

Abstract
Purely structure-based pharmacophores (SBPs) are an alternative method to ligand-based approaches and have the advantage of describing the entire interaction capability of a binding pocket. Here, we present the development of SBPs for topoisomerase I, an anticancer target with an unusual ligand binding pocket consisting of protein and DNA atoms. Different approaches to cluster and select pharmacophore features are investigated, including hierarchical clustering and energy calculations. In addition, the performance of SBPs is evaluated retrospectively and compared to the performance of ligand- and complex-based pharmacophores. SBPs emerge as a valid method in virtual screening and a complementary approach to ligand-focussed methods. The study further reveals that the choice of pharmacophore feature clustering and selection methods has a large impact on the virtual screening hit lists. A prospective application of the SBPs in virtual screening reveals that they can be used successfully to identify novel topoisomerase inhibitors.
Keywords: Structure-based pharmacophores, LUDI, Virtual screening, DNA topoisomerase I
Introduction
DNA topoisomerases are enzymes involved in the relaxation of DNA torsional strain generated during replication, transcription, recombination, repair and chromosome condensation [1], and are therefore vital to all cells undergoing division. Due to their over-expression in tumour cells, topoisomerases are important targets in cancer chemotherapy. Topoisomerase I (Top1) generates transient DNA single-strand breaks and allows a controlled rotation of the open DNA strand to relax supercoiling. During this process, an intermediate covalent protein-DNA complex is formed which is particularly vulnerable to a group of inhibitors referred to as topoisomerase poisons [2]. Topoisomerase poisons trap the intermediate complex by interacting with both protein and DNA residues, and subsequently lead to cell death [3-5]. Currently, two Top1 poisons are used clinically, topotecan and irinotecan, both derivatives of the natural product camptothecin (CPT). However, their application is limited due to side-effects and the development of drug-resistance [6]. Therefore, the search for structurally novel Top1 poisons is ongoing.
Pharmacophore models represent the spatial arrangement of chemical features of a ligand that are necessary for binding to its target protein and have been successfully applied in drug discovery [7-9]. Due to the abstract nature of pharmacophores, virtual screening of compound databases enables the identification of structurally novel potential drugs [10]. The models can be developed either based on ligand information (ligand-based), information from protein–ligand complexes (complex-based) or based on protein structural information (structure-based). The concept of purely structure-based pharmacophores (SBPs) can be applied in virtual screening when a ligand-independent approach is desired. The main characteristic of this method is that, in contrast to ligand- or complex-based pharmacophores (CBPs), the pharmacophores describe the full interaction capability of the binding pocket, thus potentially allowing the identification of potential structurally novel ligands, capable of forming novel, as yet unexploited interactions with the binding pocket.
The LUDI [11] program has been developed for de novo design of ligands starting from a protein cavity. The algorithm consists of three parts: the identification of potential interaction sites in the protein active site, the selection of molecular fragments that fit into potential interaction sites, and the search for linker fragments that enable merging of individual molecular fragments into one molecule. The calculation of potential interaction sites involves the identification of the following classes of interactions: hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), aliphatic and aromatic hydrophobic (HYD) interactions. Potential interactions are generally detected using a geometric rule-based method [12].
Apart from its use in de novo drug design, LUDI can also be used to develop SBPs which are constructed from identified potential interaction sites, as shown in several studies [13, 14]. Other approaches to develop SBPs utilising energetic calculations with the program GRID have also been suggested [15, 16]. A challenge of SBP modelling is that large numbers of pharmacophore features are generated [17]. Therefore, the selection of essential features is a non-trivial task and the best strategy might depend on the biological target. In this study, we report the development of purely structure-based DNA Top1 pharmacophores using the LUDI program and the comparison of different approaches to select essential pharmacophore features. To the best of our knowledge, this study describes the first development and evaluation of SBPs for a protein-DNA pocket. Furthermore, we report the successful use of our SBPs in the identification of novel classes of Top1 inhibitors and compare the results to those of ligand- and CBPs previously developed in our group [18].
Materials and methods
Preparation of structures
Unless stated otherwise, Discovery Studio 3.5 (DS; Accelrys, USA) was used to develop and evaluate structure-based Top1 pharmacophores. Selected crystal structures of Top1–DNA–ligand complexes were prepared as follows: Water molecules were deleted and hydrogens were added at physiological pH using the “Prepare Protein” protocol [19]. The binding site for each structure was first defined as all protein and DNA residues within 10 Å of the ligand position. To determine the shapes of the binding pockets, the binding site cavities were filled with a binding site point grid with a 0.5 Å spacing and binding points outside a 6 Å radius from the original ligand position were deleted. To detect rigid cores of different Top1 structure conformations, difference distance matrices were calculated using the PROFLEX program [20]. To allow direct comparison of protein structures, the residues identified as rigid core were used as tethers for protein superimposition in DS. Root-mean square deviations (RMSDs) between protein structures were calculated in DS using the Biopolymer Structure RMSD calculator.
Generation of interaction maps and pharmacophore features
To describe the interaction capability of each binding pocket, the LUDI program [11] was used in DS through the protocol “De Novo Receptor”. Because too many LUDI interaction sites were found with the default parameters and the binding site selected, leading to a program error, the sphere radius in LUDI was reduced to 13 Å. To further reduce the complexity of the interaction maps, the maps were generated with a polar site density of 15 instead of the default value of 25. The LUDI interaction maps containing three types of fragments (C=O, NH and C) were transformed into the corresponding HBA, HBD and HYD features using a user-generated Perl script. Whereas HYD features were defined as point features, HBD and HBA features were represented as directional features with two points, a tail representing the hypothetical ligand atom and a head representing the interacting protein atom. Only features found within the binding site grid defined previously were kept.
Hierarchical clustering of pharmacophore features
Two different approaches have been used to develop common Top1 SBPs. In the first approach, pharmacophores developed from different crystal structures were treated independently and merged after clustering, whereas in the second approach, the sequence of clustering and merging was inverted.
