

Abstract
DNA methylation is an epigenetic modification, influenced by both genetic and environmental variation, that plays a key role in transcriptional regulation and many organismal phenotypes. Although patterns of DNA methylation have been shown to differ between human populations, it remains to be determined how epigenetic diversity relates to the patterns of genetic and gene expression variation at a global scale. Here we measured DNA methylation at 485,000 CpG sites in five diverse human populations, and analysed these data together with genome-wide genotype and gene expression data. We found that population-specific DNA methylation mirrors genetic variation, and has greater local genetic control than mRNA levels. We estimated the rate of epigenetic divergence between populations, which indicates far greater evolutionary stability of DNA methylation in humans than has been observed in plants. This study provides a deeper understanding of worldwide patterns of human epigenetic diversity, as well as initial estimates of the rate of epigenetic divergence in recent human evolution.
Human evolutionary history has left a strong signature on worldwide patterns of genetic variation1–3. Principal component analyses (PCA) and related methods reveal patterns of genetic diversity within and across populations, in particular population stratification and admixture. The first two principal components of a single nucleotide polymorphism (SNP) genotype matrix are often sufficient to compare the ancestries of different human populations and to show how genetic similarity between populations varies with geographic distance2,4–7.
The relationship between the geographic patterns of ancestry in genomic and epigenomic variation has so far not been well characterized8,9; however, PCA on DNA methylation data from pairs of populations has shown partial separation10,11. Previous studies of gene expression variation have found that, unlike genotypes, expression data do not cluster by geographic location, and population ancestry cannot be determined using mRNA levels alone12,13.
The epigenome is situated at the interface between the genome and the environment14,15, and their interactions may underlie the role of epigenetics in adaptation to the environment and other complex phenotypes. However, our understanding of the global epigenomic and transcriptomic diversity across human populations is far from complete8,9,13. In particular, it remains unknown to what extent epigenetic diversity reflects human evolutionary history and genetic variation.
Results
To investigate whether human evolutionary history has shaped worldwide patterns of epigenetic variation, we analysed SNP genotypes, DNA methylation levels, and mRNA levels (using RNA sequencing (RNA-seq)) for the same 34 individuals from 5 different populations. These populations are from the Centre d’Etude du Polymorphisme Humain Human Genome Diversity Panel (CEPH-HGDP) populations16, which have revealed a great deal about human migration history4,5.
We chose these five populations to span the breadth of human worldwide migrations, and also capture differences in genetic diversity that stem from serial founder effects throughout human evolutionary history5,17. The 34 samples include lymphoblastoid cell lines (LCLs) from six Yakut, seven Cambodian, seven Pathan, seven Mozabite and seven Mayan individuals. Geographic locations of the samples were previously reported16 (Fig. 1a).
Fig. 1 |. Context of genome-wide population structure.
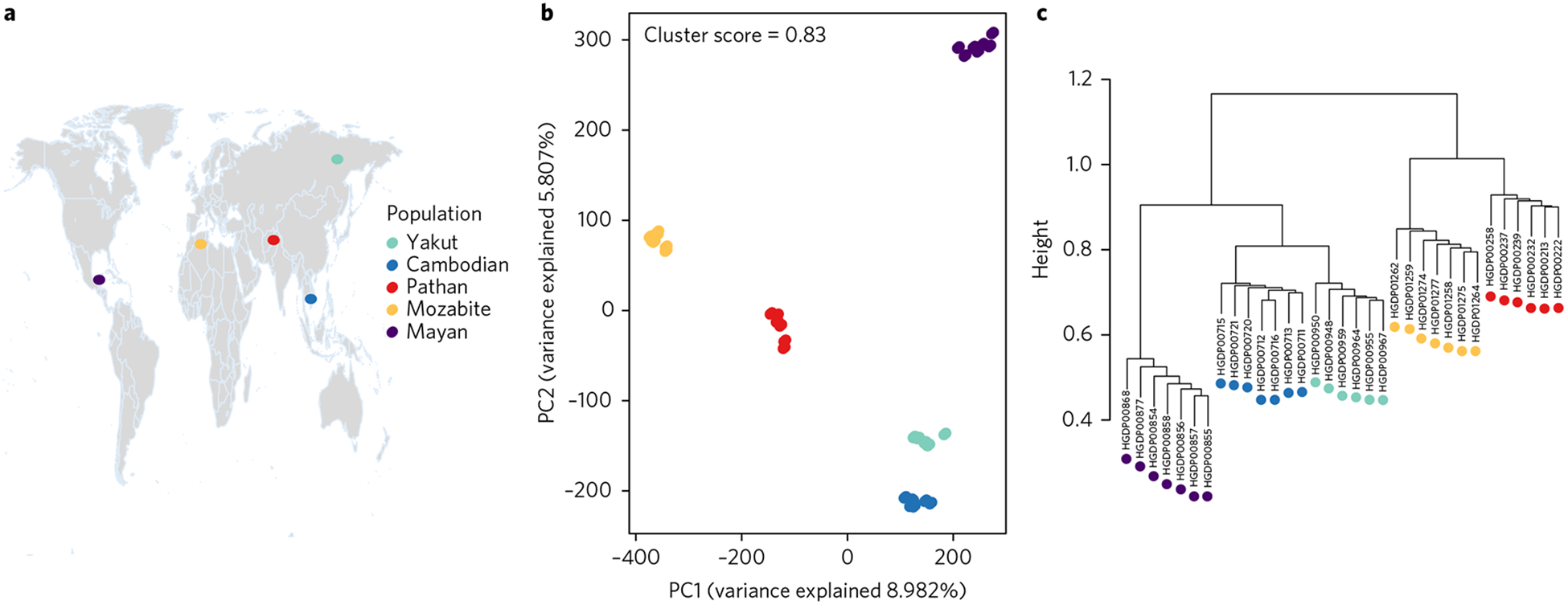
a, The geographic locations of populations in the data set, shown on a Gall–Peters projection map. b, PCA on the SNP genotype matrix. The first and second PCs explain 9% and 6% of the variation, respectively, and clearly differentiate the individuals into five well-separated clusters that correspond to the five populations sampled. c, A hierarchical clustering tree also captures the genetic relationships between the individuals and their populations.
