

 One step radiofluorination of two thymidine bioconjugates for positron emission tomography (PET) imaging; proof concept with clinical-grade 18F-labeling and preliminary mouse images.
One step radiofluorination of two thymidine bioconjugates for positron emission tomography (PET) imaging; proof concept with clinical-grade 18F-labeling and preliminary mouse images.
Abstract
In pursuit of 18F-labeled nucleosides for positron emission tomography (PET) imaging, we report on the chemical and radiochemical synthesis of two thymidine (dT) analogs, dT-C5-AMBF3 and dT-N3-AMBF3, that are radiofluorinated by isotope exchange (IEX) and studied as PET imaging agents in mice with tumor xenografts. dT-C5-AMBF3 shows preferential, and tumor-specific, uptake over dT-N3-AMBF3. This work provides a new synthetic method in order to access new nucleoside tracers for PET imaging.
Introduction
Non-invasive molecular imaging with radioisotopes, e.g. positron emission tomography (PET) and single photon emission computed tomotraphy (SPECT), now plays a pivotal role in preclinical lead validation and clinical cancer diagnosis. Whereas antibodies and peptides typically recognize extracellular targets, small molecules are designed to bind intracellular targets and/or importers that are well-known hallmarks of cancer. Among numerous small molecules, nucleosides are of enduring interest as rapidly dividing cancer cells aggressively access them to support the increased rate of DNA replication. Such interest has focused on dT analogs owing to the involvement of thymidine kinase (TK1) in the salvage pathway of DNA metabolism and historic success with 5-fluoro-uracil (5FU)/5F-dUMP as first-line antineoplastic agents that block dT synthesis.
In the thymidine salvage pathway, TK1 phosphorylates dT using ATP to form thymidine monophosphate1 (TMP). Whereas many cancers overexpress TK1 (ref. 1a and 2) they also show high proclivity for importing nutrients such as nucleosides, with preferential uptake of dT;3 dT and analogs thereof are usually imported by equilibrative nucleoside transporters (ENTs) although passive diffusion across the cell membrane is also known.4 Once phosphorylated, TMP analogs are rendered membrane impermeable, leading to intracellular retention.3 Hence, TK1 and ENTs are considered biomarkers that correlate with over-proliferation. Their visualization by non-invasive radioimaging provides a basis for positive cancer diagnosis.1a,2,5
Recently, nucleoside-derived imaging agents have been labeled, either with radiometals e.g.99mTc,668Ga,7 or with 18F.8 Radiofluorine is typically introduced directly at positions 2′–5′ to replace an OH group, and at position-5 of the nucleoside to replace a proton (notably 5FU is a drug that targets thymidylate synthase). However, in the case of chelated radiometals, chelators are conjugated via linker arms that are typically introduced on either the 5′ or 3′ hydroxyls, or on either N3 or C5 of the nucleoside itself.
Of various non-invasive radioimaging modalities, PET is known for its high sensitivity and spatiotemporal resolution.9 Of the several commonly used PET-isotopes, e.g.68Ga, 64Cu, 15O, 11C, 13N, and 18F, 18F is preferred due to its biologically relevant half-life (109.7 min), higher image resolution compared to 68Ga,10 clean decay (>95% β+), a record of FDA-approval, and scalability to GBq-level production on-demand in hospital cyclotrons.11 Typically, 18F-labeling requires punctiliously anhydrous [18F]fluoride that is used in radiosynthetic processes requiring at least two steps. For example, [18F]3′-deoxy-3′-fluorothymidine ([18F]FLT) is synthesized in a 3-step sequence at molar activity (Am) values of >37 GBq μmol–1 in radiochemical yields (RCY) of 13%.12 Preclinically, [18F]FLT was successfully used to detect response of murine colon carcinoma to lipo-Dox, a chemotherapeutic drug,1b and showed high apparent uptake in human and canine bone marrow.3
Increased interest in PET imaging has spurred development of one-step 18F-labeling methods that overcome many of the well-known challenges in 18F-labeling to make radiofluorination nearly as user-friendly as radiometallation.13 One such method uses IEX to label organosilylfluorides (SiFAs) that serve as radioprosthetic groups.14 Recently di-tertbutylphenylsilylfluoride was conjugated to gemcitabine (2′-deoxy-2′-difluorocytosine) to afford novel 18F-labeled nucleoside conjugates via IEX in one step.15
Similarly, organotrifluoroborates are readily 18F-labeled by IEX in one-step in high RCY, high purity and at Am values that are considered to be comparable if not higher than most radiotracers.16 In contrast to di-tert-butyl-bisaryl-silylfluorides, which are stabilized against solvolytic fluoride loss by virtue of sterically bulky hydrophobic groups, the ammoniomethylenetrifluoroborate (AMBF3) radio-prosthetic group17 is stabilized against solvolytic fluoride loss by virtue of its zwitterionic character and has found particularly useful applications in numerous peptide-based radiotracers; including LLP2A-DOTA,18 octreotate,19 bombesin,20 alpha-MSH,21 and PSMA-targeting peptidyl ureas.22 We have also conjugated the AMBF3 prosthetic group to smaller molecules to image hypoxia,23 carbonic anhydrase-IX activity,24 and cardiac function.25 Yet to date, this approach has not been explored to label nucleosides for PET applications. Here, we disclose the synthesis of two different dT analogs functionalized at positions N3 and C5; dT-C5-AMBF31 and dT-N3-AMBF32 (Fig. 1), successful 18F-labeling by IEX at high Am, along with correlated in vivo PET images.
Fig. 1. AMBF3-conjugated dT analogs, dT-C5-AMBF31 and dT-N3-AMBF32, used in this study.

Results
Chemistry
Two different dT-conjugates: 1 and 2, each bearing the AMBF3 group tethered to either C5 or N3 respectively were prepared according to two synthetic routes. In pursuit of these ends, a new one-pot two-step gram-scale column-free procedure was devised to synthesize 4 quantitatively (see synthetic procedure). Alkyne 4 and 5-azidomethyl-2′-deoxyuridine (5) were conjugated via CuAAC to afford 1 in 28% isolated yield (Scheme 1).26 In order to synthesize 2, 3-chloro-N,N-dimethylpropan-1-amine hydrochloride (6) was free-based, immediately alkylated with ICH2Bpin, and then fluorinated in KHF2/HCl to trifluoroborate 7 in 45% yield without optimization. Compound 7 was converted to 2 in a one-pot two-step fashion by transposing the chloride to the iodide in situ via the Finkelstein reaction, and then effecting an SN2 reaction with dT 8 (Scheme 2). Both 1 and 2 were purified by HPLC and characterized by HRMS and 1H, 13C, 11B, 19F NMR spectroscopy (available in ESI†). It is noteworthy that trifluoroborate 7 is a novel radiosynthon that should find use in installing the AMBF3 prosthetic group on a wide range of nucleophiles under relatively mild conditions (ca. pH 9 in acetone). Both quaternary ammoniums on radiosynthons 4 and 7 serve to kinetically stabilize the negative trifluoroborate against solvolysis at pH 7. The net neutral charge is thought to facilitate cell permeability.
