

Abstract
Pancreatic cancer is one of the most common causes of cancer-related deaths worldwide. The two major histological subtypes of pancreatic cancer are pancreatic ductal adenocarcinoma (PDAC), accounting for 90% of all cases, and pancreatic neuroendocrine neoplasm (PanNEN), which makes up 3–5% of all cases. PanNEN is classified into well-differentiated pancreatic neuroendocrine tumor and poorly-differentiated pancreatic neuroendocrine carcinoma (PanNEC). Although PDAC and PanNEN are commonly thought to be different diseases with distinct biology, cell of origin, and genomic abnormalities, the idea that PDAC and PanNEC share common cells of origin has been gaining support. This is substantiated by evidence that the molecular profiling of PanNEC is genetically and phenotypically related to PDAC. In the current review, we summarize published studies pointing to common potential cells of origin and speculate about how the distinct paths of differentiation are determined by the genomic patterns of each disease. We also discuss the overlap between PDAC and PanNEC, which has been noted in clinical observations.
Keywords: Pancreatic adenocarcinoma, Pancreatic neuroendocrine carcinoma, Neuroendocrine tumor, Carcinogenesis, Genomic patterns
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of malignancy found in the pancreas, with over 90% of pancreatic neoplasms diagnosed as PDAC. Epidemiologic studies of all malignant types have shown that the 5-year relative survival rate is the lowest in PDAC and that death rates from this type of cancer have been rising over the past decade [1]. Less than 5% of patients with PDAC are alive after 5 years, and in patients discovered in the early stages, the 5-year survival rate is only approximately 20% [2]. The most common driver gene mutations of PDAC include KRAS, CDKN2A, TP53, and SMAD4, which all together account for 90% of cases [3].
The second most common type of pancreatic neoplasm is pancreatic neuroendocrine neoplasm (PanNEN), which accounts for 3–5% of all cases [4]. It is important to note that poor differentiation of pancreatic neuroendocrine carcinoma (PanNEC) accounts for 10–20% of PanNEN cases. PanNEN has a prognosis similar to PDAC, with a median overall survival (OS) of only 7.5 months [5]. In contrast, well-differentiated pancreatic neuroendocrine tumors (PanNETs) are slow-growing with 5- and 15-year OS rates of 85.4% and 55%, respectively. In addition to distinguished clinicopathological characteristics and prognosis, PanNEC tumors are known to be histologically and genetically different from PanNET. For example, common somatic mutations in PanNET include MEN1 (multiple endocrine neoplasia type 1), DAXX (death-domain-associated protein)/ATRX (alpha alassemia/mental retardation syndrome X-linked), and mTOR (mammalian target of rapamycin) pathway genes. However, PanNEC tumors carry the mutations of tumor protein 53 (TP53), retinoblastoma 1 (RB1), and KRAS [6], which is similar to what is seen in PDAC, while differing completely from what occurs in PanNET [7].
The close association between adenocarcinoma and neuroendocrine carcinoma has been shown in other organs, such as the colon and rectum [8]. Transformation from non-small cell lung cancer to small cell lung cancer is commonly encountered in clinical practice, and supports the idea that these malignancies might share common cells of origin [9]. The current 2019 World Health Organization (WHO) TNM classification for NEN indicates that high-grade PanNEC should be classified according to the criteria used for classifying the carcinomas of the pancreas [10]. In addition, in clinical practice the standard chemotherapy for PDAC is also effective in treatment of PanNEC [11]. This raises the question of whether PDAC and PanNEC may have deeper connections. In this review, we summarize connections between PDAC and PanNEC including their genomic features, cells of origin, and clinical practice areas.
Embryo development and molecular regulation of pancreatic exocrine and endocrine secretions
The pancreas is an organ with dual endocrine (hormone-producing) and exocrine (enzyme-producing) functions. Exocrine secretions are formed with acini and ducts and secrete digestive proenzymes into the duodenum where they aid the digestion process. Exocrine secretion, which is represented by the islet of Langerhans, contains five distinct cell types (α-cells, β-cells, δ-cells, γ-cells, and ε-cells), and hormones, such as glucagon, insulin, and somatostatin, are secreted directly into blood circulation [12].
During the process of embryo development, however, both endocrine and exocrine cells originate from a common pool of multipotent pancreatic progenitors, which subsequently differentiate into the acing progenitors and bipotent pancreatic progenitors (bi-PPs) (Fig. 1). The former differentiate into acinar cells, while the latter give rise to ductal progenitors and endocrine progenitors which further separate into ductal cells and endocrine cells, respectively [13]. Three major epithelial cell types (islet, acinar and duct) found in the pancreas develop from the same undifferentiated pancreatic progenitors, yet have radically different functions, which indicates an interesting mechanism. The mechanism seen in the differentiation process is not synchronous, as several molecules and pathways participate in this process (Fig. 1). GATA4, GATA6, FOXA2, and PTF1A are expressed in the foregut endoderm and play important roles in the differentiation of pancreatic progenitors [14, 15]. SOX9 persists in pancreatic duct cells, but it is absent in endocrine cells and acinar cells [16]. The mechanisms that initiate the formation of specific endocrine subtypes are more sophisticated. The most effective differentiation in α-lineage is observed in Aristaless-related homeobox (ARX)expressing cells, which effectively secrete glucagon [17]. Genes for β-cell differentiation and maintenance of function are multiple. Typical genes for functional β-cell differentiation are NEUROD1, INS1, MAFA, ISL1, PDX1, and ACVR1C [18]. Genetic markers that maintain mature β-cell function and identity include MAFA, PDX1, NKX2.2 and NKX6.1 [12, 16, 19]. Ductal cell lineages and endocrine cell lineages interact and exhibit common genetic differentiation during development of the pancreas.
