

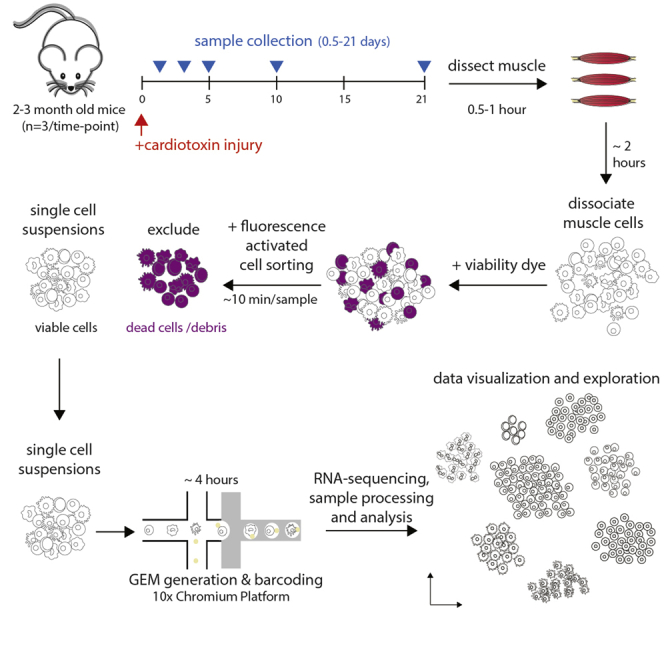
Summary
Single-cell RNA sequencing (scRNA-seq) is a powerful technique for deconvoluting and clustering thousands of otherwise intermingled cells based on their gene expression. Here, we present a complete protocol for the unbiased evaluation of regenerating murine skeletal muscle using scRNA-seq. The skeletal muscle is unique in its cellular composition as being primarily multinucleated muscle cells (myofibers). This protocol focuses on isolating mononuclear cells from muscle for subsequent scRNA-seq analysis and can be modified to assess cell populations in other tissues of interest.
For complete details on the use and execution of this protocol, please refer to Liu et al. (2015) and Oprescu et al. (2020).
Graphical Abstract
Highlights
-
•
Cardiotoxin is used to injure skeletal muscles and induce muscle regeneration
-
•
Single cells isolated from regenerating muscles are individually tagged for scRNA-seq
-
•
Unbiased transcriptional profiling of cell types involved in muscle regeneration
Single-cell RNA sequencing (scRNA-seq) is a powerful technique for deconvoluting and clustering thousands of otherwise intermingled cells based on their gene expression. Here, we present a complete protocol for the unbiased evaluation of regenerating murine skeletal muscle using scRNA-seq. The skeletal muscle is unique in its cellular composition as being primarily multinucleated muscle cells (myofibers). This protocol focuses on isolating mononuclear cells from muscle for subsequent scRNA-seq analysis and can be modified to assess cell populations in other tissues of interest.
Before You Begin
The main steps of this protocol include muscle dissection and cell dissociation, single-cell selection by fluorescence activated cell sorting, single-cell RNA library preparation using the 10x Chromium platform, sequencing, and analysis. It is intended to serve as an overview of these steps and the timing, reagents and equipment required, and potential pitfalls.
Before starting the protocol, ensure that the required reagents for single-cell RNA sequencing (scRNA-seq) are purchased and ready for use. It is important to consider the background, age, and sex of the mice such that they are matched as closely as possible across timepoints. It is recommended to pool n=3 mice/sample to control for biological variability. Ideally, researchers should process two samples per timepoint if budget allows. Mice should be 2–3 months of age at time of injury (below). Cardiotoxin is commonly used to induce muscle damage, and subsequent stem cell-mediated muscle repair (regeneration). Examination of gene expression in single cells by scRNA-seq at various regenerating time points will allow the understanding of cell populations, temporal dynamics of cell populations, and cell-cell interactions over the course of muscle regeneration.
