

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is an intestinally derived peptide that is secreted in response to feeding. The GIP receptor (GIPR) is expressed in many cell types involved in the regulation of metabolism, including α- and β-cells. Glucagon and insulin exert tremendous control over glucose metabolism. Thus, GIP action in islets strongly dictates metabolic control in the postprandial state. Loss of GIPR activity in β-cells is a characteristic of type 2 diabetes (T2D) which associates with reduced postprandial insulin secretion and hyperglycemia. Less is known about GIPR activity in α-cells or the control of glucagon secretion. GIP stimulates glucagon secretion in a glucose-dependent manner in healthy people, with enhanced activity at lower glycemia. However, GIP stimulates glucagon secretion even at hyperglycemia in people with T2D, suggesting that inappropriate GIPR activity in α-cells contributes to the pathogenesis of T2D. Here, we review the literature describing GIP action and GIPR activity in the α-cell, detailing the basic science that has shaped the view of how GIP regulates glucagon secretion. We also contrast the effects of GIP on glucagon secretion in healthy and T2D people. Finally, we contextualize these observations in light of recent work that redefines the role of glucagon in glucose homeostasis, suggesting that hyperglucagonemia per se does not drive hyperglycemia. As new medications for T2D that incorporate GIPR activity are being developed, it is clear that a better understanding of GIPR activity beyond the β-cell is necessary. This work highlights the importance of focusing on the GIPR in α-cells.
Keywords: Glucose-dependent insulinotropic polypeptide, Incretin, Alpha cell, Diabetes
1. GIP, the first incretin
Glucose dependent insulinotropic polypeptide (GIP) was the first intestinally derived peptide described to stimulate insulin secretion, fulfilling the namesake role of an incretin peptide [1]. Subsequently, glucagon-like peptide 1 (GLP-1) was identified as another incretin hormone [2], positioning GIP and GLP-1 as sister peptides in the control of glucose homeostasis for the past 30 years [3]. GLP-1 has received considerably more research interest over this time period, in part owing to the tremendous clinical success in the treatment of type 2 diabetes (T2D). Indeed, GIP is often considered the less potent, redundant peptide that fails to stimulate insulin secretion in people with T2D [4]. One differentiating factor between GIP and GLP-1 is their influence on α-cell function: whereas GIP stimulates glucagon secretion, GLP-1 reduces it [5]. Because elevated glucagon levels are often associated with T2D [6], the ability for GLP-1 to inhibit α-cell function has contributed to the therapeutic development of GLP-1 receptor (GLP-1R) agonists [7]. This narrative has also decreased the enthusiasm for developing GIP receptor (GIPR) agonists for T2D treatment, in fear of exaggerating hyperglucagonemia. This has also decreased the attention given to studying the biological relevance of GIPR in α-cells, creating a gap in knowledge relative to our extensive understanding of incretin receptor activity in β-cells. Here, we will review the available data describing the actions of GIP in α-cells and the potential implications these have for glucose homeostasis.
2. GIPR activity in β-cells
The insulinotropic actions of both GLP-1R and GIPR are dependent upon activated β-cells, which is typically defined as elevated glucose levels. GLP-1 and GIP are the only known peptides to fulfill the definition of incretin peptides, which drives insulin secretion in response to oral nutrient intake. The incretin effect accounts for up to 70 % of postprandial insulin secretion in healthy humans and decreases to about 30 % in people with T2D [8]. The expression and activity of both GIPR and GLP-1R in β-cells decreases in response to metabolic stress that mimics the environment of T2D [4,9–11], although the GIPR often appears to be the more sensitive to metabolic stress. Moreover, while physiological concentrations of either GIP or GLP-1 demonstrate reduced insulinotropic actions in people with T2D or preclinical models of hyperglycemia, pharmacological levels of GLP-1 continue to stimulate insulin secretion, whereas pharmacological levels of GIP do not [4]. Consequently, the reduced incretin effect manifested in T2D has been attributed to the reduction in GIPR activity in β-cells. Indeed, recent studies utilizing GIPR and GLP-1R antagonists in healthy subjects have concluded that GIP is the predominant physiological incretin with respect to insulin secretion [12,13], consistent with the observation that reductions in GIPR activity in β-cells correlates with reduced β-cell activity. This has led to a reemergence in interest in understanding the biological significance of GIP in both healthy states and the pathogenesis of T2D [14]. In parallel, the development of multi-receptor agonists that target the GIPR have shown promising initial results in lowering body weight in patients with T2D [15], suggesting that including GIPR as a target in diabetes interventions may exploit new potential mechanisms beyond what is achieved by GLP-1R monoagonists. Whether these mechanisms include additional activity in the β-cell beyond what is achieved by GLP-1R activity or involve non-β-cell GIPR mechanisms remains to be seen. Cell types that express the GIPR, but not the GLP-1R, include adipocytes and the α-cell. In these cell types, GIPR activity and the resulting metabolic actions have been alluded to by associations but have not been clearly defined.
3. An overview of the metabolic role of glucagon
Glucagon secretion by α-cells is most commonly ascribed to occur during hypoglycemia as a counterregulatory action to elevate glycemia. Consequently, a decrease in glucose levels is the most cited mechanism to stimulate α-cells and glucagon secretion. In addition, amino acids are often used both in vivo and ex vivo as potent glucagon secretagogues. Glucagon receptors are highly expressed on hepatocytes, where activation drives glycogenolysis and gluconeogenesis to enhance endogenous glucose production. Additionally, both fasting and postprandial glucagon levels are elevated in people with T2D relative to healthy people, and glucagon receptor antagonists lower glycemia in people with T2D [7]. These observations have subsequently positioned glucagon as a hyperglycemic agent that contributes to the pathogenesis of T2D, driving several efforts to develop therapeutic strategies that reduce glucagon activity that has spanned nearly four decades [16,17]. However, glucagon receptors have been reported in many other cell types beyond hepatocytes, including adipocytes and β-cells, and glucagon signaling in these cell types has the potential to be beneficial for T2D, through mechanisms that increase energy expenditure, induce satiety, and stimulate insulin secretion [7]. The potential metabolic actions of glucagon beyond the regulation of endogenous glucose production has sparked the development of agents that enhance glucagon receptor activity, often in combination with GLP-1R agonism [18]. AS a result, both approaches, glucagon antagonism and glucagon agonism, are being explored with the shared goal of treating diabetic hyperglycemia.
