

Summary
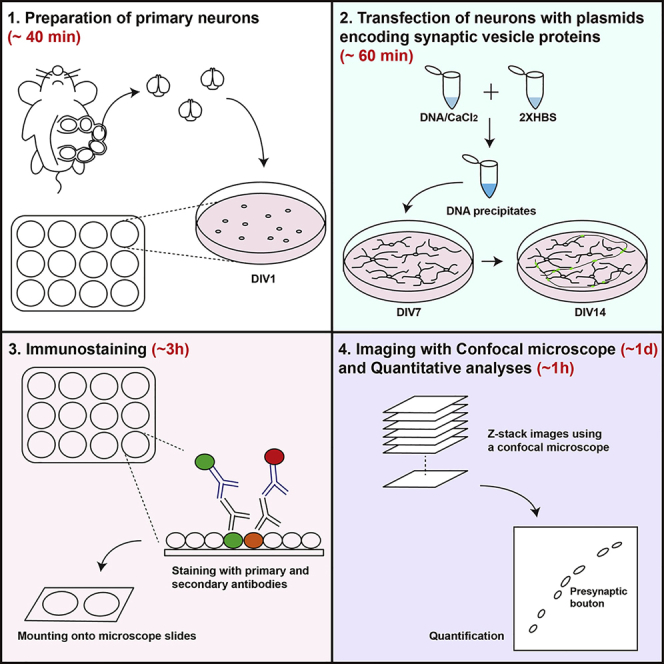
Clustering of synaptic vesicles along the neuronal axons is a critical mechanism underpinning proper synaptic transmission. Here, we provide a detailed protocol for analyzing the distribution of synaptic vesicles in presynaptic boutons of cultured neurons. The protocol covers preparation of cultured neurons, expression of synaptic vesicle-enriched proteins, and quantification procedures. Utilizing neurons from postnatal transgenic mice, this method can be applied to investigate the roles of synaptic genes in regulating vesicle dynamics at synaptic sites.
For complete details on the use and execution of this protocol, please refer to Han et al. (2020a).
Graphical Abstract
Highlights
-
•
Detailed methods for preparation of cultured neurons and transfection of plasmid DNA
-
•
Detailed guidelines for analyzing synaptic clustering in axons of presynaptic neurons
-
•
Description and troubleshooting of the most common pitfalls associated with the procedure
Clustering of synaptic vesicles along the neuronal axons is a critical mechanism underpinning proper synaptic transmission. Here, we provide a detailed protocol for analyzing the distribution of synaptic vesicles in presynaptic boutons of cultured neurons. The protocol covers preparation of cultured neurons, expression of synaptic vesicle-enriched proteins, and quantification procedures. Utilizing neurons from postnatal transgenic mice, this method can be applied to investigate the roles of synaptic genes in regulating vesicle dynamics at synaptic sites.
Before You Begin
This protocol describes the reagents, equipment, and experimental steps required for preparing dissociated primary cultured neurons and quantitatively analyzing synaptic vesicle clustering, distribution, and/or localization in presynaptic axons. We have performed these procedures for more than a decade, but have never published a full detailed protocol as a separate article. Unless otherwise stated, we describe preparation of rat cultured neurons and concomitant analyses, although we would note that this protocol is equally applicable to the preparation of cultured mouse neurons. C57BL/6 mice (2–8 months old) were used as breeders for the purposes of obtaining pregnant female mice. Successful mating can be confirmed by detection of a vaginal plug in the female, by palpitation, or by visual confirmation of pregnancy. Coated coverslips and various media should be prepared in advance (i.e., the day before preparation of neuron cultures). For preparation of neuron culture media, please refer to “Materials and Equipment”.
Prepare Coated Coverslips
Timing: 2 days
-
1.
Incubate coverslips in concentrated nitric acid in ceramic racks for at least 24 h at 24°C–26°C.
-
2.
Discard the nitric acid in a chemical fume hood in accordance with institutional guidelines.
-
3.
Rinse coverslips in a Petri dish by incubating with deionized water for 30 min, then discard the rinse solution.
-
4.
Repeat Step 3 five times.
-
5.
Sterilize coverslips in a muffle furnace (JEIO Tech; Cat# MF-G) at 240°C for 4 h.
-
6.
Transfer coverslips to a sterilized Petri dish in a culture hood and store them at 24°C–26°C.
-
a.
Prepared coverslips can be stored in a culture hood after exposure to ultraviolet light for 30 min and used for up to ∼1 month.
-
b.
Perform steps 7 and 8 one day prior to dissection procedures.
-
7.
Place a sterilized coverslip in each well of a 12-well plate.
-
8.