Among the genotype data2, 644,258 SNPs passed our quality control filters and were kept for subsequent analyses. We measured DNA methylation levels with the Illumina 450K Methylation array18, which quantifies methylation at 485,000 CpG sites genomewide. After extensive filtering and quality control (see Methods), the data used in the analyses here consisted of 310,289 CpG sites. mRNA abundance levels were previously determined using cufflinks-2.0.219, reported as FPKM (fragments per kilobase of exon per million mapped reads) estimates for each transcript13.
Worldwide patterns of human allele frequencies reflect population-specific evolutionary histories and adaptation to local environments, and correspond to self-identified groups or to geographically and linguistically similar populations4,5,20–22. This general agreement between genetic variation and geographic location has also been found in the HGDP data set2,23.
To characterize genetic divergence between these five populations, we carried out PCA on the SNP genotype matrix. The first and second principal components explained 9% and 6% of the genetic variation, respectively, and clearly differentiated the individuals into five well-separated clusters that correspond to the five populations sampled (Fig. 1b). Even with the limited sample size, the population structure revealed by the SNP genotypes was extremely robust. To facilitate comparison between the genetic and epigenetic data sets, we quantified the strength of the genomic PCA clustering by computing the silhouette cluster scores (SCS)24 (see Methods) for the individuals in the five populations as well as the average SCS for the entire data set (Supplementary Fig. 1). The SCS of an individual measures how similar it is to its own predefined population cluster, relative to individuals in other clusters, while the average SCS across all individuals is a measure of how tightly the data correspond to their known populations. For the genetic clustering presented in Fig. 1b, this average score is 0.83, with a median of 0.9. A tree generated using hierarchical clustering also captures the genetic relationships between the individuals and their populations (Fig. 1c). The branching pattern of this tree agrees with the accepted order of ancestral human expansion, consistent with the ‘out of Africa’ hypothesis5,25,26.
As an initial measure of population specificity, we used the nonparametric Kruskal–Wallis (K–W) test to identify CpG sites that were differentially methylated between the five different populations. Comparing the observed distribution of P values to the uniform distribution expected by chance (black line in Fig. 2a), we found a significant excess of population-specific sites characterized by a shift towards low P values (Fig. 2a). We identified 6,901 CpG sites with K–W P < 0.01 (24% false discovery rate (FDR)), 312 CpG sites with K–W P < 0.001 (12% FDR), and three CpG sites with K–W P < 0.0001 (3% FDR). We observed more CpG sites passing each of these three P value cutoffs than 99.2–99.6% of matched randomized data sets, suggesting significant levels of population differentiation. Of the 312 CpG sites with K–W P < 0.001, 79 overlap sites of population-specific DNA methylation previously identified in a study of three populations8 (Supplementary Table 4).