Hierarchical clustering using the UPGMA algorithm was performed with the DS Pharmacophore Tools. Different clustering distances were found optimal for different feature types and approaches: In the first approach, HBD and HBA features were clustered using a distance of 1 and 1.4 Å, respectively. Selection of features was performed by comparing the individual Top1 pharmacophores and superimposition with the corresponding crystal structures. In the second approach, clustering distances of 1.5 and 1.6 Å were used for HBD and HBA features, respectively. For the clustering of HYD features, all HYD residues (Ala, Cys, Met, Ile, Leu, Val, Phe, Tyr and Trp residues) in the binding pocket were selected and HYD point features outside a 4 Å radius of the carbon atoms of the selected residues were deleted. Hierarchical clustering of HYD features was performed using a distance of 2 and 3 Å for approach 1 and 2, respectively. After the clustering, either cluster centres were kept (approach 1) or average features of the clusters were calculated (approach 2). A cyclic π-interaction (CYPI) feature developed in our previous study [18] was added manually to the pharmacophores in order to represent stacking interactions with the DNA. The feature location was determined using Top1 crystal structure ligands as guidance and a large sphere radius (3 Å) was chosen to enable variability of the feature location.
Selection of favourable features based on energetic calculations
The electrostatic maps for each crystal structure were calculated using the “Electrostatic potential with focussing” protocol based on the DelPhi program [21]. Prior to the calculations, the atomic charges and radii for all protein-DNA complexes were assessed with the CFF [22] and CHARMm [23] force fields and partial charges were calculated using the Momany-Rone method [23]. Default parameters were used for electrostatic calculations with one exception. The number of grid points per axis was set to 251 and the grid centre was defined as the centroid of the DNA cleavage site. With these settings, a fine grid with a spacing of approximately 0.5 Å was obtained for the ligand binding site. Electrostatic maps were overlaid with the pharmacophore and the electrostatic potentials were calculated for each HBD and HBA feature head using a user-generated Perl script. The script determines the electrostatic potential of a pharmacophore point by calculating the average potential of the surrounding 27 grid points. The nearest grid point and the surrounding 26 points were obtained by changing the x, y and z coordinates by −1 or +1. The following feature selection scheme was used: In case of HBA, features were kept if the average potential ± standard error (SE) of the potential was positive for both force fields used or positive and ambiguous (average ± SE positive for one and negative for other force field). On the other hand, HBD features were kept if average potentials ± SE were negative for both force fields or negative and ambiguous. Furthermore, different feature weights were defined dependent on the type of feature. Pharmacophore features representing interactions with the DNA or the protein backbone, or features with ambiguous grid potentials, received a feature weight of 0.5, whereas all other features received a weight of 1.
Lennard-Jones potentials were calculated using the protocol “Calculate Interaction Energy” for the CHARMm and CFF force field. Each HYD point feature was converted into a carbon atom and the van der Waals energy to the surrounding atoms was calculated. A 10 Å cut-off for non-bonded interactions was used for the calculations.
Virtual database screening
Pharmacophore subqueries were generated using a Perl script which generates all possible feature combinations for a specified number of features. The compound database of the National Cancer Institute (NCI, USA), as implemented in Discovery Studio (NCI2000), was screened with pharmacophores using the “3D Database Screening” protocol. Screening with subqueries was performed sequentially. All hits obtained were combined and, if duplicates were found, only the compounds with the higher pharmacophore fit value were retained. Hit lists obtained from screening were filtered using Lipinski’s “Rule of Five” [24] using the “Filter by Lipinski and Veber rules” protocol and only applying the Lipinski filter. Up to one violation of the Lipinski rules was allowed.
To compare the performance of the SBP approaches to a simple virtual screening method, a two-dimensional similarity search was performed using the Top1 ligands from the four crystal structures utilised for pharmacophore development as input. Two-dimensional molecular fingerprints were calculated for the Top1 ligands as well as all compounds of the NCI2000 database. The ECFP_6 fingerprint developed for the modelling of structure–activity relationships was used for this purpose [25]. Compounds similar to the Top1 ligands were retrieved using the protocol “Find Similar Molecules by Fingerprints”, using a Tanimoto similarity cut-off of 0.5.
Chemical similarity and diversity
To determine the chemical similarity between two hit lists, the “Compare Libraries” protocol was used. A global fingerprint was calculated for each hit list using ECFP_6 fingerprints and the similarity between hit lists was determined using a Tanimoto Index. To determine the most diverse compounds of a hit list, the “Find Diverse Molecules” protocol was used which calculates similarities based on molecular fingerprints and determines a specified number of most diverse hits.
Ligand-pharmacophore mapping
Mappings of known Top1 ligands to the LUDI pharmacophores were performed using the “Ligand Profiler” protocol. Prior to the mapping, ligand conformations were generated with the “Generate Conformations” protocol using the “best” option. During the ligand-pharmacophore mapping, a rigid search was used and a maximum of 3 omitted features was allowed. To compare the mapped poses to the crystal structure poses, the ligands were overlayed and the heavy atom RMSD was calculated in DS.
Docking validation study
A set of 40 known active and inactive Top1 inhibitors was selected from the literature (see Online Resource 1) and used to validate the docking procedure. The set contained 22 active compounds for which activity has been measured semi-quantitatively in a DNA cleavage assay (see below). Compounds were selected based on chemical and functional diversity to represent the major classes of currently known Top1 poisons.
Docking of selected compounds was performed with the GOLD [26] program version 5.0 accessed through the DS protocol “Dock Ligands (GOLD)”. The crystal structure of the topotecan-Top1-DNA complex (PDB code: 1K4T [27]) was used for this purpose. Ligands were deleted and hydrogens were added. The SH-group at the DNA cleavage site was mutated to OH. The binding site was defined as the cavity 7.5 Å around the initial ligand position. Docking was performed with flexible side chains which were defined according to the protein residues observed to interact with topotecan in the crystal structure (Asn352, Glu356, Arg364, Lys425, Lys532, Asp533 and Thr718). 10 docking runs were performed for each ligand and the use of different scoring functions (GOLD score, ChemPLP score) was evaluated. Water molecules were either deleted or selected waters were kept and allowed to spin, translate and disappear during the docking. The docking poses were clustered based on a 2.0 Å RMSD of heavy atoms. For the evaluation of compounds, the best-scored pose of the largest cluster as well as the mean score of the largest cluster were considered.
Receiver-operating-characteristic (ROC) curves were calculated for docking scores using the protocol “Calculate ROC curve”. This protocol outputs the ROC curve as well as the area under the curve (AUC) and a ROC evaluation. For AUC values below 0.6, the ROC analysis is considered as failed, whereas AUC values between 0.6 and 0.7, 0.7 and 0.8 and higher than 0.8 are considered to describe a poor, fair or good model, respectively.
