

Abstract
Osteoblastoma is a locally aggressive tumour of bone. Until recently, its underlying genetic features were largely unknown. During the past two years, reports have demonstrated that acquired structural variations affect the transcription factor FOS in a high proportion of cases. These rearrangements modify the terminal exon of the gene and are believed to stabilise both the FOS transcript and the encoded protein, resulting in high expression levels. Here, we applied in‐depth genetic analyses to a series of 29 osteoblastomas, including five classified as epithelioid osteoblastoma. We found recurrent homozygous deletions of the NF2 gene in three of the five epithelioid cases and in one conventional osteoblastoma. These events were mutually exclusive from FOS mutations. Structural variations were determined by deep whole genome sequencing and the number of FOS‐rearranged cases was less than previously reported (10/23, 43%). One conventional osteoblastoma displayed a novel mechanism of FOS upregulation; bringing the entire FOS gene under the control of the WNT5A enhancer that is itself activated by FOS. Taken together, we show that NF2 loss characterises a subgroup of osteoblastomas, distinct from FOS‐rearranged cases. Both NF2 and FOS are involved in regulating bone homeostasis, thereby providing a mechanistic link to the excessive bone growth of osteoblastoma.
Keywords: FOS, FOSB, NF2, WNT5A, osteoblastoma, osteosarcoma
Introduction
Osteoblastoma is a bone‐forming tumour that harbours mutations affecting FOS, or more rarely its paralogue FOSB, in a high proportion of cases [1]. The exact frequency of osteoblastomas with FOS or FOSB mutations varies depending on the methodology applied. By fluorescence in situ hybridisation (FISH) analysis, around 90% of investigated cases have been reported to harbour structural rearrangement of the FOS gene [1, 2]. By deep sequencing of a limited number of cases, the FOS rearrangements were shown to affect the terminal exon of the gene [1]. The functional outcome is increased FOS expression, likely due to reduced degradation of both FOS mRNA and its protein product [3]. By immunohistochemical analysis, 60–80% of osteoblastomas show increased FOS expression, which can be used as a reliable marker in routine clinical diagnostics [2, 4]. Less than 5% of cases display rearrangements of the FOSB gene [1, 4]. One case confirmed by deep sequencing analysis harboured a PPP1R10‐FOSB fusion gene, in which the coding parts of FOSB were placed under the control of the PPP1R10 promoter. Other FOSB positive cases were found by immunohistochemical or FISH analyses.
Although FOS and FOSB mutations are frequent findings in osteoblastoma, they do not underlie all cases. We have previously reported complex genome rearrangements including recurrent chromosome 22q12 deletions in osteoblastoma [5]. These complex alterations were found in so‐called epithelioid (previously referred to as aggressive) osteoblastoma. Epithelioid osteoblastoma has the same clinical behaviour as conventional osteoblastoma, i.e. surgery cures most cases, but tumours that are inaccessible or recur can cause considerable morbidity [6]. In addition, osteoblastoma can be diagnostically challenging because its histological features may overlap with those of high‐grade osteosarcoma. Here, we report genetic data from a series of conventional and epithelioid osteoblastomas. Our main finding was complete loss of the NF2 gene in a subgroup of non‐FOS‐rearranged, preferentially epithelioid osteoblastomas.
Materials and methods
Tumour material
Material from 29 osteoblastomas was collected from the Skåne University Hospital and the Karolinska Hospital in Sweden, the University Hospital Basel in Switzerland, the Leiden University Medical Center in the Netherlands, and the Royal Orthopaedic Hospital in Birmingham, UK. Five of the tumours were classified as epithelioid osteoblastoma, and 24 as conventional osteoblastoma. Patient age ranged from 2 to 50 years (mean and median ages 23 and 17 years, respectively), and two‐thirds were males (20/29). Detailed patient information can be found in the supplementary material, Table S1. The study was approved by the Institutional Review Boards of the participating centres.
