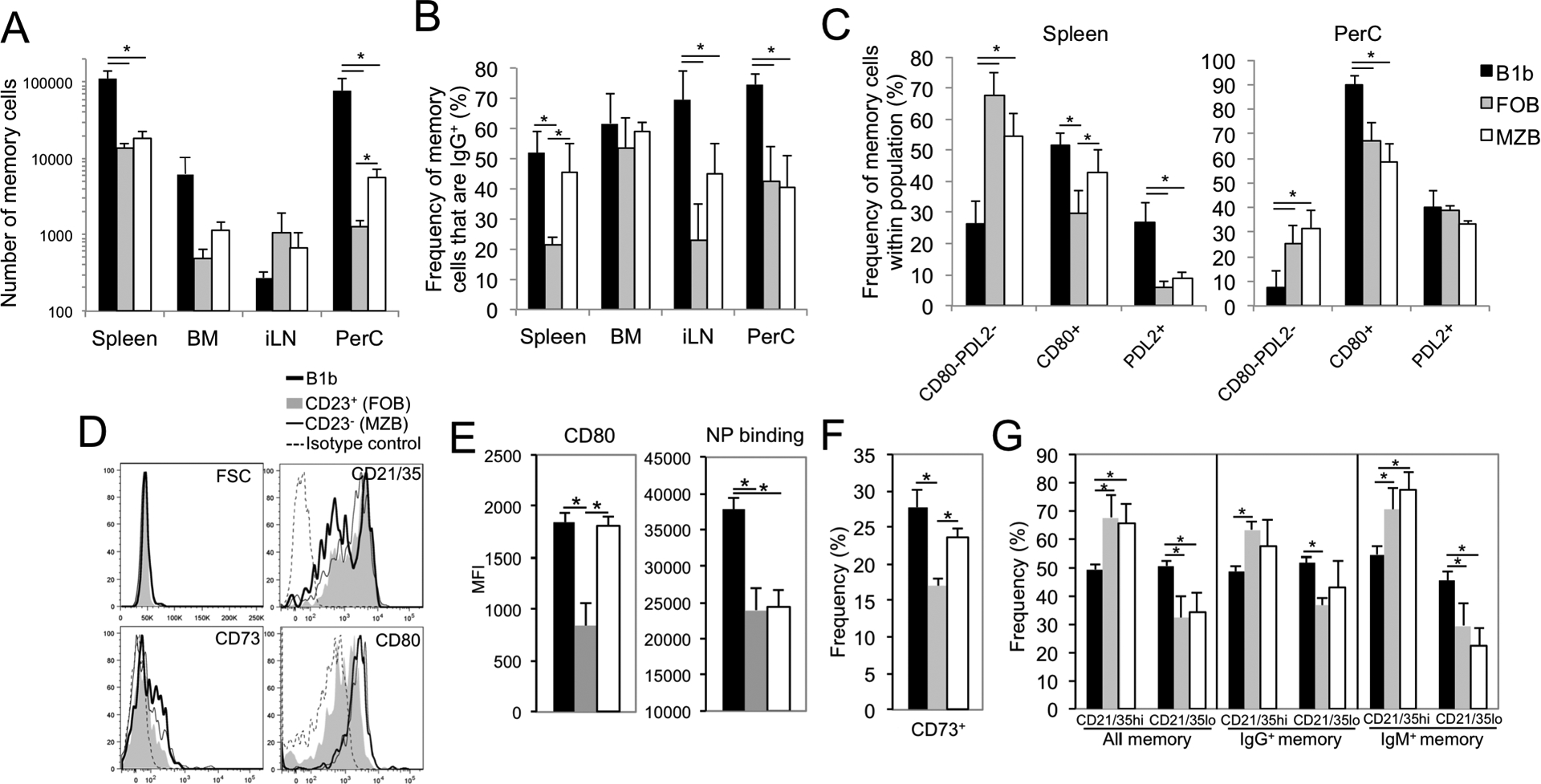
A-C) B-1b, FOB, and MZB from CD45.1+ VHB1–8 Tg mice were FACS-sorted, CFSE-labeled and transferred i.v. (5 × 105) into WT recipients. One day later, mice were immunized with 25 μg NP-Ficoll. A) Number of CD45.1+CD19+VHB1–8 Tg NP-specific memory B cells (CD138negCFSElow) in spleen, inguinal LN, peritoneal cavity, and bone marrow of recipient mice 21 days post immunization. B) Frequency of NP-specific Tg memory B cells in recipient mice that express IgG (inclusive of IgG1, IgG2b, and IgG3). C) Frequency of NP-specific Tg memory B cells in recipient mice that express a CD80−PDL2−, CD80+ or PDL2+ surface phenotype in spleen and peritoneal cavity. Asterisks (*) indicate significant differences among indicated groups (p<0.05). D-G) Sorted subsets were transferred into WT recipients and mice were immunized with 1 μg NP-Ficoll. On day 40, spleen cells were harvested and memory cells were analyzed for size (FSC) and surface marker expression (D), with NP-APC binding and CD80 MFI (E), frequency of CD73-expressing cells (F), and CD21/35 high and low-expressing population frequencies among all, IgM+, and IgG+ memory B cells (G) quantified. Asterisks (*) indicate significant differences among indicated groups (p<0.05, A-C, n=5–9 recipients/group from 2–3 independent experiments, and D-G, n=3–4 recipients/group).