Docking for virtual screening
Compounds were selected for docking based on their pharmacophore fit value (top 20 and top 50 compounds for LUDI1 and LUDI2 list, respectively) and their chemical diversity (20 and 50 most diverse compounds for LUDI1 and LUDI2 hit lists, respectively). Flexible docking was performed as described in the previous section. GOLD was used to score poses and all but two water molecules were deleted prior to docking. Two water molecules were kept in the binding pocket because an analysis of the complex using the ViewContacts software [28] revealed that those two water molecules are capable of forming water-mediated hydrogen bonds. The water molecules were allowed to rotate, translate and disappear during the docking.
DNA cleavage assay
The Top1 inhibitory activity was measured in a DNA cleavage assay as described previously [29]. Briefly, 3′-radiolabeled DNA substrates are incubated with the Top1 enzyme and the drug to be tested, allowing the formation of ternary enzyme–DNA-drug complexes. The use of a strong protein denaturant, sodium dodecyl sulphate, leads to a denaturation of Top1 covalently bound to DNA, and polyacrylamide gel electrophoresis enabled the visualisation of cleavage products. The activity of a drug is measured semi-quantitatively, by comparison to the activity of 1 μM camptothecin (CPT). The scoring of the activity is defined as follows: 0: no activity; +: 25–50 % CPT activity; ++: 50–75 % CPT activity; +/++: 25–75 % CPT activity; +++: 75–100 % CPT activity; ++++: compound is equipotent or more potent than CPT.
Results and discussion
Generation of ligand binding pocket interaction maps and pharmacophores
DNA Top1 is an interesting drug target as the ligand binding pocket consists of both protein and DNA atoms. Furthermore, the ligand binding pocket is not fully present in the apo complex. To investigate the pocket size in apo and holo structures, two crystal structures of a Top1-DNA (PDB code: 1K4S) [27] and a Top1-DNA-ligand complex (PDB code: 1K4T) [27] were superimposed as described in the Methods section. The analysis revealed that although the protein exhibits a similar conformation in both crystal structures and the RMSD between the pocket amino acids is low (main chain RMSD = 1.04 Å, heavy atom RMSD = 1.69 Å), differences are observed for the DNA molecules. As shown in Fig. 1, when the ligand is absent, the DNA strand at the cleavage site is broken, however, without forming a binding cavity. Therefore, only experimental structures of ternary Top1-DNA-ligand complexes could be used for this study.
Fig. 1.
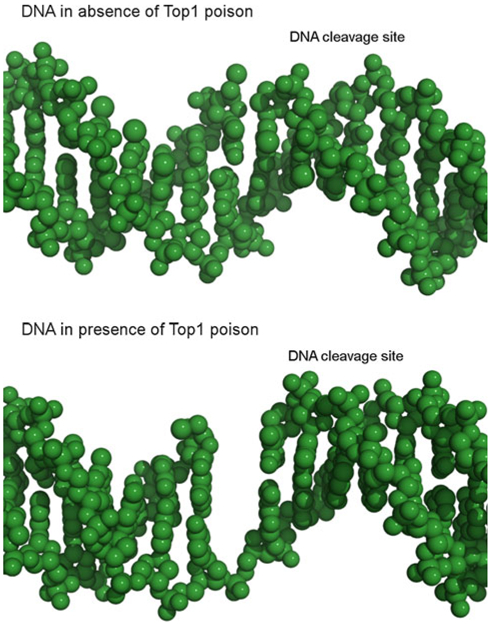
Comparison of DNA cleavage sites in the absence and presence of a Top1 poison. DNA is shown as green spheres. DNA structures obtained from crystal structures of covalent Top1-DNA complexes: PDB 1K4S (top panel, no ligand), and 1K4T (bottom panel, ligand bound) [27]
In order to develop structure-based Top1 pharmacophores, only structures with wild-type ligand binding pocket sequences were selected from the available crystal structures of ternary Top1 complexes. This resulted in the selection of four crystal structures containing the inhibitors camptothecin (PDB code: 1T8I) [30], topotecan (PDB code: 1K4T) [27], the indenoisoquinolines MJ-II-38 (PDB code: 1SC7) [30] and AI-III-52 (PDB code: 1TL8) [31]. Differences in DNA sequence at the cleavage site between the crystal structures were observed. Whereas most structures contain a thymine-guanine sequence, the thymine-cytosine sequence is present in one of the structures (PDB code: 1TL8). Moreover, although the protein structures are relatively similar in the pocket regions (heavy atom RMSD between 0.772 and 1.529 Å in pairwise comparisons), even small conformational changes might lead to different interaction maps. Therefore, four instead of a single structure were chosen to account for flexibility as observed in the experimental structures of ternary complexes.
The structures were prepared and superimposed as explained in the Methods section. To describe the interaction capabilities of the binding pockets, interaction maps were generated for all structures using the LUDI software. These interaction maps contain three types of fragments representing three types of atoms, namely HBA, HBD and hydrophobic atoms (HYD). All fragments of the LUDI interaction maps were converted into pharmacophore features using a script developed in-house, thereby creating a negative image of the binding pocket. HBD and acceptor (HBA and HBD) features normally consist of a feature tail, located on the heavy atom of the ligand, and a projection ending in the feature head, representing the location of the interacting atom. Therefore, HBA and HBD features were placed so that the feature head was located on a protein atom and the feature tail represented the position of the hypothetical ligand. In the case of HYD features, which are non-directional, the point feature simply represents the position of the atoms of the hypothetical ligand. Each LUDI pharmacophore consisted of more than 1,000 features and was thus unsuitable for virtual screening. Therefore, two distinct approaches to reduce the number of pharmacophore features were explored.
Generation of common structure-based pharmacophores: approach 1
In the first approach to generate structure-based Top1 pharmacophores, LUDI maps were generated for all input structures. After overlaying of the pharmacophores with the respective crystal structures, it was noted that many pharmacophore features represented the same interaction site and a clustering of the features was necessary. Hierarchical clustering was therefore performed to determine the clusters of features that represent specific protein–ligand and DNA-ligand interactions. For HBD and acceptor features as well as HYD features in close proximity to HYD protein residues, clustering distances were adjusted manually to match the visible clusters of features representing specific protein/DNA interactions. On the other hand, HYD features representing interactions with the DNA were deleted and replaced with a CYPI feature which was developed previously [18]. This was due to the fact that Top1 poisons interact with the DNA bases via stacking interactions; however, this interaction type is not recognized in the LUDI software. Hierarchical clustering and deletion of HYD features representing DNA-interactions resulted in a 40-fold reduction in the amount of pharmacophore features (reduced LUDI pharmacophores).