Genome‐wide copy number and structural analyses of bulk tumour DNA
DNA was extracted from fresh frozen tumour biopsies according to standard procedures [7]. SNP array analysis was performed using the CytoScan HD arrays and Chromosome Analysis Suite v 4.0 (Thermo Fisher Scientific, Waltham, MA, USA), and the Illumina Human Omni‐Quad BeadChips and GenomeStudio software (Illumina, San Diego, CA, USA) [5, 7]. SNP array findings in seven of the cases have previously been published, including two aberrant and five normal profiles (see supplementary material, Table S1) [5]. Whole genome mate pair sequencing was carried out using the Nextera Mate Pair Library Preparation Kit (Illumina) [7]. Sequencing depth was 2.4× on average (mapping coverage 1.7×) and the mean insert size was 3.0 kb, resulting in a median spanning coverage of 45.2× of the human genome (mean 48.1×, range 25.7–100.1×). Sequencing reads were trimmed using NxTrim v 0.4.2 and then aligned against the GRCh37/hg19 build using the Borrows‐Wheeler Aligner v 0.7.15 [8]. To identify structural rearrangements, the sequence data were analysed using Integrative Genomics Viewer, as well as the structural variant callers TIDDIT v 2.7.1 [9] and Delly2 v 0.7.8 [10]. Case 1 and a matched normal control sample were analysed by whole genome paired‐end sequencing using the Complete Genomics platform. Sequencing depth was approximately 100× and 30×, respectively. Case 2 and a matched normal control sample were analysed by whole exome sequencing, as previously described [11]. Copy number plots based on whole genome and whole exome data were created using CNVkit [12].
Whole genome low‐pass sequencing of individual cells
Whole genome sequencing of cryopreserved primary osteoblastoma cells was performed using a modified single cell whole genome sequencing protocol and 77 base pair single reads were generated using a NextSeq 500 sequencing instrument (Illumina) [13]. Copy number analysis was performed using AneuFinder [14].
Transcriptome sequencing
RNA was extracted from fresh frozen tumour biopsies according to standard procedures and sequenced using the TruSeq RNA Sample Preparation Kit v2 (Illumina) [7]. Sequencing reads were aligned to the GRCh37/hg19 build using STAR v 2.5.2b [15]. For comparison of relative gene expression levels, gene counts were FPKM (fragments per kilobase per million mapped reads) normalised using Cufflinks with default settings [16], and visualised using the Qlucore Omics Explorer version 3.5 (Qlucore AB, Lund, Sweden). In total, gene expression data from 13 osteoblastomas and, as control, 69 osteosarcomas were available.
FISH and immunohistochemical analyses
FISH analyses on formalin‐fixed paraffin‐embedded slides and metaphase spreads were performed as described previously [2], using break‐apart probes for FOS. Immunohistochemical analysis was performed using a rabbit polyclonal antibody against the N‐terminal region of FOS and a rabbit monoclonal antibody for FOSB [2].
Results and discussion
Genomic copy number status was determined in all 24 conventional and five epithelioid osteoblastomas using SNP array analysis. Out of the 29 cases, seven displayed acquired DNA copy number alterations (see supplementary material, Table S2). Five cases harboured hemizygous deletions of either whole or parts of chromosome arm 6q (Figure 1A), of which four also had hemi‐ and homozygous deletions in chromosome arm 22q (Figure 1B). The latter clustered to a minimal deleted region in chromosome band 22q12 (Figure 1C). Combined with copy number and structural variant information from whole exome and whole genome sequencing analyses, we could confirm that the NF2 gene in 22q12 was affected by intragenic homozygous deletions in all four cases (Figure 1C and see supplementary material, Tables [Link], [Link]). In line with this, the relative expression level of NF2 was low in affected cases compared with other osteoblastomas and osteosarcomas (Figure 2A). Three out of the four cases with homozygous NF2 deletions showed intragenic homozygous deletions affecting the ZNRF3 gene. However, in the remaining NF2‐deleted case (Case 2), we detected neither homozygous loss nor low relative expression level of ZNRF3, arguing against ZNRF3 as a target for the deletions (Figures 1C and 2B). The number of ascertained NF2‐deleted osteoblastomas is still too low to make any definite correlations with clinical features. However, we noted that three out of four cases with intragenic homozygous NF2 loss were classified as epithelioid osteoblastoma (see supplementary material, Table S1). We found no acquired genetic alterations in the remaining two epithelioid osteoblastomas. In one of the epithelioid osteoblastomas with 6q and NF2 losses, we subjected individual cryopreserved cells to whole genome sequencing. Out of 178 individual cells sequenced, 15 (8%) showed acquired copy number variations in agreement with those detected in bulk tumour DNA, i.e. loss of 6q and regions harbouring NF2 in 22q12 (Figure 3A,B and see supplementary material, Figure S1). Presumably, these cells represent the neoplastic clone and the remaining cells constitute admixed normal cells.