Scheme 1. Reagents and conditions: 5-azidomethyl-2′-deoxyuridine (5), CuSO4, NaAsc, K2CO3, 3 : 2 MeCN : H2O, r.t., 15.3 h, 28% yield.

Scheme 2. Reagents and conditions: 1. NaI, acetone, 56 °C, 17 h; 2. 8, K2CO3, acetone, 56 °C, 6 h, quantitative yield, using dT in limiting quantities.

Radiochemistry
Radiolabeling proceeded by one-step IEX with no-carrier-added (NCA) [18F]fluoride, as recently reported.18,22,27 Briefly, [18F]fluoride was eluted from a QMA light cartridge in PBS and allowed to mix with either 1 or 2 dissolved in DMF (Scheme 3). Following heating for 10–20 min, the reaction was quenched, HPLC purified, retained by a C18 Sep-Pak and eluted with 50% EtOH. Evidence of radiochemical purity is given in Fig. 2.
Scheme 3. Radiolabeling of 1 and 2; R = nucleoside-linker-ammonio-CH2.
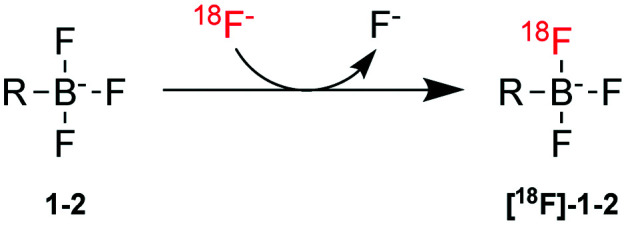
Fig. 2. Characteristic radioactivity and UV chromatograms for 1 (left) and 2 (right).

RCYs and Am values were calculated (see ESI†) and reported in Table 1. We note that ca. 15% [18F]1 or [18F]2 is lost on Sep-Pak purification and thus isolated yields are lower. In addition, whereas we report decay-corrected yields, we should note that the radiosynthesis is complete within 30–40 min; as such isolated RCYs are at most 23% lower than reported with decay correction. An explanation as to why the Am values for [18F]2 are nearly twice those of [18F]1 is not immediately forthcoming (see discussion on Am). For both 1 and 2, water–octanol partition coefficients (log P) were less than –3, indicating significant hydrophilicities and generally poor retention on Sep-Pak.
Table 1. Summary of molar activities (Am), isolated radiochemical yield (RCY) and log P7.4 for 18F-radiolabeling of 1 and 2. RCYs are decay corrected.
| Compound | A m a (GBq μmol–1) | RCY (%) | log P7.4 b |
| 1 | 106.1 ± 57.6 c | 28.6 ± 22.6 | –3.01 ± 0.04 |
| 2 | 252.2 ± 52.7 | 37.9 ± 28.6 | –4.29 ± 0.09 |
a P < 0.01.
b P < 0.0001.
c n = 7 for Am of 1, else n = 3.
Imaging
To image the uptake of [18F]1 and [18F]2, we attempted PET imaging in mice bearing U87M tumor xenografts as this tumor had been previously imaged with [18F]FLT.28 On first glimpse, [18F]1 provided images that identified the tumor with an apparent uptake value of 1.5 %ID g–1 while [18F]2 failed to provide any tumor images (Fig. 3).
Fig. 3. A) Two mice with U87M xenografts imaged with [18F]1 (left); B) one mouse imaged with [18F]2, 1 h post injection. Red circles provide an approximate outline of rather large U87M tumors used in these mice.

It was noted that for this pilot imaging study (Fig. 3), tumors were particularly large; whereas uptake was observed, particularly in the case of the mice pair in Fig. 3A, only a fraction of the tumor was actually visualized with [18F]1 while there was little contrast observed in images obtained with [18F]2. It is noted that much of the tumor, outlined by the red circle, is not observed. To better assess this uptake, tumors were regrown to a smaller size and mice were injected with tracers for ex vivo biodistribution analysis. Impressively, both tracers cleared to the bladder showing uptake values of 81 ± 34 %ID g–1 for [18F]1 and 279 ± 91 %ID g–1 for [18F]2. Yet uptake values in all other organs were extremely low, and tumor uptake values were on the order of 0.1 %ID g–1 for both [18F]1 and [18F]2 making it difficult to draw clear conclusions as to the specificity of uptake. Not surprisingly, a blocking experiment did not show significant blocking (see Discussion). Nevertheless, we calculated tumor : non-tumor ratios for select organs (Table 2). Both tracers showed somewhat higher uptake in tumor over other key organs.
Table 2. Ex vivo biodistribution results of [18F]1 and [18F]2 1 h post injection. All tumor-to-normal tissue uptake ratios are expressed in mean ± s.d., n = 3.
| 1 h p.i. | [18F]1 | [18F]2 |
| Tumor : bone | 2.02 ± 0.65 | 1.84 ± 0.43 |
| Tumor : muscle | 4.13 ± 0.90 | 3.76 ± 0.29 |
| Tumor : blood | 1.11 ± 0.16 | 0.78 ± 0.07 |
| Tumor : kidney | 0.30 ± 0.04 | 0.19 ± 0.07 |
Discussion
This work is the first report of dT-AMBF3 conjugates that are 18F-labeled in a single step by IEX. Primarily, we rationalized the design of 1 and 2 based on antecedent reports of functionalized dTs. In regards to designing 1, we followed literature reports26 describing ready access of 5-azido-dT (5). Notably, 5 has found important applications for intracellular uptake and DNA incorporation and thus compelled us to examine applications of a duly AMBF3-conjugated precursor that is formed by CuAAC reaction.29 In designing 2, we appreciated that dT and thia-dT had been conjugated at N3 with an alkyne for Cu-mediated cycloaddition with [18F]fluorethylazide to give triazolyl-linked dTs.30 Similarly, dT had been conjugated at N3 to an alkyl-isocyanate or MAMA chelator for 99mTc-labeling6 or to a DOTA-linker for labeling with 68Ga or 111In.7b In the case of the 18F-labeled triazolyl-dT analogs, these proved to be poor substrates for thymidine kinase. In the case of 99mTc-labeled dT conjugates, by 60 min p.i., tumor uptake fell to <1 %ID g–1. Similarly, the 68Ga-DOTA-labeled dT (also conjugated at N3), showed little uptake in lymphoma cells. Notwithstanding the lack of uptake with these antecedent N3-conjugated radiometallated chelates, it is noteworthy that sterically demanding carboranes have also been conjugated to N3 for use as candidate therapeutics for boron neutron capture therapy (BNCT).31 Hence, we felt that it would be worthwhile to investigate an N3-conjugated AMBF3. As these metal chelates are zwitterions, we postulated that the somewhat smaller zwitterionic AMBF3 radioprosthetic group might have provided more encouraging results. Taken together, the above formed the rationale for which we investigated 1 and 2 in terms of 18F-labeling and in vivo activity.