In order to determine common structural features of Top1 binding pockets, the pharmacophores developed for each complex were combined into a common Top1 SBP. Superimposition of the reduced LUDI pharmacophores with their respective crystal structures allowed the identification of residues involved in hypothetical ligand interactions. Based on this information, the individual reduced LUDI pharmacophores were combined and features common to all models were kept. Moreover, all interactions with protein backbone and DNA bases, excluding the CYPI interaction, were deleted as interactions with the protein backbone are unspecific and interactions with the DNA bases depend on the DNA sequence in the crystal structures. A summary of the first approach is given in Fig. 2. The resulting common Top1 pharmacophore contained 3 HBD, 1 HBA, 2 HYD and 1 CYPI feature (Fig. 3a).
Fig. 2.

Overview of approaches to generate SBPs
Fig. 3.

Common structure-based Top1 pharmacophores. Pharmacophore models have been generated using the first (a) and the second (b) approach and are shown superimposed onto a Top1-DNA complex (PDB code: 1K4T) [27]. Features represent interactions with the protein side chains (upper panels) and with the DNA nucleotides (lower panels). Pharmacophore features are represented as points with surrounding tolerance spheres, HBA in green, HBD in pink, HYD features in cyan and the CYPI feature in orange. The protein is shown in cartoon representation in dark cyan and the side chains involved in interactions are indicated as sticks and labelled. The DNA is shown as green sticks and the + and − end of the cleaved DNA is indicated
Generation of common structure-based pharmacophores: approach 2
In the second approach, pharmacophores resulting from LUDI maps for each crystal structure were merged first and then reduced by hierarchical clustering as described above. This is in contrast to the first approach and was chosen because it was noted that information from all crystal structures might be important for clustering. After generation of a reduced pharmacophore with hierarchical clustering and superimposition of the pharmacophore with all crystal structures, features representing interactions with DNA bases were removed to create a pharmacophore independent of the DNA sequence. However, features representing interactions with the DNA backbone (HBA, HBD) were kept in this approach and a CYPI feature was added as in approach 1 to represent stacking interactions. This resulted in a second common Top1 pharmacophore containing 36 features, among them 17 HBD, 12 HBA, 6 HYD and 1 CYPI feature.
Despite the large reduction in the number of pharmacophore features, the pharmacophore was too restrictive for virtual screening as no ligand would be able to satisfy all interactions and exhibit drug-like characteristics. Therefore, to further reduce the number of features, an energetic component was introduced and explored during the feature selection process. In analogy to the development of SBPs with the program GRID [32], it was decided to select features which represent energetically favourable interactions and to compare the results to the first SBP development approach. HBA and donor features were selected based on electrostatic potentials, whereas the selection of HYD features was based on calculations of van der Waals energies.
For HBD and acceptor features, average electrostatic potentials were calculated over the surrounding grid points for all crystal structures. HBA features should point to the hydrogen atom of a HBD and therefore feature heads should be located close to positive charge. Similarly, HBD features should point to an electronegative atom, a HBA, and thus feature heads should be located in proximity to negative charge. The application of those two rules and the selection scheme summarised in the Methods section led to the elimination of 11 and the selection of 18 favourable HBA/HBD features. Furthermore, different feature weights were introduced to distinguish between clearly favourable features (feature weight = 1 for non-ambiguous electrostatic potentials, see Methods section) and possibly favourable features (feature weight = 0.5 for ambiguous electrostatic potentials). Interactions with the protein backbone received a feature weight of 0.5 due to their non-specificity. To account for van der Waals interaction energies at the HYD feature points, average Lennard-Jones potentials over all crystal structures were calculated and two features with favourable potentials were selected for the common Top1 pharmacophore. To further reduce the number of pharmacophore features and enable virtual screening, a distance constraint was added to eliminate features representing interactions which might not be able to be satisfied by a small molecule. The CYPI feature, representing important stacking interactions, was defined as the central feature of the pharmacophore and features too far from the central feature were removed. The distance cut-off of 9.1 Å was determined from known Top1 ligands present in crystal structures. A summary of the second approach is given in Fig. 2. The resulting second common Top1 pharmacophore consisted of 9 HBD, 2 HBA, 1 HYD and 1 CYPI feature (Fig. 3b).
Comparison of pharmacophores obtained with both approaches
In order to approximate the shape of the binding pocket in the common structure-based Top1 pharmacophores, excluded volumes were placed on Cα-atoms of the binding site residues and all heavy atoms of the DNA cleavage site nucleotides using the Top1-DNA-topotecan crystal structure [27] as it is the structure with the highest resolution. It was noted that features representing several protein interactions were common to both pharmacophores. These included the HBDs interacting with Glu356, Asp533 and Asn722, the HBA interacting with Arg364 as well as the HYD feature interacting with Tyr426. However, the spatial arrangement of these common features is different between the two pharmacophores. Interestingly, Arg364 has been found to interact with all classes of Top1 inhibitors for which crystal structures have been solved [3]. Asp533, Glu356 and Asn722 have also been found to interact with some known Top1 poisons [27, 30, 31], confirming that important interactions can be identified with both SBP-development approaches.
Virtual screening with common structure-based pharmacophores
The application of both common Top1 pharmacophores in virtual database screening of the National Cancer Institute (NCI, USA) compound database resulted in no hits, suggesting that further reduction of the number of pharmacophore features was necessary for screening. Therefore, subqueries of the pharmacophores were generated, representing subsets of the large common Top1 pharmacophores. All subqueries were required to have a CYPI feature in accordance with the importance of π-π interactions in known topoisomerase poisons.
From the common SBP generated in approach 1 (LUDI1), all combinations of subqueries containing 5 features including the CYPI feature were generated. This resulted in 15 pharmacophore models to which excluded volumes were added. Their subsequent application in virtual screening led to the identification of 1,057 hit compounds, 392 of which passed a druglikeness filter. On the other hand, the common SBP generated in the second approach (LUDI2) consisted of more features than the LUDI1 model and hence, 7-feature subqueries including CYPI were generated. This resulted in 924 subqueries. To decrease computing time, a second distance cut-off was introduced to decrease the number of subqueries based on inter-feature distances, eliminating subqueries with features which were too far apart. To determine a suitable inter-feature distance cut-off, the ligands present in Top1-DNA crystal structures were extracted and the largest interatomic distance was identified as 14.1 Å. The use of this inter-feature distance cut-off resulted in a reduction of the number of subqueries to 455, which were subsequently used in database screening. Screening with the LUDI2 subqueries resulted in more than 4,000 hits. However, the number of hits was reduced to 815 upon application of a druglikeness filter and addition of excluded volumes.