Fig. 2 |. Population specificity of Cpg methylation.
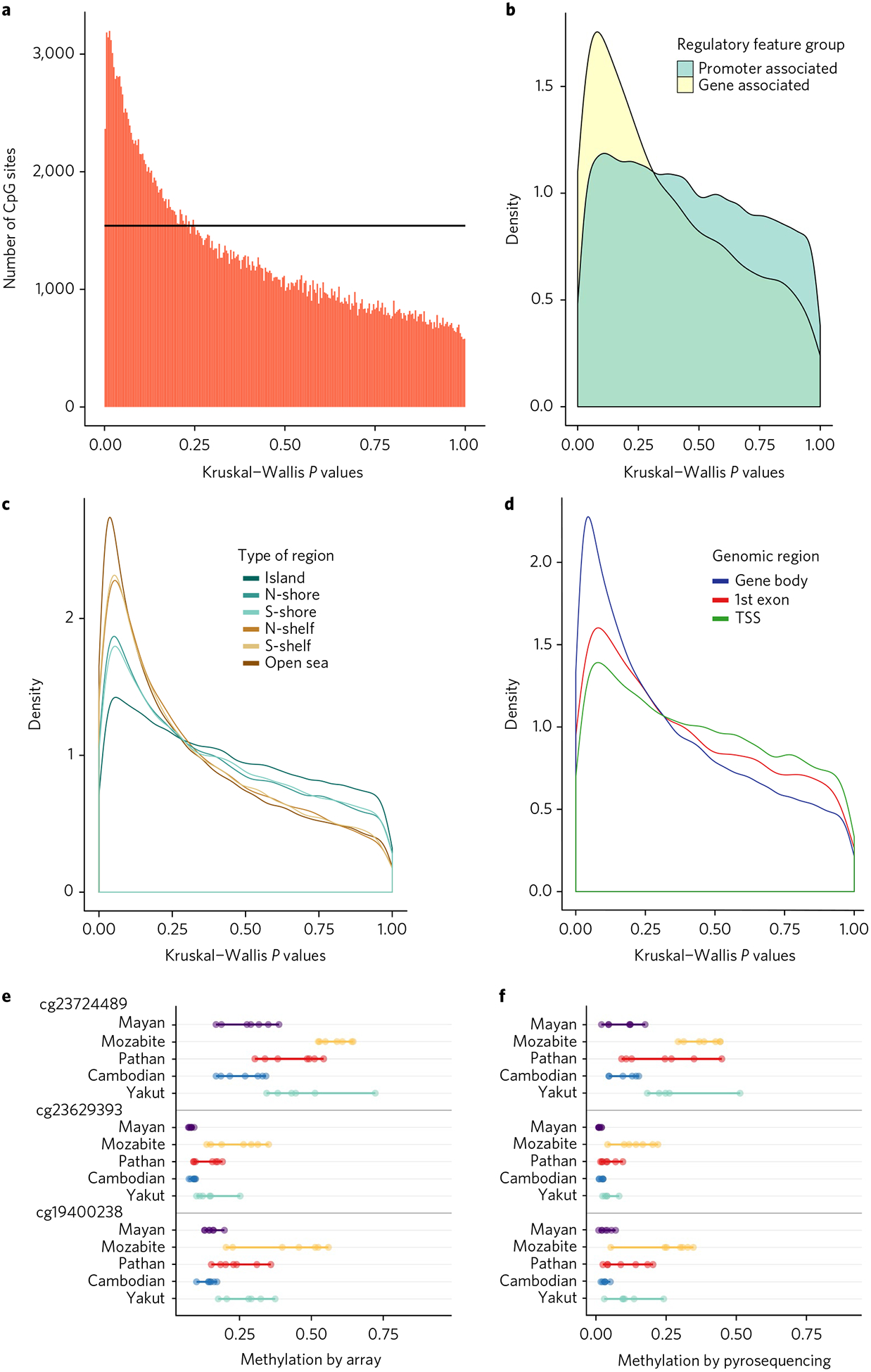
a, A graph of Kruskal–Wallis P values for all CpG sites across all individuals in the five different populations. The black horizontal line corresponds to the uniform P value distribution expected by chance. b–d, Differences based on different types of CpG regions. The CpG sites that exhibit population differentiation are enriched in regions that are gene-associated, outside of CpG islands, and inside gene bodies (TSS, transcription start sites). e,f, Comparison of percentage methylation by array (e) and by pyrosequencing (f) for the top three CpG sites with highest population specificity.
We next investigated how population differences in DNA methylation patterns vary across different genomic regions. Comparing sites within gene bodies versus promoters, we found greater divergence within genes (Fig. 2b). Further separation of the promoter-associated sites revealed that population differences are enriched outside CpG islands, which are genomic regions with high CpG content but typically low levels of methylation. Population-specific sites were most frequent in regions flanking CpG islands, known as CpG shores and CpG shelves (Fig. 2c), as well as in gene bodies downstream of the first exon (Fig. 2d).
To assess the accuracy of our population-specific sites, we tested the three CpG sites with K–W P < 0.0001 for validation with another technology, pyrosequencing bisulfite-treated DNA. The results show excellent concordance with the DNA methylation levels obtained from the Illumina microarray (Fig. 2e,f), and also give similar K–W P values (Methods and Supplementary Table 1).
To estimate epigenetic divergence, we computed Pst, the phenotypic differentiation between populations27–29, for DNA methylation and mRNA levels across the genome. This metric estimates population differentiation for quantitative traits, analogous to Fst30. For a given CpG site or mRNA level, Pst = σ2b/(σ2b + 2σ2w), where σ2b is the between-population variance and σ2w is the average within-population variance (see Methods).
To investigate patterns of population differentiation in these samples, we then used these Pst values to select the most population-specific CpG sites and mRNA levels, and performed PCA on just these sites. For example, with the 200 most diverged sites/genes (highest Pst values), we found a moderate degree of population clustering, as quantified by the silhouette score (Fig. 3a,b). This general pattern persisted over a wide range of Pst cutoffs, with DNA methylation showing a slightly higher clustering score across nearly all of the range (Supplementary Fig. 2), though neither was significantly different from random (P > 0.4 for both DNA methylation and gene expression silhouette scores).
Fig. 3 |. Structure of epigenome-wide population differences.
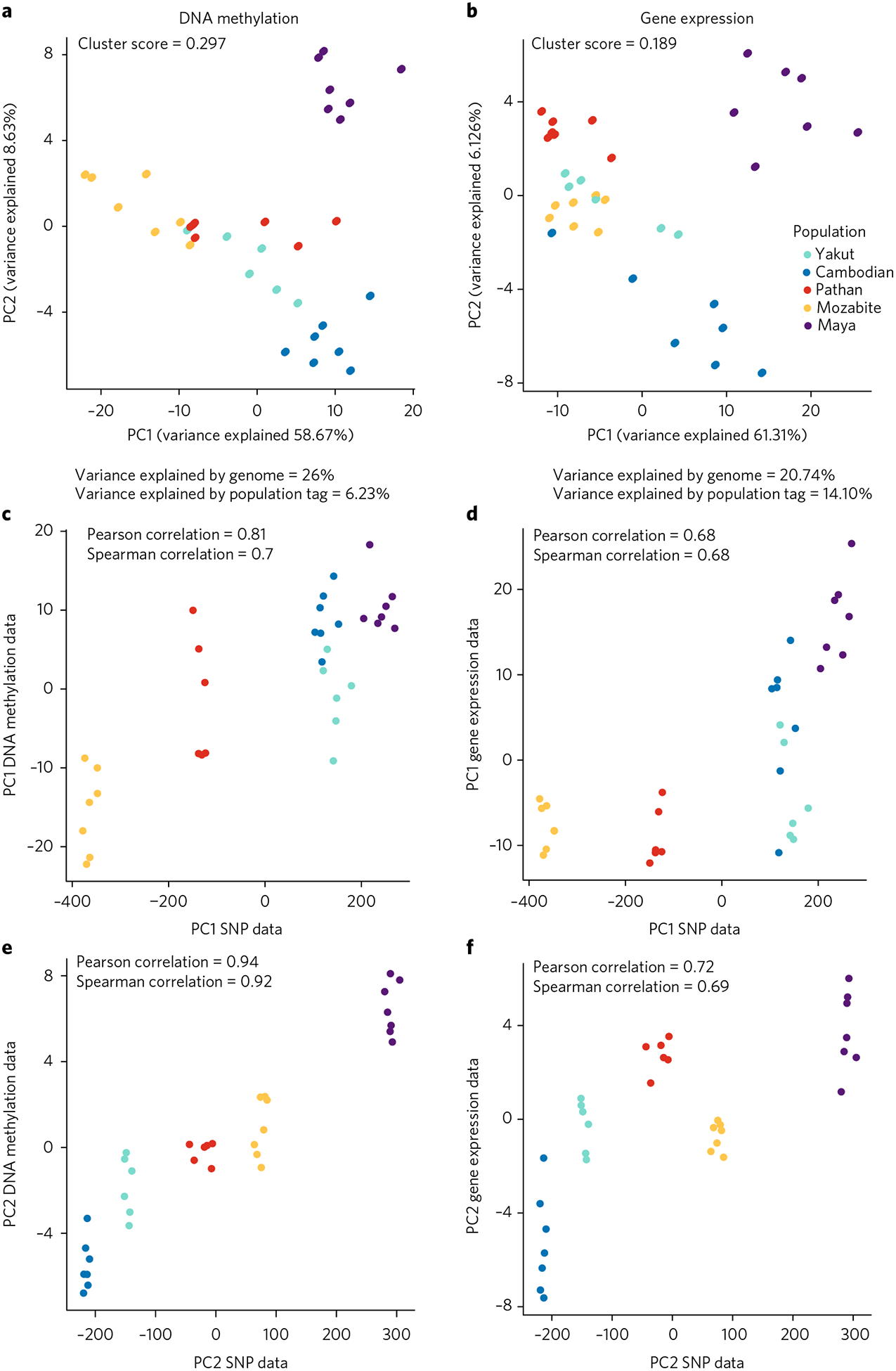
a, PCA using top 200 CpG sites with highest Pst values. b, PCA using top 200 gene expression levels with highest Pst values. Silhouette cluster scores (SCS) and percentage of variance explained by genetic variation versus the population label are as presented. c–f, Scatter plots of PC1 and PC2 SNP genotype data versus DNA methylation data (c,e) and gene expression data (d,f).
