

Summary
Primary hepatocytes are a vital tool in various biomedical research disciplines, serving as an ex vivo model for liver physiology. Obtaining high yields of viable primary mouse hepatocytes is technically challenging, limiting their use. Here, we present an improved protocol based on the classic two-step collagenase perfusion technique. The liver is washed by perfusion, hepatocytes are dissociated by collagenase, separated from other cells, and cultured. This protocol was optimized to significantly reduce procedure duration and improve hepatocyte yield and viability.
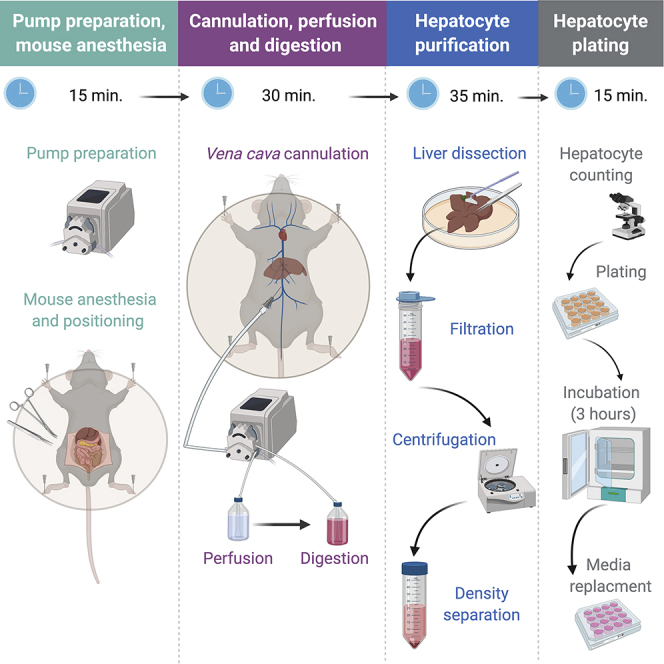
Graphical Abstract
Highlights
-
•
Primary hepatocytes are a vital research tool but their isolation is challenging
-
•
We present a protocol for quick, high-yield isolation of primary mouse hepatocytes
-
•
Vena cava cannulation increases reproducibility and requires less technical skills
-
•
Liberase digestion and portal vein clamping reduce duration and increase efficiency
Primary hepatocytes are a vital tool in various biomedical research disciplines, serving as an ex vivo model for liver physiology. Obtaining high yields of viable primary mouse hepatocytes is technically challenging, thereby limiting their use. Here, we present an improved protocol based on the classic two-step collagenase perfusion technique. The liver is washed by perfusion, hepatocytes are dissociated by collagenase, separated from other cells, and cultured. This protocol was optimized to significantly reduce procedure duration and improve hepatocyte yield and viability.
Before You Begin
Timing: 20 min. (plus incubation for 4–16 h)
Note: Steps 1–4 should be done in sterile conditions, in a biological hood (i.e., biosafety cabinet)
-
1.
Prepare cell culture plates; 12-well, 6-well, 10 cm, and 15 cm plates are suitable for hepatocyte plating.
Note: Lower diameter plates may be suitable but are less optimal due to decreased efficiency of cell dispersion across the well surface.
-
2.
Cover the bottom of the plates/wells with 0.01% rat-tail collagen solution.
-
3.
Incubate for 4–16 h at 37°C under sterile conditions (e.g., in a humidified CO2 incubator).
-
4.
Wash with PBS, aspirate PBS.
Note: Collagen-coated plates can be prepared days in advance provided they are stored under sterile conditions.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| HBSS with calcium, magnesium and phenol red | Biological industries | 02-015-1A |
| HBSS no calcium, no magnesium and no phenol red | Biological industries | 02-018-1A |
| EDTA (0.5 M) | Fisher bioreagents | BP2482-500 |
| HEPES (1 M) | Sigma-Aldrich | H0887-100ML |
| Ketamine (Clorketam) | Vetoquinol | 00 92297 43 082 |
| Xylazine (Sedaxylan) | EuroVet | SEDAXYLAN |
| DMEM low glucose | Biological industries | 01-050-1A |
| Dulbecco's Phosphate Buffered Saline (DPBS) without calcium and magnesium | Biological industries | 02-023-1A |
| Phosphate Buffered Saline (PBS) ×10 | Hylabs | BP507/500D |
| L-Glutamine Solution | Biological industries | 03-020-1B |
| Penicillin-Streptomycin Solution | Biological industries | 03-031-1B |
| William's E Medium, no glutamine | Gibco | 12551-032 |
| Fetal Bovine Serum (FBS) | Biological industries | 04-007-1A |
| Collagen | Sigma-Aldrich | C3867-1VL |
| Percoll | Santa Cruz biotechnologies | sc-500790A |
| Trypan Blue Solution | Biological industries | 03-102-1B |
| Liberase™ TM Research Grade | Sigma-Aldrich | 05401127001 |
| Experimental Models: Organisms/Strains | ||
| Mouse strain: C57BL/6JOlaHsd | Envigo | N/A |
| Other | ||
| Insulin syringe | Becton Dickinson (BD) | BD 324912 |
| Cell strainer, 70 μm | Corning | CLS431751-50EA |
| Cell lifter | Corning | CLS3008 |
| Peristaltic pump | Gilson | Miniplus 3 PVC tubing 2.06 mm diameter |
| Mouse dissection tray | N/A | N/A |
| Water bath | N/A | N/A |
| Sterile 50 mL centrifuge tubes | Corning | 430829 |
| Sterile 25 mL serological pipettes | Bio-SORFA | 315100 |
| 27 gauge needle | BD Microlance | 302200 |
| 70% ethanol | N/A | N/A |
| Head wearing magnifier eye loupe (optional) | N/A | N/A |
Materials and Equipment
Collagen Solution (50 mL)
| Reagent | Final Concentration | Stock Concentration |
|---|---|---|
| Collagen | 0.01%; 0.1 μg/mL | 100%; 1 mg/mL |
| Sterile double deionized water (DDW) | - | - |
| Total |
Note: Due to viscosity of collagen, we recommend doing a serial dilution (e.g., diluting collagen 1:100 and then diluting it again 1:100)
Anesthesia Mix
| Reagent | Final Concentration | Stock Concentration | Volume (μL) |
|---|---|---|---|
| PBS | N/A | N/A | 40 |
| Ketamine | 30 mg/mL | 100 mg/mL | 30 |
| Xylazine | 6 mg/mL | 20 mg/mL | 30 |
| Total | 100 |
Perfusion Buffer
| Reagent | Final Concentration | Stock Concentration | Volume (mL) |