Enrichment analysis and comparison of hit lists
The NCI database does not contain any information about the molecular target of the compounds it contains. It is therefore not possible to easily assess the performance of the pharmacophores in virtual screening using enrichment analysis. However, it is possible to filter the hit lists according to chemical scaffolds which have been associated with Top1 inhibition and to determine the “enrichment” of the hit lists based on this measure. This analysis was performed on the hit lists obtained with LUDI subqueries generated with both approaches. In addition, the performance of LUDI subqueries was compared to the composition of the NCI database, the results of a 2D-similarity search using the crystal structure ligands camptothecin, topotecan, MJ-II-38 and AI-III-52 and the hit lists obtained previously with ligand- and complex-based Top1 pharmacophores [18] which, to the best of our knowledge, represent the only study describing the development of Top1 pharmacophores based on enzyme inhibition data (Table 1). The presence of the following chemical structures was investigated: camptothecin derivatives, indolocarbazoles, indenoisoquinolines, flavones with a 2-phenyl-1-benzopyran-4-one scaffold, purines and 9,10-anthraquinones. Purines have been identified as novel Top1 inhibitors in our previous study [18]. Anthraquinones, for example mitoxantrone, are associated with topoisomerase II inhibition [33]. However, anthraquinone derivatives have also been found to inhibit Top1 [18].
Table 1.
Enrichment analysis of virtual screening hit lists
| Hit list | NCI 2000a | LUDI1 subqueriesa | LUDI2 subqueriesa | LBP and CBPa,b | 2D similarity searcha,c |
|---|---|---|---|---|---|
| Number of compounds | 222,734 | 392 | 815 | 746 | 61 |
| Camptothecins | 89 (0.04 %) | 3 (0.77 %) | 2 (0.25 %) | 47 (6.30 %) | 58 (95.08 %) |
| Indolo-carbazoles | 11 (0.01 %) | 0 | 2 (0.25 %) | 0 | 0 |
| Indeno-isoquinolines | 2 (0.001 %) | 0 | 0 | 0 | 0 |
| Flavones | 343 (0.15 %) | 2 (0.51 %) | 8 (0.98 %) | 22 (2.95 %) | 0 |
| 9,10-Anthraquinones | 924 (0.42 %) | 27 (6.89 %) | 38 (4.66 %) | 39 (5.23 %) | 0 |
| Purine derivatives | 3,592 (1.61 %) | 9 (2.30 %) | 208 (25.52 %) | 99 (13.27 %) | 0 |
| Enrichmentd | 2.23 % | 10.46 % | 31.66 % | 27.75 % | 95.08 % |
Filtered with Lipinski’s rule of Five, one exception allowed
Ligand-based and CBPs developed previously and used sequentially in database screening [18]
Compounds with a Tanimoto Index ≥0.5 to at least one of the Top1 X-ray structure ligands (camptothecin and indenoisoquinoline analogues) using ECFP_6 fingerprints
Based on number of compounds with scaffolds similar to known Top1 inhibitors
The analysis of chemical scaffolds revealed that the hit list obtained from screening with ligand- and complex-based Top1 pharmacophores was mainly biased towards camptothecin derivatives, but also contained a large number of the novel chemotypes. None of the LUDI hit lists retrieved as many camptothecins, despite the fact that two out of four crystal structures used for the development of SBPs contained camptothecin analogues. The bias towards known ligands observed with ligand-based pharmacophores (LBPs) and CBPs has thus been reduced. However, both LUDI1 and LUDI2 subqueries were able to retrieve chemical scaffolds associated with Top1 inhibition, with enrichment values of 10.46 and 31.66 % (approach 1 and 2, respectively). This is an encouraging result, as the enrichments observed in SBP hit lists are much higher than the Top1 scaffold enrichment of the NCI database, and this presents a positive control for the SBP approach. A simple 2D similarity search was also performed within the NCI database to serve as a control experiment. In contrast to all pharmacophore approaches, the similarity screening retrieved mostly camptothecin analogues, leading to a high enrichment, but could not identify structurally novel compounds.
To further compare the hit lists obtained with the SBP subqueries from both approaches and the hit list obtained with a ligand- and complex-based methodology, a comparison of intersecting sets was performed (Fig. 4). Only a small overlap was found between the LUDI hit lists and the list obtained with ligand- and CBPs, suggesting that the structure-based approach is different and complementary to the ligand- and complex-based approaches and that it can be used to identify potential novel inhibitors. Furthermore, only a small overlap was detected between the LUDI1 and LUDI2 hit lists, implying that the two approaches produce distinct results. These findings were further supported by calculations of chemical similarity between the hit lists. As summarised in Table 2, only very low similarities were observed between the hit lists, suggesting that the approach used to select SBP features has a large influence on the hit list obtained.
Fig. 4.

Intersection sets between different hit lists. LUDI1 and LUDI2 represent the hit lists obtained in virtual screening of the NCI2000 database with the first and second approach of SBPs. The LBP + CBP list has been obtained previously with ligand- and complex-based Top1 pharmacophores [18]
Table 2.
Similarity scores of pharmacophore hit lists based on Tanimoto similarity of a global ECFP_6 fingerprint
| Hit list | LUDI1 subqueriesa |
LUDI2 subqueriesa |
LBP and CBPa,b |
|---|---|---|---|
| LUDI1 subqueriesa | 1 | ||
| LUDI2 subqueriesa | 0.2036 | 1 | |
| LBP and CBPa,b | 0.1510 | 0.1625 | 1 |
Filtered with Lipinski’s rule of Five, one exception allowed
Ligand-based and CBPs developed previously and used sequentially in database screening [18]
Performance of structure-based pharmacophores in mapping known topoisomerase I inhibitors
In order to determine the ability of the LUDI pharmacophores to map known Top1 inhibitors, ligand-pharmacophore mapping to the LUDI subqueries was performed using ligands for which crystal structures of the ternary complex have been solved [27, 30, 31]. It was noted that the Top1 control ligands could only be mapped to the pharmacophore subqueries if feature omissions were allowed. The compounds camptothecin, topotecan, MJ-II-38, AI-III-52 and SA315F were able to map to subsets of the LUDI1 and LUDI2 pharmacophores in many different poses. In case of LUDI1, the control compounds mapped between 2 and 4 features of the pharmacophore and up to 8 different poses were observed for each ligand (Online Resource 1). In case of LUDI2, more than ten poses were observed for topotecan and SA315F, whereas only one or two poses were found for camptothecin, MJ-II-38 and AI-III-52 (Online Resource 1). Top1 compounds mapped between 4 and 8 features of the LUDI2 pharmacophore, and the feature representing stacking interactions was mapped in all cases. Although mappings of all control ligands were possible, confirming the SBP methodology, it should be emphasized that the approach has not been optimised for pose prediction. A comparison of mapped poses of the ligands to the crystal structure poses showed that RMSD values were above 3 Å for all ligands and both approaches.