Add 1 mL of 0.1 mg/mL poly-D-lysine in 0.1 M boric acid (pH 8.5) to wells containing coverslips, and allow plates to sit for 12–16 h at 37°C in a CO2 incubator.
-
9.
After 12–16 h, wash the coverslips with autoclaved deionized water on a clean bench.
Note: It is critical to prevent coverslips from drying before use.
Preparation of Surgical Instruments
-
10.
Unfold autoclaved surgical packs (Figure 1A).
-
11.
Sterilize forceps and scissors with 70% ethanol.
-
12.
Turn on the dissection hood and clean the inside with 70% ethanol (Figure 1B).
Figure 1.

Preparation of Surgical Instruments and Dissection Hood
(A) Composition of surgical packs.
(B) Dissection hood equipped with a microscope.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bassoon | Enzo life Sciences | Cat # SAP7F407; RRID: AB_2313990 |
| Cy3-conjugated donkey anti-mouse | Jackson ImmunoResearch laboratories | Cat #715-165-150; RRID: AB_2340813 |
| FITC-conjugated donkey anti-goat | Jackson ImmunoResearch laboratories | Cat #705-095-147; RRID: AB_2340401 |
| EGFP | Rockland | Cat # 600-101-215; RRID: AB_218182 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Neurobasal medium | ThermoFisher Scientific | Cat #21103049 |
| B-27 supplement (50×) | ThermoFisher Scientific | Cat #17504-044 |
| Penicillin/streptomycin | ThermoFisher Scientific | Cat #15140122 |
| HBSS (Hank’s Balanced Salt Solution) | ThermoFisher Scientific | Cat #14065056 |
| Sodium pyruvate | ThermoFisher Scientific | Cat #11360070 |
| GlutaMax supplement | ThermoFisher Scientific | Cat #35050061 |
| Minimal essential medium (MEM) | Welgene | LM007-12 |
| Poly-D-lysine hydrobromide | Sigma-Aldrich | P0899 |
| 37% formaldehyde | Biosesang | F1012 |
| Bovine serum albumin | Sigma-Aldrich | A7906 |
| Horse serum | Gibco | 16050122 |
| EDTA | Affymetrix | 15701 |
| Calcium chloride | Affymetrix | 12531 |
| Magnesium chloride | Sigma-Aldrich | M8266 |
| Papain | Worthington | LS003126 |
| Hydrochloric acid | Reagents Duksan | Cat #1129 |
| Sodium hydroxide | DAEJUNG | 7572-3700 |
| Sucrose | Sigma-Aldrich | S7903 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| Nitric acid | Reagents Duksan | Cat #1538 |
| Boric acid | Sigma-Aldrich | B6768 |
| Critical Commercial Assays | ||
| NucleoBond ® Xtra Midi kit for transfection-grade plasmid DNA | MACHEREY-NAGEL | MN740410 |
| CalPhos Kit for neuron transfections | Takara | Cat #631312 |
| Experimental Models: Organisms/Strains | ||
| Mice: E17 pregnant females (C57BL/6J) | The Jackson Laboratory | 000664 |
| Rat: E18 pregnant females (Sprague-Dawley) | Daehan Biolink | N/A |
| Recombinant DNA | ||
| pCAGG-VGLUT1-Venus | Han et al., 2020a | N/A |
| Software and Algorithms | ||
| MetaMorph | Molecular Devices | https://www.moleculardevices.com |
| Photoshop | Adobe | https://www.adobe.com |
| Microsoft Excel | Microsoft | https://www.microsoft.com |
| Zen 2.6 | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html |
| Other | ||
| Surgical scissors | Fine Science Tools | Cat #14054-13 |
| Student Dumont #5 forceps | Fine Science Tools | Cat #91150-20 |
| Dressing forceps | KASCO | Cat #6-004 |
| Dry ice | N/A | N/A |
| CO2 incubator | ESCO | Cat # CCL-170B-8-FD |
| Centrifuge | Labogene | https://labogene.co.kr |
| Cover glasses | Marienfeld | Cat #0111580 |
| 0.2-μm syringe filters | Sartorius Stedim Biotech | https://www.sartorius.com |
| Dissecting microscope | Nikon | https://www.microscope.healthcare.nikon.com |
| Dissection hood | Bio-free | https://biofree.co.kr |
| Conical tubes | Neurex | https://saehanlab.co.kr |
| Microcentrifuge tubes | Axygen | https://www.corning.com |
| 12-well plates, 6-well plates | SPL | https://www.spllifesciences.com |
| Furnace | JEIO Tech | Cat #MF-G |
| Petri dishes | SPL | https://www.spllifesciences.com |
| Confocal microscope (LSM800) | Zeiss | http://www.zeiss.com |
Materials and Equipment
Prepare Neuron Culture Media before Starting the Procedure
Complete Neurobasal (CNB) Media
| Reagent | Final Concentration | Volume (mL) |
|---|---|---|
| Neurobasal Medium | 1× | 472.5 |
| B-27 Supplement | 2% | 10 |
| Sodium pyruvate | 1 mM | 5 |
| GlutaMax | 1× | 5 |
| Penicillin Streptomycin | 50,000 U/mL | 5 |
| Fetal Bovine Serum | 0.5% | 2.5 |
| Total | 500 |
Papain Solution
| Reagent | Final Concentration | Volume (μL) |
|---|---|---|
| HBSS | 1× | 4,910 |
| Papain | 14 U/mL | 80 |
| 0.5M EDTA | 0.5 μM | 5 |
| 1M CaCl2 | 1 μM | 5 |
| Total | 5,000 |
Note: After mixing all reagents for the papain solution, vortex and filter the solution using a 0.2-μm syringe filter.