To determine the major drivers of variation, we further examined the first two PCs, which explained ~60% of the variance in both DNA methylation and mRNA levels. Interestingly, these corresponded well to the first two PCs for the genotype data from these same samples, with Pearson correlations of 0.81 for PC1 and 0.94 for PC2 of the DNA methylation data, and 0.68 and and 0.72 for gene expression (Fig. 3c–f). All four correlations were highly significant, as assessed by randomization of sample labels (P ≤ 3 × 10−4 for Pearson correlations and P ≤ 8 × 10−4 for Spearman correlations). This suggested that a common underlying factor may be associated with a substantial portion of the population specificity that we observed. In addition, these patterns are not consistent with this divergence being driven by batch effects (for example, from taking blood samples separately from each population), as these would not be expected to correlate with genotype PCs.
To further characterize this population specificity, we computed the divergence in DNA methylation and gene expression as pairwise Manhattan distances between every pair of individuals, and then compared these with the pairwise genetic distance, as estimated by the number of alleles that differ across all genotyped SNPs (see Methods). We observed a strong correlation between genetic and epigenetic divergence (Pearson’s r = 0.6 and Spearman’s ρ = 0.6 using the 200 CpG sites with highest Pst; Fig. 4a), but a much weaker one between genetic and gene expression divergence (r = 0.16 using the 200 genes with highest Pst; Fig. 4b). This suggests that DNA methylation changes accumulate in a more clock-like fashion than gene expression changes, analogous to the ‘molecular clock’ that has been observed for protein sequences31.
Fig. 4 |. epigenetic divergence as a linear function of genetic distance.
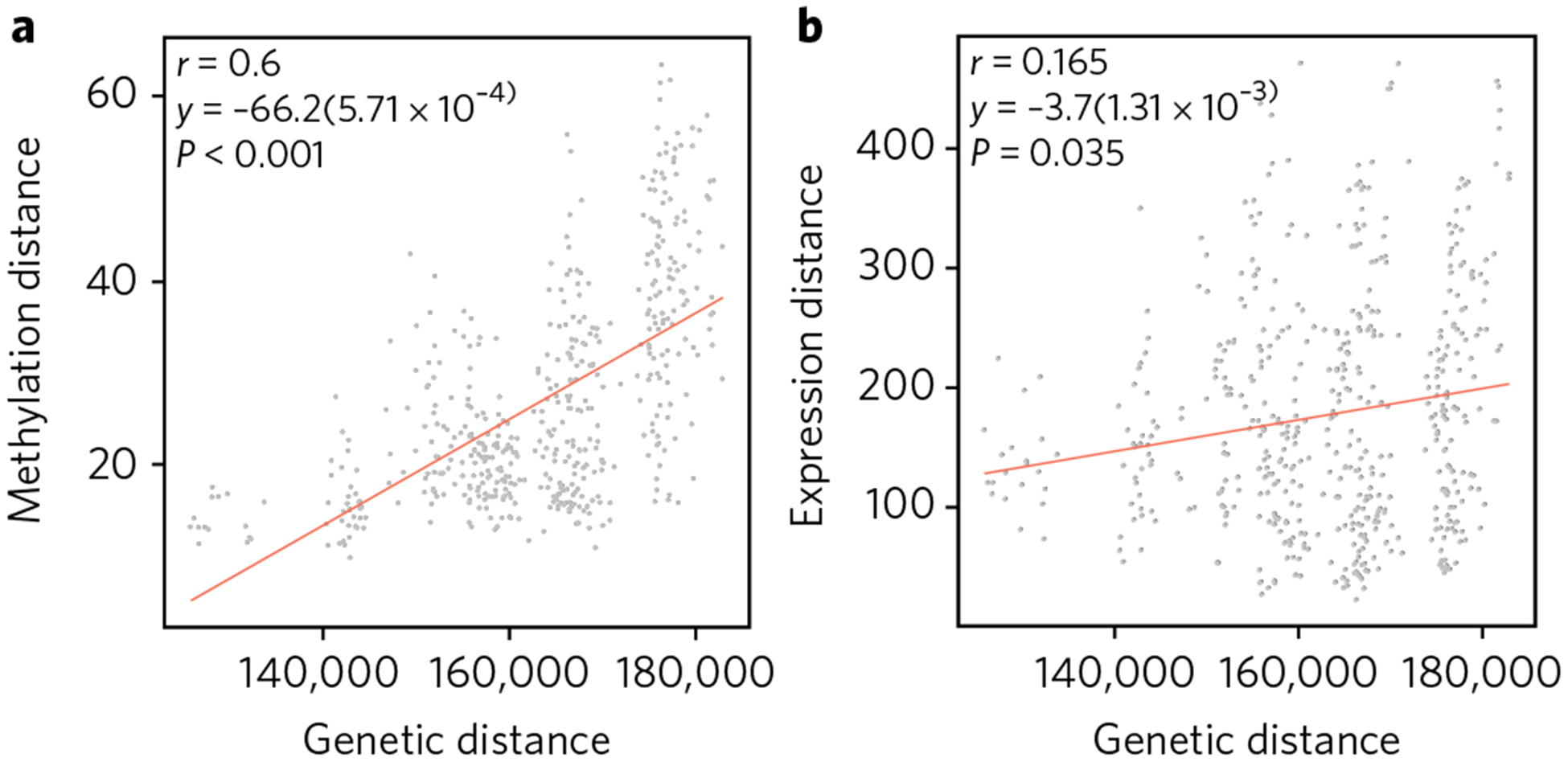
a,b, The x axis represents sequence divergence, measured as number of allele differences. The y axes are the genome-wide CpG methylation Manhattan distance (a; using the CpG sites with top 200 Pst values, between every pair of individuals across the five populations) and the genome-wide mRNA Manhattan distance (b; using the expression levels with top 200 Pst values, between every pair of individuals across the five populations). The linear regression lines are shown, together with the correlation coefficients and permutation P values using 1,000 randomizations of the data.
The strong relationship between genetic and epigenetic divergence (Fig. 4a) allowed us to estimate an approximate rate of DNA methylation divergence in humans. We regressed the epigenetic distance of all 310,289 CpG sites against genetic distance for every pair of samples, yielding a genome-wide relationship between these two measures of divergence. To express this relationship as the per-generation rate of DNA methylation change, we converted genetic distances into generations using several estimates of average human generation time and divergence times between our five populations (Methods). The median rate estimate for change in each site’s methylation level was 6.8 × 10−6 per generation (95% confidence interval: 3.0 × 10−6−1.1 × 10−5). This is substantially higher than the germline genetic mutation rate in humans (even at hypermutable CpG sites), but much lower than the DNA methylation ‘epimutation rate’ in Arabidopsis thaliana (see Discussion).