Cardiotoxin-Induced Skeletal Muscle Injury
Timing: ~1 month, depending on availability of mice and regenerative stages to be evaluated
Note: It is recommended to pool at least three mice per sample to control for biological variability. Ideally, each timepoint should contain two samples to assess for potential batch effects if budge allows. If this is not feasible, using a balanced design such that if samples must be processed on different days, they are not processed in the order of their regeneration timepoint.
-
1.
Prepare ketamine/xylazine and cardiotoxin (CTX) solutions.
-
a.
Ketamine/xylazine preparation.
-
i.
Mix 0.9 mL ketamine (10 mg/mL) with 0.1 mL xylazine (20 mg/mL) and 9 mL saline solution (can be stored at room temperature, 15°C–25oC).
-
b.
Cardiotoxin preparation.
-
i.
Dilute cardiotoxin with saline to 10 μM CTX working solution (can be stored at −20oC in 1 mL aliquots, 50 μL will be injected per TA muscle).
-
2.
Muscle injury.
-
a.
Anesthetize mice using ketamine/xylazine solution.
-
i.
Administer 0.01 mL/g of body weight via intraperitoneal (IP) injection.
-
b.
Shave hair over left and right tibialis anterior (TA) muscle prior to injury.
-
c.
Using a 27-gauge needle, insert the needle parallel to the fibula and slowly inject 50 μL of 10 μM CTX per TA (left and right) to induce injury.
Note: It is it difficult to assess the efficiency of muscle injury prior to dissection. Upon dissection, injured muscle will be stiff at < 3 days post injury compared to non-injured muscle and should easily pull away from the tendon and at the knee (see muscle dissection and Figure 1). At later timepoints (3–7 days), well-injured muscle will appear red as the tissue is healing. Past 10 days, the injured muscle may appear smaller than non-injured but should recover in size by 21–30 days.
Figure 1.

Step-By-Step Dissection of TA Muscles
-
3.
Repeat muscle injury for the regeneration timepoints of interest. Users should consider both early (e.g., 0.5, 2, 3.5, 5 days) and late (e.g., 10, 21 days) timepoints to best capture the immediate response to muscle injury and resolution.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 10% Tween 20 | Bio-Rad | 1662404 |
| 50% glycerol | Ricca Chemical Company | 3290-32 |
| Bovine Serum Albumin | Gemini Bio | 700-105P |
| Buffer EB | Qiagen | 19086 |
| Cardiotoxin (CTX) | Sigma | 11061-96-4 |
| Collagenase II | Worthington | LS004177 |
| Dispase | Roche | 04942078001 |
| F10 medium | Gibco | 11550-043 |
| Fetal bovine serum | HyClone | SH30088 |
| Ketamine HCl | Akron | 59399-114-10 |
| Low TE buffer | Thermo Fisher Scientific | 12090-015 |
| Penicillin-Streptomycin | ThermoFisher | 15070063 |
| Phosphate-buffered saline | Gibco | 21600-069 |
| Red blood cell lysis solution | Promega | Z3141 |
| Xylazine | Akron | 59399-110-20 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3′ GEM Library & Gel Kit v3 | 10x Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10x Genomics | PN-1000153 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| SPRI-select Reagent Kit | Beckman Coulter | B23318 |
| Zombie Violet Fixable Viability Dye | BioLegend | 423113 |
| Deposited Data | ||
| scRNA-seq regenerating muscle data | GEO | GSE138826 |
| Experimental Models: Organisms/Strains | ||
| C5Bl6/ WT mice | Purdue TG Core | N/A |
| Software and Algorithms | ||
| Cell Ranger Software | 10x Genomics | v3.1 |
| Seurat | Satija lab: satijalab.org/seurat/ | v3 |
| Other | ||
| 10 mL syringe | BD Falcon | 302995 |
| 10x Magnetic Separator | 10x Genomics | 230003 |
| 18G1 1/2 needles | BD | 305196 |
| 40 μm filters | BD Falcon | 352340 |
| 27-gauge insulin syringes | Exel Int | 26028 |
| PCR tubes, 0.2 mL 8-tube strips, sterile | Eppendorf | 951010022 |
| Syringe filter | CellTreat | 229751 |
| 2 mL low binding conical-bottom microcentrifuge tube, nuclease free | VWR | 16466-042 |
| Centrifuge | ThermoFisher | IEC Centra CL2 |
| 15 mL conical tube | Fisher Scientific | 14-959-53A |
| PCR machine | N/A | N/A |
| Vortex genie | VWR | SI-0236 120V |
| Normal saline solution | Vedco | 50989-885-17 |
| Low retention 10 μL filtered pipette tips | Fisher Scientific | 02-707-000 |
| Low retention 200 μL filtered pipette tips | Fisher Scientific | 02-707-006 |
| Low retention 1,000 μL filtered pipette tips | Fisher Scientific | 02-707-008 |
Note: Microcentrifuge tubes and pipette tips must be DNase and RNase free.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shihuan Kuang, skuang@purdue.edu.