4. Repositioning glucagon action in the control of glucose homeostasis
Numerous recent reports have described the insulinotropic properties of glucagon through paracrine interactions between α- and β-cells. The glucagon receptor (GCGR) is expressed in β-cells at comparable levels to GIPR and GLP-1R, and all three receptors are class B G-protein coupled that utilize Gαs to stimulate the production of cAMP. Thus, glucagon would be expected to stimulate insulin secretion through mechanisms that are similar to incretin peptides. In fact, glucagon-stimulated insulin secretion was reported well before the discovery of incretin peptides [19]. Notably, the insulinotropic actions of glucagon are predominately mediated through the GLP-1R, although some activity is manifested by the GCGR [20–22]. The close proximity of α- and β-cells positions α-cells to influence β-cell function, evident by the observation that paracrine interactions between these two cell types dictates the glycemic set point of an organism [23]. Impairing α-cell input, facilitated by proglucagon peptide action, to the β-cell by using glucagon null mice reduces overall β-cell tone, reducing insulin secretion in response to glucose, amino acids, and depolarizing agents in isolated islets [21]. Preventing α- to β-cell communication in vivo also reduces insulin secretion in response to glucose or amino acids and results in impaired glucose homeostasis [21,24,25]. Interestingly, there is limited evidence for increased susceptibility to fasting- or insulin-induced hypoglycemia when glucagon signaling is interupted [24,26], demonstrating that impairment of α-cell function has a greater impact on postprandial conditions. These observations have forced the field to reconsider how α-cell activity influences postprandial glucose metabolism and question the dogma that enhanced α-cell activity exclusively causes hyperglycemia.
5. GIPR expression and function in α-cells
The GIPR is expressed in all three major endocrine cells of the pancreatic islet: α-cells, β-cells, and δ-cells [27]. GIPR expression in rodent and human α-cells has been demonstrated by detection of mRNA [28] and protein [28,29], and through single cell RNA sequencing [30,31]. Functionally, GIP has been shown to increase cAMP concentrations in isolated α-cells [28] and the αTC1 cell line [29], and to activate cAMP/PKA sensitive pathways, increase calcium concentrations, and enhance depolarization-evoked glucagon secretion [32]. Similar to insulin secretion in β-cells, dynamic changes in cAMP in α-cells is acutely linked to glucagon secretion [33,34]. Thus, there is substantial evidence that both rodent and human α-cells express a functional GIPR capable of modulating α-cell activity.
6. GIP stimulation of glucagon; evidence from preclinical models
The suppression of glucagon levels is lower in response to oral glucose rather than intravenous glucose administration [35], suggesting that a gut-derived factor prevents the ability for glucose to suppress α-cell activity. Since GLP-1 suppresses glucagon secretion [36], and GIP stimulates glucagon secretion, that gut-derived factor has been speculated to be GIP. However, this hypothesis has not been formally tested. Surprisingly, there is little preclinical evidence to demonstrate that GIPR activation in α-cells stimulates glucagon secretion. GIP has been shown to stimulate glucagon release in αTC1 cells [29] and in perfused rat pancreas models [37–39]. The latter utilized a GIPR antagonist (GIP (3–30)NH2) to demonstrate that glucagon secretion in response to GIP can be prevented, providing some level of specificity. While the available evidence points to a direct effect of GIP on α-cells, studies utilizing α-cell GIPR knockouts have not yet been conducted. Interestingly, in the perfused rat pancreas, GIP only stimulated glucagon secretion at low glucose (4.4 mM) and not at postprandial glucose concentrations (8.9 mM) [38]. These data suggest that GIPR activity in the α-cell is glucose-dependent, similar to what is described for incretin receptor activity in β-cells. However, the key difference is that in α-cells, GIP is more effective at stimulating glucagon secretion when glucose concentrations are lower, whereas in β-cells, GIP is more effective at stimulating insulin secretion when glucose concentrations are higher. Elevated glucose facilitates β-cell activity, while lowered glucose levels facilitate α-cell activity, indicating that GIPR action in both cell types is dependent upon readiness of the cell, more so than the glucose levels alone. It is also possible that the signaling mechanisms of GIPR are different in α- versus β-cells. However, GIPR activity in α-cells is thought to signal through cAMP/PKA pathways [28,32], a pathway that is glucose dependent in β-cells [40]. Thus, an α-cell specific mechanism for GIPR activity would involve a cAMP/PKA signaling pathway that differs from that of β-cells. While this is possible, it seems unlikely. A more likely explanation is that GIPR uses cAMP/PKA to potentiate activated α-cells, which occurs when glucose levels are low enough. Indeed, elevating glucose levels increases the overall tone of the both β-cells and δ-cells, both of which are known to impair α-cell activity through either β-to-α cell interactions [41–43] or δ-to-α cell interactions [44,45]. Pharmacological doses of GIP (1–10 nM) have been shown to stimulate somatostatin secretion from the δ-cells in the islet [37,46] and somatostatin secretion is marginally increased by hyperglycemia [46]. In turn, somatostatin inhibits GIP secretion from the gut [47], with mixed reports on its effect on glucagon secretion from neighboring α-cells [48]. Overall, the paracrine effect would imply that GIPR activity in α-cells is subject to the net inhibitory tone from neighboring β- and δ-cells, and that elevated glucose levels provide sufficiently high inhibitory tone to prevent GIP-stimulated glucagon secretion.