Fixing Solution
| Reagent | Final Concentration | Amount |
|---|---|---|
| 37% formaldehyde | 3.7% | 50 mL |
| 10× PBS | 1× | 50 mL |
| Sucrose | 4% | 20 g |
| Deionized water | n/a | 400 mL |
| Total | 500 mL |
Permeabilization Solution
| Reagent | Final Concentration | Volume (mL) |
|---|---|---|
| 10 % Triton X-100 | 0.2% | 4 |
| 10× PBS | 1× | 20 |
| Deionized water | n/a | 176 |
| Total | 200 |
PBS-BH Solution
| Reagent | Final Concentration | Amount |
|---|---|---|
| 10× PBS | 1× | 50 mL |
| Bovine serum albumin | 0.1% | 0.5 g |
| Horse serum | 3% | 15 mL |
| Deionized water | n/a | 435 mL |
| Total | 500 mL |
Note: After mixing all reagents for the phosphate-buffered saline–bovine serum albumin/horse serum (PBS-BH) solution, filter the solution using a 0.2-μm syringe filter.
Poly-D-lysine Solution
| Reagent | Final Concentration | Volume (mL) |
|---|---|---|
| 2 mg/mL Poly-D-lysine | 0.1 mg/mL | 2.5 |
| 0.1 M Boric acid | 0.1 M | 47.5 |
| Total | 50 mL |
Step-By-Step Method Details
Tissue Harvesting
-
1.
When using late embryonic stage (E17–18) rat fetuses, euthanize the dam, then remove the uterus and free individual fetuses from the embryonic sack (Figure 2).
-
2.
Place fetuses into a sterile Petri dish and continue as outlined below.
-
3.
Decapitate rat fetus or pup, following guidelines approved by the researcher’s Institutional Animal Care and Use Committee.
-
4.
Remove skin and skull and place each brain in a 60-mm Petri dish containing ice-cold Hank’s Balanced Salt Solution (HBSS) onto an ice pack.
-
5.
Under the dissecting microscope, dissect hippocampi or cortices one at a time in a 100-mm Petri dish containing enough dissection solution to cover the tissue.
-
6.
Once meninges are removed and the hippocampus or cortex is freed from the brain, cut out the extra tissue and transfer the hippocampus or cortex to a 35-mm Petri dish containing HBSS.
Figure 2.

Preparation of Rat Fetuses for Primary Neuron Culture
(A) Euthanized pregnant female rat.
(B) Opening in the mid-ventral side of the rat.
(C) Prenatal pups in the uterus.
(D) Opening the uterus and removing pups using autoclaved sterile forceps.
Tissue Dissociation
-
7.
After all hippocampi or cortices have been collected, transfer them to a 15-mL conical tube containing papain solution and allow them to incubate at 37°C for 15 min.
Note: Prepare papain solution freshly in a 15-mL conical tube immediately before starting dissection.
Note: During the papain incubation step, wash the coverslips three times with deionized water, place in wells of a 12-well plate, and add 1.5 mL warmed CNB medium to each well.
-
8.
Stop the digestion by removing papain solution using a 1-mL pipette.
-
9.
Add 5 mL of HBSS, close the tube and invert it gently a few times. Let the hippocampi or cortices settle for about 2 min and then remove the HBSS.
-
10.
Repeat Step 9 two more times.
-
11.
Add 1 mL of CNB medium and triturate tissues with a fire-polished Pasteur pipette 40 times. Try not to introduce air bubbles during this step.
Plating Dissociated Cultured Neurons on Coated Coverslips
-
12.
Dilute 20 μL of cell suspension into 180 μL of plating media and count cells on a hemocytometer.
-
13.
Plate 3.0 × 105 cells onto 18-mm poly-D-lysine-coated coverslips placed in wells of a 12-well plate containing warmed CNB medium.
-
14.
Maintain cultures at 37°C and 5% CO2.