Materials Availability
Materials used in this protocol are commercially available and no mouse lines were generated for this protocol.
Data and Code Availability
Data generated in this study is available at GSE138826.
Materials and Equipment
The 10x Genomics Chromium System is required for this protocol. Alternative scRNA-seq platforms may be used – however, this protocol has not been tested with other platforms. It is also critical so use nuclease-free pipette tips, nuclease-free microcentrifuge tubes, and work under sterile conditions.
Alternatives: BD FACS Aria II or similar is required to perform fluorescence activated cell sorting (FACS) to exclude debris/dead cells and generate single-cell suspensions.
Alternatives: To assess cDNA quality during the generation of barcoded single-cell libraries, an Agilent High Sensitivity Chip or equivalent Bioanalyzer is required.
Alternatives: Although incubating muscle digestions at 37°C with gentle shaking (100 rpm) is recommended, if a sterile incubator is not available incubating in a water bath at 37°C with intermittent shaking should suffice.
Wash Medium
| Reagent | Final Concentration | Volume (mL) |
|---|---|---|
| Ham’s F10 Nutrient Mix | n/a | 445 mL |
| Pen/Strep (100×) | 1× | 5 mL |
| Fetal bovine serum | 10% | 50 mL |
| Total | n/a | 500 mL |
Muscle Dissociation Buffer
| Reagent | Final Concentration | Amount |
|---|---|---|
| Wash medium | n/a | 20 mL |
| Collagenase type II | 725 U/mL | 50 mga |
| Total | n/a | 20 mL |
Units/mg dry weight varies per batch.
Stock Collagenase Type II Solution (10 mL)
| Reagent | Final Concentration | Amount |
|---|---|---|
| Collagenase type II | 1,000 U/mL | 40 mga |
| 1× PBS | n/a | 10 mL |
| Total | n/a | 10 mL |
Units/mg dry weight varies per batch.
Stock Dispase Solution (10 mL)
| Reagent | Final Concentration | Amount |
|---|---|---|
| Dispase | 11 U/mL | 140 mga |
| 1× PBS | n/a | 10 mL |
| Total | n/a | 10 mL |
Units/mg dry weight varies per batch.
Prepare 0.1% BSA Solution by Dissolving BSA Powder into 1× PBS
| Reagent | Final Concentration | Amount |
|---|---|---|
| Bovine Serum Albumin (BSA) powder | 1 mg/mL (0.1%) | 10 mg |
| 1× PBS | n/a | 10 mL |
Dilute to 0.05% BSA solution in 1× PBS with equal volumes of 1× PBS: 0.1% BSA solution.
Note: Stock collagenase and dispase solutions should be filtered through a 0.22 μm filter and stored at −20°C in 1 mL aliquots. BSA solution should also be filter sterilized and can be stored at 4°C for 2 weeks.
Step-By-Step Method Details
This section lists the major steps, provides step-by-step details and timing for each major step. Please note that there are stopping points designated in the 10x Chromium protocol but for the purpose of reproducibility, suggested stopping points are noted based on previous successful runs.