7. GIP stimulation of glucagon in healthy people
GIP has consistently been demonstrated to increase circulating glucagon concentrations in healthy people [49,50] (Fig. 1). In subjects fasted overnight, GIP increased glucagon levels in a dose-dependent manner, up to three-fold at a dose of 60 pmol/kg [49]. Similar to the preclinical evidence, the ability for GIP to stimulate glucagon secretion in healthy humans is glucose-dependent. GIP infusion (0.8 pmol/kg/min) failed to stimulate glucagon secretion during a hyperglycemic clamp [4]. This was clearly established through cross-over studies in healthy subjects where GIP was infused (4 pmol/kg/min) during a hypoglycemic, euglycemic, or hyperglycemic clamp [50]. Insulin secretion in response to GIP increased proportionally with elevated glycemia, fitting with the expected outcomes for β-cell function and serving as a positive control. GIP stimulated glucagon secretion at hypoglycemia (2.5 mM glucose) and euglycemia (5 mM glucose) but failed to do so at hyperglycemia (12.5 mM glucose). Interestingly, GIP-stimulated glucagon secretion was modest at both euglycemia and hypoglycemia, increasing circulating levels by ~3 pM versus saline control. For context, the hypoglycemia stimuli in these studies increase glucagon levels by ~30 pM, while hyperglycemia decrease levels by ~5 pM. Thus, the physiological significance of GIP-stimulated glucagon secretion remains to be established.
Fig. 1.
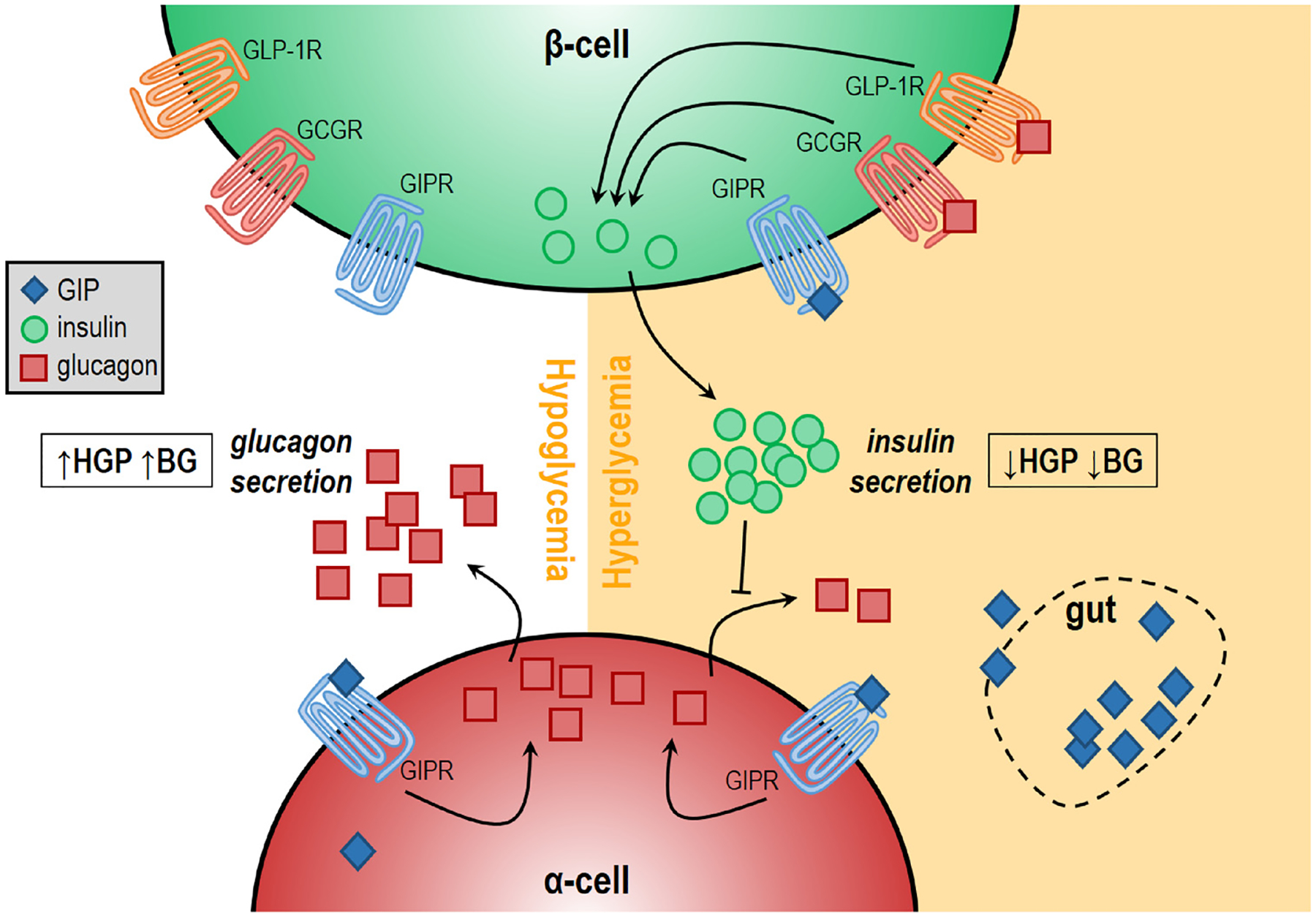
GIP stimulates glucagon secretion in a glucose-dependent manner. Stimulation of glucagon secretion by GIP is more potent at lower glucose concentrations than at higher glucose concentrations. On the other hand, GIP-stimulated insulin section only occurs at high glucose concentrations. The suppressed ability for GIP to stimulate glucagon secretion at high glucose may be due to increased inhibitory tone from β-cells, which suppress α-cell activity and thereby limit GIP-stimulated glucose secretion. Abbreviations: HGP = hepatic glucose production; BG = blood glucose.