|---|---|---|---|
| HBSS no Ca2+ no Mg2+ no phenol red | - | - | 487 |
| EDTA | 0.5 mM | 0.5 M | 0.5 |
| HEPES | 25 mM | 1 M | 12.5 |
| Total | 500 |
Notes:
-
•
The final pH at 37°C should be 7.4
-
•
EGTA can be used as alternative to EDTA. We have found no difference in yield between the two chelating agents.
Digestion Buffer
| Reagent | Final Concentration | Stock Concentration | Volume (mL) |
|---|---|---|---|
| HBSS with Ca2+, Mg2+ and phenol red | - | - | 487.5 |
| HEPES | 25 mM | 1 M | 12.5 |
| Total | 500 |
Note: The final pH at 37°C should be 7.4
Maintenance Media
| Reagent | Final Concentration | Stock Concentration | Volume (mL) |
|---|---|---|---|
| Williams E media | - | - | 490 |
| Glutamine | 1%; 2 mM | 100%; 200 mM | 5 |
| Penicillin-Streptomycin Solution | 1% Pen-100 units/mL Strep-0.1 mg/mL |
100% Pen-10,000 units/mL Strep- 10 mg/mL |
5 |
| Total | 500 |
Note: Many protocols add dexamethasone, insulin, transferrin and selenium to maintenance media. We found that this is not needed in short-term culturing. Importantly, these reagents profoundly affect hepatocyte biology (Batista et al., 2019; Goldstein et al., 2013; Lin et al., 2007; Weiller et al., 2004) and thus may affect experiment outcome.
Plating Media
| Reagent | Final Concentration | Stock Concentration | Volume (mL) |
|---|---|---|---|
| DMEM low glucose | - | - | 470 |
| FBS | 5% | 100% | 25 |
| Penicillin-Streptomycin Solution | 1% Pen-100 units/ mL Strep-0.1 mg/ mL |
100% Pen-10,000 units/ mL Strep- 10 mg/ mL |
5 |
| Total | 500 |
Liberase Stock Solution
| Reagent | Concentration | Amount |
|---|---|---|
| Liberase | 1 mg/mL | 50 mg |
| Digestion buffer | - | 50 mL |
| Total | 50 mL |
Notes:
-
•
The preparation of Liberase solution detailed here relates to preparation of stock concentration from powder. The stock is further diluted to a final concentration of 25 μg/mL during the procedure (step 5).
-
•
We found that Liberase, a specific type of collagenase is significantly more reproducible than other commercial collagenases.
-
•
Aliquot and store at −80°C. We have found that using aliquoted and frozen Liberase (with one or two freeze-thaw cycles) does not substantially affect enzyme activity (in contrast to other types of collagenase). Thus, there is no need to freshly prepare a Liberase solution.
Percoll Solution
| Reagent | Final Concentration | Stock Concentration | Volume (mL) |
|---|---|---|---|
| Percoll | 90% | 100% | 9 |
| PBSX10 | 1× | 10× | 1 |
| Total | 10 |
Note: Percoll solution should be prepared fresh during the procedure.
Step-By-Step Method Details
This protocol is aimed at isolating hepatocytes from mouse liver. Following anesthesia, the vena cava is cannulated and the liver is perfused to chelate calcium and wash out blood. Then, collagenase is perfused to the liver in order to dissociate extracellular matrix. Finally, the liver is dissected and hepatocytes are purified by density-based separation. This protocol presents several advances over similar protocols (Berry and Friend, 1969; Casciano, 2000; Klaunig et al., 1981; Li et al., 2010; Renton et al., 1978; Seglen, 1976; Severgnini et al., 2012). The main improvements of this protocol are better reproducibility, shortened duration, reduced technical challenge, increased yield and higher viability. These are achieved by several steps we altered or optimized. For example: (a) We found that retrograde perfusion through the vena cava permits easier cannulation as opposed to portal vein cannulation. (b) Periodical clamping of the portal vein provides a visible checkpoint for proper perfusion and greatly facilitates efficient washing and digestion. (c) The type of collagenase used (Liberase) shows quicker digestion and better reproducibility compared to other collagenases. (d) Percoll-based density separation results in a population of purified hepatocytes of high viability. Some of these protocol improvements were already implemented in our previous publications (Goldstein et al., 2017a; Goldstein et al., 2017b) where we isolated hepatocytes for experiments demanding a high yield of cells (such as chromatin immunoprecipitation sequencing – ChIP-seq).
Pump Preparation and Mouse Anesthesia
Here, the pump is washed and primed with perfusion buffer. The mouse is anesthetized and positioned on the dissection tray.
This section is shown in Methods Video S1.
-
1.
Warm water bath to 42°C.
-
2.
Place perfusion buffer in the water bath.
-
3.
Prepare the peristaltic pump:
-
•
Run 70% ethanol through the tubing.
-
•
Run air through the tubing for 30–60 s.
-
•
Wipe the end of the tubing with a paper towel.
-
•
Connect 27-gauge needle to the outlet end of the tubing using a luer lock (Figure 1).
Figure 1.