The observed epigenetic differences between populations could be caused by genetic or environmental variation, or a combination of both. Since these data are from lymphoblastoid cell lines (LCLs) that were grown in a controlled laboratory environment, the more likely driver of the observed differences is the genetic background. For example, both CpG methylation and mRNA levels could be influenced by inter-population differences in allele frequencies at genetic variants that affect these molecular traits (known as methylation and expression quantitative loci (meQTLs and eQTLs), respectively). To investigate how much of the observed population specificity can be explained by genetic variation, we first identified the local SNP (in a 200 kb window from the CpG site, or the transcription start site (TSS) for mRNA) most strongly associated with each of the 200 most population-specific CpGs or mRNAs across all of our samples. We then performed an analysis of variance including these single SNP genotypes for each of the genes/CpG sites used in Fig. 3 to assess whether the SNP genotype or population was a stronger predictor of DNA methylation or expression (Methods). We compared the average variance explained by genotype with that of the population label across all the genes/CpG sites used for the PCA in Fig. 3.
We found that the CpG sites with the highest degree of population specificity were more strongly associated with the local SNP than with population, and this local SNP explained a much higher percentage of the variance than the population label (the SNP genotype explained 26% of the variance, whereas the population label explained 6.2%). The population-specific mRNA levels showed weaker association with local SNPs (20% of the variance), but stronger association with population (14% of the variance). These results suggest that population-specific DNA methylation patterns are explained more by local genetic variants than are population-specific expression levels, and also indicate that cell line artifacts or batch effects are most likely not responsible for the population specificity we observed, as they would be unlikely to correlate with the SNP genotypes9.
Discussion
Characterization of human epigenomic variation is essential for investigating the mapping from genotype to phenotype as well as the role of the epigenome in diseases. Our analysis of five worldwide populations revealed a strong correspondence between population-specific DNA methylation, mRNA levels, and genotypes. The correlation with genetic divergence was stronger for DNA methylation, and, consistent with this, our results suggest stronger local genetic control of population-specific DNA methylation levels than of mRNA expression levels. This could be due to differences in the genetic architectures of these molecular traits, although we cannot exclude the possibility that mRNA levels could be more susceptible to batch effects than DNA methylation measurements. In any case, our results suggest that population-specific batch effects are not driving our results for DNA methylation (we also note that although cell culture can induce epigenetic changes in LCLs, existing variation between different individuals is typically preserved32).