Fig. 1.

Endocrine and exocrine cell differentiation during pancreas development
Genomic features shared among PDAC, PanNET, and PanNEC
There are major differences in driver gene landscapes and related pathways among PDAC, PanNET and PanNEC (Fig. 2, Table 1). The most common driver genes found in PDAC are KRAS (88–100% of cases), TP53 (85% of cases), CDKN2A (90% of cases) and SMAD4 (55% of cases) [20, 21]. These four driver genes fulfill key roles in pancreatic tumorigenesis including the transition of pancreatic precursors such as PanIN and IPMN to PDAC [22]. In addition, about 10% of PDAC manifestations are inherited, and the best-studied germline mutations linked to familial pancreatic cancer risk are the components of DNA double-strand break repair machinery, including BRCA1, BRCA2, PALB2 (partner and localizer of BRCA2), the Fanconi anemia genes FANCC and FANCG, and ATM (ataxia telangiectasia mutated) [23].
Fig. 2.
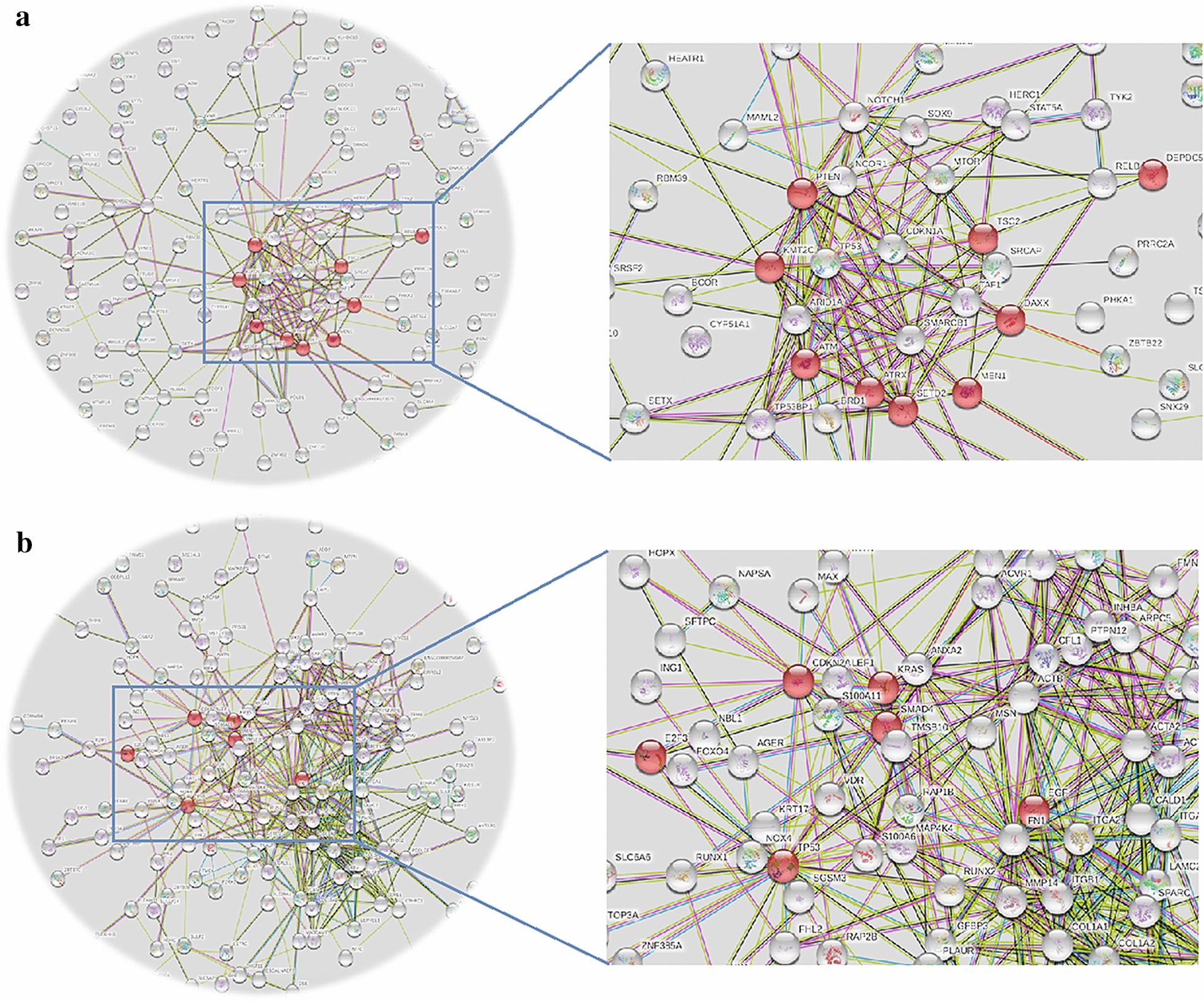
Driver gene association network between PanNET (a) and PDAC (b)
Table 1.
