

Abstract
The aim of the present study was to explore the potential anticonvulsant effects of β-hydroxybutyrate (BHB) in a kainic acid (KA)-induced rat epilepsy model. The KA-induced rat seizure model was established and BHB was administrated intraperitoneally at a dose of 4 mmol/kg 30 min prior to KA injection. Hippocampal tissues were then obtained 1, 3 and 7 days following KA administration, following which the expression levels of neuron-specific enolase (NSE) and glial fibrillary acidic protein (GFAP) were measured using a double immunofluorescence labeling method. In addition, the contents of glutathione (GSH), γ-aminobutyric acid (GABA) and ATP were measured using ELISA. Pretreatment with BHB markedly increased the expression of NSE after KA injection compared with that in the normal saline (NS) + KA group, suggesting that the application of BHB could alleviate neuronal damage in rats. The protective effect of BHB may be associated with suppressed inflammatory responses, which was indicated by the observed inhibition of GFAP expression in rats in the BHB + KA group compared with that in the NS + KA group. It was also found that GSH and GABA contents were notably increased after the rats were pretreated with BHB compared with those in the NS + KA group. To conclude, the application of exogenous BHB can serve as a novel therapeutic agent for epilepsy.
Keywords: β-hydroxybutyrate, kainic acid, epilepsy, neuron damage
Introduction
Epilepsy is a group of neurological disorders that is characterized by epileptic seizures (1,2). As of 2015, ~39 million people were suffering from epilepsy (3), and it has been reported that ~80% of cases occur in the developing world (4). Epilepsy resulted in 125,000 deaths in 2015, compared with 112,000 in 1990 (5,6). Specifically, children in the 5-9 years age group are particularly susceptible to morbidity associated with active epilepsy (7). Currently, seizures can be controlled with medication in ~70% patients (8). However, for the remaining ~30% patients with epilepsy, seizures cannot be controlled with drugs due to adverse reactions (9,10). Therefore, it remains essential to explore novel treatment strategies for epilepsy.
A number of studies have previously demonstrated that β-hydroxybutyrate (BHB) may serve an important role in epilepsy progression. Suzuki et al (11) found that BHB induced by a ketogenic diet (KD) may increase the concentration of γ-aminobutyric acid (GABA) in the epileptic brain by inhibiting astrocytic GABA degradation, which may account for its antiepileptic effects. Samoilova et al (12) showed that BHB is more suitable for treating epilepsy associated with metabolic disorders compared with that caused by KD. Additionally, BHB has been found to prevent neuronal injury induced by glutamate-mediated lipid oxidation and glycolysis inhibition (13). The administration of BHB improved glutamate transport in the brain and conferred an anticonvulsant effect (14). Abdelmalik et al (15) found that pretreatment with BHB reduced the frequency of seizures induced by acute hypoglycemia. BHB has also been reported to exhibit anticonvulsant effects on epileptic models induced by pilocarpine, flurothyl and 4-aminopyridine (16-19).
A previous study demonstrated that exogenous BHB administration at a dose of 4 mmol/kg served as an alternative to KD in exerting protective effects in a kainic acid (KA)-induced the epilepsy model (20). Therefore, in the present study, the potential antiepileptic effects of exogenous BHB on KA-induced epilepsy were explored further. The expression levels of neuro-specific enolase (NSE) and glial fibrillary acidic protein (GFAP) were evaluated using double immunofluorescence labeling, whilst the contents of glutathione (GSH), GABA and ATP were measured using ELISA.
Materials and methods
Animals
A total of 60 male Wistar rats (age, 3 weeks; weight, 60±10 g) were obtained from The Shandong University Animal Center. Rats had free access to food and tap water and were housed at a standard temperature (22±1˚C) and humidity (50±5%) under a 12-h light/dark cycle. The rats were maintained under standard housing conditions until the time of the experiment. The present study was approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine (Shanghai, China). All experimental procedures were conducted according to the National Institute of Health Guidelines (21).