The rates at which methylation is gained or lost at CpG sites across the human genome is an open question that we have started to address. The rate of epigenetic evolution has been explored using the model plant A. thaliana33–35. Populations descended from a single seed for 30 generations were used to examine the extent of naturally occurring variation in DNA methylation and the frequency of epimutation over time. The estimated epimutation rate was 4.46 × 10−4 per CpG site per generation — about 5 orders of magnitude higher than the genetic mutation rate of 7 × 10−9 estimated for the same lines33,35.
Our epigenetic divergence rate estimate is about two orders of magnitude lower than the epimutation rates in A. thaliana, but over two orders of magnitude greater than the rate of the germline genetic mutation rate in humans36. It is important to note that our human epigenetic divergence rates are not the same as epimutation rates, since natural selection may have acted to promote or suppress changes in DNA methylation. Nevertheless, we infer that the epimutation rate is probably far higher in A. thaliana than in humans, since natural selection is unlikely to account for the ~100-fold difference that we observed between these two species (for comparison, selection in human/chimpanzee protein-coding regions leads to only a ~5-fold slower rate of evolution at nonsynonymous sites than synonymous sites37).
Because of the limited sample size, we could not estimate how much of the population-specific DNA methylation we observed is due to allele frequency changes in global meQTLs9,38, population-specific meQTLs, or differences in environment. Classifying the contribution of these different factors to worldwide epigenetic diversity is an important step for future studies.
Through the accumulation of small allele-frequency differences across many loci, previous studies have identified geographic patterns from allele frequency variation among human populations5,6. Similarly, understanding patterns of human epigenetic diversity and evolutionary stability will be essential for understanding how population structure can shape the architecture of phenotypic traits. This epigenetic structure of human populations could be particularly relevant in various medical contexts. Variation in both genetic and epigenetic disease phenotypes, risk factors for different environmental exposures or differences in drug response may depend on ancestry and could be population specific14,39. Further thorough characterizations of worldwide human epigenetic variation will prove to be informative for understanding the origins of human phenotypic variation.
Methods
Samples.
The data set comprises SNP, CpG methylation and gene expression (RNA-seq) information for individuals from five of the Human Genome Diversity Cell Line Panel populations: 6 Siberian Yakut individuals, 7 Cambodian individuals, 7 Pakistani Pathan individuals, 7 Algerian Mozabite individuals, and 7 Mexican Mayan individuals. The SNP data set is comprised of only 6 Pathan individuals, for a total of 33 individuals. The geographic locations of these populations were previously reported16.
Silhouette cluster scores (SCS).
The silhouette value for each point is a measure of how similar that point is to points in its own cluster, when compared to points in other clusters. In our case, the clusters are the populations. The silhouette value for the i-th point, Si, is defined as Si = (bi−ai)/max{ai,bi}, where ai is the average distance from the i-th point to the other points in the same cluster as i, and bi is the minimum average distance from the i-th point to points in a different cluster, minimized over clusters. The silhouette value ranges from −1 to 1, and a high silhouette value indicates that i is well matched to its own population, and poorly matched to neighbouring populations. If most individuals have a high silhouette value, then the clustering solution is appropriate. If many individuals have a low or negative silhouette value, then the clustering solution may have either too many or too few populations. The silhouette clustering evaluation criterion was used with the Euclidean distance, but can be used with any distance metric.
Genome-wide human DNA methylation data.
DNA methylation measurements of bisulfite-treated genomic DNA were performed with the HumanMethylation450 BeadChip assay (Illumina), quantifying methylation at 485,000 sites per sample at single-nucleotide resolution, using experimental procedures recommended by the manufacturer. The bisulfite-converted DNA is subjected to a whole-genome amplification step, followed by fragmentation and hybridization to probes on the microarray. Following hybridization, allele-specific single-base extension of the probes incorporates a fluorescent label (ddNTP) for detection. Using the Illumina GenomeStudio software provided by the manufacturer, methylation levels (β values) were then computed by dividing the methylated probe signal intensity by the sum of methylated and unmethylated probe signal intensities. These β values range from 0 (completely unmethylated) to 1 (completely methylated), and provide a quantitative readout of relative DNA methylation for each CpG site within the whole cell population. Samples from the five populations were run together in a randomized order to avoid confounding batch effects with population differences. Technical replicates across different runs had correlations r > 0.99. All our samples passed internal controls included on the HumanMethylation450 array, including controls for array background, hybridization quality, target specificity and bisulfite conversion. Furthermore, all samples passed the quality control check of having detection P > 0.05. Subsequent cluster analysis indicated the absence of any outlier samples.
Normalization of β values across individuals.
The data were colour corrected, background corrected, quantile normalized and SWAN normalized to correct for type I and type II difference40. To perform the background normalization, background intensity (as measured by negative background probes present on the array) was subtracted from the raw intensities to adjust for varying background signals across different samples. This background adjustment was done separately for raw data from the green and red channels to adjust for Cy3 and Cy5 differences. All negative intensities were assigned values of zero before further normalizations were performed. To minimize batch effects across different sets of arrays, background-adjusted raw data from both channels were quantile normalized separately. The quantile normalization is done at the intensity level, whereas the SWAN normalization is done at the m value level and includes a step that randomly chooses a subset of type II probes to normalize to type I probes and then normalizes the rest of the type II probes to the normalized type II probes. This randomization step results in slightly different result every time SWAN normalization is done, so in comparing β values created from one normalization run to those in another, it is usual to see slight differences. The β values were obtained after obtaining the m values, using the formula β = 2m/(1 + 2m). In all of our analyses, we used β values since we saw no differences in the genome-wide trends or the top sites when using m values. We prefer β values because they seem easier to interpret.