Summary of the core pathways and genes in PDAC, PanNET and PanNEC
| Pathways | PDAC | PanNET | PanNEC |
|---|---|---|---|
| RAS/MAPK signaling | KRAS, MAP2K4 | KRAS, BRAF | |
| NF-kB signaling | NF-κB, IKK | ||
| JAK/STAT signaling | MAP4K3, TNF, BCL2 | BCL2 | |
| TGFb/SMAD signaling | TGF-bR2, SMAD4 | ||
| Wnt/b-catenin signaling | DKK1, HMGA2 | WNT | |
| Hedgehog signaling | SOX3,GLI1,GLI3, TBX5, LRP2 | LRP2 | |
| Hippo signaling | YAP, TAZ | ||
| SWI/SNF | ARID1A | ||
| Small GTPase-dependent signaling | DEPDC2 | ||
| Notch signaling | MYC, GATA6 | NOTCH1, HES1 | |
| DNA damage repair | TP53, BRCA1, BRCA2 | TP53, MDM2, MDM4 | TP53 |
| Cell cycle | CDKN2A, APC2, FBXW7, CHD1 | CDK4, p16,p27 | RB1, APC, CDKN2A, FBXW7 |
| c-MET | MET | ||
| PI3K/AKt signaling | PTEN, PI3KCA, AKT, mTOR, TSC1/2 | PTEN, PI3KCA, mTOR | |
| Chromatin remodeling | DAXX,ATRX, MDM2 | ||
| Angiogenesis pathway | HIF-1, BNIP3 | HIF-1a, VEGF, PDGF, PHDs,Kit, Tie-2 | |
| Menin pathway | MEN1, MLL |
Comparisons between genetic landscapes of PanNEC and PDAC indicate that almost half of PanNEC instances are genetically and phenotypically related to PDAC [24]. Whole exon analysis shows that TP53 and RB are the most common altered genes in NEC [25]. TP53 mutations are present in approximately 70–95% of PanNEC cases [26], and Rb markers have been identified in 74% of the instances of PanNEC [27]. Mutations in TP53 and RB1 genes are the pivotal drivers, and related pathways are central features of PanNEC development and are associated with poor survival [28]. Other predominantly mutated genes involve APC, CDKN2A, BRAF, KRAS, PTEN and PIK3CA [28, 29]. FBXW7, WNT, BCL2, and CTNNB1, which are mutated in partial PDAC, are also related to PanNEC oncogenesis but at a lower rate of occurrence [30]. Most KRAS-positive PanNEC also express MUC1 and carcinoembryonic antigen (CEA) as the markers of ductal differentiation. Exocrine lineage markers may help to reveal the potential relationship of some PanNEC occurrences with conventional PDAC. Activating KRAS mutations in PanNEC expression show the same hot spot areas in PDAC, such as exon 2 and exon 3 ++− [27]. The most common gene mutations in mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs) are TP53, KRAS, and BRAF [31], which have been found to overlap between PanNEC and PDAC. One study has shown that in MiNEN, the mutations of CDKN2A, GNAS, ERBB2, and BRAF found in the PanNEC section were located next to the PDAC section [32].
The driver genes in PanNETs are totally different from those found in PanNEC [33], PanNEC has a much higher mutation burden when compared with PanNETs. Next generation sequencing analysis has shown that the most common gene mutations in PanNETs include MEN1 (37%), DAXX/ATRX (22% and 10%), and mTOR pathway genes, particularly PTEN(7%), tuberous sclerosis complex 2 (TSC2, [4%]) and PIK3CA [7]. MEN1 acts as a hub gene and interacts with all core pathways, and DAXX/ATRX also cooperates during tumorigenesis [34, 35]. The NET G3 subgroup, defined in a recent WHO classification, has the similar genetic mutations as PanNET G1/2, namely ATRX(19%), SF3B1(19%), and MEN1(12%) [29]. However, MEN1 and DAXX/ATRX genes mutations seem to occur very rarely in PanNEC [36]. Whereas mutation rates in RB and TP53 appear to be very low in PanNETs. In general, 80% of PanNETs do not have mutations in the Rb protein [37]. Even in PanNET G3, RB abnormalities were lower than those in PanNEC G3 (42% to 71.4%) [26, 33]. Overexpression of the Bcl-2 protein was observed in 50–100% of PanNECs, higher than that found in PanNETs (18%). Furthermore, Bcl-2 overexpression was significantly correlated with higher Ki67 in PanNEC [38]. In addition, the PanNEC expression levels of mTOR were higher than those found in PanNETs [39]. Some genes that are often seen to mutate in PDAC can be found to be abnormal in PanNETs, but this always is a more aggressive characteristic, leading to poor prognosis. For example, high expression of Cdk4 has been shown to lead to inactivation in PanNETs and is associated with a higher grade [40].
Twelve classical signaling pathways accompany PDAC tumorigenesis. Among them, the Hedgehog pathway, Notch pathway, Wnt pathway, RAS/MAPK/PI3K pathway, and JAK-STAT pathway are recognized as main contributors to PDAC progression [41, 42]. Unlike PDAC, DNA damage repair, chromatin remodeling, telomere alteration, and the PI3K/mTOR pathway are main pathways in the PanNET process.