Performance of structure-based pharmacophores in identifying novel topoisomerase I inhibitors
While the enrichment analysis presents encouraging results for the SBP approaches, it cannot give an indication on how well the methods will perform in virtual screening to retrieve new hit compounds. To evaluate the performance of both pharmacophore development approaches in a prospective manner, a number of compounds was selected from the NCI virtual screening hit lists and tested in a Top1 inhibition assay [29]. The selection of compounds was based on chemical diversity as well as a docking study confirming their ability to fit into the Top1-DNA pocket. From the LUDI1 and LUDI2 hit lists, all compounds with scaffolds associated with Top1 inhibition (see Table 1) were removed. Then, the top-scored compounds as well as the most-diverse compounds in each hit list were identified and subjected to docking using GOLD [26] as described in the Methods section. This ensures that compounds are not only selected based their pharmacophore fit values which have not been optimised to match experimental binding affinities and might therefore not correlate with biological activity at all.
Based on a docking validation study with known Top1 inhibitors and Top1 inactive compounds, it was found that, despite benchmarking studies suggesting the superiority of ChemPLP in pose prediction and virtual screening [34], the GOLD scoring function outperforms the ChemPLP scoring function in Top1 docking in a ROC analysis (Online Resource 1). This might be due to the fact that scoring functions have not been optimised nor validated for protein-DNA targets such as topoisomerases. Furthermore, an analysis of the topotecan-Top1-DNA crystal structure used for docking with the recently developed ViewContacts software [28] suggested that two water molecules located in the binding pocket might have an impact on ligand binding and the ROC analysis revealed that GOLD docking with those waters leads to a small improvement of performance (Online Resource 1). Therefore, the water molecules were used in docking.
The docking poses for each compound were clustered and evaluated based on the largest cluster of solutions. Based on the docking validation study (Online Resource 1), two score thresholds, a score of 85 and higher for the best-scoring pose of the largest cluster and a mean score of 75 and higher for all poses of the largest cluster, were found to result in good ratio of true positives to true negatives (TN; Online Resource 1) and were therefore used to select compounds for biological testing. The selection was also based on chemical diversity to already selected compounds as well as limited by the availability of compounds. In the end, 6 compounds from each LUDI hit list were tested for Top1 inhibition (Table 3; Figs. 5, 6). Of the six compounds from the LUDI1 list, two (NSC 68788 and 114378, Fig. 6) showed mild Top1 inhibition (+, 20–50 % of CPT activity). Interestingly, all of the tested compounds selected from the LUDI2 list showed Top1 inhibitory activity, with the two most active compounds (NSC 34237 and 372074, Fig. 6) ranked as “++” (50–75 % of CPT activity). To the best of our knowledge, none of the tested compounds has been previously associated with Top1 inhibition. Furthermore, two of the active compounds display mild cytotoxic activity in the NCI 60 cell lines assay (publicly available data, see Table 3). Therefore, both of our approaches to develop SBP for the Top1-DNA pocket are successful in identifying novel classes of Top1 inhibitors which could be investigated further in the development of anticancer treatments.
Table 3.
Top1 inhibition of selected compounds
| List | Compound | CAS- RNa |
Top1 Inhibitionb |
Cytotoxic activityc |
|---|---|---|---|---|
| LUDI1 | NSC 68788 | 6949-30-0 | + | No data |
| NSC 114378 | 958835-11-5 | + | No data | |
| NSC 319992 | 200263-20-3 | 0 | No data | |
| NSC 356818 | 906625-92-1 | 0 | No data | |
| NSC 371684 | 6298-31-3 | 0 | No data | |
| NSC 649351 | 133476-19-4 | 0 | GI50 between 2.18 and 100 μM | |
| LUDI2 | NSC 34237 | 17051-80-8 | ++ | No data |
| NSC 83217 | 3905-92-8 | + | GI50 between 52.4 and 100 μM | |
| NSC 162537 | 907177-90-6 | + | No data | |
| NSC 302569 | 904222-18-0 | + | No data | |
| NSC 359465 | 677334-31-5 | + | No data | |
| NSC 372074 | 908826-95-9 | ++ | GI50 between 10 and 100 μM |
Chemical Abstracts Registration Number
Top1 inhibition ranking: 0 (no activity); +(20–50 % of 1 μM CPT activity); ++(50–75 % of 1 μM CPT activity)
Publicly available data (http://dtp.nci.nih.gov) measured in the US National Cancer Institute 60 human tumour cell line anticancer drug screen [38]; GI50 corresponds to the concentration of the drug which results in a 50 % growth inhibition
Fig. 5.
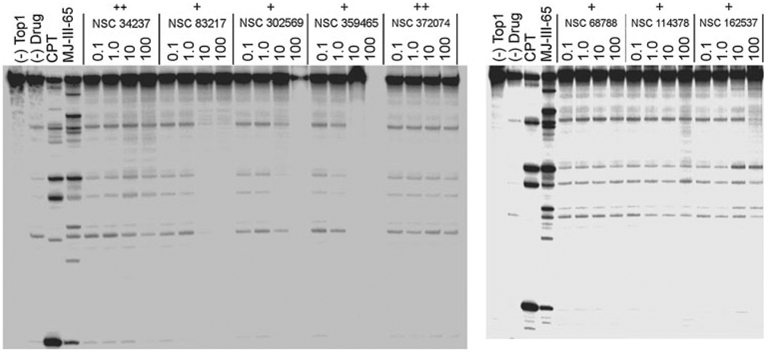
Top1-mediated DNA cleavage induced by tested compounds. The two gels indicate: DNA alone (lane 1, (−) Top1); Top1 + DNA (lane 2, (−) Drug); Top1 + DNA + 1 μM CPT (lane 3, CPT), Top1 + DNA + 1 μM MJ-III-65 (lane 4, MJ-III-65); Top1 + DNA + compounds tested at a concentration of 0.1, 1, 10 and 100 μM (other lanes). The scoring of the activity is defined as follows: +: 25–50 % CPT activity; ++: 50–75 % CPT activity
Fig. 6.