Figure 1.
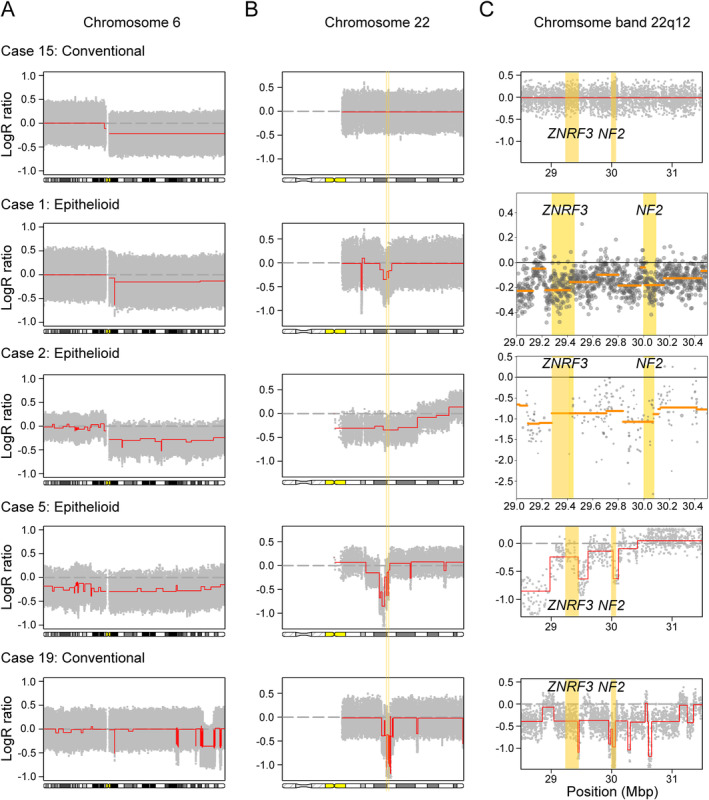
High‐resolution genomic copy number analyses reveal a subgroup of osteoblastoma harbouring recurrent deletions in 6q and 22q12. (A) SNP array analysis detects recurrent deletions in chromosome arm 6q in five osteoblastomas. (B) Four of them harbour concomitant deletions in chromosome arm 22q. (C) Whole genome paired‐end sequencing (Case 1), whole exome sequencing (Case 2), and SNP array analysis (Cases 5 and 19) show that these deletions cluster to the ZNRF3 and NF2 genes in 22q12. The latter gene is affected by intragenic homozygous deletions in all four cases. Case 15 did not harbour any deletions affecting chromosome 22. Yellow lines mark the positions of the ZNRF3 and NF2 genes.
Figure 2.
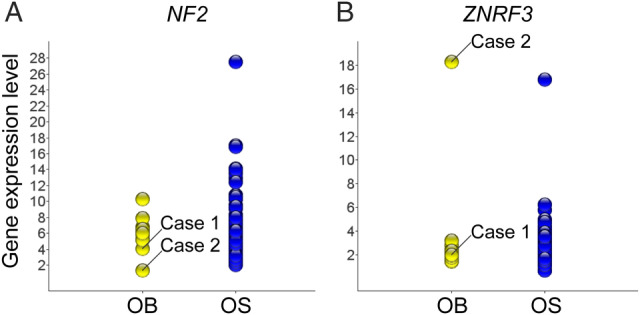
Transcriptome analysis consolidates NF2 as the most likely target for 22q12 deletions in osteoblastoma. (A) Cases 1 and 2 harbour intragenic homozygous NF2 deletions and concomitant low NF2 expression levels, compared with osteoblastomas without detected NF2 deletion (n = 11) and osteosarcomas (n = 69). (B) Case 1 harbours an intragenic homozygous deletion and concomitant low expression level of the ZNRF3 gene. Case 2 harbours a hemizygous loss and relative high expression level of ZNRF3. OB, osteoblastoma; OS, osteosarcoma.