8. GIP stimulation of glucagon in people with T2D
The circulating concentrations of GIP in people with T2D are reported to be elevated after fasting and in response to oral or mixed nutrient stimuli [51]. The ability for GIP to stimulate insulin secretion in people with T2D is reduced compared to healthy controls [52]. On the other hand, GIP retains the ability to stimulate glucagon secretion in people with T2D [51,53,54]. Furthermore, unlike what is reported in healthy people, subjects with T2D demonstrate increased glucagon secretion in response to GIP when glucose concentrations are elevated [4]. During intravenous glucose administration to achieve plasma glucose concentrations of ~15 mM, GIP infusion (4 pmol/kg/min) increased glucagon levels by ~2 pM [53]. Interestingly, GLP-1 reduced glucagon levels in these studies, and the combination of GIP and GLP-1 counteracted each other to produce no change in glucagon levels. Subsequent studies reiterated the combined effects of GIP and GLP-1 in fasting, hyperglycemic T2D subjects, showing that GIP elevates glucagon concentrations, GLP-1 reduces glucagon concentrations, while the combination yielded glucagon concentrations comparable to control conditions [54]. A comparison of the glucagonotropic properties of GIP at hypoglycemia, euglycemia, and hyperglycemia in patients with T2D demonstrated that GIP is able to produce comparable increases in glucagon concentrations at all glycemic levels [55]. Similar to healthy subjects, the effect sizes were modest (< 5 pM increases) relative to the dynamic changes induced by glycemia alone. Still, these collective data sets indicate that the glucose-dependency of GIP-stimulated glucagon secretion does not hold in people with T2D.
One potential explanation of why GIP stimulates glucagon secretion at hyperglycemia in people with T2D and not healthy subjects is the difference in GIP-stimulated insulin secretion between these two populations (Fig. 2). Indeed, it is well documented that GIP is significantly less insulinotropic in people with T2D [4,54], owing to the decrease in GIPR expression in β-cells that results from chronic hyperglycemia and metabolic stress [4,9–11]. Activation of β-cells increases in the inhibitory tone on α-cells, reducing the amount of glucagon secretion basally and in response to α-cell secretagogues [56]. Thus, it is plausible that GIPR activity in β-cells in healthy people provides sufficient inhibitory tone on the α-cell to prevent GIPR stimulated glucagon secretion. Loss of GIPR β-cell activity in T2D would dampen this inhibitory tone and permit GIP-stimulated glucagon secretion. This hypothesis is amendable to testing in patients with type 1 diabetes (T1D), where β-cell GIPR activity cannot contribute to overall islet tone. During a hypoglycemic clamp in c-peptide negative T1D subjects, GIP infusion (4 pmol/kg/min) elevated glucagon levels, increased endogenous glucose production, and decreased the glucose infusion rate [57]. However, when GIP was infused (4 pmol/kg/min) at euglycemia (~7 mM) or hyperglycemia (~11 mM) in T1D patients, GIP failed to increase glucagon levels [58]. Thus, the glucose-dependency of GIP-stimulated glucagon secretion is retained in people with c-peptide negative T1D. This suggests that factors beyond impaired β-cell function or reduced β-cell GIPR expression dictate the ability for GIP to stimulate glucagon secretion at elevated glycemia in T2D. T2D is manifested by both α-cell dysfunction [59] and extrapancreatic metabolic dysfunction [60], suggesting the both direct and indirect factors could contribute to alter GIPR α-cell activity in T2D.
Fig. 2.
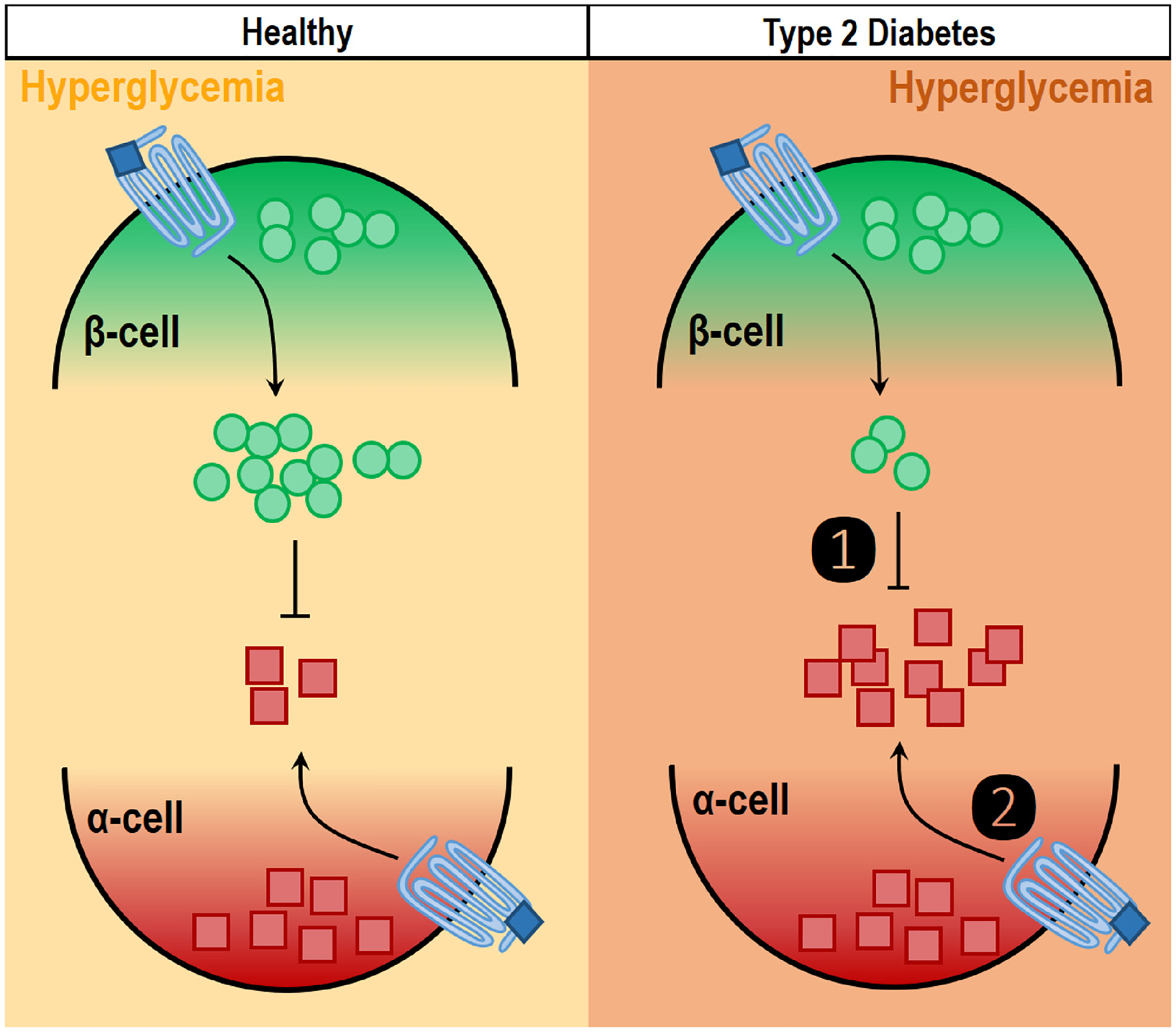