-
15.
The next day, replace the medium with fresh CNB medium (Figure 3).
Note: It is important to monitor cultures daily for potential problem; otherwise, planned experiments could be compromised. However, observations should be as brief as possible to minimize stress (e.g., light exposure, pH and temperature variations) on the cells.
Figure 3.
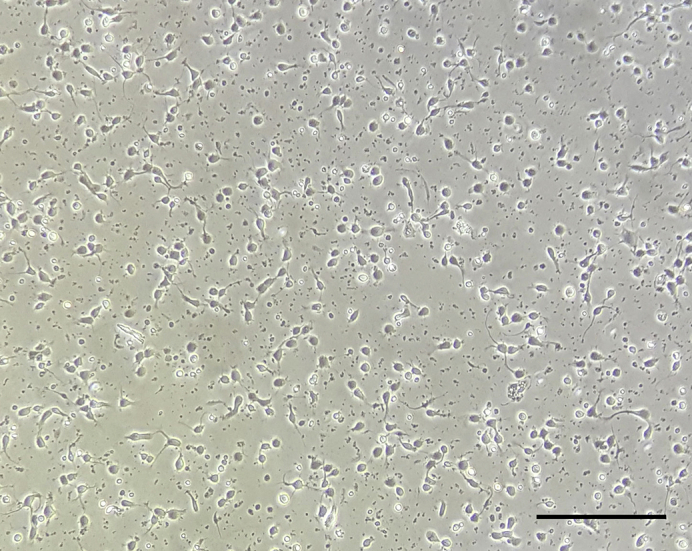
Micrograph of Primary Hippocampal Neurons at 1 Day In Vitro (DIV1)
Scale bar, 0.2 mm.
Transfection of Cultured Neurons with Plasmids
-
16.
At about DIV7, check the quality of the plated neurons by observing them under a microscope (Figure 4).
-
17.
For each transfection, prepare Solution A and Solution B in separate sterile microcentrifuge tubes according to the manufacturer’s instructions (CalPhos Mammalian Transfection Kit). For each ∼3.5 × 105 neurons plated in a 12-well plate, make Solution A by mixing 8 μg of pCAGG-VGLUT1-Venus plasmid DNA with 9 μL of a 2 M CaCl2 solution and sufficient deionized water to bring the total volume to 75 μL. Solution B consists of 75 μL of 2× HBS.
Note: Prepare pCAGG-VGLUT1-Venus plasmids by DIV7 using an endotoxin-free DNA Midi Kit.
-
18.
Carefully vortex Solution A while adding Solution B dropwise (usually takes 30 s∼1 min per reaction).
-
19.
Incubate the mixed A+B Solution at 24°C–26°C for 30 min.
-
20.
While the A+B solution incubates, use forceps to transfer a coverslip with plated neurons to another 12-well plate containing 2 mL of MEM and 10 μL of 1 M MgCl2.
Note: Retain the 12-well plate containing CNB medium conditioned by the original cultures of coverslip-plated neurons.
-
21.
Gently vortex transfection (A+B) solution and then add the solution dropwise to wells of a 12-well plate containing coverslip-plated neurons.
-
22.
Gently move the plate back and forth to evenly distribute the transfection solution.
-
23.
Incubate the plate at 37°C in a 5% CO2 incubator for 15 min.
CRITICAL: it is imperative that precipitates can be clearly observed under a microscope after the 15-min incubation period. If precipitates are not detected, incubate for an additional 5–10 min.
-
24.
Prepare 6-well plates containing 2 mL of HBSS per well (three wells are normally needed to process each coverslip).
-
25.
Transfer the coverslip to the first well of a 6-well plate containing 2 mL of HBSS and gently shake the plate.
-
26.
Transfer the coverslip to the second well of a 6-well plate containing 2 mL of HBSS and gently shake the plate.
-
27.
Transfer the coverslip to the third well of a 6-well plate containing 2 mL of HBSS and gently shake the plate.
-
28.
Transfer the coverslip to the 12-well plate containing the original neuron-conditioned CNB medium.
-
29.
Twenty-four hours later, assess the success of transfection by monitoring VGLUT1-Venus fluorescence in neurons under a fluorescence microscope.
Figure 4.