In both cases, we performed well-established radiolabeling via IEX. In terms of radiosynthesis, [18F]1 was obtained in a higher isolated yield than [18F]2 because of better sample recovery from Sep-Pak purification likely owing to its relatively greater hydrophobicity reflected by log P. Nevertheless, the apparent RCY of [18F]2 is higher than that of [18F]1 when calculated from the crude HPLC trace. In both cases there was considerable variation in terms of RCY. Whereas an explanation for such variation is not immediately forthcoming, such observation cohere with previous reports on certain tracers from our lab. We suspect that aggregates may form upon concentration in vacuo during the IEX reaction. If such were to happen, this may lead to irreproducible and/or depressed yields as well as Am values. Of note, other peptide–AMBF3 conjugates are labeled in higher RCYs with less variability; here we caution that these radiosyntheses are currently unoptimized and work is ongoing to address the nature of this variability.
It is worth noting that in the case of IEX reactions, the use of a large molar excess of precursor (in this case natF present in 1 and 2) still exceeds the considerable amounts of [19F]fluoride, which commonly contaminates all preparations of NCA [18F]fluoride, caps Am values to 740 GBq μmol–1 or less.32 Hence, the source of such error is unlikely to be due to variation in the Am value of the NCA [18F]fluoride, but rather the possibility that the IEX reaction may not have reached total completion. While it recognized that IEX reactions are necessarily carrier-added labeling reactions (carrier is added in the form of the AMBF3-precursor), nevertheless, Am values are still found to be high by most standards. For instance, in comparison to reports of multi-step labeling of [18F]FLT that require 90–140 min,33 radiosynthesis here is complete in 40 min, and Am and RCY values obtained are competitive as if not higher than, values obtained for [18F]FLT. Notwithstanding these concerns, IEX on 1 and 2 enables one-step access to corresponding radiotracers. Both were highly polar with log P values less than –3. Such polarity likely explains the considerable accumulation in bladder and very rapid clearance such that there is little accumulation elsewhere by 60 min p.i.
Preliminary PET images showed differential uptake of [18F]1 over [18F]2 when examined with large tumors (ca. 10 mm in diameter). While images suggest that there is more tumor uptake with [18F]1 than with [18F]2, uptake was not confluent in larger tumors, which defies explanation. Unfortunately, images acquired with [18F]1 were discordant with the biodistribution data that were acquired using mice with considerably smaller tumors. An explanation for this discordance is not immediately forthcoming although larger tumors are expected to contain necrotic regions that may cause tracer to accumulate therein giving the appearance of uptake. While the uptake values are low for both tracers, tumor : muscle and tumor : bone ratios were consistent with other reports on labeled nucleosides. Ideally there should be uniform uptake of [18F]1 or [18F]2, but what we observed was a size dependent uptake.
Low tumor uptake for [18F]2 is consistent with previous reports of dTs labeled with 68Ga and 99mTc, all of which are lower than the current standard imaging agent [18F]FLT. These low uptake values are somewhat surprising since carboranes have been conjugated the N3 position and these conjugates showed promising action for BNCT.31,34 In general, sterically demanding prosthetic groups are conjugated to N3 as it is thought that TK1 will more readily accept these sterically encumbered dTs. In contrast, conjugation to C5 would likely interfere with the TK's ability to phosphorylate the nucleoside, a processing even that would be needed to ensure retention following uptake. Although [18F]1 appeared to show somewhat greater tumor uptake, it is unlikely that uptake is mediated by ENTs and may be more attributable to passive diffusion or otherwise an unknown uptake pathway. It is currently unknown whether [18F]1 or [18F]2 is being phosphorylated in cells. In our hands, an in vitro assay of phosphorylation with [32P]-γ-ATP showed minimal phosphorylation at early time-points but not continued phosphorylation (data not shown). Such would be consistent with other 18F-labeled dT analogs, which are shown to be poor substrates for TK.30 Although blocking was unsuccessful, it is not surprising as other studies similarly failed to show significant blockable uptake of other 18F- and 99mTc-labeled tracers and would suggest that any observed uptake is likely due to non-specific processes or passive diffusion.
Whereas tumor uptake values are low, this work features facile, one-step radiosynthesis with reasonable yields at high Am. Despite the relative convenience of this labeling method compared to the synthesis of [18F]FLT, images are inferior to those obtained with [18F]FLT. Taken together, the imaging and biodistribution results are consistent with other radiometallated dTs and despite the radiosynthetic ease, the in vivo data highlight the challenges associated with developing imaging agents that comprise both ease-of-radiosynthesis and high tumor uptake. It is suggested that other sites of modification and/or alternate linkers may improve image quality and tumor uptake while providing for a robust labeling manifold.
Conclusions
We report on two novel dT conjugates for 18F-labeling and their application to PET imaging. The application of 7 along with CuAAC conjugation represent two attractive synthetic routes for developing new 18F-labeled nucleoside conjugates. This work validates the use of the AMBF3 prosthetic group with dT for labeling in good yield (>25%) and at high Am (>100 GBq μmol–1). Given the large number of nucleosides that have been investigated for diagnosis, therapy and further applications in synthetic biology, this work demonstrates facile 18F-labeling of a new class of nucleoside conjugates that may find use in imaging aspects of nucleoside uptake by PET. It is anticipated that this work paves the way for synthesizing other AMBF3–nucleoside conjugates for use in PET.