We next examined the distribution of CD80 and PDL2 expression by memory B cells formed from B-1b, FOB and MZB donor cells 21 days post NP-Ficoll (25 μg) immunization. A higher frequency of B-1b cell memory cells expressed CD80 in spleen and peritoneal cavity as well as PDL2 in the spleen relative to FOB and MZB memory cells, and the B-1b memory population had the lowest frequency of double negative (CD80−PDL2−) cells in spleen and peritoneal cavity (Fig. 5C). With the exception of an increased frequency of CD80-expressing cells among the MZB cell-derived splenic memory pool over FOB-derived memory B cells, MZB-derived and FOB-derived memory B cells had a similar distribution among CD80−PDL2−, CD80+, and PDL2+ subpopulations.
Analysis of splenic memory B cells originating from B-1b, FOB, and MZB cells 40 days post immunization using low dose (1 μg) NP-Ficoll also revealed differences among subsets (Fig. 5D–G). Despite similar average size (FSC) among memory cells (B-1b, 46K; FOB, 44K; MZB, 45K), B-1b memory cells had significantly higher levels of NP-binding (1.6-fold based on MFI; Fig. 5E). In addition, B-1b memory cells in the spleen had a significantly increased frequency of CD21/35low-expressing cells relative to FOB and MZB memory cells (Fig. 5D and G), which was most evident among IgM+ memory cells (Fig. 5G). Relative to FOB, memory B-1b and MZB cells had significantly increased CD80 expression as well as increased frequencies of CD73-expressing cells (Fig. 5D–F). Thus, TI-2 Ag-induced B-1b, MZ and FO memory B cells exhibit phenotypic differences, with B-1b memory cells having significantly increased capacity for Ag binding, with increased skewing to a CD21/35low phenotype and along with MZB memory cells, higher frequencies of CD80- and CD73-expressing memory cells.
Examination of adoptively transferred Tg memory B cells 60 days post NP-Ficoll immunization revealed fewer memory B cells (5 to 10-fold) were present relative to d21 values. B-1b memory cells remained significantly higher than FOB- and MZB-derived memory B cells in both spleen and peritoneal cavity (Fig. 6A). We examined the effect of boosting in recipient mice of Tg B-1b and FOB cells 10 weeks post primary immunization. At this time point NP-specific serum IgG levels had fallen to low levels (<10 μg/ml). In contrast to the lack of boosting observed with d30 secondary immunization (Fig. 4), boosting at d70 elicited a 30-fold increase in IgG production in boosted B-1b recipients whereas a moderate ~2-fold increase was observed in boosted FOB recipients (Fig. 6B). IgMa levels also increased 8-fold over d70 (pre-boost) values in boosted B-1b cell recipients, but not in boosted FOB recipients. There was a significant increase in NP-specific total (largely endogenous) IgM levels, indicating little to no recall response was made by transgenic FOB memory cells. B-1b recipients which were not boosted showed no change in Ab levels. Thus, relative to FOB and MZB cells, B-1b cells more effectively seed IgM and IgG memory B cell populations with increased frequencies expressing CD80 and/or PDL2 and primary localization in the spleen and peritoneal cavity. Importantly, memory B-1b cells can be effectively activated to produce significant quantities of NP-specific IgM and IgG following boosting when IgG levels fall below a certain threshold.
Figure 6. Generation of long-term memory by VHB1–8 Tg B cell subsets in response to NP-Ficoll.

B-1b, FOB, and MZB from CD45.1+ VHB1–8 Tg mice were FACS-sorted, CFSE-labeled and transferred i.v. (5 × 105) into WT recipients. One day later, mice were immunized with 25 μg NP-Ficoll. A) Number of CD45.1+CD19+VHB1–8 Tg NP-specific memory B cells (CD138negCFSElow) in spleen and peritoneal cavity of recipient mice 60 days post immunization. Asterisks (*) indicate significant differences among indicated groups (p<0.05, n=4–5/group). Results representative of 2 independent experiments. B) Recipient mice of FACS-sorted peritoneal B-1b and splenic FOB and were immunized with 25 μg NP-Ficoll on d0 and boosted 10 weeks later. NP-specific serum IgM, IgG, and IgMa concentrations were measured by ELISA pre-boost (d70) and post-boost. Asterisks (*) indicate significant differences at the indicated time points compared to preimmune (d70) levels of the same group.
CD80+ memory B-1b cells divide, class switch and differentiate into ASC upon Ag re-encounter, but are inhibited in the presence of Ag-specific IgG
To specifically determine whether CD80+ memory B-1b cells can be reactivated by Ag to divide, class switch, and differentiate into ASC, we adoptively transferred CD80+ VHB1–8 Tg memory IgM+ B-1b cells into naïve secondary WT recipient mice i.p. and assessed their responses following immunization (Fig. 7A). In these experiments, we also examined the extent to which memory B cells responded to Ag in the presence or absence of NP-specific IgG1 administered 1 hour prior to immunization. CD80+ memory B-1b cells isolated from both the peritoneal cavity and spleen of control Ab-treated recipient mice divided extensively, class-switched to IgG, and differentiated into CD138+ ASC 4 days following immunization (Fig. 7B). Memory B-1b cells in recipient mice that received NP-specific IgG1 expanded in the peritoneal cavity to the same extent as control Ab-treated mice (Fig. 7B). Thus, NP-Ficoll-IgG complexes within the peritoneal cavity may remain somewhat stimulatory to memory B-1b cells, at least with respect to division. However, few cells were present in the spleen when NP-specific IgG1 was administered (Fig. 7B). In fact, the numbers in the spleen of NP-specific IgG1-treated mice were similar to the number of NP-specific Tg memory B-1b cells that had migrated to the spleen in non-immunized recipient mice (no Ag control group). The numbers of IgG+, CD138+, and IgG+CD138+ cells in anti-NP IgG1-treated mice were significantly decreased in both the peritoneal cavity and spleen relative to control Ab-treated mice.