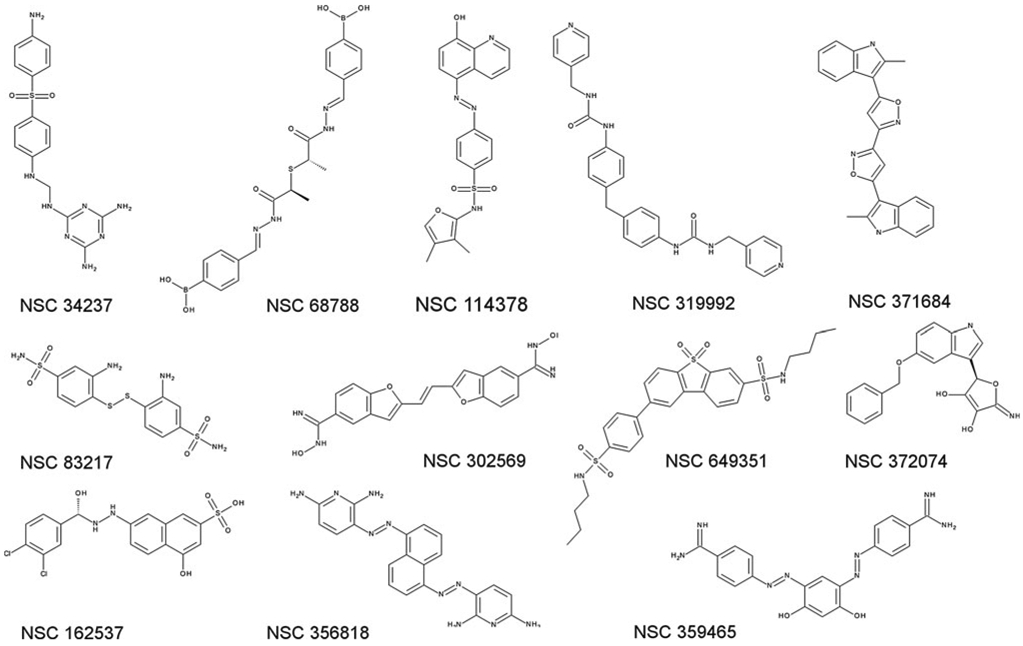
Chemical structures of all tested compounds
Conclusions
The selection of essential pharmacophore features is a well-known challenge in the development of SBPs [35]. Here, we describe two approaches to develop purely SBPs for Top1. Both approaches are based on LUDI interaction maps describing the interaction capability of the Top1 binding pocket. However, the approaches explore different methods to combine information from multiple structures, to cluster and to select pharmacophore features. An interesting finding of this study is that the choice of feature clustering and selection methods has a large impact on the resulting virtual screening hit list. Indeed, the two approaches developed in this study retrieve hit lists with only little overlap and chemical similarity, emphasising that the feature selection is a crucial step. When assessing their performance retrospectively, both approaches were able to retrieve compounds associated with Top1 inhibition, although the hit list obtained with the second, energy-based approach displayed a higher enrichment in known Top1 scaffolds. Biological testing of compounds selected from the two hit lists revealed that the hit rate obtained with the second approach is considerably higher (100 % for the second approach as compared to 33 % for the first). However, the hit rate might be influenced by compound unavailability as well as the small number of compounds tested. Whether the methodology of the second approach described here is also superior for other targets cannot be inferred and more benchmarking studies comparing different feature selection procedures, such as the recently published study examining the effect of different clustering distances and interaction ranges on the reproducibility of known protein-ligand interactions [36], are required to draw general conclusions.
A limitation of the current approach is that no information about water molecules in the binding pockets is included. This is mainly due to the fact that the LUDI program is not able to identify interactions with water. However, another reason is that only one of the Top1 ternary complex crystal structures contains water molecules. In a recent study, Hu and Lill suggest a new approach to select pharmacophore features based on a hydration-site analysis [37]. Assuming that the binding affinity of a ligand is increased when water molecules gain free energy upon being displaced from the protein pocket, pharmacophore features are selected based on proximity to hydration sites with favourable energies. Future studies could investigate the application of a similar hydration-site analysis with energetic calculations to take into account interaction strength. Another limitation consists of the limited sampling of the Top1 conformational space through the use of only four structures. The development of fully dynamic SBPs based on a structural ensemble could be helpful to select common features or to introduce feature weights based on the relative occurrence of specific interaction sites. However, due to the absence of a ligand-binding cavity in the apo protein-DNA complexes, simulations could be problematic and tend to structures with pockets too small to accommodate a ligand. Simulations would thus need to be performed in the presence of a small molecule, which would introduce bias towards that ligand.
Despite the challenges and limitations, purely SBPs can be successfully generated and applied in virtual screening, even for unusual ligand binding pockets such as the protein-DNA pocket of topoisomerase. They represent a computationally inexpensive alternative to docking and, because they are not biased towards known ligands, they represent a complementary approach to ligand- and CBPs. Biological testing confirmed that the pharmacophores can be used to identify structurally novel Top1 inhibitors which could be investigated as lead compounds for the development of novel anticancer treatments. Apart from the use of SBPs in virtual screening, future applications could involve the comparison of ligand binding pockets of homologous proteins.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The authors gratefully acknowledge the NCI Developmental Therapeutics Program (http://dtp.cancer.gov) for providing compound samples. The authors acknowledge Tom Dupree for the use of his script to superimpose proteins by tethers. M.D. acknowledges financial assistance from the University of New South Wales, Australia, in providing a PhD scholarship in the form of a University International Postgraduate Award (UIPA), as well as the Translational Cancer Research Network (TCRN) Australia for providing a Postgraduate Research Scholarship Top-up in 2012.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10822-013-9695-x) contains supplementary material, which is available to authorized users.
Contributor Information
Malgorzata N. Drwal, Department of Pharmacology, School of Medical Sciences, University of New South Wales, Sydney, NSW 2052, Australia
Keli Agama, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA.
Yves Pommier, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA.
Renate Griffith, Department of Pharmacology, School of Medical Sciences, University of New South Wales, Sydney, NSW 2052, Australia.