Experimental section
Chemicals and reagents
5-Azidomethyl-2′-deoxyuridine 5 was synthesized according to literature procedure.26 Concentrated ammonium hydroxide and sodium chloride were purchased from BDH Chemicals. Anhydrous copper (ii) sulfate, formic acid, potassium carbonate, 2-propanol, concentrated hydrochloric acid, sodium iodide were purchased from Fisher Scientific. 3-Chloropropyldimethylamine hydrochloride 6, acetonitrile, dichloromethane, diethyl ether, N,N-dimethylformamide, N,N-dimethylpropargylamine 3, methanol, thymidine 8 and trifluoroacetic acid were purchased from Sigma-Aldrich. Iodomethylboronic acid pinacol ester was purchased from Frontier Scientific. Anhydrous magnesium sulfate and potassium hydroxide were purchased from Anachemica. Potassium bifluoride was purchased from Acros Organics. Sodium ascorbate was purchased from Spectrum Quality Products. Sep-Pak cartridges were purchased from Waters. Silver nitrate was purchased from Mallinckrodt Pharmaceuticals. Thin layer chromatography (TLC) were performed on precoated aluminum or glass-backed plates containing Silica Gel 60 F254 from EMD Chemicals. Chemical preparative HPLC purification was performed on Agilent 1100 series with Agilent Eclipse XDB-C18 5 mm 9.2 × 250 mm semipreparative column. Conditions for radiochemical HPLC purification can be found in the radiosynthesis section. N,N-Dimethylformamide was distilled under reduced pressure and stored in 3 Å molecular sieves under Ar. The storage vessel was degassed by a water aspirator for 1 h prior to usage. Acetone was dried with anhydrous Na2SO4 or MgSO4 prior to use. Unless otherwise stated, other chemicals and solvents were used and stored under manufacturer guidelines. 1H, 11B, 13C and 19F 300–600 MHz NMR were taken by Bruker Avance series NMR spectra are available in the ESI.† LCMS spectra were taken by Waters 2695 Separation module and Waters-Micromass ZQ.
5-Hydroxymethyl-2′-deoxyuridine
The synthesis was adapted from previous literature.12 2.26 g of white crystals were synthesized in 58% yield. Rf = 0.18 (43 : 7 CH2Cl2 : MeOH); 1H NMR (300 MHz, CD3OD) δ (ppm): 7.96 (s, 1H), 6.30 (t, J = 6.0 Hz, 1H), 4.38–4.42 (m, 1H), 4.32 (s, 2H), 3.93 (s, J = 3.0, 1H), 3.76 (qd, J1 = 3.0, J2 = 9.0, 1H), 2.17–2.31 (m, 2H); 13C NMR (75 MHz, CD3OD) δ (ppm): 165.15, 152.25, 139.43, 115.28, 88.92, 86.50, 72.29, 62.92, 57.98, 41.26; LCMS [M + Na]+, calcd. 281.2, measured 281.2.
5-Azidomethyl-2′-deoxyuridine, 5
The synthesis was adapted from previous literature.26 291 mg of yellow oil was synthesized in 61% yield. Rf = 0.32 (1 : 1 CH2Cl2 : acetone); 1H NMR (300 MHz, CD3OD) δ (ppm): 8.13 (s, 1H), 6.26 (t, J = 6.0 Hz, 1H), 4.39–4.43 (m, 1H), 4.09 (s, 2H), 3.95 (s, J = 3.0, 1H), 3.77 (qd, J1 = 3.0, J2 = 9.0, 2H), 2.18–2.36 (m, 2H); 13C NMR (75 MHz, CD3OD) δ (ppm): 165.14, 152.03, 141.34, 110.35, 89.01, 86.70, 72.05, 62.80, 48.29, 41.48; LCMS [M – H]–, calcd. 282.2, measured 282.2.
N-((Difluoroboraneyl)methyl)-N,N-dimethylprop-2-yn-1-aminium fluoride, 4
Diethyl ether (37 mL), N,N-dimethylprop-2-yl-1-amine (3, 1.12 g, 1.45 mL, 13.44 mmol, 2.0 eq.) and 2-(iodomethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.8 g, 1.2 mL, 6.7 mmol, limiting) were charged into a 50 mL Falcon tube and was allowed to react for 30 min with occasional stirring by inverting the tube 5 times in 10 minute intervals. Glass vessels must be avoided during acidic fluorination in order to prevent etching in the later step. The white solid was filtered and air dried for 5 minutes and directly used for fluorination. Assuming quantitative yield during alkylation, the quaternary ammonium (9, 13.44 mmol, limiting), 4 M KHF2 (6.95 mL, 27.82 mmol, 4.14 eq.) and 4 M HCl (5.38 mL, 21.50 mmol, 3.20 eq.) were introduced to a 50 mL Falcon tube and was allowed to react for 75 min with occasional stirring by inverting the tube 5 times in 10 minute intervals. The reaction was quenched by concentrated NH4OH until the pH reaches 8 and lyophilized to give a white powder. The powder was resuspended in minimal amount of water and injected into a C18 Sep-Pak cartridge. 6 column volumes (“CV”) of H2O were used to wash the column, followed by 4 CVs of 10% MeCN/H2O to recover AMBF3 from the cartridge. Silver nitrate (1.26 g, 14.78 mmol, 1.1 eq.) was added to the pooled eluents, and subsequently vortexing to give a yellow precipitate. Sodium chloride (0.862 g, 14.78 mmol, 1.1 eq.) was then introduced to the mixture, followed by vortexing and sonication to give more white precipitates. The mixture was filtered through a coarse glass frit to give a colorless solution. The solvent was removed in vacuo, followed by 3 × 100 mL MeCN extraction. The solvent was removed in vacuo and resuspended in water. The aqueous solution was lyophilized to yield 1.19 g AMBF34 (Quantitative yield over 2 steps) as a fine white powder. Rf = 0.70 (100% acetone); 1H NMR (300 MHz, CD3CN) δ (ppm): 4.10 (d, 2H, J = 3 Hz), 3.08–3.10 (m, 7H), 2.45 (br, 7H); 11B NMR (96 MHz, CD3CN) δ (ppm): 1.57; 13C NMR (75 MHz, CD3CN) δ (ppm): 81.07, 73.62, 57.32, 53.49; 19F NMR (282 MHz, CD3CN) δ (ppm): 139.16 (1 : 1 : 1 : 1, J = 54 Hz); LCMS [M + Na]+, calcd. 183.1, measured 183.3.