Mechanisms of altered GIP-stimulated glucagon secretion in T2D hyperglycemia. GIP does not stimulate glucagon secretion when glucose levels are elevated in a healthy individual. However, in people with T2D, GIP does stimulate glucagon secretion at hyperglycemia. The observation that GIP stimulates glucagon secretion even at hyperglycemia in people with T2D is possibly due to (1) reduced inhibitory tone via paracrine effects of insulin from β-cells, as people with T2D have reduced β-cell function. Alternatively, GIP may stimulate hyperglycemia in people with T2D because of (2) intrinsic changes in the α-cell that permit GIPR activity at all glucose levels. The phenomenon would suggest impaired α-cell activity is the characteristic of T2D. These are not mutually exclusive explanations, and both may contribute to the altered GIPR α-cell activity in T2D.
9. Lessons from GWAS studies
Genome wide association studies (GWAS) have identified relationships between genetic variation in the GIPR and changes in glucose homeostasis, insulin secretion, and body mass index (BMI) [61,62]. A detailed characterization of one single nucleotide polymorphism (SNP) (rs10423929) demonstrated an association between the A-allele carrier and impaired GIP-stimulated insulin secretion, indicating the SNP leads to loss of GIPR function [63]. GIPR gene expression was decreased in islets from carriers of the A allele, to the same degree seen in GIPR expression in islets from T2D relative to nondiabetic donors. This SNP is also associated with reductions in BMI, lean body mass, and waist circumference. Consequently, it is difficult to determine if the reductions in insulin secretion are primary or secondary to the reduced body weight. Twelve individual studies were conducted in populations that contain this SNP [61–73], of which one reported glucagon levels [63]. In Supplementary Table 1 of this paper, fasting glucagon levels (n = 1018) and glucagon concentrations 120 min following an oral glucose tolerance test (n = 989) did not differ between genotypes. However, carriers of the A allele had lower fasting glucagon levels with an adjusted p value of 0.053 [63]. Whether a sufficiently powered sample size would demonstrate reduced glucagon values in people that have the loss of function GIPR allele remains to be seen. Moreover, understanding the physiological significance of this for glucose tolerance and/or weight is unknown.
10. Conclusions
The biological actions of incretin hormones are centered on their contribution to glucose-stimulated insulin secretion. However, it has become abundantly clear that GLP-1 is involved in many processes beyond stimulation of β-cell activity [3]. As more attention is being given to GIP, a similar number of non-β-cell actions are being described. Activity of GIPR in tissues types like white adipocytes [74], brown adipocytes [75], cardiomyocytes [76], and bone [77] is still being investigated within the framework of metabolic disease. While the phenomenon of GIP-stimulated glucagon secretion is well established, the mechanisms that regulate this process are still unclear. Moreover, the contribution of GIPR activity in α-cells to overall metabolism is altogether unknown. It is imperative to elucidate these contributions in both healthy individuals and in the setting of metabolic dysfunction. As discussed above, the metabolic role of glucagon is being reevaluated and expanded beyond its counterregulatory role and the prevention of hypoglycemia [56]. This transitional period has produced a phase marked by equal efforts aiming to both antagonize and enhance glucagon action for the treatment of metabolic dysfunction. It is remarkable to note that the parallel situation has emerged for therapeutic efforts centered on GIP: whereas antagonism of the GIPR improves glucose tolerances and prevents weight gain [78–80], so do GIPR agonists [15,81,82]. Thus, the only conclusion from this dichotomy is that a strong need exists to better understand how GIPR signaling influences metabolism. Investigating GIPR activity in the α-cell presents an opportunity to target the conundrums of both glucagon and GIP simultaneously.