Establishment of the KA-induced rat epilepsy model
On postnatal day 21, 60 Wistar rats were randomly assigned into the following four groups (n=15 rats in each group): i) normal saline (NS); ii) NS + KA; iii) BHB + KA; and iv) BHB groups. Rats in the BHB and NS groups were injected with 4 mmol/kg BHB (1 mmol/ml, cat. no. H6501; Sigma-Aldrich, Merck KGaA) or 4 ml/kg NS, respectively. Rats in the BHB + KA group were pretreated with 4 mmol/kg BHB (1 mmol/ml) that was administered intraperitoneally 30 min prior to KA (10 mg/kg; cat. no. K0250; Sigma-Aldrich, Merck KGaA) injection intraperitoneally. Rats in the NS + KA group were administered NS intraperitoneally 30 min prior to KA injection. Selection of the BHB dose was based upon a previous study (20). Seizure behavior of rats was analyzed for 2 h, 1 h after KA administration according to the scale previously devised by Racine (22): i) stage I, facial clonus; ii) stage II, head nodding or wet dog shaking; iii) stage III, forelimbs clonus; iv) stage IV, rearing forelimbs; and v) stage V, rearing, jumping or falling. Rats which presented with seizure behaviors of stages ≥IV were considered to be epileptic. If the status epilepticus continued for >90 min, 10% chloral hydrate (400 mg/kg; Sigma-Aldrich; Merck KGaA) was injected intraperitoneally to stop seizure behavior. No rat exhibited any sign of peritonitis after chloral hydrate injection.
NSE and GFAP expression
At 1, 3 and 7 days after KA administration (n=5 rats at each time point), rats were anesthetized with 10% chloral hydrate (400 mg/kg, intraperitoneal injection, n=5 rats at each time point in each group) and decapitated before their skulls were immediately cut open. No rats exhibited signs of pain after the administration of chloral hydrate. The left hemisphere of the brain was then obtained and immediately fixed in 4% paraformaldehyde for 24 h at 4˚C, which was embedded in paraffin and 4-µm thick coronal paraffin sections were prepared for staining. The expression levels of NSE and GFAP in the hippocampal tissues were assessed using a double immunofluorescence labeling method. Coronal paraffin sections were dewaxed successively in xylene for 10 min twice and then rehydrated using a descending ethanol gradient before 0.3% Triton X-100 was added for 15 min at 37˚C. After blocking with 5% bovine serum albumin (Beijing Zhongshan Jinqiao Biotechnology Co. Ltd; OriGene Technologies, Inc.) for 1 h at 37˚C, the coronal paraffin sections were incubated with a rabbit NSE antibody (1:100 dilution; cat. no. ab79757; Abcam) and goat GFAP antibody (1:100 dilution; cat. no. ab53554; Abcam) overnight at 4˚C. After washing three times, the sections were incubated with DyLight® 488-conjugated AffiniPure donkey anti-rabbit IgG H + L (1:1,000 dilution; cat. no. ab96919; Abcam) and Alexa Fluor 647-conjugated AffiniPure donkey anti-goat IgG H + L (1:1,000 dilution; cat. no. A21447; Life Technologies; Thermo Fisher Scientific, Inc.) secondary antibodies for 2 h at room temperature. DAPI (100 ng/ml; Beijing Solarbio Science & Technology Co., Ltd.) was used to stain the nucleus for 15 min at room temperature. After washing for a further three times, the sections were observed under a fluorescence microscope at x400 magnification (Olympus Corporation), with three view fields of view taken per section. From the images, the mean optical density of NSE- and GFAP-positive fibers was measured using the ImageJ (version 1.49; National Institute of health) program to assess changes in neuron and astrocyte content in the rat brains, respectively.
GSH and GABA content
Hippocampal tissues were removed from the right hemisphere 1, 3 and 7 days after KA administration and immediately stored at -80˚C. The frozen hippocampal tissues were defrosted to room temperature and 9X weight of cold NS was added to the tissues and grind was done in ice-cold NS. After the cells were fragmented, 10% homogenized hippocampal tissue (the ratio of tissue:NS was 1:9) was centrifuged for 15 min at 510 x g at 4˚C. The supernatant was then obtained for subsequent experimentation. Using a Bio-Rad Model 450 microplate reader (Bio-Rad Laboratories, Inc.), GSH (cat. no. CEA294Ge) and GABA (cat. no. CEA900Ge) contents were measured using the corresponding ELISA kits (Cloud-Clone Corp.) according to the manufacturer's protocols.
Statistical analysis
Statistical analyses were performed using the SPSS software 20.0 (IBM Corp.). All data are presented as the mean ± standard error of the mean. Two-way analysis of variance was used to analyze the main effect of treatment, the main effect of time, and the interaction between treatment and time. Significant differences between specific groups were analyzed using Bonferroni corrections. In the present study, P<0.05 was considered to indicate a statistically significant difference. All experiments were performed in triplicate.