Note: For details on the execution of the single-cell suspensions from muscle, please refer to Liu et al. 2015. For complete details regarding the generation of single-cell droplets, cDNA synthesis, amplification and library preparation for scRNA-seq, please refer to the 10x Chromium protocol which can be found at: https://assets.ctfassets.net/an68im79xiti/4tjk4KvXzTWgTs8f3tvUjq/2259891d68c53693e753e1b45e42de2d/CG000183_ChromiumSingleCell3__v3_UG_Rev_C.pdf
For details regarding the Seurat package used from exploration of single-cell data, please refer to satijalab.org/seurat/ and the “Quantification and Statistical Analysis” section.
Muscle Dissection and Digestion—Day 1
Injured and non-injured tibialis anterior (TA) muscles are dissected and digested to generate single-cell suspensions.
Note: This protocol is adapted from Liu et al., 2015.
Note: If there are >9 mice (>3 samples), it is recommended to have multiple people working together as sample processing can be time-consuming and this may affect the digestion times of other samples.
Before starting, prepare muscle dissociation buffer and have dishes with 1× PBS on ice ready for each sample. Reagents should be ice cold and samples kept on ice unless otherwise specified It is also important to maintain sterile techniques throughout the entirety of this procedure.
-
1.
Dissect TA muscles from injured and/or non-injured mice (Figure 1).
-
a.
Spray mice with 70% EtOH.
-
b.
Gently remove skin to reveal TA muscle.
-
c.
Remove membrane (epimysium) that covers the TA muscle by gently poking along the fibula (without disrupting the muscle) with tweezers until membrane can be removed.
-
d.
Cut the distal tendon of the TA using a pair of micro surgical spring scissors.
-
e.
Using tweezers, hold the severed tendon and gently pull the TA muscle up toward the knee.
-
f.
Cut the proximal tendon as close to the knee as possible and place the muscle in 1× PBS on ice.
CRITICAL: keeping the tendon intact will prevent muscle hypercontraction and make it easier to cut the muscles for digestion.
-
2.
Using a vacuum, aspirate 1× PBS and rinse dissected muscles three more times with 5 mL 1× PBS to remove any additional fur.
-
3.
To cut the muscle, hold the tendon with a pair of tweezers and cut the TA parallel to the muscle fibers (such that the TA is cut into small fascicles and held at the tendon).
-
a.
The tendon can be cut away after the TA has been cut.
-
4.
Place the TA muscles in a 15 mL conical tube with 5 mL muscle dissociation buffer.
-
5.
Wrap the cap with paraffin to prevent leaking and incubate all samples at 37°C for 1 h with gentle shaking (100 rpm).
-
6.
Fill tube with ice cold wash medium to stop digestion.
-
7.
Centrifuge samples at 525 × g at room temperature (22°C–25°C) for 5 min to pellet sample and remove debris.
-
8.
Remove supernatant to leave 4 mL.
-
9.
Add 0.5 mL stock collagenase type II (stock at 1,000 U/mL; final at 100 U/mL) and 0.5 mL stock dispase (stock at 11 U/mL; final at 1.1 U/mL) to each tube and gently resuspend pellet.
-
10.
Wrap cap with paraffin and incubate samples at 37°C for 30 min with gentle shaking (100 rpm).
-
11.
Gently run sample through 18-gauge needle on a 10 mL syringe 10–12 times to break up any remaining muscle pieces.
-
12.
Fill tube with ice cold wash medium to stop reaction.
-
13.
Centrifuge samples at room temperature (15°C–25°C) for 5 min at 525 × g.
-
14.
Remove supernatant leaving ~4 mL medium and gently resuspend pellet.
-
15.
Run sample through pre-wet 40 μm filter and rinse filter with additional 10 mL ice cold wash medium.
-
16.
Centrifuge samples at room temperature (15°C–25°C) for 5 min at 525 × g.
-
17.
Red blood cell lysis:
-
a.
Completely remove supernatant and resuspend pellet in 2 mL red blood cell lysis solution at room temperature (15°C–25°C).
-
b.
Incubate at room temperature (15°C–25°C) for a maximum of 3 min.
-
c.
Fill tube with 1× PBS to stop red blood cell lysis.
-
d.
Centrifuge samples at room temperature (15°C–25°C) for 5 min at 525 × g and proceed with staining.