Funding sources
K.E. is supported by a training grant from NIH/NIDDK (T32 DK007012-41). J.E.C is supported by the American Diabetes Association (1-18-JDF-017), NIH/NIDDK (R01 DK123075), and is a Borden Scholar.
References
- [1].Baggio LL, Drucker DJ, Biology of incretins: GLP-1 and GIP, Gastroenterology 132 (2007) 2131–2157. [DOI] [PubMed] [Google Scholar]
- [2].Kreymann B, Williams G, Ghatei MA, Bloom SR, Glucagon-like peptide-1 7–36: a physiological incretin in man, Lancet 2 (1987) 1300–1304. [DOI] [PubMed] [Google Scholar]
- [3].Drucker DJ, Mechanisms of action and therapeutic application of glucagon-like Peptide-1, Cell Metab 27 (2018) 740–756. [DOI] [PubMed] [Google Scholar]
- [4].Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W, Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus, J. Clin. Invest 91 (1993) 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Campbell JE, Drucker DJ, Pharmacology, physiology, and mechanisms of incretin hormone action, Cell Metab 17 (2013) 819–837. [DOI] [PubMed] [Google Scholar]
- [6].Dunning BE, Gerich JE, The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications, Endocr. Rev 28 (2007) 253–283. [DOI] [PubMed] [Google Scholar]
- [7].Campbell JE, Drucker DJ, Islet alpha cells and glucagon–critical regulators of energy homeostasis, Nat. Rev. Endocrinol 11 (2015) 329–338. [DOI] [PubMed] [Google Scholar]
- [8].Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S, Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes, Diabetes Care 34 (Suppl. (2)) (2011) S251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Piteau S, Olver A, Kim SJ, Winter K, Pospisilik JA, Lynn F, et al. , Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat, Biochem. Biophys. Res. Commun 362 (2007) 1007–1012. [DOI] [PubMed] [Google Scholar]
- [10].Zhou J, Livak MF, Bernier M, Muller DC, Carlson OD, Elahi D, et al. , Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets, Am. J. Physiol. Endocrinol. Metab 293 (2007) E538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xu G, Kaneto H, Laybutt DR, Duvivier-Kali VF, Trivedi N, Suzuma K, et al. , Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes, Diabetes 56 (2007) 1551–1558. [DOI] [PubMed] [Google Scholar]
- [12].Nauck MA, Meier JJ, GIP and GLP-1: stepsiblings rather than monozygotic twins within the incretin family, Diabetes 68 (2019) 897–900. [DOI] [PubMed] [Google Scholar]
- [13].Gasbjerg LS, Helsted MM, Hartmann B, Jensen MH, Gabe MBN, Sparre-Ulrich AH, et al. , Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals, Diabetes 68 (2019) 906–917. [DOI] [PubMed] [Google Scholar]
- [14].Finan B, Muller TD, Clemmensen C, Perez-Tilve D, DiMarchi RD, Tschop MH, Reappraisal of GIP pharmacology for metabolic diseases, Trends Mol. Med 22 (2016) 359–376. [DOI] [PubMed] [Google Scholar]
- [15].Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. , Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial, Lancet 392 (2018) 2180–2193. [DOI] [PubMed] [Google Scholar]
- [16].Lefebvre PJ, Paquot N, Scheen AJ, Inhibiting or antagonizing glucagon: making progress in diabetes care, Diabetes Obes. Metab 17 (2015) 720–725. [DOI] [PubMed] [Google Scholar]
- [17].Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D, Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist, Science 215 (1982) 1115–1116. [DOI] [PubMed] [Google Scholar]
- [18].Capozzi ME, DiMarchi RD, Tschop MH, Finan B, Campbell JE, Targeting the Incretin/Glucagon system with triagonists to treat diabetes, Endocr. Rev 39 (2018) 719–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Samols E, Marri G, Marks V, Promotion of insulin secretion by glucagon, Lancet 2 (1965) 415–416. [DOI] [PubMed] [Google Scholar]
- [20].Chepurny OG, Matsuokas MT, Liapakis G, Leech CA, Milliken BT, Doyle RP, et al. , Correction: nonconventional glucagon and GLP-1 receptor agonist and antagonist interplay at the GLP-1 receptor revealed in high-throughput FRET assays for cAMP, J. Biol. Chem 294 (2019) 8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, et al. , Beta Cell tone is defined by proglucagon peptides through cAMP signaling, JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, et al. , Insulin secretion depends on intra-islet glucagon signaling, Cell Rep 25 (2018) 1127–1134 e1122. [DOI] [PubMed] [Google Scholar]
- [23].Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, et al. , Paracrine interactions within the pancreatic islet determine the glycemic set point, Cell Metab 27 (2018) 549–558 e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, et al. , Intra-islet glucagon signaling is critical for maintaining glucose homeostasis, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, et al. , Glucagon lowers glycemia when beta-cells are active, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, et al. , The role of pancreatic preproglucagon in glucose homeostasis in mice, Cell Metab 25 (2017) 927–934 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. , Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets, Mol. Metab 5 (2016) 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, Ling Z, et al. , Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells, Diabetes 45 (1996) 257–261. [DOI] [PubMed] [Google Scholar]
- [29].Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS, et al. , Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes, Diabetes 58 (2009) 1342–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xin Y, Kim J, Ni M, Wei Y, Okamoto H, Lee J, et al. , Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 3293–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, et al. , Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes, Cell Metab 24 (2016) 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ding WG, Renstrom E, Rorsman P, Buschard K, Gromada J, Glucagon-like peptide I and glucose-dependent insulinotropic polypeptide stimulate Ca2+-induced secretion in rat alpha-cells by a protein kinase A-mediated mechanism, Diabetes 46 (1997) 792–800. [DOI] [PubMed] [Google Scholar]