Cells of origin that are shared among PDAC, PanNET, and PanNEC
The origins of PDAC and PanNEN are complex. PDAC can arise either from a precursor cell of intralobular ducts or acinar cells of exocrine secretion. The process of PDAC tumorigenesis is initiated by oncogenic KRAS and requires the repression of the epithelial differentiation program and activation of factors that are normally expressed during the embryonic development of the pancreas. Precursors of pancreatic cancer, namely pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN), participate in this process. Indeed, up to 33% of pancreatic tissue from autopsy series contains PanIN [43]. Duct-like cell lineages with a mutation in the driver gene KRAS, can develop into PanIN and progress to PDAC after additional multi-genetic (CDKN2A, SMAD4, and TP53) processes. The genetic features associated with IPMN and MCN are less well characterized [44]. Acinar cells transform to a duct-like phenotype during an acinar-to-ductal metaplasia (ADM) process. With a mutation in the driver gene KRAS, ADM cells stay locked in a ductal stage, and progress to PanIN and PDAC during additional multi oncogenic signaling [45, 46].
PanNETs are heterogeneous, and their differences can be explained partly by the PanNET cells of origin. Specifically, at least two different progenitor cells can give rise to non-functional PanNETs, the α-cells lineage (ARX +) or β-cells lineage (PDX +) [19]. Global DNAme profiles of the early stage of PanNET show strong similarities with both α- and β-cell DNAme profiles, while late stage tumors show a lower degree of similarities with normal cell types [47]. An additional study confirmed that different molecular subtypes of PanNET arise from different cellular origins, or from mature β-cell lineage, or islet cell precursors [48]. Loss of MEN1 is an early event in PanNET tumorigenesis, while ATRX/DAXX loss and ALT are relatively late events [49]. ACVR1C plays a key role in islet cell differentiation and also works as a suppresser during PanNET development. ACVRIC also contributes to a higher Ki67 index [50].
PanNEC cell of origin is more sophisticated. PanNEC may in fact originate from a separate, potentially non-neuroendocrine lineage, as seen in the growing body of evidence showing that the genomic origination of PanNEC is different from that of PanNETs. PanNEC may originate from undifferentiated progenitor cells [51], which is the same in the occurrence of PDAC (Fig. 3). A mouse model of PanNEC has shown a loss of many markers during beta-cell, alpha-cell, and delta-cell differentiation, including markers of endocrine progenitors, such as MAFA, PDX1, and NKX6.1. RB and/or the p53 signaling network are associated with this process. Furthermore, Id1, a marker of neural stem cells, was identified as specifically expressed in PanNEC [52]. This raises the possibility that PanNEC may have stem cell-like properties or arise from pancreatic progenitor cells.
Fig. 3.

Hypothesis of the evolution of PanNET, PDAC, and PanNET cells of origin
Tumor microenvironment among PDAC, PanNET, and PanNEC
In addition to genetic landscape, the tumor microenvironments (TME) of these neoplasms also have distinct characteristics. PDAC has a unique desmoplastic TME, which generates a large stromal component and accounts for 15–90% of the tumor [53]. This desmoplastic stroma compromises immune cells, tumor blood perfusion and oxygen delivery, in turn generating an obstacle for angiogenesis. This identifies another feature of PDAC, impaired tumor vasculature and hypoxia [54]. Desmoplastic stroma causes obstacles in chemotherapy and anti-angiogenesis target therapy in PDAC. Most PanNEC cases exhibit hypo vascular patterns similar to PDAC. In contrast, PanNETs have extraordinary tumor vascularization with high expression of several pro-angiogenic molecules, including platelet-derived growth factor receptors and, vascular endothelial growth factor and its receptors [55]. The hyper vascular characteristics of PanNET provide a way to target angiogenesis therapy [56].
It has been observed that profiles of tumor-infiltrating lymphocytes (TILs) in PanNEN and PDAC samples form three distinct clusters and are concordant with histological types PanNEC, PDAC, and PanNETs [57]. This indicates a correlation between immune profile and tumor histology. PanNEC and in some instances, PDAC have revealed a hot immune TME when compared to PanNETs. TIL and its PD-1/PD-L1 + subsets have been found less abundant in PanNETs compared to those found in PanNEC and PDAC [57]. The stroma of PDAC is rich in CD3 + CD4 + helper T (Th) cells, CD8 + T cells, and CD4 + CD25 + fork-head box P3 regulatory T cells. PD-1 and PD-L1 are associated with poor prognosis in patients with PDAC [58]. PanNEC shows the intensive accumulation of CD3 +/CD4 T cells, which is comparable with that found in PDAC [57]. There is a clear shift at the NET–NEC transition in immune-related profiles of GEP-NEN. Reduced CD8 + infiltration and enhanced PD-L1 expression in tumor cells are associated with higher tumor grading [59]. Among PanNET cases, 32–65% have a high degree of T cell infiltrates (CD3 + , CD45RO + , and CD8 +) in intratumoral cases and 50–70%, in extratumoral cases [60]. CD3 +/PD-1high and CD204 +/PD-L1high populations were significantly higher in PanNETs with a higher grade [57]. Furthermore, the expression of PD-L1(Stroma) was higher in PanNEC than in PanNETs [59]. Since immune checkpoints such as PD-1/PD-L1 are mainly expressed in high-grade NEN, the KEYNOTE-028 trial evaluated treatment with pembrolizumab in solid tumors and was able to show that partial response and progression-free survival in patients with PanNETs and PDAC were 6.3% at 4.5 months and 0% at 1.7 months, respectively [61, 62]. Case series have also reported that patients with PanNEC respond to nivolumab and have a long survival of 8 years following standard chemotherapy [63]. Equivalent trials are now in progress for GEP-NETs. However, most phase I and II clinical trials have failed to show any clinical efficacy in the case of PDAC [64].