Figure 7. Ag-induced expansion and ASC differentiation in CD80+ memory B-1b cells is regulated by CD80 and Ag-specific IgG.

A-F) Peritoneal cells enriched for CD19+VHB1–8 Tg memory B-1b cells (CD45.1+ NP-APC+CD11b+CD80) were harvested from donors 2–3 months post NP-Ficoll immunization and either transferred into recipient mice (A-E) or cultured in vitro (F) to assess Ag reactivation. A) Phenotype of CD19+ NP-specific memory B-1b cells (CD45.1+IgM+CD80+CD138negCD11b+) used for transfer experiments. Shaded histograms depict isotype control staining. B-E) CD80+ memory B-1b cells (5 × 105) were transferred into WT (B-D) or mumt (E) mice i.p. as depicted in (A). The next day, mice received 250 μg mouse anti-NP IgG1 or control mouse IgG i.p., and 1 hour later, were immunized with 5 μg NP40-Ficoll i.p. One group received cells without immunization and another received no cells but were immunized. VHB1–8 Tg CD45.1+CD19+NP-APC+ numbers (B) and surface phenotype (C) were assessed by flow cytometry 4 days post immunization. NP-specific serum IgM and IgG (IgG2a, 2c, and 3) concentrations were measured by ELISA for WT (d4; D) and mumt recipients (d6 and 12; E). Asterisks indicate significant differences between immunized mice that received control IgG and naïve or NP-specific IgG1 recipients (p<0.05, n=4 mice/group). F) CFSE-labeled unfractionated peritoneal cells containing VHB1–8 Tg CD45.1+CD19+NP-APC+CD11b+CD80+ memory B-1b cells (as shown in A) or naïve VHB1–8 Tg CD45.1+CD19+NP-APC+ B cells, were cultured in media alone or in the presence of NP-Ficoll and control rat IgG or a CD80 blocking mAb (1G10). On day 4, cell number, division, and phenotype were assessed by flow cytometry. Asterisks indicate significant differences between mean cell numbers (p<0.05). Results are representative of 3 independent experiments.
Phenotypic differences were also found among transferred memory B-1b cells from naïve, immune plus control IgG-treated, and immune plus anti-NP IgG1-treated recipients (Fig. 7C). CD21/35 expression was significantly lower in immune recipients, potentially indicative of complement receptor engagement and internalization. CD11b expression was significantly decreased on peritoneal memory B-1b cells in immune control IgG-treated mice, suggesting activation-induced downregulation had occurred, enabling migration out of the cavity. By contrast, CD11b was only partially decreased in recipients that had received anti-NP-specific IgG1. Nonetheless, increased cell size (FSC), CD86 and PD1 expression indicated significant activation of memory B-1b cells had occurred in both control and anti-NP IgG1 treated recipients, despite significantly lower cell numbers in spleens of anti-NP IgG1 treated recipients.
NP-specific Ab levels (d4) in treated mice reflected findings obtained with cell analysis, with control Ab-treated recipients producing significantly more NP-specific IgM and IgG (IgG2a, IgG2b, and IgG3) than recipients treated with NP-specific IgG1 (Fig. 7D). Memory B-1b cells did not make Ab in the absence of immunization and WT mice that did not receive memory cells produced relatively little Ab (Fig. 7D). Similar results were obtained when B cell-deficient (mumt) mice served as recipients of CD80+ memory B-1b cells (Fig. 7E). Thus, CD80+ memory B-1b cells recirculate in the resting state and behave like bona fide memory B cells in that they have the capacity to divide, class switch, and produce Ab upon antigen re-encounter. However, Ag-specific IgG significantly reduces the ability of memory B-1b cells to expand and differentiate to Ab-producing cells.
CD80+ memory B-1b cells divide and differentiate into ASC in response to Ag in vitro and CD80 blockade increases ASC differentiation
We next assessed whether VHB1–8 Tg memory B-1b B cells were responsive to Ag in vitro and whether CD80 regulated their responsiveness by using the CD80 blocking mAb, 1G10. As shown in Fig. 7F, NP-Ficoll alone was sufficient to induce not only memory B-1b cell division, but also substantial differentiation to CD138+ ASC (20-fold increase over medium-only cultures) by day 4 in cultures of unsorted peritoneal cells incubated with control IgG. By comparison, naïve VHB1–8 Tg peritoneal B-1b cells divided extensively but only a small proportion differentiated into ASC (~2 fold increase over medium-only cultures). Cultures of memory B-1b cells treated with the CD80 blocking mAb had significantly increased numbers of NP-specific total B cells and IgG+ B cells relative to those with control mAb (Fig. 7F), albeit a somewhat modest (~25%) increase. The CD80 blocking mAb had the most significant effect on differentiation in that it doubled the number of CD138+ NP-specific B cells. By contrast, the CD80 mAb had no measurable effect on the number of NP-specific, IgG+, or CD138+ B cells recovered in cultures of naïve VHB1–8 Tg B cells. Thus, CD80 blockade supports increased ASC differentiation in cultures of Ag-stimulated CD80+ memory B-1b cells.