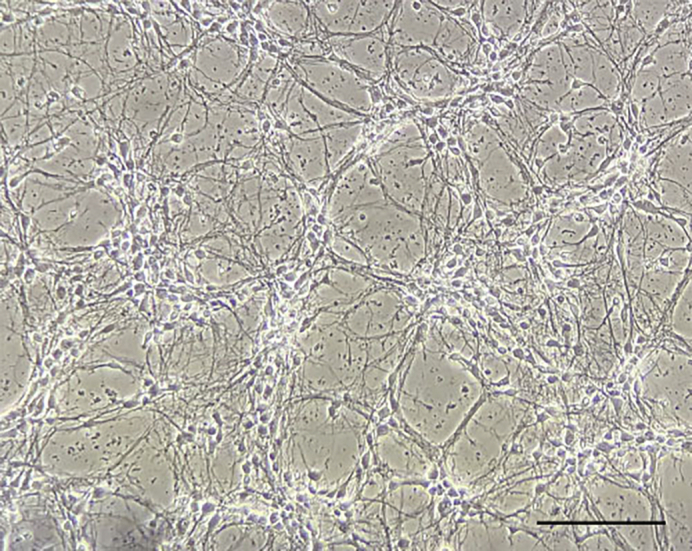
Micrograph Showing the Morphology of Primary Hippocampal Neurons at DIV7
Scale bar, 0.2 mm.
Immunocytochemistry
-
30.
At DIV14, aspirate CNB medium from 12-well plates and wash coverslip-plated neurons by adding ice-cold PBS.
Note: Before immunocytochemistry procedures, inspect all transfected neurons under a fluorescence microscope to ensure their viability and health.
-
31.
Add fixing solution and incubate for 10 min at 4°C.
-
32.
Aspirate the fixing solution and wash neurons by adding ice-cold PBS.
-
33.
Repeat Step 32 twice more.
-
34.
Add permeabilization solution to 12-well plates and incubate at 24°C–26°C for 10 min.
-
35.
Aspirate the permeabilization solution and incubate with PBS-BH solution at 24°C–26°C for 15 min to block nonspecific binding of antibodies.
-
36.
Incubate with primary antibodies as follows:
-
a.
Prepare 100 μL of primary antibody solution (anti-Bassoon [diluted 1:100] and anti-GFP [diluted in 1:1,000] in PBS-BH solution) per coverslip.
Note: Antibodies that label other active zone proteins (e.g., Piccolo, RIM1, or Munc13-1) would serve the same purpose as the anti-Bassoon antibody and could be substituted for it in this step.
-
b.
Place a 100-μL drop of primary antibody solution on a parafilm-coated plate for each coverslip.
-
c.
Retrieve the coverslips using forceps, and place onto drops of primary antibody solution, and incubate at 24°C–26°C for 1 h.
-
37.
Wash coverslip-plated neurons by transferring coverslips to wells of a 12-well plate filled with 1× PBS.
-
38.
Repeat Step 37 once more.
-
39.Incubate with secondary antibodies as follows:
-
a.Prepare 100 μL of secondary antibody solution (anti-mouse-Cy3 [diluted 1:500] and anti-goat-FITC [diluted in 1:150] in PBS-BH solution) for each coverslip.
-
b.Place 100-μL drop of secondary antibody solution on a parafilm-coated plate for each coverslip.
-
c.Retrieve coverslips using forceps, place onto drops of secondary antibody solution, and incubate at 24°C–26°C for 1 h. Cover plates with foil to minimize light exposure.
-
a.
-
40.
Wash coverslip-plated neurons by transferring coverslips to wells of a 12-well plate filled with 1× PBS.
-
41.
Repeat Step 40 once more.
-
42.
Prepare microscope slides (Superfrost Plus; Fisher Scientific) and place a drop of VECTASHIELD antifade mounting medium (H-1200; Vector Laboratories) on the surface of the slide.
-
43.
Retrieve coverslips from the 12-well plate using forceps and place on slide glasses with neurons facing down.
-
44.
Seal each coverslip with transparent nail polish.
Confocal Imaging
-
45.
Acquire Z-stack images in standard mode using a laser-scanning confocal microscope (Zeiss LSM800 with Airyscan module) equipped with a 63× objective (0.156 μm/pixel; X-Y dimension 1,024 × 1,024) and Zen2.6 Software (Zeiss).
-
46.
Configure Z-stack images using Acquisition Dimensions and four slices with a 1.2-μm interval setting.
-
47.
Generate maximum intensity projections for all captured Z-stack files (Figure 5).
-
48.
Save max intensity projection files in 16-bit Tiff file format.
Figure 5.

Fluorescence Image Processed Using Maximum Intensity Projections
Red puncta, Bassoon immunostaining; green puncta, fluorescence of transfected VGLUTI-Venus protein.
Quantitative Analyses
-
49.
Separate RGB colors in images using Photoshop (Adobe) and save each image in gray mode inTiff file format.
-
50.
Open gray image files corresponding to transfected neurons expressing VGLUT1-mVenus into MetaMorph software (Molecular Devices) (Figure 6).
-
51.
Calibrate pixel length in images using the Length Calibration Function (Figure 7).
-
52.