Figure 3.
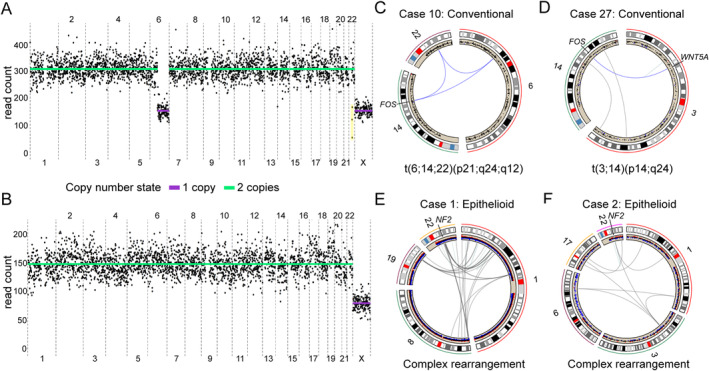
NF2 deletions and FOS structural variations are mutually exclusive in osteoblastoma. (A) Whole genome sequencing of an individual cell from Case 1 shows hemizygous loss of chromosome arm 6q. The patient is male, and a single copy of the X chromosome is detected (the Y chromosome is not shown). The low number of reads mapping to the three copy number bins highlighted in yellow suggest a combination of hemi‐ and homozygous losses across this region (27.7–31.1 Mb, according to genome assembly GRCh37/hg19). (B) A representative cell with no acquired copy number alterations, presumably a non‐neoplastic cell.(C) A balanced three‐way translocation results in structural rearrangement of the 3′ part of the FOS gene in Case 10. (D) A balanced two‐way translocation juxtaposes the complete coding region of FOS and the enhancer region of WNT5A in Case 27. (E,F) Complex structural variations affecting 22q12 and many other chromosomal regions result in genomic copy number imbalances, including intragenic homozygous losses of NF2 in Cases 1 and 2.
Out of the 23 osteoblastomas for which we could evaluate structural chromosome alterations by whole genome sequencing, FOS rearrangements were found in 10 (43%), and FOSB rearrangement was not detected (see supplementary material, Tables S1 and S5). This proportion of FOS and FOSB rearranged cases is lower than what has been found by FISH analyses [1, 2]. Our whole genome sequencing analysis of bulk tumour DNA, with a median spanning coverage of 45× of the human genome, showed only few sequencing reads that supported the FOS rearrangements even in positive cases. Thus, deep sequencing analyses of DNA from bulk tumour tissue and individual cells suggest that there is a high proportion of normal cells admixed with the neoplastic clone, and this may obscure the detection of acquired genetic alterations in osteoblastoma.
The FOS rearrangements detected here were generated by balanced two‐ or three‐way translocations (Figure 3C,D and see supplementary material, Tables S1 and S5). Few or no other genomic alterations were detected in these cases. Two of our FOS rearranged cases are particularly noteworthy. In Case 20, we detected a FOS‐KIAA1199 fusion (see supplementary material, Table S5), very similar to the FOS‐KIAA1199 fusion previously reported in osteoblastoma [1]. The competitive advantage, if any, of this particular rearrangement is unknown. In Case 27, we detected a t(3;14)(p14;q24) that did not affect the terminal exon of FOS. Instead, the breakpoint in 14q24 was located in one of the FOS promoter/enhancer regions, 23 kb upstream of the gene. The translocation placed the complete coding region of the FOS gene under the enhancer region of WNT5A (Figure 3D and see supplementary material, Table S5). WNT5A is a WNT ligand involved in bone metabolism and is paradoxically induced by FOS [17]. Upregulation of FOS in this case was confirmed by immunohistochemistry (see supplementary material, Table S1). In the present series of osteoblastomas, FOS rearrangement was mutually exclusive from NF2 deletion (see supplementary material, Table S1). While the FOS‐rearranged cases displayed balanced chromosome alterations, NF2‐deleted cases were characterised by unbalanced, complex rearrangements that affected chromosome band 22q12 and several other chromosomal regions (Figure 3E,F).