Results
NSE and GFAP expression
The expression levels of NSE and GFAP were evaluated using double immunofluorescence (Figs. 1-3). For NSE, the interaction between time and treatment revealed no statistically significant difference, whilst the main effect of the treatment factor was statistically significant (P<0.01). After KA administration, NSE expression was found to be significantly lower in the NS + KA group compared with that in the NS group (D1, P<0.01; D3, P<0.05) and the BHB group (D1, D3, P<0.01; D7, P<0.05; Fig. 4A and Table SI). By contrast, the expression of NSE was revealed to be significantly higher in the BHB + KA group compared with that in the NS + KA group (P<0.05) after 1 day of KA administration (Fig. 4A and Table SI). No significant differences in NSE expression among different time points were observed, suggesting that time exerted little influence on NSE expression.
Figure 1.

Immunofluorescence staining of NSE and GFAP in the hippocampus of rats 1 day after KA injection. Green signals represent NSE, red signals represent GFAP and the blue signals represent the cell nuclei stained with DAPI. Scale bar, 50 µm. NSE, neuron specific enolase; GFAP, glial fibrillary acidic protein; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline.
Figure 2.

Immunofluorescence staining of NSE and GFAP in the hippocampus tissues of rats 3 days after KA injection. Green signals represent NSE, red signals represent GFAP and blue signals represent the cell nuclei stained with DAPI. Scale bar, 50 µm. NSE, neuron specific enolase; GFAP, glial fibrillary acidic protein; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline.
Figure 3.
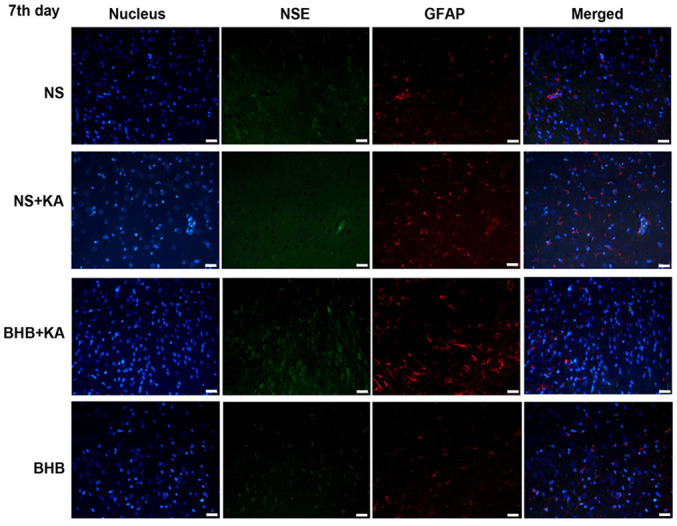
Immunofluorescence staining of NSE and GFAP in the hippocampus tissues of rats 7 days after KA injection. Green signals represent NSE, red signals represent GFAP and blue signals represent the cell nuclei stained with DAPI. Scale bar, 50 µm. NSE, neuron specific enolase; GFAP, glial fibrillary acidic protein; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline.
Figure 4.
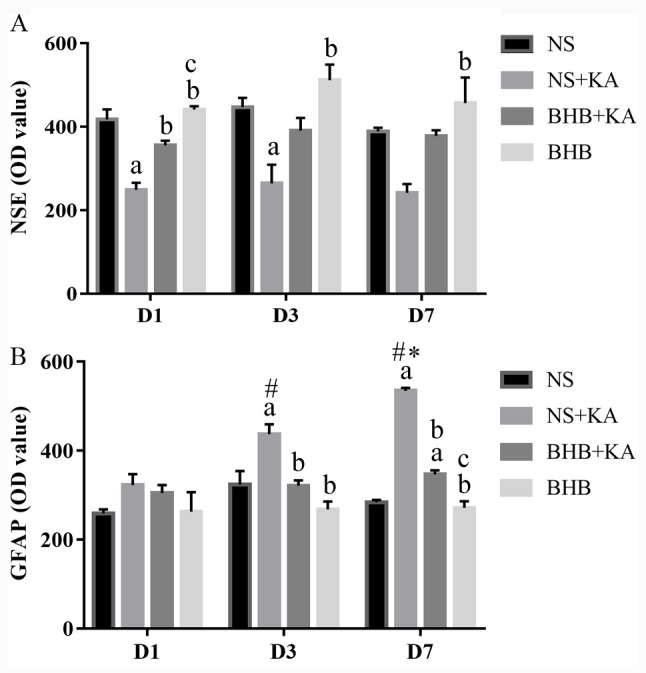
Expression levels of NSE and GFAP in the hippocampal tissue. (A) NSE expression 1, 3 and 7 days after different treatments. KA administration decreased the NSE expression (D1 and D3), whilst BHB alleviated this reduction (D1). (B) GFAP expression 1, 3 and 7 days after different treatments. KA administration increased the GFAP expression but pretreatment with BHB significantly reduced GFAP expression (D3 and D7). aP<0.05 vs. NS; bP<0.05 vs. NS+KA; cP<0.05 vs. BHB+KA; #P<0.05 vs. D1 NS + KA; *P<0.05 vs. D3 NS + KA; OD, optical density; NSE, neuron specific enolase; GFAP, glial fibrillary acidic protein; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline.