- [33].Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A, Glucose controls glucagon secretion by directly modulating cAMP in alpha cells, Diabetologia (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hughes JW, Ustione A, Lavagnino Z, Piston DW, Regulation of islet glucagon secretion: beyond calcium, Diabetes Obes. Metab 20 (Suppl. (2)) (2018) 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meier JJ, Deacon CF, Schmidt WE, Holst JJ, Nauck MA, Suppression of glucagon secretion is lower after oral glucose administration than during intravenous glucose administration in human subjects, Diabetologia 50 (2007) 806–813. [DOI] [PubMed] [Google Scholar]
- [36].de Heer J, Rasmussen C, Coy DH, Holst JJ, Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas, Diabetologia 51 (2008) 2263–2270. [DOI] [PubMed] [Google Scholar]
- [37].Sparre-Ulrich AH, Gabe MN, Gasbjerg LS, Christiansen CB, Svendsen B, Hartmann B, et al. , GIP(3–30)NH2 is a potent competitive antagonist of the GIP receptor and effectively inhibits GIP-mediated insulin, glucagon, and somatostatin release, Biochem. Pharmacol 131 (2017) 78–88. [DOI] [PubMed] [Google Scholar]
- [38].Pederson RA, Brown JC, Interaction of gastric inhibitory polypeptide, glucose, and arginine on insulin and glucagon secretion from the perfused rat pancreas, Endocrinology 103 (1978) 610–615. [DOI] [PubMed] [Google Scholar]
- [39].Opara EC, Go VL, Influence of gastric inhibitory polypeptide (GIP) and glucose on the regulation of glucagon secretion by pancreatic alpha cells, Regul. Pept 32 (1991) 65–73. [DOI] [PubMed] [Google Scholar]
- [40].Tengholm A, Cyclic AMP dynamics in the pancreatic beta-cell, Ups. J. Med. Sci 117 (2012) 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, et al. , Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system, Cell Metab 3 (2006) 47–58. [DOI] [PubMed] [Google Scholar]
- [42].Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB, Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release, Diabetes 54 (2005) 1808–1815. [DOI] [PubMed] [Google Scholar]
- [43].Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB, Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells, Nat. Cell Biol 5 (2003) 330–335. [DOI] [PubMed] [Google Scholar]
- [44].Elliott AD, Ustione A, Piston DW, Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic alpha-cell by lowering cAMP, Am. J. Physiol. Endocrinol. Metab 308 (2015) E130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schuit FC, Derde MP, Pipeleers DG, Sensitivity of rat pancreatic A and B cells to somatostatin, Diabetologia 32 (1989) 207–212. [DOI] [PubMed] [Google Scholar]
- [46].Szecowka J, Grill V, Sandberg E, Efendic S, Effect of GIP on the secretion of insulin and somatostatin and the accumulation of cyclic AMP in vitro in the rat, Acta Endocrinol (Copenh) 99 (1982) 416–421. [DOI] [PubMed] [Google Scholar]
- [47].Moss CE, Marsh WJ, Parker HE, Ogunnowo-Bada E, Riches CH, Habib AM, et al. , Somatostatin receptor 5 and cannabinoid receptor 1 activation inhibit secretion of glucose-dependent insulinotropic polypeptide from intestinal K cells in rodents, Diabetologia 55 (2012) 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rutter GA, Regulating glucagon secretion: somatostatin in the spotlight, Diabetes 58 (2009) 299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Meier JJ, Gallwitz B, Siepmann N, Holst JJ, Deacon CF, Schmidt WE, et al. , Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia, Diabetologia 46 (2003) 798–801. [DOI] [PubMed] [Google Scholar]
- [50].Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK, Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans, Diabetes 60 (2011) 3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L, Egan JM, GIP contributes to islet trihormonal abnormalities in type 2 diabetes, J. Clin. Endocrinol. Metab 99 (2014) 2477–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Vilsboll T, Krarup T, Madsbad S, Holst JJ, Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients, Diabetologia 45 (2002) 1111–1119. [DOI] [PubMed] [Google Scholar]
- [53].Lund A, Vilsboll T, Bagger JI, Holst JJ, Knop FK, The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes, Am. J. Physiol. Endocrinol. Metab 300 (2011) E1038–1046. [DOI] [PubMed] [Google Scholar]
- [54].Mentis N, Vardarli I, Kothe LD, Holst JJ, Deacon CF, Theodorakis M, et al. , GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes, Diabetes 60 (2011) 1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Christensen MB, Calanna S, Holst JJ, Vilsboll T, Knop FK, Glucose-dependent insulinotropic polypeptide: blood glucose stabilizing effects in patients with type 2 diabetes, J. Clin. Endocrinol. Metab 99 (2014) E418–426. [DOI] [PubMed] [Google Scholar]
- [56].Finan B, Capozzi ME, Campbell JE, Repositioning glucagon action in the physiology and pharmacology of diabetes, Diabetes (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Christensen M, Calanna S, Sparre-Ulrich AH, Kristensen PL, Rosenkilde MM, Faber J, et al. , Glucose-dependent insulinotropic polypeptide augments glucagon responses to hypoglycemia in type 1 diabetes, Diabetes 64 (2015) 72–78. [DOI] [PubMed] [Google Scholar]
- [58].Christensen M, Knop FK, Vilsboll T, Aaboe K, Holst JJ, Madsbad S, et al. , Glucagon-like peptide-2, but not glucose-dependent insulinotropic polypeptide, stimulates glucagon release in patients with type 1 diabetes, Regul. Pept 163 (2010) 96–101. [DOI] [PubMed] [Google Scholar]