(((3-Chloropropyl)dimethylammonio)methyl)trifluoroborate, 7
3-Chloropropyldimethylamine hydrochloride (6, 227.8 mg, 1.44 mmol, 1.07 equiv.) was suspended in H2O (2.5 mL) in a 15 mL Falcon tube. Glass vessels must be avoided during acidic fluorination in order to prevent etching in the later step. The tube was chilled in 1 : 1 EtOH : H2O dry ice bath, and Et2O (4 mL) and KOH (424.4 mg, 7.56 mol, 5.59 eq.) were introduced, followed by vortexing. The layers were separated and the aqueous layer was extracted with Et2O (4 mL). The combined organic layer was washed with H2O (2.5 mL × 1) and brine (2.5 mL × 1) in a 30 mL separatory funnel. The organic phase was dried over anhydrous Na2SO4. The Et2O filtrate was topped up to 22 mL, followed by introduction of 2-(iodomethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (250 μL, 362.5 mg, 1.35 mmol, limiting reagent) and a Teflon stir bar. The solvent is removed after 30 minutes to give a viscous yellow oil, and the crude mixture was resuspended, in order, H2O (1 mL), 3 M KHF2 (1.45 mL, 340 mg, 4.35 mmol, 3.21 eq.), 4 M HCl (1.45 mL, 5.80 μmol, 4.29 eq.), and allowed to fluorinate for 2 hours. The mixture was neutralized to pH 8 by concentrated NH4OH, followed by lyophilization to give an off-white powder. The crude was resuspended in minimal amount of water, followed by 3 M AgNO3 (7.3 mL, 3.72 g, 21.90 μmol, 16.2 equiv.) and then 6 M NaCl (1.00 mL, 351 mg, 6.00 μmol, 4.43 equiv.) to give a yellow-grey precipitate. After centrifugation, the supernatant was lyophilized to give a white powder, followed by extraction with MeCN (100 mL × 3). The filtrate was dried and resuspended in minimal amount of water for C18 Sep-Pak cartridge injection. The cartridge was washed with 6 column volumes (“CV”) of H2O, followed by 4 CVs of 10% MeCN/H2O. 10 CVs of MeCN was used to recover product 7 from the cartridge in the 100% MeCN fractions as a colorless oil (124.1 mg, 610 μmol, 45%). TLC (9 : 1 CH2Cl2 : acetone): Rf = 0.40; 1H NMR (300 MHz, CD3CN) δ (ppm): 3.64 (t, 2H), 3.31–3.36 (m, 2H), 2.98 (s, 6H), 2.33 (br, 2H), 2.13–2.26 (m, 2H); 11B NMR (96 MHz, CD3CN) δ (ppm): 1.61 (J = 159 Hz); 13C NMR (75 MHz, CD3CN) δ (ppm): 65.17, 54.14, 42.75, 26.90; 19F NMR (282 MHz, CD3CN) δ (ppm): –139.58. (J = 54 Hz). HRMS ESI-TOF, [M + Na]+, [C6H1410B35ClF3NNa]+, calcd. for 225.0977; measured 225.0797.
Trifluoro((((1-((1-((2R,4S,5R)-4-hydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)dimethylammonio) methyl)borate, dT-C5-AMBF3, 1
Alkyne 4 (5.39 mg, 32.7 μmol, 3.08 eq.) was suspended in 3 : 2 CH3CN : H2O (53.0 μL). 32.8 μL 1 M Cu(ii)SO4(aq) (32.8 μmol, 3.10 eq.) followed by 65.7 μL 1 M sodium ascorbate (65.7 μmol, 6.20 eq.) were introduced into the reaction vessel. 3.00 mg of azide 5 (10.6 μmol, 1 eq.) was then added to the solution and immediately neutralized to pH 7 by 2.48 mg of K2CO3 (17.9 μmol, 1.69 eq.). The mixture was stirred for 15.3 h at 45 °C. The mixture was quenched by concentrated NH4OH in excess, followed by acidification by 0.1% formic acid to pH 3. The crude reaction was preliminarily purified by a Sep-Pak column in 0–100% MeCN : H2O. Subsequent HPLC purification (solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in MeCN; 0–18 min, 0–50% B; 18–21 min, 50–100% B; 21–26 min, 100% B; 26–31 min, 100–0% B; 31–36 min, 0% B; flow rate, 2.0 mL min–1; column temperature, 19–21 °C) afforded triazole 1 as an white-brown lyophilized powder (1.07 mg, 2.39 μmol, 23%). TLC (7 NH4OH : 11 iPrOH : 2 H2O): Rf = 0.67; 1H NMR (600 MHz, CD3CN) δ (ppm): 9.08 (br, 1H), 8.11 (s, 1H), 8.10 (s, 1H), 6.14 (t, 1H), 5.23 (q, 2H), 4.44 (s, 2H), 4.33–4.35 (m, 2H), 3.87 (dd, J = 4 Hz, 1H), 3.70 (tq, J = 12, 6 Hz, 2H), 3.35 (d, 1H), 3.17 (dd, J = 6 Hz, 1H), 2.95 (d, 6H), 2.23–2.29 (m, 3H), 1.11 (t, 1H). 13C NMR (151 MHz, CD3CN) δ (ppm): 163.42, 151.01, 142.13, 137.36, 128.90, 108.76, 88.50, 86.43, 71.45, 62.31, 61.51, 53.46, 47.69, 41.26. 19F NMR (282 MHz, CD3CN) δ (ppm): –138.66 (1 : 1 : 1 : 1, J = 60 Hz). HRMS ESI-TOF: [M + Na]+, [C16H12BF3N6NaO5]+ calcd. 470.1787, measured 470.1786; retention time: 11.5 min; λmax = 264 nm.
Trifluoro(((3-(3-((2R,4S,5R)-4-hydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-5-methyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl)propyl)dimethylammonio)methyl)borate, dT-N3-AMBF3, 2
3-Chloropropyldimethylammoniomethylenetrifluoroborate (7, 116.79 mg, 574.08 μmol, 33.2 eq.) was suspended in acetone (4 mL). Sodium iodide (91.21 mg, 608.52 μmol, 35.2 eq.) was introduced to the solution and allowed to reflux for 17 hours. The crude product was subjected to gravitational filtration and solvent removal in vacuo, and then resuspended in 250 μL acetone. In a separate flask, thymidine (5, 4.20 mg, 17.3 μmol, limiting agent), potassium carbonate (7.91 mg, 57.2 μmol, 3.30 eq.) was dissolved under Ar to give a colorless solution. The crude 3-iodopropyldimethyl-ammonio-methylenetrifluoroborate in acetone was cannulated into the colorless solution and was refluxed for 6 hours. The solvent was removed and resuspended in 1 mL water. It was transferred to a 15 mL Falcon tube and, in order, 1 M silver nitrate (300 μL, 300 μmol, 17.3 eq.) and brine (33 μL) were introduced to give a pale yellow precipitate. After centrifugation, the supernatant was lyophilized and then washed with MeCN (5 mL) three times. The white solids were resuspended in water and resolved in a C18 purification cartridge (H2O/MeCN), followed by preparative TLC (9 : 1 DCM : MeOH) to give the desired product 2 as a white powder (6.6 mg, 96%). 2 mg of 2 was further resolved by HPLC for radiolabeling studies (solvent A, 0.1% trifluoroacetic acid (TFA) in MilliQ-filtered H2O; solvent B, 0.1% TFA in MeCN; 0–15 min, 0–42% B; 15–16 min, 42–100% B; 16–21 min, 100% B; 21–22 min, 100–0% B; 22–28 min, 0% B; flow rate, 2.0 mL min–1; column temperature, 19–21 °C). TLC (2 iPrOH : 1 NH4OH : 1 H2O): Rf = 0.13; 1H NMR (300 MHz, CD3OD) δ (ppm): 7.86 (d, J = 1 Hz, 1H), 6.29 (t, J = 7 Hz, 1H), 4.39 (dt, J = 6.1, 3.8 Hz, 1H), 4.02 (td, J = 6.5, 1.4 Hz, 2H), 3.92 (q, J = 3.5 Hz, 1H), 3.77 (qd, J = 12.0, 3.6 Hz, 2H), 3.35 (s, 1H), 3.02 (s, 6H), 2.30–2.39 (m, 2H), 2.21–2.29 (m, 2H), 2.07–2.16 (m, 2H), 1.92 (d, J = 1 Hz, 3H); 11B NMR (96 MHz, CD3OD) δ (ppm): 1.55; 13C NMR (75 MHz, CD3OD) δ (ppm): 165.58, 152.42, 136.69, 110.65, 88.89, 87.27, 71.98, 65.21, 62.72, 54.14, 41.35, 39.28, 22.94, 13.17; 19F NMR (282 MHz, CD3OD) δ (ppm): –141.47; HRMS ESI-TOF, [M + Na]+, [C16H2710BF3NaO5]+, calcd. for 431.1924; measured 431.1931; λmax = 264 nm.