The interaction between treatment and time on GFAP expression showed significant differences (P<0.01), whilst the main effect of treatment on GFAP expression was also found to be significant (P<0.01). After 3 and 7 days of KA administration, GFAP expression was significantly higher in the NS + KA group compared with that in the NS (D3, P<0.05; D7, P<0.01) and BHB groups (both P<0.01, Fig. 4B), whilst the expression of GFAP was significantly decreased in the BHB + KA group compared with that in the NS + KA group (D3, P<0.05; D7, P<0.01; Fig. 4B and Table SII). However, after 7 days of KA administration, GFAP expression was significantly higher in the BHB + KA compared with that in BHB group (P<0.01; Fig. 4B and Table SII). In addition, time was also found to significantly exert influence on GFAP expression (P<0.01). Within the NS + KA groups, GFAP expression increased along with time (P<0.05, D1 vs. D3, D3 vs. D7; P<0.01, D1 vs. D7). By contrast, there was no significant difference among the different time points in NS, BHB+KA and BHB groups.
These results suggested that KA can cause neuron damage and compensatory astrocyte hyperplasia, which can be reversed by BHB treatment. There were no differences in NSE and GFAP expression between the BHB and NS groups at any point in time, indicating that BHB did not exert toxic effects on the brain tissues.
GSH content
There was no difference in the interaction between time and treatment on GSH contents, where the main effect of time also did not reveal significant influence. At 1 and 7 days after KA administration, GSH content was found to be significantly lower in the NS + KA groups compared with that in the NS groups (both P<0.01) and the BHB groups (both P<0.01; Fig. 5 and Table SIII). GSH levels were also revealed to be significantly higher in the BHB + KA group compared with those in the NS + KA groups after 1 and 7 days (both P<0.01; Fig. 5 and Table SIII). These results suggested that BHB can alleviate the reduction in GSH caused by KA administration in rats. In addition, no differences in the GSH contents were observed between the BHB and NS groups, implicating the safety of BHB.
Figure 5.
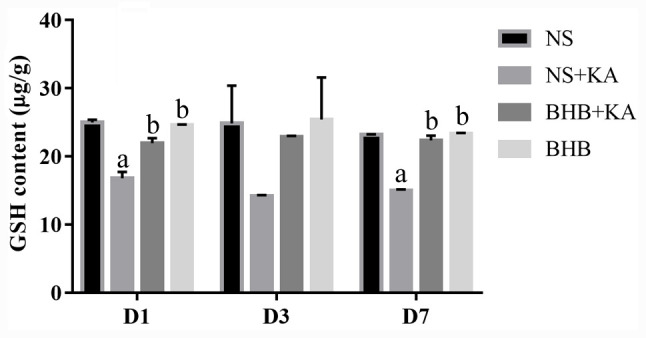
GSH contents in the hippocampal tissues as measured using ELISA after 1, 3 and 7 days of different treatments. GSH contents were decreased after KA administration whilst pretreatment with BHB alleviated this reduction (D1 and D7). aP<0.05 vs. NS; bP<0.05 vs. NS+KA; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline; GSH, glutathione.
GABA contents
The interaction between time and treatment showed no significant influence on GABA levels, where the main effect of time also did not reveal statistical influence. After 1, 3 and 7 days of KA administration, GABA levels were significantly reduced in the NS + KA groups compared with those in the NS groups (all P<0.01) and the BHB groups (all P<0.01; Fig. 6 and Table SIV). At all three time points, following pretreatment with BHB, the GABA contents were found to be significantly higher in the BHB + KA group compared with those in the NS + KA group (D1 and D7, P<0.01; D3, P<0.05; Fig. 6 and Table SIV). However, in the BHB + KA group, GABA contents remained significantly decreased compared with those in NS (P<0.01) and BHB groups (P<0.01) 1 day after KA administration (Fig. 6 and Table SIV). These results demonstrated that KA administration reduced GABA levels whilst BHB alleviated this decrease in GABA caused by KA treatment in rats. No differences were found between BHB and NS groups in terms of GABA levels.
Figure 6.