Needle and Tubing Preparation
The needle is connected to the outlet end of the tubing by using a luer lock. The tubing and needle fixture is supported by a heavy object (here a media bottle ring weight) to elevate it from the dissection tray. This elevation facilitates proper cannulation.
The anesthetized mouse is positioned on the dissection tray. The mouse fur and skin are cut in a “U” shape. Mouse intestine and the rest of viscera are moved to the right and both the portal vein and vena cava are revealed. Steps: 7–10.
-
4.
Prime the tubing with warm perfusion buffer (pump speed 3 mL/min).
Note: Purge some perfusion buffer to wash residual ethanol.
Note: Avoid bubbles in the tubing.
Note: The optimal pump speed varies greatly depending on the tubing diameter, needle gauge and other specifications. In this protocol, pump speed was optimized to the detailed tubing specifications and needle gauge (see Key Resources Table).
-
5.
Prepare two 50 mL tubes (labeled “DB-chilled” and “DB-Liberase”), each with 10 mL digestion buffer. Keep the tube labeled “DB-chilled” on ice. Add 250 μL Liberase solution to the tube labeled “DB-Liberase” and warm it in the water bath.
-
6.
Anesthetize mouse by intraperitoneal injection of anesthesia mix (3.75 μL/g body weight. Final concentration for ketamine = 112.5 mg/kg; for xylazine =22.5 mg/kg).
Note: Make sure the mouse is completely anesthetized by the pedal reflex.
Note: Mice should be 8–10 weeks old, younger mice have smaller veins, older mice have more fat lining the vena cava. Both these attributes make cannulation more challenging.
-
7.
Place mouse on the edge of the dissection tray and secure limbs using needles (Figure 2).
Figure 2.