After quality control check, normalization and filtering probes overlapping known SNPs in the Phase 3 1,000 Genome database and probes on the sex chromosomes, the CpG methylation data consisted of β values for 310,289 CpG sites.
Probe annotation.
Probe annotations are provided by Illumina and have been discussed extensively in other publications describing the array18. For example, proximal promoters were defined as the CpG sites located within 200 bp or 1,500 bp upstream of the described transcription start site and in the 5’-untranslated region and exon 1. The CpG shores were defined as regions located within 2 kb of CpG islands, while the CpG Shelves were defines as regions located between 2 and 4 kb from the CpG islands.
Calculation of false discovery rates.
The FDRs were computed by permutation, which preserves aspects of the data that might affect the results of the analyses. For the population-specific methylation analysis, the FDRs were estimated using 1,000 randomizations where the population tags were assigned randomly to every individual and the Kruskal–Wallis P values were recomputed on this randomized data. We chose to use the nonparametric Kruskal–Wallis test because it makes no assumptions about normality of the data, in contrast to ANOVA methods that have been used in some studies of DNA methylation across human populations.
Validation of population-specific CpG sites through bisulfite pyrosequencing.
Bisulfite PCR-pyrosequencing assays were designed with PyroMark Assay Design 2.0 (Qiagen). The regions of interest were amplified by PCR using the HotstarTaq DNA polymerase kit (Qiagen) as follows: 15 minutes at 95 °C (to activate the Taq polymerase), 45 cycles of 95 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s, and a 5 minute 72 °C extension step. For pyrosequencing, a single-stranded DNA was prepared from the PCR product with the Pyromark Vacuum Prep Workstation (Qiagen) and the sequencing was performed using sequencing primers on a Pyromark Q96 MD pyrosequencer (Qiagen). The quantitative levels of methylation for each CpG dinucleotide were calculated with Pyro Q-CpG software (Qiagen). Primer sequences are available upon request.
Concordance between array and pyrosequencing percentage methylation.
For the three most differentiated sites, the K–W P values using the Illumina array and the ones obtained by pyrosequencying are presented in Supplementary Table 1.
The Pst values.
Pst is a measure of the proportion of variance explained by between-population divergence. It is the phenotypic analog of the population genetics parameter Fst27,29. For a single probe, Pst was calculated as σ2b/(σ2b + 2σ2w), where σ2b is the between population variance and σ2w is the average within population variance. Pst values range from 0 to 1, with values near 1 signifying that the majority of epigenetic variance for a probe is between populations rather than within populations.
Analysis of variance using local SNPs.
For every CpG and mRNA level, the strongest-associated local SNP was defined as the SNP within a 200 kb window from the CpG site or the transcription start site (TSS) of the gene with the largest correlation with the methylation or mRNA levels across all individuals. We restricted our analysis to the 200 CpGs and genes in Fig. 3. An analysis of variance was performed to obtain the variance explained by the population tag and the SNP for every population-specific CpG or expression level: level ~ population + SNP + ε, where ‘level’ denotes the methylation or expression level of that CpG or mRNA, ‘population’ denotes the population tag of the individual and ‘SNP’ denotes the genotypes of the individuals. We then averaged the variances across the CpGs/mRNAs to obtain the average variance presented in Fig. 3. For the top 200 mRNAs we also identified the local CpG site within a 200 kb window from the transcription start site with the largest correlation with the expression levels across all individuals. We then performed an analysis of variance including methylation as a covariate: level ~ population + SNP + CpGmethyl + ε where ‘level’ denotes the expression level of that mRNA, ‘population’ denotes the population tag of the individual, ‘SNP’ denotes the genotypes of the individuals, and ‘CpGmethyl’ denotes their methylation levels.
Calculation of P values of the PC1 and PC2 correlations between genetic and epigenetic data.
The P values were computed by permutation, which preserves aspects of the data that might otherwise affect the results of the analyses. The P values were estimated using 10,000 randomizations where the population tags were assigned randomly to every individual and Pst values were recomputed on this randomized data. For every randomization, using the top 200 Pst values, we computed the PCs and determined both the Pearson and Spearman correlations with the PCs for the genotype data (Fig. 1b). We then counted the number of times the correlation from these 10,000 randomizations is greater, in absolute value, than the true correlation.
Pairwise genetic and epigenetic distances.
For each pair of individuals, the methylation and mRNA distances were computed using the Manhattan distance between the epigenetic vectors corresponding to each individual. The epigenetic vectors were comprised of the top 200 population-specific values (filtering by either the K–W P value or the Pst measure). The Manhattan distance was used to recapture the Hamming distance when the data are binary. One Mayan individual was an outlier and was excluded for the CpG methylation analysis. The genetic distance between each pair of individuals was computed as the number of alleles that are different between the two individuals. For example, if the genome is encoded using 0, 1, 2 at a site, this distance is an extension of the Hamming distance, taking into account shared alleles. The reported correlation P values were determined using 1,000 permutations of the data.
Population divergence times and computation of an approximate epigenetic divergence rate.
For each pair of populations, we computed a range of predicted separation times, using previous studies based on both archaeological and genetic evidence17,41. We tested a generation time of both 20 and 30 years. With a generation time of 20 years, the minimum and maximum hypothesized separation times for each pair of populations in our study, in generations, are shown in Supplementary Tables 2 and 3.