Pathological diagnosis and serum detection
The pathology diagnosis of PanNEC is sometimes mistaken for PDAC, as both often have nuclear atypia, necrosis and a high mitotic rate; furthermore, PanNEC may have an adenocarcinoma component [10, 65]. PanNEC is composed of highly atypical neoplastic cells and can be further classified into small and large cell carcinoma. Large cell (61%) is more common than small cell (39%) [66]. In general, PanNEC cells are pleomorphic and its classic salt-and-pepper chromatin pattern is not as evident. Geographic necrosis, as well as perineural and angiolymphatic invasion, are often present in PanNEC. Microscopically, PDAC often has glandular architecture and mucin-producing glands, but can have single-cell infiltration. In addition, the neoplastic cells have irregular nuclear chromatin and prominent nucleoli. Overall, cells in PDAC tend to be more pleomorphic than those in PanNENs. IHC is useful for distinguishing these malignancies. In PanNET, chromogranin A (CgA) and Synaptophysin (Syn) are the most sensitive biomarkers, and staining is generally found to be diffuse and strong; however, CgA and Syn are less sensitive in the diagnosis of PanNEC compared with PanNETs [67]. Well-differentiated morphology and lower Ki67 proliferation index were found to be correlated with the strong expression of SSTR2A and CgA, and highly positive SSTR2A was associated with longer survival in PanNETs [68].
There is overlap in serum biomarkers between PanNEC and PDAC. CA199 and CEA are sensitive and specific markers for PDAC diagnosis and prognosis. Abnormal CA199 and CEA levels have been found in PanNEC, while serum CA199, CEA, and AFP levels above the upper limit have been observed in 23.8%, 28.6% and 19.0% of PanNEC cases, respectively [69]. Abnormal CA199 and CA125 levels are more commonly found in PDAC than in PanNEC. AFP levels in PanNEC are higher than those found in PDAC. Serum CgA has been shown to have the highest sensitivity for diagnosing patients with PanNET G1 and G2. Elevated serum CgA levels have been found in 50–100% of patients with PanNET and have been shown to be correlated with tumor burden and metastasis in PanNETs [70]. However, CgA is less useful for detecting PanNEC tumors, especially in small cell carcinoma [71]. Instead, neuron-specific enolase has been proven to have higher sensitivity; however, its use is limited by its low specificity [72].
Convergence of clinical management
Standard system therapies vary widely among PDAC, PanNEC and PanNET (Table 2). National Comprehensive Cancer Network treatment guidelines for PanNEC recommend cisplatin and etoposide chemotherapy, with an objective response rate of 41–67% and a median survival of 15–19 months [73]. The standard therapy for PDAC is FOLFIRINOX (5-fluorouracil, irinotecan, and oxaliplatin). FOLIFIRINOX treatment has acceptable side effect profiles and rapid objective response in patients with PanNEC [11]. Somatostatin analogue is standard therapy for PanNETs with dual effects—inhibiting hormone secretion as well as anti-tumor proliferation [56]. The mTOR inhibitor Everolimus is a standard target therapy for PanNETs [74]. The mTOR signaling pathway has also been demonstrated to be active in a small number of PDAC cases, and preclinical data have supported mTORC1 inhibitors in a subgroup of PDAC patients with the hyper-activation of the PI3K-mTOR pathway [75]. However, clinical trials have had disappointing results in PDAC patients treated with Everolimus as a single agent (ORR 0%–6%), or in combination with other agents [76, 77]. The deregulation of the mTOR pathway is not of crucial importance in treatment of PDAC.
Table 2.
Clinical practices compared between PDAC, PanNEC and PanNET
| PDAC | PanNEC | PanNET | |
|---|---|---|---|
| Diagnosis | |||
| Serum test | CA199, CEA,CA125 |
NSE CA199, CEA, AFP |
CgA, 5-HIAA Functional hormone |
| Imaging |
Computer tomography Magnetic resonance imaging F18-FDG PET/CT |
Computer tomography Magnetic resonance imaging F18-FDG PET/CT |
Computer tomography Magnetic resonance imaging Ga68-DOTATATE PET/CT Somatostatin receptor scintigraphy |
| Therapy | |||
| Localized |
Resection Neoadjuvent chemo + resection |
Resection |
Active surveillance Resection |
| Advanced and metastasis |
SYSTEM TREATMENT Irinotecan + Oxaliplatin + fluorouracil Gemcitabine + Albumin-Paclitaxel Clinical trials OTHERS Radiotherapy |
SYSTEM TREATMENT Etoposide + cisplatin Irinotecan + cisplatin Clinical trials OTHERS Radiotherapy |
LOCAL TREATMENT Debulking surgery Transarterial chemoembolization, Ablation of liver metastasis SYSTEM TREATMENT Somatostatin analogs Sunitinib and everolimus Temozolomidecapecitabine |
| Follow-up | 3 months | 3 months | Localized 6-12 months; Metastasis 3-6 months |
Conclusions
Current data support the idea that the tumorigenesis of PanNEC and PDAC malignancies might overlap. The genetic profile of PanNEC is distinct from that of PanNETs, while PanNEC harbors TP53 and RB1 alterations, which overlap in PDAC. The genetic profile of PanNEC also lacks neuroendocrine-related genetic changes, such as mutations in MEN1 and ATRX/DAXX. Additionally, a growing body of evidence suggests that there are overlaps in treatments for PDAC and PanNEC, as well as in their prognoses. Our review was able to show an obvious relationship between conventional PDAC and PanNEC in a subset of cases. These preliminary observations raise practical issues for the proper care of patients, which could become the basis for improved therapy. More studies are needed to further discover the still unknown factors surrounding the intersection of PDAC and PanNEC biology and pathology.