- [59].Moon JS, Won KC, Pancreatic alpha-cell dysfunction in type 2 diabetes: old kids on the block, Diabetes Metab. J 39 (2015) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stehouwer CDA, Microvascular dysfunction and hyperglycemia: a vicious cycle with widespread consequences, Diabetes 67 (2018) 1729–1741. [DOI] [PubMed] [Google Scholar]
- [61].Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, et al. , Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge, Nat. Genet 42 (2010) 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. , Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index, Nat. Genet 42 (2010) 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lyssenko V, Eliasson L, Kotova O, Pilgaard K, Wierup N, Salehi A, et al. , Pleiotropic effects of GIP on islet function involve osteopontin, Diabetes 60 (2011) 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Arora GP, Almgren P, Brons C, Thaman RG, Vaag AA, Groop L, et al. , Association between genetic risk variants and glucose intolerance during pregnancy in north Indian women, BMC Med. Genomics 11 (2018) 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Han L, Li Y, Tang L, Chen Z, Zhang T, Chen S, et al. , IGF2BP2 rs11705701 polymorphisms are associated with prediabetes in a Chinese population: a population-based case-control study, Exp. Ther. Med 12 (2016) 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Javorsky M, Gotthardova I, Klimcakova L, Kvapil M, Zidzik J, Schroner Z, et al. , A missense variant in GLP1R gene is associated with the glycaemic response to treatment with gliptins, Diabetes Obes. Metab 18 (2016) 941–944. [DOI] [PubMed] [Google Scholar]
- [67].Berglund LM, Lyssenko V, Ladenvall C, Kotova O, Edsfeldt A, Pilgaard K, et al. , Glucose-dependent insulinotropic polypeptide stimulates osteopontin expression in the vasculature via Endothelin-1 and CREB, Diabetes 65 (2016) 239–254. [DOI] [PubMed] [Google Scholar]
- [68].Garg G, McGuigan FE, Kumar J, Luthman H, Lyssenko V, Akesson K, Glucose-dependent insulinotropic polypeptide (GIP) and GIP receptor (GIPR) genes: an association analysis of polymorphisms and bone in young and elderly women, Bone Rep 4 (2016) 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ono S, Suzuki Y, Fukui N, Sawamura K, Sugai T, Watanabe J, et al. , GIPR gene polymorphism and weight gain in patients with schizophrenia treated with olanzapine, J. Neuropsychiatry Clin. Neurosci 27 (2015) 162–164. [DOI] [PubMed] [Google Scholar]
- [70].Ahlqvist E, Osmark P, Kuulasmaa T, Pilgaard K, Omar B, Brons C, et al. , Link between GIP and osteopontin in adipose tissue and insulin resistance, Diabetes 62 (2013) 2088–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sonestedt E, Lyssenko V, Ericson U, Gullberg B, Wirfalt E, Groop L, et al. , Genetic variation in the glucose-dependent insulinotropic polypeptide receptor modifies the association between carbohydrate and fat intake and risk of type 2 diabetes in the Malmo Diet and Cancer cohort, J. Clin. Endocrinol. Metab 97 (2012) E810–818. [DOI] [PubMed] [Google Scholar]
- [72].Windholz J, Kovacs P, Tonjes A, Dittrich K, Bluher S, Kiess W, et al. , Effects of genetic variants in ADCY5, GIPR, GCKR and VPS13C on early impairment of glucose and insulin metabolism in children, PLoS One 6 (2011) e22101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ono S, Suzuki Y, Fukui N, Sugai T, Watanabe J, Tsuneyama N, et al. , Association between the GIPR gene and the insulin level after glucose loading in schizophrenia patients treated with olanzapine, Pharmacogenom. J 12 (2012) 507–512. [DOI] [PubMed] [Google Scholar]
- [74].Joo E, Harada N, Yamane S, Fukushima T, Taura D, Iwasaki K, et al. , Inhibition of gastric inhibitory polypeptide receptor signaling in adipose tissue reduces insulin resistance and hepatic steatosis in high-fat diet-fed mice, Diabetes 66 (2017) 868–879. [DOI] [PubMed] [Google Scholar]
- [75].Beaudry JL, Kaur KD, Varin EM, Baggio LL, Cao X, Mulvihill EE, et al. , Physiological roles of the GIP receptor in murine brown adipose tissue, Mol. Metab (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ussher JR, Campbell JE, Mulvihill EE, Baggio LL, Bates HE, McLean BA, et al. , Inactivation of the glucose-dependent insulinotropic polypeptide receptor improves outcomes following experimental myocardial infarction, Cell Metab 27 (2018) 450–460 e456. [DOI] [PubMed] [Google Scholar]
- [77].Schiellerup SP, Skov-Jeppesen K, Windelov JA, Svane MS, Holst JJ, Hartmann B, et al. , Gut hormones and their effect on bone metabolism. Potential drug therapies in future osteoporosis treatment, Front. Endocrinol. (Lausanne) 10 (2019) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Killion EA, Wang J, Yie J, Shi SD, Bates D, Min X, et al. , Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models, Sci. Transl. Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- [79].Gasbjerg LS, Gabe MBN, Hartmann B, Christensen MB, Knop FK, Holst JJ, et al. , Glucose-dependent insulinotropic polypeptide (GIP) receptor antagonists as anti-diabetic agents, Peptides 100 (2018) 173–181. [DOI] [PubMed] [Google Scholar]
- [80].Kaneko K, Fu Y, Lin HY, Cordonier EL, Mo Q, Gao Y, et al. , Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition, J. Clin. Invest 130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Frias JP, Bastyr EJ 3rd, Vignati L, Tschop MH, Schmitt C, Owen K, et al. , The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090–2746, in patients with type 2 diabetes, Cell Metab 26 (2017) 343–352 e342. [DOI] [PubMed] [Google Scholar]
- [82].Mroz PA, Finan B, Gelfanov V, Yang B, Tschop MH, DiMarchi RD, et al. , Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism, Mol. Metab 20 (2019) 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]