Discussion
Identifying factors regulating TI-2 Ab responses is critical for improving vaccines against pathogens and cancers, as well as for modulating responses that contribute to autoimmunity and allergy. Not unexpectedly, the generation and maintenance of B cell memory to TI-2 Ags is somewhat distinct from that to TD Ags. There is potential for both long-lived renewing plasmablasts and non-secreting Ag-experienced memory B cells to contribute to increased Ab production upon TI Ag re-encounter (7, 10, 11, 17, 19). However, in the case of Ags that lack additional stimuli, and/or in the presence of Ag-specific IgG, this capacity is constrained. In the current study, we used the VHB1–8 knock-in model to investigate the contribution of Ag dose, Ag affinity, and B cell subsets to the generation of resting (non-Ab secreting) TI-2 memory. We demonstrate that: 1) high affinity, high Ag dose drives IgG+ memory formation, but formation of overall memory B cell numbers is comparable between a low (1 μg) and high (25 μg) Ag dose; 2) high dose, low affinity Ag elicits fewer memory B cells overall but generates the same frequency of IgG+ memory cells as low dose high affinity Ag; and 3) IgM and IgG production levels are similarly modulated by Ag dose and affinity. Our findings also reveal naïve VHB1–8 Tg NP-specific B-1b cells have significantly increased Ag binding capacity, signaling potential, and Blimp1 expression, and due to these and perhaps other differences, generate significantly greater Ab responses and memory formation to TI-2 Ag relative to MZB and FOB cells, despite sharing the same Ag receptor. For the first time we also demonstrate: 1) TI-2 memory B cells express markers shared by TD memory B cells, including CD80, PDL2, CD73, as well as bimodal CD21/35 expression; 2) that B-1b, FO, and MZ B cells all contribute to the TI-2 Ag memory B cell pool, although the generation of IgG+ memory, CD80/PDL2 expression, and tissue distribution differs significantly among subsets as does the potential for producing Ab upon secondary Ag exposure, 3) that CD80+ memory B-1b cells have the capacity to robustly respond to Ag by dividing and effectively differentiating into ASC, and 4) that CD80 and Ag-specific IgG1 regulate the ability of CD80+ memory B-1b cells to divide and produce Ab upon Ag reencounter. In summary, our study provides novel insight into the regulation of TI-2 memory by Ag dose, B cell subsets, Ag-specific IgG, and CD80, all of which are critical factors to consider for vaccine strategies targeting these types of Ags.
Our work highlights the importance of Ag dose and affinity in driving TI-2 memory B cell formation. Previous work comparing responses between VHB1–8hi and VHB1–8lo mice, in which B cells have a 40-fold lower affinity for NP-Ficoll, demonstrated moderate differences (2–3 fold) in the proliferative responses of B cells to NP-Ficoll which was paralleled by similar differences in Ab responses (22). In that study, a greater number of high affinity B cells were stimulated to divide in the very early response relative to the low affinity B cells. Using the VHB1–8hi model, we found IgM and IgG responses were significantly enhanced (7 to 10-fold increase) in response to NP-Ficoll (the high affinity Ag) relative to the low affinity Ag (TNP-Ficoll). Immunization with a 25-fold lower dose of the high affinity Ag resulted in significantly lower IgG, but similar IgM production. Memory formation paralleled findings with Ab responses in that overall memory B cell generation was nearly equivalent between low and high dose NP-Ficoll, but high dose Ag significantly skewed B cells to form IgG+ memory. In contrast, low affinity Ag led to formation of a significantly lower number of memory B cells. Interestingly, the frequency of these Tg memory B cells that had class-switched was equivalent to that found for the low dose high affinity Ag condition. Collectively, these results demonstrate a role for TI-2 Ag dose and affinity in driving high level IgG production and IgG+ memory B cell generation. Although moderate to excessive Ag receptor signaling on its own does not appear to support AID expression and subsequent class switch recombination on spleen B cells in vitro (35, 36), increased BCR signaling (as a result of higher TI-2 Ag doses or Ag receptor expression) in select populations, such as B-1b or MZB cells, may have different effects on AID expression and hence, IgG switching. Additional accessory signals present in vivo may also support dose-dependent BCR-induced AID expression by these or other cells (35). Indeed, our work demonstrates a B cell-activating adjuvant significantly increases TI-2 Ab production to NP-Ficoll and other TI-2 Ags (13). Further work is necessary to determine the extent to which Ag dose and epitope display, subset targeting, and accessory signals can be modulated in order to enhance vaccine responses to TI-2 Ags.