GABA contents in the hippocampal tissues as measured using ELISA after 1, 3 and 7 days of different treatments. GABA contents were reduced by KA administration whilst BHB pretreatment relieved this reduction (D1, D3 and D7). aP<0.05 vs. NS; bP<0.05 vs. NS + KA; cP<0.05 vs. BHB + KA; GABA, gamma-aminobutyric acid; BHB, β-hydroxybutyrate; KA, kainic acid; NS, normal saline.
Discussion
Epilepsy is one of the most prevalent serious neurological disorders, for which it is important to develop novel effective therapies. Previous studies have documented exogenous BHB to be an anticonvulsant that exerts neuroprotective effects both in vitro and in vivo (12,23). In the present study, the antiepileptic effects of BHB in a KA-induced epilepsy rat model were explored. Neuronal damage in the hippocampus was demonstrated to be alleviated after rats were pretreated with BHB. Additionally, the present study revealed that BHB was capable of blocking the activation of astrocytes whilst preserving the expression of GSH and GABA after KA administration.
NSE levels have been previously reported to be applicable for determining seizure durations and to estimate the prognosis of brain injuries (24). The NSE contents were found to be significantly higher in the serum of children with epilepsy compared with those in unaffected children, suggesting that elevated serum NSE after epileptic seizures may be associated with brain damage (25,26). GFAP is a glial cell marker in the development of the central nervous system that is mainly expressed in activated astrocytes (27,28). After epileptic seizure attacks, GFAP expression was previously revealed to be significantly elevated in astrocytes (29). In the present study, NSE and GFAP were used to stain neurons and activated astrocytes respectively, where KA injection resulted in an inflammatory environment in rat brains, as indicated by the extensive activation of glial cells. In addition, the number of neurons was found to be increased in the BHB + KA group compared with that in the NS + KA group whilst the degree of astrocyte activation was reduced. These results indicated that neuronal damage induced by KA was alleviated after the rats were pretreated with BHB, which may be due in part to its ability to inhibit the activation of glial cells.
It has been previously shown that oxidative stress is one of the main pathological mechanisms of epilepsy (30,31). During the progression of epilepsy, reactive oxygen species (ROS) can damage the cell membrane, proteins, enzymes and DNA components within the nucleus and the mitochondria (32). GSH is a part of the main antioxidant system that neutralizes the excessive ROS. GSH is capable of preventing damage to important cellular components caused by ROS, including free radicals, peroxides, lipid peroxides and heavy metals (33). Previous studies have demonstrated that the elimination of GSH is closely associated with a number of human diseases, including neurodegenerative diseases, diabetes and acquired immune deficiency syndrome (34,35). The present study showed that GSH levels in the hippocampal tissues were significantly reduced after rats were treated with KA, suggesting that the ability to eliminate free radicals is reduced in epilepsy. Results from the present study also revealed that administration of BHB reversed the reduction in GSH caused by KA administration in the rat hippocampus. Therefore, it can be potentially concluded that BHB can diminish ROS damage caused by KA by preserving GSH levels. This is in accordance with a previous study that also showed that BHB treatment can reduce the overproduction of ROS and activate GSH further in the epileptic hippocampus (36).
GABA is the main inhibitory neurotransmitter in the central nervous system that serves a critical role in the development of epilepsy (37,38). Increased GABA synaptic activity can reduce the excitability of neurons (39), whilst a reduced GABA level can enhance the excitability of neurons (37). GABAA receptors are ligand-gated ion channels that hyperpolarize neurons by increasing inward chloride conductance (38). Since the activation of these receptors results in a rapid inhibitory effect, they serve a principal role in nerve transmission processes in the central nervous system (38). GABAB receptors can reduce calcium entry and mainly mediate slow synaptic inhibition, which is involved with numerous types of epilepsy and cognitive impairment (37,38). It was demonstrated that ketones can alter glutamate metabolism by increasing GABA synthesis, which would in turn dampen seizure activity (40). It has also been previously demonstrated that BHB can reduce the incidence of seizure-like activity in a GABAB-dependent manner (41). In the present study, GABA levels in the hippocampal tissues were markedly reduced after rats were treated with KA. This reduction in GABA can increase the excitability of the neurons, thereby reducing the threshold of epileptic seizures. GABA levels in the hippocampus tissue were significantly increased after the rats were pretreated with BHB. These results suggest that elevations in the levels of GABA following the application of BHB can dampen seizure activity. In the present study, a correlation analysis between the GABA content and the number of neurons and astrocytes was not performed, which would be of significance for understanding the mechanism of BHB further. This is a limitation of the present study.