Note: Pelleted cells should now appear white, with little to no red (i.e., blood cells) present.
Staining for Cell Viability—Day 1
Live/dead viability stain to ensure live cells are sorted for downstream processing.
-
18.
Remove supernatant and resuspend sample in 3 mL 1× PBS.
-
a.
Add 0.5 mL cell suspension to fresh tube (for no-stain control) and 0.5 mL 0.1% BSA (to final concentration of 0.05% BSA) and leave on ice.
-
19.
To remaining sample, add 2.5 μL Zombie Violet viability dye (dilution of 1:1,000) and wrap with aluminum foil to protect from light.
-
20.
Incubate sample at room temperature (15°C–25°C) for 20 min on orbital shaker at 50 rpm.
-
21.
After 20 min, fill tube with 1× PBS and centrifuge sample at room temperature (15°C–25°C) for 5 min at 525 × g.
-
22.
Remove supernatant, resuspend sample in 2 mL 0.05% BSA solution and proceed with FACS.
Fluorescence Activated Cell Sorting—Day 1
Fluorescence activated cell sorting (FACS) to exclude debris, clumped cells, and generate single-cell suspensions for scRNA-seq.
Note: This is generally performed by a core facility or with prior expertise. The general gating strategy is described below.
-
23.
Use the forward and side scatter to select based on size of cells (Figure 2).
Figure 2.

Representative Fluorescence Activated Cell-Sorting Data
(A) No stain control sample. FSC-A and SSC-A are used to initially gate cells (Gate 1, red outline). Cells are further refined based on SSC-H vs. SSC-W (Gate 2, red outline) and subsequently FSC-H vs. FSC-W (Gate 3, red outline). Baseline fluorescence of cells (no stain) in live/dead Violet-A channel vs SSC-A is used to select cells for stained sample (No stain, black outline).
(B) Live/dead stained sample. Same gating strategy as outlined for (A). Note: no stain and Gate 4 (live cells) are the same but appearance is affected by the automatic scaling of the X-axis. This representative sample is from mice at 5 days post injury.
SSC, side scatter; FSC, forward scatter (-A, area; -H, height; -W, width).
-
24.
Determine gate for live/dead viability stain based on no-stain control (Figures 2A and 2B).
-
a.
Live/dead viability dye will be excluded from the cytoplasm of live cells but will bind the membrane of both live and dead cells. Therefore, dead cells and debris will appear much brighter than live cells (Figure 2B).
-
25.
Collect 100,000 live cells into a 2 mL sterile, low binding microcentrifuge tube containing 450 μL 0.05% BSA solution. Keep it on ice.
Generation of Single-Cell Suspensions and cDNA Synthesis—Day 1
Note: This portion outlines general steps that are detailed in the 10x Chromium Single Cell 3′ v3 kit. This is not meant to replace the protocol but rather highlight ideal stopping points, outline tips, and provide quality control examples from successful runs.
Generation of single-cell droplets, in-drop reverse transcription, and initial cDNA amplification.
-
26.
Centrifuge sorted cells for 5 min at 525 × g at room temperature (22°C–25°C) and resuspend in 200 μL 0.05% BSA solution.
-
27.
Use a hemocytometer or automated cell counter to confirm the density of cell suspension and viability.
-
28.
The 10x protocol outlines the targeted cell recovery and the respective cell stock density; for example, for a desired 10,000-target cell recovery, the cell density ranges from 900–1,200 cells/μL (see protocol for specific details).
-
29.
Follow the manufacturer’s instructions for the generation of Gel Bead-In Emulsions (GEMs) using the 10x Chromium system.
-
a.
The specialized 10x vortex Adapter (Product # 330002) attachment to vortex the Single Cell 3′ Gel Beads is not necessary; using a 3-inch platform and holding the GEMs while vortexing is sufficient.
-
30.
Proceed with the in-GEM reverse transcription (RT) per the manufacturer’s instructions.
-
a.
It is recommended to use a nearby PCR machine, as excessive movement with the GEMs may comprise their integrity and affect overall data quality.