Acknowledgements
None.
Authors’ contributions
HLG Article writing and funding acquisition, HLG and WQW Data collection, XJY and LL Funding acquisition, review, and editing. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Science Foundation for Distinguished Young Scholar of China (81625016), the National Natural Science Foundation of China (81802675, 81871941, 81872366, 81827807, 81701630 and 81702341), the Outstanding Academic Leader Program of the “Technological Innovation Action Plan” of the Shanghai Science and Technology Commission (18XD1401200), the Scientific Innovation Project of the Shanghai Education Committee(2019-01-07-00-07-E00057), and the Natural Science Foundation of Shanghai (19ZR1410800).
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xian-Jun Yu, Email: yuxiangjun@fudanpci.org.
Liang Liu, Email: liuliang@fudanpci.org.
References
- 1.Siegel RL, et al. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Christenson ES, et al. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: a bright future. Lancet Oncol. 2020;21(3):e135–e145. doi: 10.1016/S1470-2045(19)30795-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makohon-Moore A, et al. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16(9):553–565. doi: 10.1038/nrc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cives M, et al. Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J Clin. 2018;68(6):471–487. doi: 10.3322/caac.21493. [DOI] [PubMed] [Google Scholar]
- 5.Sorbye H, et al. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer. 2014;120(18):2814–2823. doi: 10.1002/cncr.28721. [DOI] [PubMed] [Google Scholar]
- 6.Fraenkel M, et al. Epidemiology of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol. 2012;26(6):691–703. doi: 10.1016/j.bpg.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Jiao Y, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jesinghaus M, et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod Pathol. 2017;30(4):610–619. doi: 10.1038/modpathol.2016.220. [DOI] [PubMed] [Google Scholar]
- 9.Bar J, et al. Transformation to small cell lung cancer as a mechanism of resistance to immunotherapy in non-small cell lung cancer. Lung Cancer. 2019;138:109–115. doi: 10.1016/j.lungcan.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 10.Gill, A.J., et al., Pancreatic neuroendocrine neoplasms, 5th edition. The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Vol. 1. IARC, Lyon, pp. 343–372 Digestive System Tumours., 2019.
- 11.Zhu J, et al. Treatment of High-Grade Metastatic Pancreatic Neuroendocrine Carcinoma with FOLFIRINOX. J Gastrointest Cancer. 2015;46(2):166–169. doi: 10.1007/s12029-015-9689-0. [DOI] [PubMed] [Google Scholar]
- 12.Jennings RE, et al. Human pancreas development. Development. 2015;142(18):3126–3137. doi: 10.1242/dev.120063. [DOI] [PubMed] [Google Scholar]
- 13.Mamidi A, et al. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature. 2018;564(7734):114–118. doi: 10.1038/s41586-018-0762-2. [DOI] [PubMed] [Google Scholar]
- 14.Bastidas-Ponce A, et al. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic beta-cells. Mol Metab. 2017;6(6):524–534. doi: 10.1016/j.molmet.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, K., et al., FOXA2 Is Required for Enhancer Priming during Pancreatic Differentiation. Cell Rep. 2019; 28(2): p. 382-393 e7. [DOI] [PMC free article] [PubMed]
- 16.Jennings RE, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62(10):3514–3522. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezania A, et al. Production of functional glucagon-secreting alpha-cells from human embryonic stem cells. Diabetes. 2011;60(1):239–247. doi: 10.2337/db10-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen MBK, et al. Recapitulating and deciphering human pancreas development from human pluripotent stem cells in a dish. Curr Top Dev Biol. 2018;129:143–190. doi: 10.1016/bs.ctdb.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Cejas P, et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med. 2019;25(8):1260–1265. doi: 10.1038/s41591-019-0493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waddell N, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witkiewicz AK, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igarashi T, et al. Evaluation of KRAS mutation status in a patient with concomitant pancreatic neuroendocrine neoplasm and intraductal papillary mucinous neoplasm. Pancreas. 2019;48(5):e34–e35. doi: 10.1097/MPA.0000000000001286. [DOI] [PubMed] [Google Scholar]
- 23.Petersen GM. Familial Pancreatic Adenocarcinoma. Hematol Oncol Clin North Am. 2015;29(4):641–653. doi: 10.1016/j.hoc.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konukiewitz B, et al. Pancreatic neuroendocrine carcinomas reveal a closer relationship to ductal adenocarcinomas than to neuroendocrine tumors G3. Hum Pathol. 2018;77:70–79. doi: 10.1016/j.humpath.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, L.M., Zhang Y, Guo Y et al., Genomic landscape of neuroendocrine neoplasms from gastrointestinal tract, pancreas, lung, rare and unknown primary locations. Neuroendocrinology. 2020. 17th ENETS conference Abstract.