Radiochemistry and pharmacology
Radiosynthesis
The radiolabeling procedure was adapted from previous work;10 37–55 GBq [18F]fluoride in water was retained by a QMA trap and then eluted with saline (100 μL) into the radiolabeling vessel containing 90–100 nmol of precursor 1 or 2 dissolved in 1 : 1 1 M pyridazine hydrochloride in water : DMF (15–30 μL, pH 2.5). The mixture was incubated on a 80 °C sand bath for 10 min and the solvent was removed in vacuo for 10–15 min at 80 °C. The reaction residue was quantitatively transferred by 1.5 mL 5% NH4OH (v/v) for HPLC purification with method A or B. Analytical traces were run by method C or D. Batch numbers of [18O]-H2O used to generate [19F]fluoride can be found in ESI.† Method A (semi-preparative) for 1: Phenomenex Luna C18 semi-preparative column (5 μm, 100 Å, 250 × 10 mm). Solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in MeCN; 0–8 min, 5% B; 8–18 min, 10% B; 18–28 min, 25% B; flow rate, 4.5 mL min–1. Method B (semi-preparative) for 2: solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in MeCN; 0–14 min, 5% B; 14–28 min, 25% B; 28–38 min, 50% B; flow rate, 4.5 mL min–1. Method C (analytical) for 1: Agilent HPLC systems 1200 series equipped with Phenomenex Jupiter C18 analytical column (10 μm, 300 Å, 250 × 4.6 mm). Solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in MeCN; 0–10 min, 0–10% B; 10–11 min, 10–100% B; 11–14 min, 100% B; 14–15 min, 100–0% B; 15–18 min, 0% B; flow rate, 2.0 mL min–1. Method D (analytical) for 2: solvent A, 0.1% trifluoroacetic acid (TFA) in water; solvent B, 0.1% TFA in MeCN; 0–9 min, 0–20% B; 9–10 min, 20–100% B; 10–15 min, 100% B; 15–16 min, 100–0% B; 16–22 min, 0% B; flow rate, 2.0 mL min–1. Radiochemical yield RCY was calculated using the integrated area of free fluoride I18F– and that of radiolabeled compound 1 or 2Ic by 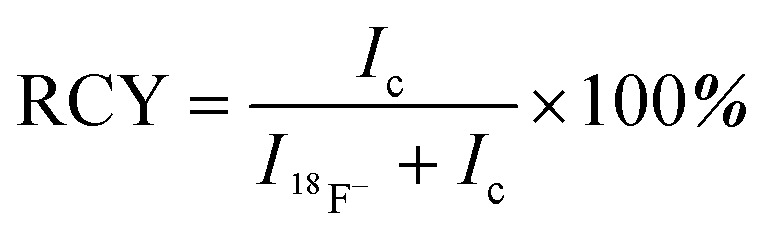 .
.
Cell culture
The U87 cell line was obtained commercially from ATCC (HTB-14), and confirmed pathogen-free by the IMPACT1 test (IDEXX BioResearch). The cells were cultured in RPMI 1640 medium (StemCell Technologies), supplemented with 10% FBS, 100 U mL–1 penicillin and 100 μg mL–1 streptomycin at 37 °C in a humidified incubator supplied with 5% CO2.
Tumor inoculation
All animal experiments were conducted according to the guidelines established by Canadian Council on Animal Care and approved by Animal Ethics Committee of the University of British Columbia. Male immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were acquired from an in-house breeding colony at the BC Cancer Research Centre. For tumor inoculation, mice were sedated by inhalation with 2% isoflurane in 2.0 L min–1 O2, 5 million U87 cells were inoculated subcutaneously at the right dorsal flank.
PET/CT imaging and biodistribution studies
PET/CT imaging and biodistribution studies were performed according to previously published procedures.11 Briefly, PET/CT imaging was performed on a μPET/CT scanner (Inveon, Siemens). The tumor-bearing mice were injected with 4–6 MBq of the 18F-labeling compound via tail vein. After injection, the mice were allowed to recover and roam in their cages. Upon reaching the time point, the mice were sedated again and positioned on the scanner for a baseline CT scan followed by a 10 min static PET scan. For biodistribution studies, the tumor-bearing mice were injected with 1–2 MBq of the 18F-labeling compound as described above. At 1 h post-injection, the mice were euthanized by CO2 inhalation, blood was promptly withdrawn, and the organs of interest were harvested and weighed. The radioactivity was measured on a WIZARD 2480 gamma counter (PerkinElmer).
In vitro uptake assay
In vitro uptake assays were performed using U87 cells; 50 000 cells per well were seeded onto a 24 well poly-d-lysine coated plate (Corning) four days before the experiment and allowed to grow to confluence. Growth media was removed, and reaction buffer containing 4.8 mg mL–1 HEPES, 100 U mL–1 penicillin, 1000 μg mL–1 streptavidin and 2 mg mL–1 BSA was added, and allowed to incubate with cells at 37 °C for at least an hour. Approximately 500 kcpm of [18F]-radiotracer was added to each well, without or with 0.1, 1, 10, 100 and 1000 μM of thymidine (n = 4). The reaction mixture was incubated at 37 °C with gentle agitation for 1 h. After the incubation, the reaction mixture was removed, and cells were washed with ice-cold PBS twice. 0.25% trypsin solution was used to harvest the cells and radioactivity was measured on a WIZARD 2480 gamma counter (PerkinElmer).
log P7.4
10 μL (ca. 40 μCi) of the tracer was charged into a 15 mL Falcon tube containing PBS (pH 7.4, 3.0 mL) and n-octanol (3.0 mL). With a 1 minute pause in between, the tube was vortexed for 20 s twice, and then phase separated by centrifugation at 3 krpm. Aliquots from each layer, 2 mL from n-octanol and 50 μL from PBS, were used for scintillation counting by a NaI scintillation detector (Bioscan). After volumetric adjustment, log P7.4 was evaluated by the activity in n-octanol phase, ao, and PBS phase, ap, by 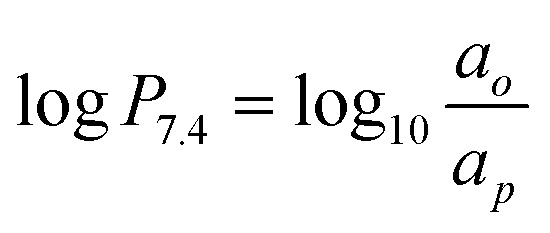 .