-
31.
After RT, follow the 10x protocol instructions for cDNA clean-up and initial amplification.
-
a.
The 10x protocol contains pictures of what the samples should look like after the addition of the recovery reagent. There is sometimes a cloudy appearance to the aqueous phase. If brief centrifugation does not clear it completely, it is OK to proceed.
-
b.
When removing the recovery reagent, only remove the 125 μL recommended by 10x Chromium even if this leaves pink recovery reagent.
-
32.
Proceed with the final clean-up after the initial cDNA amplification and send samples for quality control (QC) analysis via TapeStation or Bioanalyzer.
-
a.
This is an ideal stopping point. Representative traces from successful runs are shown in Figures 3A and 3B.
Figure 3.

Representative Quality Control Results
(A–D) Traces were generated from an Agilent DNA High Sensitivity Chip. (A) Initial cDNA library QC trace for 10 DPI sample, average size 1,793 bp. (B) Initial cDNA library QC trace image for 21 DPI, average size 1,684 bp. (C) Post-library construction QC trace for 10 DPI sample, average size 447 bp. (D) Post-library construction QC trace for 21 DPI sample, average size 463 bp. Note: peaks at 35 and 10,380 bp are spike-in controls.
QC, quality control; DPI, days post injury; FU, fluorescence units; bp, base pairs.
Pause Point: The 10x Chromium protocol also suggests that GEMs can be stored at 4°C for 12 h or for 72 h at −20°C after the RT if necessary.
Library Preparation and Sequencing—Variable Timing
Note: Again, this portion outlines general steps that are detailed in the 10x Chromium Single Cell 3′ v3 kit. This is not meant to replace the protocol.
Note: The timing of this is variable. Library preparation is relatively quick (< 2 h), but the turn-around time for sequencing will depend on the respective core or company the samples are sent to.
Library Preparation and Next-Generation Sequencing
-
33.
Refer to 10x Chromium Single Cell 3′ Reagents Kits User Guide for details on amplification, library construction and QC. Representative traces from successful runs are shown in Figures 3C and 3D.
-
34.
Samples are ready for high-throughput sequencing and the 10x Chromium protocol outlines specific requirements depending on sequencing platform used. Generally, 50,000 reads/cell yields best results.
Expected Outcomes
Images for expected outcomes at various stages of the protocol are described below.
Step-by-step procedure for dissection of TA is illustrated in Figure 1. After dissociation of cells from non-injured and injured muscles, cell viability is assessed via FACS. Figure 2 is an example of our FACS data to highlight gating strategy and expected results. Cells are first gated based on forward and side scatter area (FSC-A, SSC-A) and further refined to eliminate aggregated cells and debris based on forward and side scatter, respectively (Figure 2). Live cells (i.e., cells that do not or minimally stain with viability dye) are selected based on a no-stain control (Figure 2A, right panel no stain, Figure 2B, right panel + viability stain) and sorted into collection tube containing 0.05% BSA solution. After sorting, cells are centrifuged, re-counted, loaded onto the 10x Chromium platform and processed according to the manufacturer’s instructions. The 10x Chromium 3′ gene expression kit has two quality control (QC) points where it is recommended to analyze the samples by TapeStation or Bioanalyzer. After the initial amplification, QC traces from an Agilent DNA Hypersensitivity Chip should be relatively clean with a curve ranging from ~600–2,000 base pairs (Figures 3A and 3B). After fragmentation and library preparation, QC traces from an Agilent DNA Hypersensitivity Chip should be clean with a curve now ranging from ~300–600 base pairs (Figures 3C and 3D). Finally, after the samples are sequenced and the data are initially processed, batch effects can indirectly be evaluated by principal component analysis (PCA) of all the regeneration timepoints pooled together and colored by batch. Ideally, cells and clustering results should not separate by batch (Figure 4).
Figure 4.

Examination of Potential Batch Effects
(A) Principal component analysis and embedding of all cells evaluated by scRNA-seq from six regeneration stages and non-injured muscle, colored by batch.
(B) UMAP embedding of cells colored by batch.