It has been previously demonstrated that BHB is a more efficient energy source compared with glucose and that the presence of BHB can reduce ATP production from glycolysis (42,43). Glycolytic ATP is the primary source of energy that supports plasma membrane functions, including ATP-sensitive potassium (KATP) channels. Lower glycolytic ATP levels would lead to higher KATP channel opening probability, which would cause membrane hyperpolarization and reduce the influx of calcium via voltage-gated calcium channels. This would in turn reduce the release of excitatory amino acids and decreased neuron excitability (44-46). KATP channels, which are widely distributed in the hippocampus, would open with higher probability in the presence of BHB, which may underlie the anticonvulsive effects of ketone bodies.
In the present study, only the ATP contents in the BHB group were found to be greater compared with that of the detection threshold on day 7 (46.26±0.81 ng/g). This experiment could not detect ATP in other experimental groups. Considering the rapid degradation of ATP during the tissue preparation process, the frozen hippocampus tissues might have been the main cause of this. Due to the significant elevations in ATP production, ATP could still be detected in the BHB group despite its rapid degradation.
Recently, several studies have demonstrated that BHB confers neuroprotective effects on the central nervous system against oxygen toxicity, Alzheimer's and Parkinson's disease (47-49). Although a series of studies have demonstrated that BHB has protective effects in various epileptic models (9,20,36,50), it remains necessary to verify the effects of BHB in other epileptic models. Additionally, it is difficult to maintain stable BHB concentrations in the blood, which limits the efficacy of BHB administration for clinical application (45). Therefore, further studies focused on BHB treatment for antiepileptic therapy are required to confirm its efficacy and explore the underlying mechanisms.
Taken together, the similarity between the results mediated by BHB and KD in epileptic models suggest that exogenous BHB could replace KD as an anticonvulsant treatment for epilepsy. In particular, there are some limitations of KD applications, including nausea, constipation and abdominal pain (51). By contrast, BHB administration has not been reported to cause adverse effects, which may improve the patients' quality of life. Therefore, the application of exogenous BHB may serve as a novel therapeutic technique in treating epilepsy. However, it is essential to explore the therapeutic effect of exogenous BHB further in the future.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The present study was supported by a project of the Shandong Province Science and Technology Program (grant no. 2014GSF118179) and the Special Foundation for Taishan Scholars (grant no. ts20110814).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
JW designed the study and supervised the project. JS performed the experiments and completed the manuscript. YW contributed to the acquisition, analysis and interpretation of data for the study. JX performed the statistical analyses, helped with supervising the whole project and was accountable for revision of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chang BS, Lowenstein DH. Epilepsy. N Engl J Med. 2003;349:1257–1266. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- 2.Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- 3.Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/S0140-6736(16)31678-6. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kernich CA. Patient and family fact sheet Epilepsy. Neurologist. 2003;9:265–266. doi: 10.1097/01.nrl.0000087837.81229.b8. [DOI] [PubMed] [Google Scholar]
- 5.Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1459–1544. doi: 10.1016/S0140-6736(16)31012-1. GBD 2015 Mortality and Causes of Death Collaborators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. GBD 2013 Mortality and Causes of Death Collaborators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodie MJ, Elder AT, Kwan P. Epilepsy in later life. Lancet Neurol. 2009;8:1019–1030. doi: 10.1016/S1474-4422(09)70240-6. [DOI] [PubMed] [Google Scholar]
- 8.Eadie MJ. Shortcomings in the current treatment of epilepsy. Expert Rev Neurother. 2012;12:1419–1427. doi: 10.1586/ern.12.129. [DOI] [PubMed] [Google Scholar]