References
- 1.Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3(6):430–440 [DOI] [PubMed] [Google Scholar]
- 2.Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y (2006) A novel norindenoisoquinoline structure reveals a common interfacial inhibitor paradigm for ternary trapping of the topoisomerase I-DNA covalent complex. Mol Cancer Ther 5(2):287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pommier Y (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6(10):789–802 [DOI] [PubMed] [Google Scholar]
- 4.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20(11):3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang XW, Qing C, Xu B (1999) Apoptosis induction and cell cycle perturbation in human hepatoma hep G2 cells by 10-hydroxycamptothecin. Anticancer Drugs 10(6):569–576 [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y (2009) DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 109(7):2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langer T, Hoffmann RD (2006) Pharmacophore modelling: applications in drug discovery. Expert Opin Drug Discov 1(3):261–267 [DOI] [PubMed] [Google Scholar]
- 8.Leach AR, Gillet VJ, Lewis RA, Taylor R (2010) Three-dimensional pharmacophore methods in drug discovery. J Med Chem 53(2):539–558 [DOI] [PubMed] [Google Scholar]
- 9.Yang SY (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 15(11–12):444–450 [DOI] [PubMed] [Google Scholar]
- 10.Hessler G, Baringhaus K-H (2010) The scaffold hopping potential of pharmacophores. Drug Discov Today: Technol 7(4):e263–e269 [DOI] [PubMed] [Google Scholar]
- 11.Böhm HJ (1992) The computer-program Ludi: a new method for the denovo design of enzyme-Inhibitors. J Comput Aided Mol Des 6(1):61–78 [DOI] [PubMed] [Google Scholar]
- 12.Böhm HJ (1992) LUDI: rule-based automatic design of new substituents for enzyme inhibitor leads. J Comput Aided Mol Des 6(6):593–606 [DOI] [PubMed] [Google Scholar]
- 13.Schuller A, Fechner U, Renner S, Franke L, Weber L, Schneider G (2006) A pseudo-ligand approach to virtual screening. Comb Chem High T Scr 9(5):359–364 [DOI] [PubMed] [Google Scholar]
- 14.Barillari C, Marcou G, Rognan D (2008) Hot-spots-guided receptor-based pharmacophores (HS-Pharm): a knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J Chem Inf Model 48(7):1396–1410 [DOI] [PubMed] [Google Scholar]
- 15.Löwer M, Geppert T, Schneider P, Hoy B, Wessler S, Schneider G (2011) Inhibitors of Helicobacter pylori protease HtrA found by ‘virtual ligand’ screening combat bacterial invasion of epithelia. PLoS ONE 6(3):e17986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tintori C, Corradi V, Magnani M, Manetti F, Botta M (2008) Targets looking for drugs: a multistep computational protocol for the development of structure-based pharmacophores and their applications for hit discovery. J Chem Inf Model 48(11):2166–2179 [DOI] [PubMed] [Google Scholar]
- 17.Löwer M, Proschak E (2011) Structure-based pharmacophores for virtual screening. Mol Inf 30(5):398–404 [DOI] [PubMed] [Google Scholar]
- 18.Drwal MN, Agama K, Wakelin LPG, Pommier Y, Griffith R (2011) Exploring DNA topoisomerase i ligand space in search of novel anticancer agents. PLoS ONE 6(9):e25150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spassov VZ, Yan L (2008) A fast and accurate computational approach to protein ionization. Prot Sci 17(11):1955–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller PA, Leach SP, Luu TT, Titmuss SJ, Griffith R (2000) Development of computational and graphical tools for analysis of movement and flexibility in large molecules. J Mol Graph Model 18(3):235–241, 299 [DOI] [PubMed] [Google Scholar]
- 21.Nicholls A, Honig B (1991) A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J Comput Chem 12(4):435–445 [Google Scholar]
- 22.Maple JR, Hwang MJ, Jalkanen KJ, Stockfisch TP, Hagler AT (1998) Derivation of class II force fields: V. Quantum force field for amides, peptides, and related compounds. J Comput Chem 19(4):430–458 [Google Scholar]
- 23.Momany FA, Rone R (1992) Validation of the general purpose QUANTA ®3.2/CHARMm® force field. J Comput Chem 13(7):888–900 [Google Scholar]
- 24.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1–3):3–25 [DOI] [PubMed] [Google Scholar]
- 25.Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50(5):742–754 [DOI] [PubMed] [Google Scholar]
- 26.Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267(3):727–748 [DOI] [PubMed] [Google Scholar]
- 27.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L (2002) The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 99(24):15387–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuhn B, Fuchs JE, Reutlinger M, Stahl M, Taylor NR (2011) Rationalizing tight ligand binding through cooperative interaction networks. J Chem Inf Model 51(12):3180–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dexheimer TS, Pommier Y (2008) DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc 3(11):1736–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB (2005) Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 48(7):2336–2345 [DOI] [PubMed] [Google Scholar]
- 31.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M (2005) Synthesis and mechanism of action studies of a series of norindenoisoquinoline topoisomerase I poisons reveal an inhibitor with a flipped orientation in the ternary DNA-enzyme-inhibitor complex as determined by X-ray crystallographic analysis. J Med Chem 48(15):4803–4814 [DOI] [PubMed] [Google Scholar]
- 32.Goodford PJ (1985) A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 28(7):849–857 [DOI] [PubMed] [Google Scholar]
- 33.Liang H, Wu X, Guziec LJ, Guziec FS, Larson KK, Lang J, Yalowich JC, Hasinoff BB (2006) A structure-based 3D-QSAR study of anthrapyrazole analogues of the anticancer agents losoxantrone and piroxantrone. J Chem Inf Model 46(4):1827–1835 [DOI] [PubMed] [Google Scholar]
- 34.Liebeschuetz JW, Cole JC, Korb O (2012) Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J Comput Aided Mol Des 26(6):737–748 [DOI] [PubMed] [Google Scholar]
- 35.Sanders MPA, McGuire R, Roumen L, de Esch IJP, de Vlieg J, Klomp JPG, de Graaf C (2012) From the protein’s perspective: the benefits and challenges of protein structure-based pharmacophore modeling. Med Chem Comm 3(1):28–38 [Google Scholar]
- 36.Hu B, Lill MA (2013) Exploring the potential of protein-based pharmacophore models in ligand pose prediction and ranking. J Chem Inf Model 53(5):1179–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu B, Lill MA (2012) Protein Pharmacophore Selection Using Hydration-Site Analysis. J Chem Inf Model [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoemaker RH (2006) The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6(10):813–823 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.