Using the same model system, Obukhanych and Nussenzweig reported TI-2 memory B cells (at day 14 post immunization) display a phenotype that is distinct from conventional memory cells (7). However, our results suggest TI-2 Ag memory B cells express markers shared by TD memory B cells, including CD80, PDL2, and CD73, as well as biomodal CD21/35 expression at day 21 post-immunization and beyond. Our work in non-human primates showed a similar result for blood and splenic TNP-specific memory B cells following TNP-Ficoll immunization (27, 28). Importantly, there is significant heterogeneity in expression of these markers, as is observed in TD memory populations in mice (25, 26, 37). Previous work by Shlomchik and colleagues, in which a TD model of NP-CGG immunization was used, elegantly showed the expression of these markers bears significance with respect to levels of somatic hypermutation as well as the ability of the cells to re-enter germinal center (GC) reactions versus rapidly differentiate into ASC (37, 38). For example, CD80+PDL2+ TD memory B cells occur more frequently in the IgG+ pool and harbor more somatic hypermutations. PDL2- and CD80-expressing cells are also more likely to undergo ASC differentiation upon restimulation whereas double negative (CD80−PDL2−) TD memory B cells (typically IgM+) are more likely to reseed germinal centers (37, 38). It is interesting that a substantial fraction (at least 30%) of TI-2 memory B cells develop into CD80-expressing memory B cells and that similar frequencies were generated with high vs. low dose Ag conditions, despite striking differences in the generation of IgG+ memory B cells. Although we observed higher levels of CD80 expression on IgG+ B cells, the significant expression on both IgM+ and IgG+ memory B cells appears to represent a difference that exists between TD and TI-2 memory B cells. At present, it is not clear as to whether differential signals guide their differentiation.
B-1b cells differentiated into Ab-secreting cells more robustly than MZ B and FO B cells following NP-Ficoll immunization. This may be due in part, to their higher Ag receptor expression and increased BCR signaling capacity, as was evidenced by striking increases in intracellular calcium levels following Ag exposure relative to the modest increases displayed by splenic B cells. This was unexpected as Ab-mediated surface IgM crosslinking does not reproduce these differences in intracellular calcium flux among these subsets (15, 39). These differences may be explained by higher Ag receptor expression by VHB-18 Tg B-1b cells and the differential capacity of haptenated Ficoll to aggregate BCRs (IgM and IgD) relative to the divalent and selective IgM crosslinking triggered by anti-IgM Ab. In contrast to the VHB1–8 Tg B-1b cells analyzed in our study, CD5+B220loCD23− peritoneal B-1a cells expressing the VH11/Vκ9 Tg Ag receptor specific for phosphatidylcholine (PtC) exhibit negligible receptor-mediated calcium mobilization when either anti-IgM or PtC liposomes are used as a stimulus—a finding that is dependent on Ag receptor specificity, recent Ag encounter, localization within the peritoneal cavity, and elevated Lck expression (39, 40). Thus, peritoneal B-1b cells expressing a Tg BCR specific for non-self Ag exhibit distinct signaling properties relative to peritoneal B-1a cells expressing a Tg BCR specific for self -Ag. Nonetheless, NP-specific peritoneal B-1b cells from naïve VHB1–8 Tg mice were also found to express low levels of Blimp1, similar to WT B-1 cells (34). Its expression in naïve B-1b cells may have further contributed to their increased capacity for Ab production. Increased access to Ag localized in the peritoneal cavity may have also contributed to increased B-1b cell Ab responses. Despite being delivered i.v., B-1b cells (non-Ag-specific) accumulated in the peritoneal cavity to a greater extent (~4-fold) than FOB and MZB cells 1 week post transfer (Supplemental Fig. 1C). Nonetheless, the vast majority of non-Ag-specific Tg B-1b cells were localized in the spleen at this time point. Thus, it is perhaps not unexpected that intramuscular NP-Ficoll immunization also resulted in VHB1–8 Tg B-1b cells producing significantly more NP-specific IgM and IgG than CD23+ spleen B cells (Supplemental Fig. 1D).
Consistent with the recirculating characteristics of B-1b cells (10, 41), FACS-sorted VHB1–8 Tg CD11b+B220lowCD5− and CD11b+B220hiCD5− (B-1b) cells were localized to some degree in all tissues 7 days post transfer, although their surface phenotype differed depending on the tissue (Supplemental Fig. 2), indicating the B-1b phenotype is tissue-dependent and may overlap with that of other subsets, including FOB and MZB cells. TI-2 memory B-1b cells, like naïve B-1b cells, were also found to recirculate, and were more enriched in the spleen relative to FO- and MZ-derived memory B cells. Notably, memory B-1b cells recirculated to the spleen even if adoptively transferred into the peritoneal cavity. This may bear significance with regard to the preferential nature of TI-2 ASC responses to form in the spleen as opposed to other tissues, such as the lymph nodes (42, 43).