(C) UMAP embedding of cells colored by regeneration timepoints.
(D) UMAP embedding of cells, colored by cluster. The results suggest that there is no obvious separation based on batch. Batch 1, 0.5 and 3.5 DPI; Batch 2, 10 and 21 DPI; Batch 3, NI; Batch 4, 2 and 5 DPI.
DPI, days post injury; NI, non-injured.
Quantification and Statistical Analysis
This section provides a brief overview of the workflow for single-cell data analysis using CellRanger version 3.1 and the R package Seurat (Satija et al., 2015).
Note: For initial data analysis, the R Seurat package is an excellent tool to explore single-cell data. Tutorials on the use of Seurat can be found at https://satijalab.org/seurat/.
-
1.
Download the 10x Chromium Cell Ranger software and follow the instructions outlined on their website tutorial to align reads and generate the cell ID × gene count matrix.
-
2.
Download and install the most recent version of Seurat.
-
a.
This requires the R computing environment so install R if it is not already installed.
-
3.
A workbook on how to load data and perform the initial analysis with Seurat can be found via the link from the Satija lab provided above. Below are aspects to consider when performing the data analysis but are not meant to limit the scope of the analysis or usage of Seurat.
-
a.
If there are multiple samples from the same timepoint, researchers can use the IntegrateData function or the CCA-based anchors method (Butler et al., 2018, Stuart et al., 2019) to minimize differences across samples from the same timepoint that may be due to batch. These methods were primarily developed to integrate scRNA-seq data from common sources generated in different batches, from different species or by different scRNA-seq platforms. These methods can therefore be used to combine samples from the same timepoint to minimize batch effects.
-
b.
If each sample is a different timepoint (or after samples from the same timepoint have been integrated from step 3a), the data can be merged and processed together. Details can be found in Seurat workbook.
Note: Batch effects are difficult to directly quantify between samples from different timepoints, as both biological and technical variability may contribute to the observed differences between samples. Nonetheless, PCA can be helpful to survey any outstanding batch effects across different timepoints, and ideally timepoints should not separate purely based on batch (Figure 4A).
-
c.
We suggest altering various parameters such as the number of neighbors, dimensions, resolution, when running the FindClusters(), FindNeighbors(), RunUMAP() functions to determine the parameter settings best suited for the data, as there are no set methods that work uniformly for all datasets.
Limitations
Highlighted below are some potential limitations to consider during experimental design and data analysis.
Both the cellular and extracellular environment of the muscle are highly dynamic throughout the course of regeneration. Thus, at various timepoints there is no method to guarantee that all cells are uniformly released during the digestion procedure. Although the stringency of gating for FACS is minimal, the forward and side scatter of (for example) various immune populations can depend on the immune cell stage and function. If there is any intramuscular adipogenesis, it would be difficult to capture adipocytes using FACS and centrifugation. Data analysis and exploration are therefore imperative for evaluating the presence of previously reported cell populations and their characteristics.
Capture of muscle stem cells (MuSCs) at early (0.5- and 2-days post injury) timepoints may also be less efficient than at other timepoints. This may be due to significant changes in the regenerating environment and/or the relative abundance of MuSCs to infiltrating cells. Nonetheless, researchers can consider using a MuSC reporter to ensure that MuSCs are sorted for scRNA-seq profiling at these early timepoints.
Finally, this protocol is intended for the scRNA-seq of mononuclear cells and is not optimized to profile myonuclei. Although we detected a population of myonuclei (Figure 4D), this was most likely due to FACS sorting these as individual cells. Other methods (not discussed here) will be needed to perform single-nuclei transcriptomic profiling.
Troubleshooting
Described below are some potential problems and recommendations for troubleshooting.
Generally, this protocol leaves little room for troubleshooting as the 10x Chromium protocol is designed to address potential issues but also to minimize those arising. However, it is advisable that researchers perform FACS analysis on muscle samples to ensure the isolation protocol is effective and cell viability is >50% (which may also vary regeneration timepoint, as early timepoints may contain more debris).
Problem
Cell viability < 50%. It is not recommended to proceed with samples that have low viability as it may introduce unexpected biases in the results.