To remove background signals, set the threshold using the Threshold Function (minimum, 100; maximum, 255) such that pixels above the minimum threshold take on an orange coloration (Figure 7).
-
53.Measure presynaptic bouton length as follows:
-
a.Draw a straight line across each orange region (Figure 8A).
-
b.Open a Region Measurement window (Figure 8B).
-
c.Open Log to link to Microsoft Excel (Figure 8C).
-
d.Click Log Data to export data to a Microsoft Excel sheet. The values of all parameters of the Region Measurement window are exported to the Excel sheet. The average of Distance values corresponds to the length of a presynaptic bouton (Figure 8; red box).
-
a.
-
54.Determine the percentage of VGLUT1-postive Bassoon puncta as follows:
-
a.Draw a line surrounding an axonal bouton labeled with VGLUT1-Venus to define the region of interest (ROI) (Figure 9A).
-
b.Save each ROI in Rgn file format (Figure 9B).
-
c.Open gray images files corresponding to Bassoon puncta in MetaMorph software.
-
d.Calibrate pixel length in images using the Length Calibration Function (Figure 10A).
-
e.To remove background signals, set the threshold using Threshold Function (minimum, 100; maximum, 255) such that pixels above the minimum threshold take on an orange coloration (Figure 10B).
-
f.Load saved ROIs for the gray image corresponding to Bassoon puncta (Figures 10C and 11A).
-
g.Open a Region Measurement window (Figure 11B)
-
h.Click Open Log to link to Microsoft Excel (Figure 11C).
-
i.Click Log Data to export data to an Excel sheet. The values of all parameters of the Region Measurement window are exported to the Excel sheet. Average intensity indicates the intensity of VGLUT1-positive Bassoon (Figure 11; red box).
-
a.
Note: Instead of using the commercial software packages described in this protocol (i.e., MetaMorph, Photoshop and Microsoft Excel), free, open-source programs with similar functions could be used (e.g., ImageJ, GNU Image Manipulation Program [GIMP], and LibreOffice).
Figure 6.

An Opened Gray Image in MetaMorph Software
Figure 7.

Calibrating Distances and Thresholding a Gray Image in MetaMorph Software
Figure 8.

Measurement of Presynaptic Bouton Length in MetaMorph Software
(A) Drawing a line across each presynaptic bouton.
(B and C) Measuring the bouton length and exporting it to a Microsoft Excel sheet.
Figure 9.

Drawing Lines Defining ROIs Corresponding to VGLUT1-Venus-Positive Puncta
(A) Drawing a line surrounding a presynaptic bouton visualized by VGLUT1-Venus fluorescence.
(B) Saving each ROI in Rgn file.
Figure 10.

Analysis Example
Gray image for visualizing Bassoon puncta (A), calibrating distances (B), setting the threshold, and (C) defining ROIs.
Figure 11.

Measuring the Percentage of VGLUT1-Venus and Bassoon Colocalization
(A) Loading the ROI.
(B) Opening a Region Measurements window.
(C) Exporting the files to the Microsoft excel.
Expected Outcomes
A non-stressed female normally has 8–12 embryos. Embryos that are too small or too large will result in a decrease in the survival rate of plated cells within the first few days after culturing. Acid-washed coverslips, neuron culture media, and plasmid DNAs should be prepared in advance (i.e., at least 1 day before) based on precise information about mating date.
In a typical imaging experiment, cultures should be plated at a density of 3.5 × 105 cells per 12-well plate. Obtaining high-quality cultures is highly skill-dependent, but is easily achievable with care and practice. Several points should be carefully considered prior to culturing (Troubleshooting 1).
Cultured neurons can be infected with recombinant lentiviruses expressing the gene of interest. In our recent studies (Han et al., 2020a; Han et al., 2020b), we produced lentiviruses expressing Cre recombinase (using inactive Cre recombinase as a negative control) and used them to infect cultured (∼DIV3–4) neurons derived from mice harboring a floxed allele of the targeted presynaptic adhesion molecules. Lentiviruses are produced in HEK293T cells by transfections of the lentivirus backbone vectors, psPAX2 and pMD2.G, followed 72 h later by replacement of the supernatant with neuron culture media (i.e., complete neurobasal media). In our experience, a 7-day incubation of cultured neurons with lentiviral supernatant works well in most cases (>95% infection rate), although we observe a sharp decrease in virus titer for lentiviral vectors with large inserts (>8 kb).
Cultured neurons can also be transfected with any mammalian expression vector(s) using the Ca2+-phosphate method. Successful transfections using this method rely on experience with pipetting to generate precipitates. Note that, in our hands, lentiviral infections work well using DIV2–6 cultured neurons, whereas plasmid transfections work well using DIV3–14 cultured neurons. If the lentiviral vector or the expression plasmid harbors fluorophore moiety, neurons expressing exogenously introduced protein can be easily recognized using fluorescence microscopy, which, in our case, is set up in the tissue culture room.