We have previously shown recurrent deletions in 22q12 in osteoblastoma, affecting genes that are linked to WNT signalling and bone homeostasis [5]. In the present study, we analysed more cases with a higher resolution. This enabled us to pinpoint the NF2 gene as the most likely target for the 22q12 deletions in osteoblastoma. In line with this, loss of NF2 disrupts the Hippo signalling pathway, a key component in osteoclast formation and bone homeostasis [18], and Nf2‐deficient mice show increased bone volume [19]. This does not rule out the possibility that additional genes in 22q12, such as MN1, ZNRF3 and/or KREMEN1, play a role in osteoblastoma development. However, in support for NF2, there is cross talk between NF2 and FOS signalling pathways. More specifically, loss of NF2 will lead to decreased activation of the Hippo pathway, which normally inhibits the activity of the transcriptional co‐activators YAP and TAZ, allowing them to have a longer‐lasting effect in the nucleus [18]. YAP and TAZ also cross activate the AP‐1 transcription factor complex, of which FOS is a main component. YAP/TAZ and AP‐1 can synergistically activate downstream target genes, and a prolonged effect of YAP and TAZ due to loss of NF2 may thereby lead to increased FOS activity and a continued deregulation of signalling pathways.
In summary, we have found mutually exclusive FOS rearrangements and intragenic homozygous NF2 deletions in osteoblastoma. The latter were associated with additional genomic losses, complex structural variations, and clearly clustered to the epithelioid subtype of osteoblastoma. Hitherto, defects in genes that regulate bone homeostasis are common to osteoblastomas.
Author contributions statement
KHS, ES and KHN conceived and designed the study. KHS, LC, JH, LM, JN, HvdB, DCJS, FF, JS, SWL, KS, JVMGB, MK, DB, ES and KHN generated, analysed or interpreted data. OB, VPS, KS, JVMGB, DB and ES provided tumour material and clinical information. KHS and KHN wrote the manuscript with contributions from all other authors.
Supporting information
Figure S1. Whole genome sequencing of cryopreserved primary osteoblastoma cells from case 1
Table S1. Clinical and genetic features of osteoblastoma
Table S2. Genomic copy number alterations detected by SNP array analysis
Table S3. Genomic copy number state of genes in 22q12
Table S4. Structural chromosome alterations affecting the NF2 gene detected by whole genome paired‐end sequencing and whole genome mate pair sequencing
Table S5. Structural chromosome alterations affecting the FOS gene detected by whole genome DNA mate pair sequencing
Acknowledgements
KHS was supported by the Royal Physiographic Society (Lund, Sweden) and the Faculty of Medicine at Lund University. KHN was supported by the Swedish Childhood Cancer Fund, the Swedish Cancer Society, the Swedish Research Council, the Åke Wiberg Foundation, the Royal Physiographic Society (Lund, Sweden) and the Crafoord Foundation. ES was supported by the Maggie Stephens Foundation, Erik and Angelica Sparres Research Foundation, Greta and Johan Kocks Foundation, and by grants from the Swedish state under the agreement between the Swedish Government and the county councils, the ALF‐agreement (F 2014/413). MK and DB were supported by the Swiss National Science Foundation, the Foundation of the Basel Bone Tumour Reference Centre, the Gertrude von Meissner Stiftung, and the Stiftung für krebskranke Kinder, Regio Basiliensis. JVMGB is financially supported by the Netherlands Organization for Scientific Research (ZON‐MV VICI 170.055).