Potential Solution
Low viability may arise due to over-digestion of muscle and potential solutions to this include (1) cut muscle into larger pieces or (2) reduce digestion times in 5-min increments.
Cell death may also arise from keeping cells at room temperature or excessive processing time. To minimize cell death after FACS, slowing down the sorting speed and coating the collection tube with FBS can help to preserve viability. Ensuring the samples are maintained on ice unless otherwise specified and gentle handling of the cell suspensions are important as forceful pipetting and agitation will impact cell viability. Finally, it is critical to work quickly to load cells onto the 10x Chip post FACS.
Problem
Batch effects.
Potential Solution
Experimental design prior is imperative for mitigating batch effects. Either include multiple samples per regeneration stage, perform all experiments on the same day (depending on sample number) or set up a balanced design (such that sample processing does not occur in the order of regeneration stage). If batch effects are present across samples from the same timepoints, Seurat outlines a CCA anchors-based method or has an IntegrateData function that can be used to minimize the effects batch have on data interpretation (Butler et al., 2018, Stuart et al., 2019). However, these methods may mask biological diversity if performed across samples from different timepoints (Figure 5), and the presence of batch effects are difficult to quantify across samples with anticipated biological heterogeneity. While PCA can provide some insight, it is at the discretion of the researcher to determine if batch effects pose a significant challenge to data interpretation. Alternatively, researchers can consider clustering each timepoint individually and analyzing them separately.
Figure 5.

Integration of Samples from Different Timepoints May Mask Biological Heterogeneity
(A) Merged data from the six regeneration timepoints and non-injured skeletal muscle, colored by timepoints.
(B) Data from the regeneration timepoints processed using Seurat’s CCA anchors-based method, colored by timepoint.
(C) Data from the regeneration timepoints processed using Seurat’s integration method, colored by timepoint. These methods are commonly used to correct for batch effects across similar samples but may mask the biological diversity across dynamic and heterogenous samples. For example, progressive changes in the immune cell populations during muscle regeneration are anticipated as the regenerative environment changes from a pro- to anti-inflammatory environment. Correction for batch effects yields super-imposition of immune cells from all timepoints (largest cluster). Clustering and analysis of each timepoint individually also recapitulated the observed trend from (A) (data not shown). It is thus up to the discretion of the researcher to determine which methods are appropriate.
DPI, days post injury; NI, non-injured.
Acknowledgments
This work was supported by grants from the US National Institutes of Health (R01AR071649), National Institute of Food and Agriculture (NC-1184), United States Department of Agriculture, and Purdue University Center for Cancer Research (P30CA023168). Funding for open access charge: NIH R01AR071649. We would like to acknowledge the Purdue Genomics Core Facility for quality control and sequencing, Purdue University’s Flow Cytometry and Cell Separation Facility for assistance with fluorescence-activated cell sorting, and Purdue’s Center for Cancer Research for support with the 10x Chromium platform. We would also like to thank Dr. Gregory Cresswell and the Ratliff lab at Purdue University for assistance with the 10x Chromium scRNA-seq platform; Drs. Nadia Atallah, Luis F. Brito, and Jun Wan for cluster access and advice on data analysis; Kun Ho Kim for critical reading of the protocol; and Jun Wu for technical support.
Author Contributions
S.N.O. wrote the protocol, F.Y. assisted with the protocol development, and S.K. conceived the protocol.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Stephanie N. Oprescu, Email: soprescu@purdue.edu.
Shihuan Kuang, Email: skuang@purdue.edu.
References
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Cheung T.H., Charville G.W., Rando T.A. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc. 2015;10:1612–1624. doi: 10.1038/nprot.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprescu S.N., Yue F., Qiu J., Brito L.F., Kuang S. Temporal dynamics and heterogeneity of cell populations during muscle regeneration. iScience. 2020;23:10093. doi: 10.1016/j.isci.2020.100993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Stoeckius M., Smibert P., Satija R., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y. Comprehensive integration of single-cell data resource comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data generated in this study is available at GSE138826.