We computed both the CpG methylation divergence and genetic distance as pairwise Manhattan distances between every pair of individuals. We then regressed the epigenetic distance on genetic distance. To express genetic distance in number of generations, we also regressed this against published estimates of divergences times for our populations, converted into number of generations. With two possible generation times and two estimates of the time since divergence, we obtained four estimates of the epigenetic rate of evolution. The four resulting estimates of fraction methylation change per site per generation (and 95% confidence intervals) were: for 30 years generation length, minimum separation time was 1.2 × 10−5 (95% confidence interval: 5.2 × 10−6−1.9 × 10−5) and maximum separation time was 6.2 × 10−6 (95% confidence interval: 2.7 × 10−6−9.7 × 10−6); for 20 years generation length, minimum separation time was 7.4 × 10−6 (95% confidence interval: 3.2 × 10−6−1.2 × 10−5) and maximum separation time was 4.1 × 10−6 (95% confidence interval: 1.8 × 10−6−6.3 × 10−6).
Data availability.
Raw data have been deposited in the Gene Expression Omnibus database under accession number GSE101431.
Supplementary Material
Acknowledgements
O.C. and M.W.F. acknowledge support from the Morrison Institute for Population and Resource Studies at Stanford and the Stanford Centre for Computational, Evolutionary and Human Genomics. B.M.H. acknowledges support from NIH grant 3R01HG003229 to C. B. Bustamante. M.S.K. is a Senior Fellow of the Canadian Institute for Advanced Research and the Canada Research Chair in Social Epigenetics. We thank members of the Feldman Laboratory, in particular N. Creanza and J. Granka, for helpful discussions, and M. Jones for comments. This research was done using resources provided by the Open Science Grid, which is supported by the National Science Foundation award 1148698, and the US Department of Energy’s Office of Science.
Footnotes
Competing interests
The authors declare no competing financial interests.
Supplementary information is available for this paper at doi:10.1038/s41559-017-0299-z.
References
- 1.Cavalli-Sforza LL, Menozzi P & Piazza A The History and Geography of Human Genes (Princeton Univ. Press, Princeton, NJ, 1994). [Google Scholar]
- 2.Li JZ et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Novembre J et al. Genes mirror geography within Europe. Nature 456, 98–101 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakobsson M et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451, 998–1003 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran S et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl Acad. Sci. USA 102, 15942–15947 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg NA et al. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 10, e70 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Heyn H et al. DNA methylation contributes to natural human variation. Genome Res. 23, 1363–1372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser HB, Lam LL, Neumann SM & Kobor MS Population-specificity of human DNA methylation. Genome Biol. 13, R8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moen EL et al. Genome-wide variation of cytosine modifications between European and African populations and the implications for complex traits. Genetics 194, 987–996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fagny M et al. The epigenomic landscape of african rainforest hunter-gatherers and farmers. Nat. Commun 6, 10047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stranger BE et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8, e1002639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin AR et al. Transcriptome sequencing from diverse human populations reveals differentiated regulatory architecture. PLoS Genet. 10, e1004549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jirtle RL & Skinner MK Environmental epigenomics and disease susceptibility. Nat. Rev. Genet 8, 253–262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feil R & Fraga MF Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet 13, 97–109 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Cann HM et al. A human genome diversity cell line panel. Science 296, 261–262 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Henn BM, Cavalli-Sforza LL & Feldman MW The great human expansion. Proc. Natl Acad. Sci. USA 109, 17758–17764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandoval J et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6, 692–702 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Trapnell C et al. Differential gene and transcript expression analysis of rna-seq experiments with tophat and cufflinks. Nat. Protoc 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalli-Sforza LL, Piazza A, Menozzi P & Mountain J Reconstruction of human evolution: bringing together genetic, archaeological, and linguistic data. Proc. Natl Acad. Sci. USA 16(85), 6002–6006 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conrad DF et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet 38, 1251–1260 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Creanza N et al. A comparison of worldwide phonemic and genetic variation in human populations. Proc. Natl Acad. Sci. USA 112, 1265–1272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg NA et al. Genetic structure of human populations. Science 298, 2381–2385 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Lovmar L, Ahlford A, Jonsson M & Syvänen A-C Silhouette scores for assessment of SNP genotype clusters. BMC Genom. 6, 35 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavalli-Sforza LL & Feldman MW The application of molecular genetic approaches to the study of human evolution. Nat. Genet 33, 266–275 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Henn BM, Cavalli-Sforza LL & Feldman MW The great human expansion. Proc. Natl Acad. Sci. USA 109, 17758–17764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pujol B, Wilson AJ, Ross RIC & Pannell JR Are Qst-Fst comparisons for natural populations meaningful? Mol. Ecol 17, 4782–4785 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Edelaar P, Burraco P & Mestre IG Comparisons between Qst and Fst — how wrong have we been? Mol. Ecol 20, 4830–4839 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Leinonen T, McCairns RJS, O’Hara RB & Merilä J Qst–Fst comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat. Rev. Genet 14, 179–190 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Weir BS & Cockerham CC Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984). [DOI] [PubMed] [Google Scholar]
- 31.Zuckerkandl E & Pauling L Horizons in Biochemistry. (Academic, New York, 1962). [Google Scholar]
- 32.Caliskan M, Cusanovich DA, Ober C & Gilad Y The effects of EBV transformation on gene expression levels and methylation profiles. Human Mol. Genet 20, 1643–1652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van der Graaf A et al. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl Acad. Sci. USA 112, 6676–6681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker C et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480, 245–249 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Schmitz RJ et al. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334, 369–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch M Rate, molecular spectrum, and consequences of human mutation. Proc. Natl Acad. Sci. USA 107, 961–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikkelsen TS et al. The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Bell JT et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 120, R10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feinberg AP Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Maksimovic J, Gordon L & Oshlack A SWAN: Subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scally A & Durbin R Revising the human mutation rate: implications for understanding human evolution. Nat. Rev. Genet 13, 745–753 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data have been deposited in the Gene Expression Omnibus database under accession number GSE101431.