.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This work was funded by Natural Sciences and Engineering Research Council (NSERC). AAWLW was a recipient of NSERC-Undergraduate Student Research Awards (NSERC-USRA).
Footnotes
†Electronic supplementary information (ESI) available. See DOI:10.1039/d0md00054j
References
- (a) Svobodova S., Topolcan O., Holubec L., Treska V., Sutnar A., Rupert K., Kormunda S., Rousarova M., Finek J. Anticancer Res. 2007;27:1907–1909. [PubMed] [Google Scholar]; (b) Lee W. C., Chang C. H., Ho C. L., Chen L. C., Wu Y. H., Chen J. T., Wang Y. L., Lee T. W. J. Biomed. Biotechnol. 2011;7:535902. doi: 10.1155/2011/535902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolayirli M., Papila C., Korkmaz G. G., Papila B., Aydogan F., Karatas A., Uzun H. J. Clin. Lab. Anal. 2013;27:220–226. doi: 10.1002/jcla.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields A. F., Grierson J. R., Dohmen B. M., Machulla H. J., Stayanoff J. C., Lawhorn-Crews J. M., Obradovich J. E., Muzik O., Mangner T. J. Nat. Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- Plotnik D. A., Emerick L. E., Krohn K. A., Unadkat J. D., Schwartz J. L. J. Nucl. Med. 2010;51:1464–1471. doi: 10.2967/jnumed.110.076794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. H., Huang S. Q., Wang Y. D., Yang A. Z., Wen J., Xu X. H., Chen Y., Chen Q. B., Wang Y. H., He E. L., Zhou J., Skog S. Sensors. 2011;11:11064–11080. doi: 10.3390/s111211064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Duan X. J., Zhang X. R., Gan Q. Q., Fang S. A., Ruan Q., Song X. Q., Zhang J. B. MedChemComm. 2018;9:705–712. doi: 10.1039/c7md00635g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Celen S., de Groot T., Balzarini J., Vunckx K., Terwinghe C., Vermaelen P., Van Berckelaer L., Vanbilloen H., Nuyts J., Mortelmans L., Verbruggen A., Bormans G. Nucl. Med. Biol. 2007;34:283–291. doi: 10.1016/j.nucmedbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- (a) Pulido J., de Cabrera M., Sobczak A. J., Amor-Coarasa A., McGoron A. J., Wnuk S. F. Bioorg. Med. Chem. 2018;26:5624–5630. doi: 10.1016/j.bmc.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmid M., Neumaier B., Vogg A. T. J., Wczasek K., Friesen C., Mottaghy F. M., Buck A. K., Reske S. N. Nucl. Med. Biol. 2006;33:359–366. doi: 10.1016/j.nucmedbio.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Ghosh P., Gelovani J. G., Alauddin M. M. J. Labelled Compd. Radiopharm. 2007;50:1185–1191. [Google Scholar]
- (a) Rahmim A., Zaidi H. Nucl. Med. Commun. 2008;29:193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]; (b) Hicks R. J., Hofman M. S. Nat. Rev. Clin. Oncol. 2012;9:712–720. doi: 10.1038/nrclinonc.2012.188. [DOI] [PubMed] [Google Scholar]
- (a) Kemerink G. J., Visser M. G. W., Franssen R., Beijer E., Zamburlini M., Halders S., Brans B., Mottaghy F. M., Teule G. J. J. Eur. J. Nucl. Med. Mol. Imaging. 2011;38:940–948. doi: 10.1007/s00259-011-1732-1. [DOI] [PubMed] [Google Scholar]; (b) Sanchez-Crespo A. Appl. Radiat. Isot. 2013;76:55–62. doi: 10.1016/j.apradiso.2012.06.034. [DOI] [PubMed] [Google Scholar]
- Eberl S., Eriksson T., Svedberg O., Norling J., Henderson D., Lam P., Fulham M. Appl. Radiat. Isot. 2012;70:922–930. doi: 10.1016/j.apradiso.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Grierson J. R., Shields A. F. Nucl. Med. Biol. 2000;27:143–156. doi: 10.1016/s0969-8051(99)00104-3. [DOI] [PubMed] [Google Scholar]
- (a) Bernard-Gauthier V., Lepage M. L., Waengler B., Bailey J. J., Liang S. H., Perrin D. M., Vasdev N., Schirrmacher R. J. Nucl. Med. 2018;59:568–572. doi: 10.2967/jnumed.117.197095. [DOI] [PubMed] [Google Scholar]; (b) Cai H. C., Conti P. S. J. Labelled Compd. Radiopharm. 2013;56:264–279. doi: 10.1002/jlcr.2999. [DOI] [PubMed] [Google Scholar]
- (a) Schirrmacher R., Bradtmoller G., Schirrmacher E., Thews O., Tillmanns J., Siessmeier T., Buchholz H. G., Bartenstein P., Waengler B., Niemeyer C. M., Jurkschat K. Angew. Chem., Int. Ed. 2006;45:6047–6050. doi: 10.1002/anie.200600795. [DOI] [PubMed] [Google Scholar]; (b) Schirrmacher E., Wangler B., Cypryk M., Bradtmoller G., Schafer M., Eisenhut M., Jurkschat K., Schirrmacher R. Bioconjugate Chem. 2007;18:2085–2089. doi: 10.1021/bc700195y. [DOI] [PubMed] [Google Scholar]; (c) Wangler B., Quandt G., Iovkova L., Schirrmacher E., Wangler C., Boening G., Hacker M., Schmoeckel M., Jurkschat K., Bartenstein P., Schirrmacher R. Bioconjugate Chem. 2009;20:317–321. doi: 10.1021/bc800413g. [DOI] [PubMed] [Google Scholar]