- 9.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 10.Wei CX, Bian M, Gong GH. Current research on antiepileptic compounds. Molecules. 2015;20:20741–20776. doi: 10.3390/molecules201119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki Y, Takahashi H, Fukuda M, Hino H, Kobayashi K, Tanaka J, Ishii E. β-Hydroxybutyrate alters GABA-transaminase activity in cultured astrocytes. Brain Res. 2009;1268:17–23. doi: 10.1016/j.brainres.2009.02.074. [DOI] [PubMed] [Google Scholar]
- 12.Samoilova M, Weisspapir M, Abdelmalik P, Velumian AA, Carlen PL. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem. 2010;113:826–835. doi: 10.1111/j.1471-4159.2010.06645.x. [DOI] [PubMed] [Google Scholar]
- 13.Maalouf M, Rho JM. Oxidative impairment of hippocampal long-term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J Neurosci Res. 2008;86:3322–3330. doi: 10.1002/jnr.21782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Likhodii SS, Burnham WM. Ketogenic diet: Does acetone stop seizures? Med Sci Monit. 2002;8:HY19–HY24. [PubMed] [Google Scholar]
- 15.Abdelmalik PA, Shannon P, Yiu A, Liang P, Adamchik Y, Weisspapir M, Samoilova M, Burnham WM, Carlen PL. Hypoglycemic seizures during transient hypoglycemia exacerbate hippocampal dysfunction. Neurobiol Dis. 2007;26:646–660. doi: 10.1016/j.nbd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Yum MS, Ko TS, Dong WK. Anticonvulsant effects of β-hydroxybutyrate in mice. J Epilepsy Res. 2012;2:29–32. doi: 10.14581/jer.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yum MS, Ko TS, Kim DW. β-Hydroxybutyrate increases the pilocarpine-induced seizure threshold in young mice. Brain Dev. 2012;34:181–184. doi: 10.1016/j.braindev.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Minlebaev M, Khazipov R. Antiepileptic effects of endogenous beta-hydroxybutyrate in suckling infant rats. Epilepsy Res. 2011;95:100–109. doi: 10.1016/j.eplepsyres.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Thio LL, Wong M, Yamada KA. Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology. 2000;54:325–331. doi: 10.1212/wnl.54.2.325. [DOI] [PubMed] [Google Scholar]
- 20.Si J, Wang S, Liu N, Yang X, Wang Y, Li L, Wang J, Lv X. Anticonvulsant effect of exogenous β-hydroxybutyrate on kainic acid-induced epilepsy. Exp Ther Med. 2017;14:765–770. doi: 10.3892/etm.2017.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. National Research Council: Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press (US); 2011. Available from: https://www.ncbi.nlm.nih.gov/books/NBK54050/ doi: 10.17226/12910ß. [Google Scholar]
- 22.Racine RJ. Modification of seizure activity by electrical stimulation II Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 23.Xie G, Tian W, Wei T, Liu F. The neuroprotective effects of β-hydroxybutyrate on Aβ-injected rat hippocampus in vivo and in Aβ-treated PC-12 cells in vitro. Free Radic Res. 2015;49:139–150. doi: 10.3109/10715762.2014.987274. [DOI] [PubMed] [Google Scholar]
- 24.Bindra A, Kaushal A, Prabhakar H, Chaturvedi A, Chandra PS, Tripathi M, Subbiah V, Sathianathan S, Banerjee J, Prakash C. Neuroprotective role of dexmedetomidine in epilepsy surgery: A preliminary study. Neurol India. 2019;67:163–168. doi: 10.4103/0028-3886.253616. [DOI] [PubMed] [Google Scholar]
- 25.Maiti R, Mishra BR, Sanyal S, Mohapatra D, Parida S, Mishra A. Effect of carbamazepine and oxcarbazepine on serum neuron-specific enolase in focal seizures: A randomized controlled trial. Epilepsy Res. 2017;138:5–10. doi: 10.1016/j.eplepsyres.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Yardimoğlu M, Ilbay G, Dalcik C, Dalcik H, Sahin D, Ates N. Immunocytochemistry of neuron specific enolase (NSE) in the rat brain after single and repeated epileptic seizures. Int J Neurosci. 2008;118:981–993. doi: 10.1080/00207450701769232. [DOI] [PubMed] [Google Scholar]
- 27.Jacque CM, Vinner C, Kujas M, Raoul M, Racadot J, Baumann NA. Determination of glial fibrillary acidic protein (GFAP) in human brain tumors. J Neurol Sci. 1978;35:147–155. doi: 10.1016/0022-510x(78)90107-7. [DOI] [PubMed] [Google Scholar]
- 28.Venkatesh K, Srikanth L, Vengamma B, Chandrasekhar C, Sanjeevkumar A, Mouleshwara Prasad BC, Sarma PV. In vitro differentiation of cultured human CD34+ cells into astrocytes. Neurol India. 2013;61:383–388. doi: 10.4103/0028-3886.117615. [DOI] [PubMed] [Google Scholar]
- 29.Alese OO, Mabandla MV. Upregulation of hippocampal synaptophysin, GFAP and mGluR3 in a pilocarpine rat model of epilepsy with history of prolonged febrile seizure. J Chem Neuroanat. 2019;100(101659) doi: 10.1016/j.jchemneu.2019.101659. [DOI] [PubMed] [Google Scholar]