Although B-1b cells showed the greatest response to NP-Ficoll, MZB and FOB subsets divided extensively and also generated TI-2 memory. The individual roles of these memory-derived populations are not yet clear. Interestingly, work using VHJ588.3 Tg mice, which harbor B cells specific for alpha 1,3 dextran, demonstrated B-1b B cells both proliferated extensively and differentiated into ASC in response to primary and secondary Enterobacter cloacae challenge, whereas MZB cells largely committed to early ASC differentiation during the primary response, suggesting that B-1b cells may make the greatest contribution to the memory pool in this model (10). B-1b cells similarly form the memory compartment responsible for boosted Ab levels in response to secondary Borrelia hermsii infection (11). The role for B-1b cells in producing significant Ab levels in response to NP-Ficoll is not unexpected (14, 17, 44). However, their inability to support increased levels of Ab following secondary immunization with haptenated Ficoll in the presence of Ag-specific IgG suggests a critical mode of suppression that TI-2 Ags associated with live bacterial infections may in some cases overcome. Unfortunately, this does not appear to be the case with pneumococcal infections (4, 12, 45–47). Further work in this area is therefore necessary to identify optimal strategies to promote boosting with bacterial polysaccharides.
B-1b cells generated the greatest number of IgG+ memory cells as well as significantly higher frequencies of CD80- and PDL2-expressing memory cells. CD80-expressing and CD73-expressing cells were also higher among MZ memory B cells in the spleen relative to FO memory B cells. Interestingly, CD21/35 expression was bimodal in TI-2 memory cells as has been described for TD memory B cells (25), with B-1b cells having significantly higher frequencies of CD21/35low cells. Differential expression of these immune-regulatory surface markers may be critical for control of secondary responses. CD21/35 plays a key role in regulating TI-2 Ab responses through both complement-dependent regulation and regulation of CD19 function (48, 49) and its expression may therefore have a significant impact on the ability of memory B cells to participate in recall responses. The expression of CD73, an ecto-5-nucleotidase, by TD memory B cells is associated with a higher level of mutation and is indicative of a GC-dependent pathway (26, 50). It is also expressed by a fraction of B-1 cells where it may function in immune suppression (51). Finally, CD80 and PDL2 bind two potent immune-inhibitory receptors, CTLA4 and PD1, respectively, which we and others have shown inhibit Ab responses to TI-2 Ags (14, 52–54). Our results herein suggest CD80, which is highly expressed on memory B-1b cells, may in fact suppress ASC differentiation upon Ag re-encounter. Whether this is due to its interaction with CTLA4 and/or PDL1 remains to be determined. Of note, we report PD1 is also upregulated on memory B cells upon restimulation and this may further contribute to suppression via homotypic interactions among B cells expressing PDL2 and/or PDL1 (manuscript in prep). Thus, it is plausible to suspect that these and other TD memory markers (55), may differentially impact regulation of TI-2 memory B cells. This also appears to be the case for Ag-specific IgG, which often promotes TD Ab responses. The finding that memory B-1b cells localized in the peritoneal cavity divided normally in response to NP-Ficoll in the presence of NP-specific IgG but failed to divide/home to the spleen, class switch, or secrete Ab is perhaps indicative of the reduced stimulatory capacity of TI-2 Ag complexed with IgG. Although the mechanisms by which Ag-specific IgG inhibits TI-2 responses remains to be elucidated, our findings here provide clues which highlight a defect in the establishment of proper splenic B cell responses in its presence, perhaps through influencing migration and/or Ag clearance, splenic Ag localization, or epitope accessibility (masking). Further work is needed in order to determine the significance of this and other immune-regulatory mechanisms uniquely exerted on TI-2 memory B cells. Strategies aimed at overcoming Ab-dependent and –independent mechanisms of suppression exerted on these and other subsets of TI-2 memory B cells may improve future vaccines to generate improved serologic and cellular memory that could be boosted in the context of either infection or revaccination.
Supplementary Material
Key points.
B-1b cells mount superior TI-2 responses despite sharing the same BCR as B-2 cells.
B-1b and B-2 cells generate a TI-2 memory pool expressing CD80, PDL2, and CD73.
Ag-specific IgG and CD80 limit secondary Ab responses by memory B-1b cells.
Acknowledgments
This work was supported by NIAID/NIH R01AI18876 and R21AI144758 awarded to KMH. AS was supported in part by National Institutes of Health Training Grant AI007401. Shared resources support was provided by NCI-CCSG grant P30CA012197. The authors have no conflicts to disclose.