- (a) Schulz J., Vimont D., Bordenave T., James D., Escudier J. M., Allard M., Szlosek-Pinaud M., Fouquet E. Chem. – Eur. J. 2011;17:3096–3100. doi: 10.1002/chem.201003234. [DOI] [PubMed] [Google Scholar]; (b) Gonzalez C., Sanchez A., Collins J., Lisova K., Lee J. T., van Dam R. M., Barbieri M. A., Ramachandran C., Wnuk S. F. Eur. J. Med. Chem. 2018;148:314–324. doi: 10.1016/j.ejmech.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Liu Z., Li Y., Lozada J., Lin K.-S., Schaffer P., Perrin D. M. J. Labelled Compd. Radiopharm. 2012;14:491–497. [Google Scholar]; (b) Liu Z., Li Y., Lozada J., Wong M. Q., Greene J., Lin K.-S., Yapp D., Perrin D. M. Nucl. Med. Biol. 2013;40:841–849. doi: 10.1016/j.nucmedbio.2013.05.002. [DOI] [PubMed] [Google Scholar]; (c) Liu Z. B., Li Y., Lozada J., Schaffer P., Adam M. J., Ruth T. J., Perrin D. M. Angew. Chem., Int. Ed. 2013;52:2303–2307. doi: 10.1002/anie.201208551. [DOI] [PubMed] [Google Scholar]
- Liu Z. B., Pourghiasian M., Radtke M. A., Lau J., Pan J. H., Dias G. M., Yapp D., Lin K. S., Benard F., Perrin D. M. Angew. Chem., Int. Ed. 2014;53:11876–11880. doi: 10.1002/anie.201406258. [DOI] [PubMed] [Google Scholar]
- Roxin A., Zhang C. C., Huh S., Lepage M., Zhang Z. X., Lin K. S., Benard F., Perrin D. M. Bioconjugate Chem. 2019;30:1210–1219. doi: 10.1021/acs.bioconjchem.9b00146. [DOI] [PubMed] [Google Scholar]
- Liu Z. B., Pourghiasian M., Benard F., Pan J. H., Lin K. S., Perrin D. M. J. Nucl. Med. 2014;55:1499–1505. doi: 10.2967/jnumed.114.137836. [DOI] [PubMed] [Google Scholar]
- Pourghiasian M., Liu Z. B., Pan J. H., Zhang Z. X., Colpo N., Lin K. S., Perrin D. M., Benard F. Bioorg. Med. Chem. 2015;23:1500–1506. doi: 10.1016/j.bmc.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Zhang C. C., Zhang Z. X., Merkens H., Zeisler J., Colpo N., Hundal-Jabal N., Perrin D. M., Lin K. S., Benard F. Sci. Rep. 2019;9:10. doi: 10.1038/s41598-019-50014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo H. T., Lepage M. L., Lin K. S., Pan J. H., Zhang Z. X., Liu Z. B., Pryyma A., Zhang C. C., Merkens H., Roxin A., Perrin D. M., Benard F. J. Nucl. Med. 2019;60:1160–1166. doi: 10.2967/jnumed.118.216598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes P. S. G., Zhang Z. X., Kuo H. T., Zhang C. C., Rousseau J., Rousseau E., Lau J., Kwon D., Carvalho I., Benard F., Lin K. S. J. Labelled Compd. Radiopharm. 2018;61:370–379. doi: 10.1002/jlcr.3594. [DOI] [PubMed] [Google Scholar]
- Lau J., Liu Z. B., Lin K. S., Pan J. H., Zhang Z. X., Vullo D., Supuran C. T., Perrin D. M., Benard F. J. Nucl. Med. 2015;56:1434–1440. doi: 10.2967/jnumed.114.153288. [DOI] [PubMed] [Google Scholar]
- Zhang Z. X., Jenni S., Zhang C. C., Merkens H., Lau J., Liu Z. B., Perrin D. M., Benard F., Lin K. S. Bioorg. Med. Chem. Lett. 2016;26:1675–1679. doi: 10.1016/j.bmcl.2016.02.062. [DOI] [PubMed] [Google Scholar]
- Xu X. W., Yan S. Y., Hu J. L., Guo P., Wei L., Weng X. C., Zhou X. Tetrahedron. 2013;69:9870–9874. [Google Scholar]
- Zhang C., Zhang Z., Lin K.-S., Lau J., Zeisler J., Colpo N., Perrin D. M., Benard F. Mol. Pharmaceutics. 2018;15:2116–2122. doi: 10.1021/acs.molpharmaceut.7b01113. [DOI] [PubMed] [Google Scholar]
- Alexander S., Varaha S. T., John G., Vesselle H. Semin. Nucl. Med. 2007;37:429–439. doi: 10.1053/j.semnuclmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- (a) Tera M., Glasauer S. M. K., Luedtke N. W. ChemBioChem. 2018;19:1939–1943. doi: 10.1002/cbic.201800351. [DOI] [PubMed] [Google Scholar]; (b) Neef A. B., Luedtke N. W. ChemBioChem. 2014;15:789–793. doi: 10.1002/cbic.201400037. [DOI] [PubMed] [Google Scholar]
- Smith G., Sala R., Carroll L., Behan K., Glaser M., Robins E., Nguyen Q. D., Aboagye E. O. Nucl. Med. Biol. 2012;39:652–665. doi: 10.1016/j.nucmedbio.2011.12.002. [DOI] [PubMed] [Google Scholar]
- (a) Al-Madhoun A. S., Johnsamuel J., Barth R. F., Tjarks W., Eriksson S. Cancer Res. 2004;64:6280–6286. doi: 10.1158/0008-5472.CAN-04-0197. [DOI] [PubMed] [Google Scholar]; (b) Al-Madhoun A. S., Johnsamuel J., Yan J. H., Ji W. H., Wang J. H., Zhuo J. C., Lunato A. J., Woollard J. E., Hawk A. E., Cosquer G. Y., Blue T. E., Eriksson S., Tjarks W. J. Med. Chem. 2002;45:4018–4028. doi: 10.1021/jm020047q. [DOI] [PubMed] [Google Scholar]
- Bergman J., Eskola O., Lehikoinen P., Solin O. Appl. Radiat. Isot. 2001;54:927–933. doi: 10.1016/s0969-8043(00)00358-4. [DOI] [PubMed] [Google Scholar]
- Martin S. J., Eisenbarth J. A., Wagner-Utermann U., Mier W., Henze M., Pritzkow H., Haberkorn U., Eisenhut M. Nucl. Med. Biol. 2002;29:263–273. doi: 10.1016/s0969-8051(01)00289-x. [DOI] [PubMed] [Google Scholar]
- Agarwal H. K., Khalil A., Ishita K., Yang W. L., Nakkula R. J., Wu L. C., Ali T., Tiwari R., Byun Y., Barth R. F., Tjarks W. Eur. J. Med. Chem. 2015;100:197–209. doi: 10.1016/j.ejmech.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.