- 30.Puttachary S, Sharma S, Stark S, Thippeswamy T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed Res Int. 2015;2015(745613) doi: 10.1155/2015/745613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuen AWC, Keezer MR, Sander JW. Epilepsy is a neurological and a systemic disorder. Epilepsy Behav. 2018;78:57–61. doi: 10.1016/j.yebeh.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Pearson-Smith JN, Patel M. Metabolic dysfunction and oxidative stress in epilepsy. Int J Mol Sci. 2017;18(2365) doi: 10.3390/ijms18112365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–1503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 34.Lutchmansingh FK, Hsu JW, Bennett FI, Badaloo AV, McFarlane-Anderson N, Gordon-Strachan GM, Wright-Pascoe RA, Jahoor F, Boyne MS. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS One. 2018;13(e0198626) doi: 10.1371/journal.pone.0198626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sprietsma JE. Cysteine, glutathione (GSH) and zinc and copper ions together are effective, natural, intracellular inhibitors of (AIDS) viruses. Med Hypotheses. 1999;52:529–538. doi: 10.1054/mehy.1997.0689. [DOI] [PubMed] [Google Scholar]
- 36.Guo Q, Liu S, Wang S, Wu M, Li Z, Wang Y. Beta-hydroxybutyric acid attenuates neuronal damage in epilepticmice. Acta Histochem. 2019;121:455–459. doi: 10.1016/j.acthis.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Bhagat K, Singh JV, Pagare PP, Kumar N, Sharma A, Kaur G, Kinarivala N, Gandu S, Singh H, Sharma S, Bedi PMS. doi: 10.1007/s11030-020-10068-4. Rational approaches for the design of various GABA modulators and their clinical progression. Mol Divers 2020 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42 (Suppl 3):S8–S12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x. [DOI] [PubMed] [Google Scholar]
- 39.Cepeda C, Levinson S, Nariai H, Yazon VW, Tran C, Barry J, Oikonomou KD, Vinters HV, Fallah A, Mathern GW, Wu JY. Pathological high frequency oscillations associate with increased GABA synaptic activity in pediatric epilepsy surgery patients. Neurobiol Dis. 2020;134(104618) doi: 10.1016/j.nbd.2019.104618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simeone TA, Simeone KA, Rho JM. Ketone bodies as anti-seizure agents. Neurochem Res. 2017;42:2011–2018. doi: 10.1007/s11064-017-2253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, O'Leary EI, Tanner GR. The ketogenic diet metabolite beta-hydroxybutyrate (β-HB) reduces incidence of seizure-like activity (SLA) in a Katp- and GABAb-dependent manner in a whole-animal Drosophila melanogaster model. Epilepsy Res. 2017;133:6–9. doi: 10.1016/j.eplepsyres.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Mejía-Toiber J, Montiel T, Massieu L. D-beta-hydroxybutyrate prevents glutamate-mediated lipoperoxidation and neuronal damage elicited during glycolysis inhibition in vivo. Neurochem Res. 2006;31:1399–1408. doi: 10.1007/s11064-006-9189-5. [DOI] [PubMed] [Google Scholar]
- 43.Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal out-comes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care. 2016;39:1115–1122. doi: 10.2337/dc16-0542. [DOI] [PubMed] [Google Scholar]
- 44.Lund TM, Ploug KB, Iversen A, Jensen AA, Jansen-Olesen I. The metabolic impact of β-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem. 2015;132:520–531. doi: 10.1111/jnc.12975. [DOI] [PubMed] [Google Scholar]
- 45.Tanner G, Lutas A, Martínez-François JR, Yellen G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci. 2011;31:8689–8696. doi: 10.1523/JNEUROSCI.5951-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74:719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soto-Mota A, Norwitz NG, Clarke K. Why a d-β-hydroxybutyrate monoester? Biochem Soc Trans. 2020;48:51–59. doi: 10.1042/BST20190240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishnan M, Hwang JS, Kim M, Kim YJ, Seo JH, Jung J, Ha E. β-hydroxybutyrate impedes the progression of Alzheimer's disease and atherosclerosis in ApoE-deficient mice. Nutrients. 2020;12(471) doi: 10.3390/nu12020471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norwitz NG, Hu MT, Clarke K. The mechanisms by which the ketone body D-β-hydroxybutyrate may improve the multiple cellular pathologies of Parkinson's disease. Front Nutr. 2019;6(63) doi: 10.3389/fnut.2019.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yum MS, Lee M, Woo DC, Kim DW, Ko TS, Velíšek L. β-Hydroxybutyrate attenuates NMDA-induced spasms in rats with evidence of neuronal stabilization on MR spectroscopy. Epilepsy Res. 2015;117:125–132. doi: 10.1016/j.eplepsyres.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Giordano C, Marchiò M, Timofeeva E, Biagini G. Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front Neurol. 2014;5(63) doi: 10.3389/fneur.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.