Toxicity resulting from lentiviral infections and/or Ca2+-phosphate transfections frequently causes neuronal cell death, which is clearly detectable by microscopic examination prior to immunostaining experiments (Troubleshooting 2). One should avoid conducting immunostaining experiments, capturing images, and performing quantitative analyses using poor quality of neurons because it might negatively impact precise interpretation of results. Importantly, a typical confocal microscopic imaging experiment should show that a number of synaptic vesicle marker proteins (e.g., VGLUT1, VGAT, synaptophysin) are distributed and concentrated into presynaptic boutons, which in turn are identified by their immunostaining for well-established marker proteins (e.g., Bassoon, Piccolo, RIM1), used to mark anatomically defined presynaptic active zones.
Although heterogeneity is unavoidable in cultured neuron experiments, it can be reduced by repeating multiple independent experiments under the same conditions. In addition, results should be quantified by researchers who are blinded to the experimental conditions and are not involved in preparing the cultured neurons, performing the infections or transfections, or archiving fluorescence images.
Quantification and Statistical Analysis
Measurement of Presynaptic Bouton Length
-
1.
Average “Distance” values correspond to the length of a presynaptic bouton (see Table 1). This averaged value is obtained from a single image, which is considered an n-value of 1 in statistical evaluation. The mean value is the overall average of values averaged from individual experiments and should be based on at least 15 images from three or more independent experiments.
-
2.
Data are statistically evaluated using a Mann Whitney U test or analysis of variance (ANOVA) followed by Tukey’s post hoc test or Kruskal-Wallis test.
Table 1.
An Example of Quantified Data, Exported to a Microsoft Excel Sheet
| Region Label | Distance |
|---|---|
| 1 | 1.6715037 |
| 2 | 1.291841439 |
| 3 | 1.715789613 |
| 4 | 1.902360688 |
| 5 | 1.946210866 |
| 6 | 2.085730542 |
| 7 | 1.561298291 |
| 8 | 1.60278213 |
| 9 | 1.355586534 |
| 10 | 1.355586534 |
| 11 | 2.819662552 |
| 12 | 1.291841439 |
| 13 | 1.26247757 |
| 14 | 2.212251314 |
| 15 | 1.042865271 |
| 16 | 2.498827777 |
| average | 1.726038516 |
Determining the Percentage of VGLUT1-Positive Bassoon Puncta
-
3.
The percentage of VGLUT1-positive Bassoon puncta is calculated using the following formula:
% = number of regions with average intensity (> 15)/total number of regions × 100, where average intensity corresponds to the intensity of VGLUT1-positive Bassoon puncta (an average intensity <15 using our imaging settings is considered background noise).
Note: An “Auto Thresholding” Function can be used for setting background noise. Investigators should determine a fixed threshold range for their own fluorescent images and apply thresholding to all the captured images.
-
4.
This calculated value is obtained from a single image, which is considered as an n-value of 1 in statistical analyses. The mean value is the overall average of values averaged from individual experiments and should be based on at least 15 images from three or more independent experiments.
-
5.
Data are statistically evaluated using a Mann Whitney U test or ANOVA followed by Tukey’s post hoc test or Kruskal-Wallis test.
Limitations
Although dissociated primary cultured neurons have been extensively used in molecular neuroscience fields over the last three decades, they possess inherent limitations in terms of recapitulating all the characteristics of in vivo neural circuits. Despite these limitations, a number of early studies using dissociated primary cultured neurons have contributed to current models of synaptic assembly, particularly at presynaptic nerve terminals (Ahmari et al., 2000; Bamji et al., 2003; Ziv and Garner, 2004).
Although our immunostaining analyses using VGLUT1-mVenus and Bassoon (a marker for the presynaptic active zone) clearly enable us to dissect detailed molecular mechanisms responsible for clustering and confining synaptic vesicles in presynaptic bouton structures, it is impossible to discriminate “orphan” sites—in which postsynaptic apposition with the functional apparatus for neurotransmitter release is lacking—from bona fide synaptic sites (Krueger et al., 2003). Thus, implementing complementary functional imaging and/or electrophysiology approaches is critical. In addition, the magnitude of VGLUT1-mVenus fluorescence diffusion, quantified as the length of the major axis of each puncta, has been well established as a proxy for vesicle clustering. Thus, live-cell imaging analyses could be employed to precisely determine the dynamics of synaptic vesicle material influx and efflux in presynaptic structures. High-resolution electron microscopic analyses are sometimes warranted to further investigate possible anatomical defects that cannot be discerned by light microscopy, as we have done in our recent studies (Han et al., 2020a; Han et al., 2020b).