References
- 1.Astronomo RD and Burton DR. 2010. Carbohydrate vaccines: developing sweet solutions to sticky situations? Nat. Rev. Drug Discov 9: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson D and Braley-Mullen H. 1981. Antigen requirements for priming of type III pneumococcal polysaccharide-specific IgG memory responses: suppression of memory with the T-independent form of antigen. Cell Immunol. 64: 177–186. [DOI] [PubMed] [Google Scholar]
- 3.McMaster PR, Powers KG, Finerty JF and Schiffman G. 1973. Tolerance to type 3 pneumococcal polysaccharide in monkeys. Immunol. Commun 2: 361–370. [DOI] [PubMed] [Google Scholar]
- 4.Poolman J and Borrow R. 2011. Hyporesponsiveness and its clinical implications after vaccination with polysaccharide or glycoconjugate vaccines. Expert Rev. Vaccines 10: 307–322. [DOI] [PubMed] [Google Scholar]
- 5.Brodeur PH and Wortis HH. 1980. Regulation of thymus-independent responses: unresponsiveness to a second challenge of TNP-Ficoll is mediated by hapten-specific antibodies. J. Immunol 125: 1499–1505. [PubMed] [Google Scholar]
- 6.Braley-Mullen H 1985. Regulation of IgG memory responses by helper and suppressor T cells activated by the type 2 antigen, polyvinylpyrrolidone. J. Exp. Med 161: 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obukhanych TV and Nussenzweig MC. 2006. T-independent type II immune responses generate memory B cells. J. Exp. Med 203: 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haniuda K, Nojima T, Ohyama K and Kitamura D. 2011. Tolerance induction of IgG+ memory B cells by T cell-independent type II antigens. J. Immunol 186: 5620–5628. [DOI] [PubMed] [Google Scholar]
- 9.Brynjolfsson SF, Henneken M, Bjarnarson SP, Mori E, Del Giudice G and Jonsdottir I. 2012. Hyporesponsiveness following booster immunization with bacterial polysaccharides is caused by apoptosis of memory B cells. J. Infect. Dis 205: 422–430. [DOI] [PubMed] [Google Scholar]
- 10.Foote JB and Kearney JF. 2009. Generation of B cell memory to the bacterial polysaccharide alpha-1,3 dextran. J. Immunol 183: 6359–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T and Gerstein RM. 2004. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity 21: 379–390. [DOI] [PubMed] [Google Scholar]
- 12.Goldblatt D, Southern J, Andrews N, Ashton L, Burbidge P, Woodgate S, Pebody R and Miller E. 2009. The immunogenicity of 7-valent pneumococcal conjugate vaccine versus 23-valent polysaccharide vaccine in adults aged 50–80 years. Clin. Infect. Dis 49: 1318–1325. [DOI] [PubMed] [Google Scholar]
- 13.Phipps JP and Haas KM. 2019. An adjuvant that increases protective antibody responses to polysaccharide antigens and enables recall responses. J. Infect. Dis 219: 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas KM 2011. Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J. Immunol 187: 5183–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas KM, Johnson KL, Phipps JP and Do C. 2018. CD22 Promotes B-1b Cell Responses to T Cell-Independent Type 2 Antigens. J. Immunol 200: 1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haas KM, Poe JC, Steeber DA and Tedder TF. 2005. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23: 7–18. [DOI] [PubMed] [Google Scholar]
- 17.Hsu MC, Toellner KM, Vinuesa CG and Maclennan IC. 2006. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. USA 103: 5905–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall JL, Flores-Langarica A, Kingsley RA, Hitchcock JR, Ross EA, Lopez-Macias C, Lakey J, Martin LB, Toellner KM, MacLennan CA, MacLennan IC, Henderson IR, Dougan G and Cunningham AF. 2012. The capsular polysaccharide Vi from Salmonella typhi is a B1b antigen. J. Immunol 189: 5527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foote JB, Mahmoud TI, Vale AM and Kearney JF. 2012. Long-Term Maintenance of Polysaccharide-Specific Antibodies by IgM-Secreting Cells. J. Immunol 188: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunningham AF, Flores-Langarica A, Bobat S, Dominguez Medina CC, Cook CN, Ross EA, Lopez-Macias C and Henderson IR. 2014. B1b cells recognize protective antigens after natural infection and vaccination. Front. Immunol 5: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKay JT, Haro MA, Daly CA, Yammani RD, Pang B, Swords WE and Haas KM. 2017. PD-L2 Regulates B-1 Cell Antibody Production against Phosphorylcholine through an IL-5-Dependent Mechanism. J. Immunol 199: 2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shih TA, Roederer M and Nussenzweig MC. 2002. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol 3: 399–406. [DOI] [PubMed] [Google Scholar]
- 23.Gaspal FM, McConnell FM, Kim MY, Gray D, Kosco-Vilbois MH, Raykundalia CR, Botto M and Lane PJ. 2006. The generation of thymus-independent germinal centers depends on CD40 but not on CD154, the T cell-derived CD40-ligand. Eur. J. Immunol 36: 1665–73. [DOI] [PubMed] [Google Scholar]
- 24.Marshall JL, Zhang Y, Pallan L, Hsu MC, Khan M, Cunningham AF, MacLennan IC and Toellner KM. 2011. Early B blasts acquire a capacity for Ig class switch recombination that is lost as they become plasmablasts. Eur. J. Immunol 41: 3506–12. [DOI] [PubMed] [Google Scholar]
- 25.Anderson SM, Tomayko MM, Ahuja A, Haberman AM and Shlomchik MJ. 2007. New markers for murine memory B cells that define mutated and unmutated subsets. J. Exp. Med 204: 2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomayko MM, Steinel NC, Anderson SM and Shlomchik MJ. 2010. Cutting edge: Hierarchy of maturity of murine memory B cell subsets. J. Immunol 185: 7146–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yammani RD and Haas KM. 2013. Primate B-1 cells generate antigen-specific B cell responses to T cell-independent type 2 antigens. J. Immunol 190: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas KM 2015. B-1 lymphocytes in mice and nonhuman primates. Ann. N. Y. Acad. Sci 1362: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas KM, Blevins MW, High KP, Pang B, Swords WE and Yammani RD. 2014. Aging Promotes B-1b Cell Responses to Native, but Not Protein-Conjugated, Pneumococcal Polysaccharides: Implications for Vaccine Protection in Older Adults. J. Infect. Dis 209: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin F and Kearney JF. 2002. Marginal-zone B cells. Nat. Rev. Immunol 2: 323–335. [DOI] [PubMed] [Google Scholar]