- 26.Konukiewitz B, et al. Somatostatin receptor expression related to TP53 and RB1 alterations in pancreatic and extrapancreatic neuroendocrine neoplasms with a Ki67-index above 20. Mod Pathol. 2017;30(4):587–598. doi: 10.1038/modpathol.2016.217. [DOI] [PubMed] [Google Scholar]
- 27.Hijioka S, et al. Rb Loss and KRAS mutation are predictors of the response to platinum-based chemotherapy in pancreatic neuroendocrine neoplasm with Grade 3: a Japanese multicenter pancreatic NEN-G3 Study. Clin Cancer Res. 2017;23(16):4625–4632. doi: 10.1158/1078-0432.CCR-16-3135. [DOI] [PubMed] [Google Scholar]
- 28.Vijayvergia N, et al. Molecular profiling of neuroendocrine malignancies to identify prognostic and therapeutic markers: a Fox Chase Cancer Center Pilot Study. Br J Cancer. 2016;115(5):564–570. doi: 10.1038/bjc.2016.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enizelos A, E.H., Perren A, Hjortland G et al., Mutational Landscape of 109 High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms G3. Neuroendocrinology, 2020. 17th ENETS conference Abstract.
- 30.Catena L, et al. Mammalian target of rapamycin expression in poorly differentiated endocrine carcinoma: clinical and therapeutic future challenges. Target Oncol. 2011;6(2):65–68. doi: 10.1007/s11523-011-0171-z. [DOI] [PubMed] [Google Scholar]
- 31.Frizziero, M., et al., Mixed Neuroendocrine Non-Neuroendocrine Neoplasms: A Systematic Review of a Controversial and Underestimated Diagnosis. J Clin Med. 2020. 9(1). [DOI] [PMC free article] [PubMed]
- 32.Hosoda W, et al. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015;466(6):665–674. doi: 10.1007/s00428-015-1751-6. [DOI] [PubMed] [Google Scholar]
- 33.Tang LH, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin Cancer Res. 2016;22(4):1011–1017. doi: 10.1158/1078-0432.CCR-15-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corbo V, et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17(3):771–783. doi: 10.1677/ERC-10-0028. [DOI] [PubMed] [Google Scholar]
- 35.Di Domenico A, et al. Genetic and epigenetic drivers of neuroendocrine tumours (NET) Endocr Relat Cancer. 2017;24(9):R315–R334. doi: 10.1530/ERC-17-0012. [DOI] [PubMed] [Google Scholar]
- 36.Singhi AD, et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin Cancer Res. 2017;23(2):600–609. doi: 10.1158/1078-0432.CCR-16-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu W, et al. Gene amplifications in well-differentiated pancreatic neuroendocrine tumors inactivate the p53 pathway. Genes Cancer. 2010;1(4):360–368. doi: 10.1177/1947601910371979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yachida S, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36(2):173–184. doi: 10.1097/PAS.0b013e3182417d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shida T, et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother Pharmacol. 2010;65(5):889–893. doi: 10.1007/s00280-009-1094-6. [DOI] [PubMed] [Google Scholar]
- 40.Tang LH, et al. Attenuation of the retinoblastoma pathway in pancreatic neuroendocrine tumors due to increased cdk4/cdk6. Clin Cancer Res. 2012;18(17):4612–4620. doi: 10.1158/1078-0432.CCR-11-3264. [DOI] [PubMed] [Google Scholar]
- 41.Yachida S, Iacobuzio-Donahue CA. Evolution and dynamics of pancreatic cancer progression. Oncogene. 2013;32(45):5253–5260. doi: 10.1038/onc.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanda, M., et al., Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology, 2012. 142(4): p. 730-733 e9. [DOI] [PMC free article] [PubMed]
- 44.Matthaei H, et al. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8(3):141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghurburrun E, et al. Liver and pancreas: do similar embryonic development and tissue organization lead to similar mechanisms of tumorigenesis? Gene Expr. 2018;18(3):149–155. doi: 10.3727/105221618X15216414278706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guerra C, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Di Domenico A, P.C., Maire R, Bräutigam K., Epigenetic Landscape of Pancreatic Neuro-Endocrine Tumors Reveals Distinct Cells of Origin and Means of Tumor Progression. Neuroendocrinology, 2020. 17th ENETS conference Abstract. [DOI] [PMC free article] [PubMed]
- 48.Sadanandam A, et al. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, metastatic, developmental, and metabolic characteristics. Cancer Discov. 2015;5(12):1296–1313. doi: 10.1158/2159-8290.CD-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hackeng WM, et al. Aberrant Menin expression is an early event in pancreatic neuroendocrine tumorigenesis. Hum Pathol. 2016;56:93–100. doi: 10.1016/j.humpath.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 50.Benten D, et al. Establishment of the first well-differentiated human pancreatic neuroendocrine tumor model. Mol Cancer Res. 2018;16(3):496–507. doi: 10.1158/1541-7786.MCR-17-0163. [DOI] [PubMed] [Google Scholar]
- 51.Yang KC, K.S., Aird J, Lee M, Colborne S, Pancreatic Neuroendocrine Neoplasms: Dissecting the Molecular Heterogeneity. Neuroendocrinology, 2020. 17th ENETS conference Abstract.