No conflicts of interest were declared.
References
- 1. Fittall MW, Mifsud W, Pillay N, et al Recurrent rearrangements of FOS and FOSB define osteoblastoma. Nat Commun 2018; 9: 2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lam SW, Cleven AHG, Kroon HM, et al Utility of FOS as diagnostic marker for osteoid osteoma and osteoblastoma. Virchows Arch 2020; 476: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Ijzendoorn DGP, Forghany Z, Liebelt F, et al Functional analyses of a human vascular tumor FOS variant identify a novel degradation mechanism and a link to tumorigenesis. J Biol Chem 2017; 292: 21282–21290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amary F, Markert E, Berisha F, et al FOS expression in osteoid osteoma and osteoblastoma: a valuable ancillary diagnostic tool. Am J Surg Pathol 2019; 43: 1661–1667. [DOI] [PubMed] [Google Scholar]
- 5. Nord KH, Nilsson J, Arbajian E, et al Recurrent chromosome 22 deletions in osteoblastoma affect inhibitors of the Wnt/beta‐catenin signaling pathway. PLoS One 2013; 8: e80725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Andrea CE, Bridge JA, Schiller A. Osteoblastoma In WHO Classification of Tumours of Soft Tissue and Bone (4th edn), Fletcher CDM, Bridge JA, Hogendoorn PCW, et al (Eds). International Agency for Research on Cancer (IARC): Lyon, 2013; 279–280. [Google Scholar]
- 7. Saba KH, Cornmark L, Rissler M, et al Genetic profiling of a chondroblastoma‐like osteosarcoma/malignant phosphaturic mesenchymal tumor of bone reveals a homozygous deletion of CDKN2A, intragenic deletion of DMD, and a targetable FN1‐FGFR1 gene fusion. Genes Chromosomes Cancer 2019; 58: 731–736. [DOI] [PubMed] [Google Scholar]
- 8. Li H. Toward better understanding of artifacts in variant calling from high‐coverage samples. Bioinformatics 2014; 30: 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eisfeldt J, Vezzi F, Olason P, et al TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Research 2017; 6: 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rausch T, Zichner T, Schlattl A, et al DELLY: structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics 2012; 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hofvander J, Arbajian E, Stenkula KG, et al Frequent low‐level mutations of protein kinase D2 in angiolipoma. J Pathol 2017; 241: 578–582. [DOI] [PubMed] [Google Scholar]
- 12. Talevich E, Shain AH, Botton T, et al CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016; 12: e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van den Bos H, Bakker B, Taudt A, et al Quantification of aneuploidy in mammalian systems. Methods Mol Biol 1896; 2019: 159–190. [DOI] [PubMed] [Google Scholar]
- 14. Bakker B, Taudt A, Belderbos ME, et al Single‐cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 2016; 17: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobin A, Davis CA, Schlesinger F, et al STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trapnell C, Williams BA, Pertea G, et al Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhong Z, Ethen NJ, Williams BO. WNT signaling in bone development and homeostasis. Wiley Interdiscip Rev Dev Biol 2014; 3: 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang W, Han W, Qin A, et al The emerging role of Hippo signaling pathway in regulating osteoclast formation. J Cell Physiol 2018; 233: 4606–4617. [DOI] [PubMed] [Google Scholar]
- 19. Larsson J, Ohishi M, Garrison B, et al Nf2/merlin regulates hematopoietic stem cell behavior by altering microenvironmental architecture. Cell Stem Cell 2008; 3: 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Whole genome sequencing of cryopreserved primary osteoblastoma cells from case 1
Table S1. Clinical and genetic features of osteoblastoma
Table S2. Genomic copy number alterations detected by SNP array analysis
Table S3. Genomic copy number state of genes in 22q12
Table S4. Structural chromosome alterations affecting the NF2 gene detected by whole genome paired‐end sequencing and whole genome mate pair sequencing
Table S5. Structural chromosome alterations affecting the FOS gene detected by whole genome DNA mate pair sequencing