Troubleshooting
Problem 1
Poor neuron cell culture quality.
Potential Solution
There are multiple points in the procedure that could contribute and need to be assessed.
First, precisely timed pregnant female rats or mice should be used. Embryos staged at E17 (in the case of mice) or E18 (in the case of rats) are used in experiments, and 8–12 embryos are normally obtained from a single pregnant female rodent. Errors during examinations for a vaginal plug in the female, palpitation or visual confirmation of pregnancy could result in failure to obtain precisely timed pregnant females. The use of such early- or later-stage embryos could contribute to the poor quality of neuronal cultures. Although some laboratories have reported the use of P0 mouse pups, we find that cultured neurons obtained from embryos are higher quality.
Second, preparation of coverslips is a crucial step for obtaining high-quality cultured neurons. If nitric acid has been used for an extended period of time, it should be replaced with a new batch of concentrated nitric acid.
Third, poly-D-lysine solution prepared in borate buffers should be re-checked. We have had some success in adding laminin to promote the attachment and differentiation of plated neurons.
Lastly, neuron culture medium contains several components that should be carefully monitored because of lot-to-lot variability (e.g., fetal bovine serum, GlutaMAX, and B27). GlutaMAX should be aliquoted and thawed in a refrigerator for 12–16 h before use. Notably, the quality of the B27 supplement is crucial for obtaining pure and healthy neuronal populations with little or no glial contamination; thus, specific lots should be tested before using.
Problem 2
Low transfection efficiency in cultured primary neurons.
Potential Solution
An experience researcher immersed in the Ca2+-phosphate transfection method will normally obtain a fair number of transfected neurons on a single coverslip with low cell toxicity. Usually, one should expect to obtain around 100–1,000 transfected neurons on a coverslip placed in a 12-well plate (at a density of 3 × 105 neurons; transfection efficiency is around 0.033% to ∼0.33%). However, this method has been notorious for its low transfection efficiency and poor reproducibility, reflecting its sensitivity to pH, temperature and incubation time (Jiang and Chen, 2006). The key is to develop the pipetting skills necessary to successfully form homogeneous precipitates with Ca2+ ions in phosphate buffer that fall “snow-like” onto neurons during the incubation period.
The concentration, quality, and size of plasmid DNA are all capable of affecting transfection efficiency. Thus, it is often necessary to empirically optimize the transfection protocol prior to use, which may include increasing the incubation time during Ca2+ phosphate–DNA complex preparation. Other transfection methods, including lipofection, electroporation or nucleofection, could be employed as alternatives to Ca2+-phosphate transfection method in cases where low transfection efficiency continues to be problematic.
Note: Although electroporation and nucleofection yield very high transfection efficiencies, they also require specialized equipment and expensive reagents. Moreover, these methods are not ideal for adherent cell cultures.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jaewon Ko (jaewonko@dgist.ac.kr).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Not applicable.
Acknowledgments
This work was supported by a grant from the Korea Healthcare Technology R & D Project, funded by the Ministry for Health and Welfare Affairs, Republic of Korea (HI17C0080 to J.K.).
Author Contributions
J.W.U. and J.K. conceived and supervised the project. K.A.H. conducted the experiments. J.W.U., K.A.H, S.-Y.C., and J.K. analyzed data. J.W.U. and J.K. wrote the manuscript. All authors read and approved the final version of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Ji Won Um, Email: jiwonum@dgist.ac.kr.
Jaewon Ko, Email: jaewonko@dgist.ac.kr.
References
- Ahmari S.E., Buchanan J., Smith S.J. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Bamji S.X., Shimazu K., Kimes N., Huelsken J., Birchmeier W., Lu B., Reichardt L.F. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K.A., Lee H.Y., Lim D., Shin J., Yoon T.H., Lee C., Rhee J.S., Liu X., Um J.W., Choi S.Y. PTPsigma controls presynaptic organization of neurotransmitter release machinery at excitatory synapses. iScience. 2020;23:101203. doi: 10.1016/j.isci.2020.101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K.A., Lee H.Y., Lim D., Shin J., Yoon T.H., Liu X., Um J.W., Choi S.Y., Ko J. Receptor protein tyrosine phosphatase delta is not essential for synapse maintenance or transmission at hippocampal synapses. Mol. Brain. 2020;13:94. doi: 10.1186/s13041-020-00629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M., Chen G. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat. Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- Krueger S.R., Kolar A., Fitzsimonds R.M. The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron. 2003;40:945–957. doi: 10.1016/s0896-6273(03)00729-3. [DOI] [PubMed] [Google Scholar]
- Ziv N.E., Garner C.C. Cellular and molecular mechanisms of presynaptic assembly. Nat. Rev. Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.