- 52.Hunter KE, et al. Identification and characterization of poorly differentiated invasive carcinomas in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS ONE. 2013;8(5):e64472. doi: 10.1371/journal.pone.0064472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kota J, et al. Pancreatic cancer: stroma and its current and emerging targeted therapies. Cancer Lett. 2017;391:38–49. doi: 10.1016/j.canlet.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 54.Erkan M, et al. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11(5):497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sennino B, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2(3):270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pavel M, et al. ENETS Consensus Guidelines for the Standards of Care in neuroendocrine neoplasms: systemic Therapy–Biotherapy and Novel Targeted Agents. Neuroendocrinology. 2017;105(3):266–280. doi: 10.1159/000471880. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi D, et al. Profiling the tumour immune microenvironment in pancreatic neuroendocrine neoplasms with multispectral imaging indicates distinct subpopulation characteristics concordant with WHO 2017 classification. Sci Rep. 2018;8(1):13166. doi: 10.1038/s41598-018-31383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nomi T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13(7):2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 59.Milione M, et al. Microenvironment and tumor inflammatory features improve prognostic prediction in gastro-entero-pancreatic neuroendocrine neoplasms. J Pathol Clin Res. 2019;5(4):217–226. doi: 10.1002/cjp2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.da Silva A, et al. Characterization of the neuroendocrine tumor immune microenvironment. Pancreas. 2018;47(9):1123–1129. doi: 10.1097/MPA.0000000000001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mehnert, JM, et al., Pembrolizumab for the treatment of programmed death-ligand 1-positive advanced carcinoid or pancreatic neuroendocrine tumors: Results from the KEYNOTE-028 study. Cancer, 2020. [DOI] [PubMed]
- 62.Ott PA, et al. T-Cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2019;37(4):318–327. doi: 10.1200/JCO.2018.78.2276. [DOI] [PubMed] [Google Scholar]
- 63.Ugwu JK, Nwanyanwu C, Shelke AR. Dramatic response of a metastatic primary small-cell carcinoma of the pancreas to a trial of immunotherapy with nivolumab: a case report. Case Rep Oncol. 2017;10(2):720–725. doi: 10.1159/000479315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schizas D, et al. Immunotherapy for pancreatic cancer: a 2020 update. Cancer Treat Rev. 2020;86:102016. doi: 10.1016/j.ctrv.2020.102016. [DOI] [PubMed] [Google Scholar]
- 65.Klimstra DS. Nonductal neoplasms of the pancreas. Mod Pathol. 2007;20(Suppl 1):S94–S112. doi: 10.1038/modpathol.3800686. [DOI] [PubMed] [Google Scholar]
- 66.Basturk O, et al. Poorly differentiated neuroendocrine carcinomas of the pancreas: a clinicopathologic analysis of 44 cases. Am J Surg Pathol. 2014;38(4):437–447. doi: 10.1097/PAS.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lv Y, et al. Combined test of serum CgA and NSE improved the power of prognosis prediction of NF-pNETs. Endocr Connect. 2018;7(1):169–178. doi: 10.1530/EC-17-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nielsen K, et al. P53, Somatostatin receptor 2a and Chromogranin A immunostaining as prognostic markers in high grade gastroenteropancreatic neuroendocrine neoplasms. BMC Cancer. 2020;20(1):27. doi: 10.1186/s12885-019-6498-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhuge X, et al. The levels of tumor markers in pancreatic neuroendocrine carcinoma and their values in differentiation between pancreatic neuroendocrine carcinoma and pancreatic ductal adenocarcinoma. Pancreas. 2018;47(10):1290–1295. doi: 10.1097/MPA.0000000000001181. [DOI] [PubMed] [Google Scholar]
- 70.Cimitan M, et al. Somatostatin receptor scintigraphy versus chromogranin A assay in the management of patients with neuroendocrine tumors of different types: clinical role. Ann Oncol. 2003;14(7):1135–1141. doi: 10.1093/annonc/mdg279. [DOI] [PubMed] [Google Scholar]
- 71.Korse CM, et al. Chromogranin-A and N-terminal pro-brain natriuretic peptide: an excellent pair of biomarkers for diagnostics in patients with neuroendocrine tumor. J Clin Oncol. 2009;27(26):4293–4299. doi: 10.1200/JCO.2008.18.7047. [DOI] [PubMed] [Google Scholar]
- 72.Korse, C.M., et al., Choice of tumour markers in patients with neuroendocrine tumours is dependent on the histological grade. A marker study of Chromogranin A, Neuron specific enolase, Progastrin-releasing peptide and cytokeratin fragments. Eur J Cancer. 2012. 48(5): p. 662-71. [DOI] [PubMed]
- 73.NCCN, Neuroendocrine and Adrenal Tumors. NCCN Guidelines Version 4. 2018. NCCN Clinical Practice Guidelines in Oncology, 2018.
- 74.Yao JC, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. 2016;34(32):3906–3913. doi: 10.1200/JCO.2016.68.0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morran DC, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63(9):1481–1489. doi: 10.1136/gutjnl-2013-306202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Javle MM, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Okuyama, H., et al., A phase II trial of everolimus in patients with advanced pancreatic neuroendocrine carcinoma refractory or intolerant to platinum-containing chemotherapy (NECTOR trial). Neuroendocrinology. 2020. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.