- 31.Swanson CL, Wilson TJ, Strauch P, Colonna M, Pelanda R and Torres RM. 2010. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J. Exp. Med 207: 1485–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amlot PL, Grennan D and Humphrey JH. 1985. Splenic dependence of the antibody response to thymus-independent (TI-2) antigens. Eur. J. Immunol 15: 508–512. [DOI] [PubMed] [Google Scholar]
- 33.Goud SN, Kaplan AM and Bondada S. 1990. Primary antibody responses to thymus-independent antigens in the lungs and hilar lymph nodes of mice. Infect Immun. 58: 2035–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fairfax KA, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL and Tarlinton DM. 2007. Different kinetics of blimp-1 induction in B cell subsets revealed by reporter gene. J. Immunol 178: 4104–4111. [DOI] [PubMed] [Google Scholar]
- 35.Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z and Casali P. 2012. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nat. Commun 3: 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavnezer J and Schrader CE. 2014. IgH chain class switch recombination: mechanism and regulation. J. Immunol 193: 5370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weisel F and Shlomchik M. 2017. Memory B Cells of Mice and Humans. Annu. Rev. Immunol 35: 255–284. [DOI] [PubMed] [Google Scholar]
- 38.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL and Shlomchik MJ. 2014. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol 15: 631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dal Porto JM, Burke K and Cambier JC. 2004. Regulation of BCR signal transduction in B-1 cells requires the expression of the Src family kinase Lck. Immunity 21: 443–453. [DOI] [PubMed] [Google Scholar]
- 40.Chumley MJ, Dal Porto JM and Cambier JC. 2002. The unique antigen receptor signaling phenotype of B-1 cells is influenced by locale but induced by antigen. J. Immunol 169: 1735–43. [DOI] [PubMed] [Google Scholar]
- 41.Ansel KM, Harris RB and Cyster JG. 2002. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 16: 67–76. [DOI] [PubMed] [Google Scholar]
- 42.Goud SN and Bondada S. 1988. Differential responses of B cells from the spleen and lymph node to TNP-Ficoll. J. Immunol 140: 2925–2930. [PubMed] [Google Scholar]
- 43.Goud SN, Kaplan AM and Bondada S. 1990. Antibody responses to thymic independent antigens in the peripheral and mesenteric lymph nodes of mice. Reg. Immunol 3: 1–7. [PubMed] [Google Scholar]
- 44.Colombo MJ, Sun G and Alugupalli KR. 2010. T-cell-independent immune responses do not require CXC ligand 13-mediated B1 cell migration. Infect. Immun 78: 3950–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snapper CM 2012. Mechanisms underlying in vivo polysaccharide-specific immunoglobulin responses to intact extracellular bacteria. Ann. N. Y. Acad. Sci 1253: 92–101. [DOI] [PubMed] [Google Scholar]
- 46.Nuorti JP and Whitney CG. 2010. Prevention of pneumococcal disease among infants and children - use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine - recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 59: 1–18. [PubMed] [Google Scholar]
- 47.Feikin DR, Elie CM, Goetz MB, Lennox JL, Carlone GM, Romero-Steiner S, Holder PF, O’Brien WA, Whitney CG, Butler JC and Breiman RF. 2001. Randomized trial of the quantitative and functional antibody responses to a 7-valent pneumococcal conjugate vaccine and/or 23-valent polysaccharide vaccine among HIV-infected adults. Vaccine 20: 545–553. [DOI] [PubMed] [Google Scholar]
- 48.Haas KM, Hasegawa M, Steeber DA, Poe JC, Zabel MD, Bock CB, Karp DR, Briles DE, Weis JH and Tedder TF. 2002. Complement receptors CD21/35 link innate and protective immunity during Streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity 17: 713–723. [DOI] [PubMed] [Google Scholar]
- 49.Haas KM, Poe JC and Tedder TF. 2009. CD21/35 promotes protective immunity to Streptococcus pneumoniae through a complement-independent but CD19-dependent pathway that regulates PD-1 expression. J. Immunol 183: 3661–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor JJ, Pape KA and Jenkins MK. 2012. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med 209: 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaku H, Cheng KF, Al-Abed Y and Rothstein TL. 2014. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J. Immunol 193: 5904–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKay JT, Egan RP, Yammani RD, Chen L, Shin T, Yagita H and Haas KM. 2015. PD-1 Suppresses Protective Immunity to Streptococcus pneumoniae through a B Cell-Intrinsic Mechanism. J. Immunol 194: 2289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haro MA, Littrell CA, Yin Z, Huang X and Haas KM. 2016. PD-1 Suppresses Development of Humoral Responses That Protect against Tn-Bearing Tumors. Cancer Immunol. Res 4: 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boudewijns M, Jeurissen A, Wuyts M, Moens L, Boon L, Van Neerven JJ, Kasran A, Overbergh L, Lenaerts C, Waer M, Mathieu C, Ceuppens JL and Bossuyt X. 2005. Blockade of CTLA-4 (CD152) enhances the murine antibody response to pneumococcal capsular polysaccharides. J. Leukoc. Biol 78: 1060–1069. [DOI] [PubMed] [Google Scholar]
- 55.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM and Shlomchik MJ. 2010. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol 11: 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.