Mouse Positioning on the Dissection Tray
The anaesthetized mouse is placed on the edge of the dissection tray with its head protruding outside and its limbs secured with needles. Securing limbs with tape is not recommended because the area will be soaked with liquid during procedure, potentially loosening adhesion.
Note: The head should be protruding outside the tray. This facilitates easier cannulation in later steps.
-
8.
Wet the fur thoroughly with 70% ethanol.
-
9.
Make a “U”-shaped incision through the skin, secure the skin near the head using a needle (Figure 3).
Figure 3.

Incision and Preparing for Cannulation
The mouse fur and skin are cut in a “U” shape, the mouse skin is placed and secured near the head with a needle.
-
10.
Move the intestine to the right to reveal the portal vein and vena cava (Figure 4). Place a stable and heavy object adjacent to the mouse hind legs to support the tubing such that it is slightly higher than the mouse, lay the tubing and the needle on the object (Figure 1). The edge of the needle should rest on the vena cava in a flat angle (i.e., parallel to the vein, Figure 5).
Figure 4.

Portal Vein and Vena Cava exposure
Mouse intestine and the rest of the viscera are moved to the right. Both the portal vein and vena cava are revealed.
Figure 5.

Cannulation of the Vena Cava
The tip of the needle is inserted (bevel side up, in an almost flat angle) into the vena cava above the kidney. The tubing rests on the elevating object without manual support.
Note: Make sure that the tubing and needle do not move or change angle when you let go.
Cannulation and Perfusion
Here, the inferior vena cava is cannulated and the liver is perfused to wash out blood and circulating cells from the liver as well as to eliminate calcium via EDTA. Chelating calcium with EDTA facilitates loosening of cell-cell connections by perturbing calcium-dependent adhesion factors. This step, initially suggested by (Seglen, 1976) serves as a preparative step for liver digestion in the next section. The original protocol was developed for rat and therefore, perfusing the liver through the portal vein was amenable. We found that in mice, retrograde perfusion via the inferior vena cava is significantly less challenging and therefore more reproducible.
This section is shown in Methods Video S2.
-
11.
Turn on the pump and let the warm perfusion buffer reach the needle (the buffer within the tubing has already cooled down to 20°C–25°C).
Note: Before proceeding, make sure you clearly see the portal vein and vena cava.
-
12.
While buffer is running through the needle, insert the needle into the vena cava above the kidney (Figure 5).
CRITICAL: After 1–2 s, you should see white spots forming in the liver and/or expansion/swelling of the portal vein , this means perfusion buffer is indeed flowing through the liver (Troubleshooting 1).
Note: Insert the needle in a flat angle relative to the vein.
Note: The needle bevel should be facing upwards.
-
13.
Immediately upon appearance of white spots and/or portal vein swelling (occurs 2–3 s after cannulation), cut the portal vein with scissors. The liver should clear of blood instantly; you’ll observe plenty of blood rushing out of the portal vein and the liver will turn yellow-white within a few seconds.
-
14.
Clamp the portal vein with forceps for 7–10 s (Figures 6A and 6B). Make sure no fluid is passing through.
Figure 6.

Clamping the Portal Vein
(A and B) The portal vein is clamped with forceps for 7–10 s to stop fluid exiting the liver. Liver swells upon clamping (compare A to B).
The inferior vena cava is cannulated and the liver is perfused to wash out blood and circulating cells as well as to eliminate calcium via EDTA. Steps: 11–15.
Note: Clamping serves two purposes: (a) allowing perfusion buffer to reach all liver vasculature. (b) a visible checkpoint (liver swelling and relaxation) validating liver is indeed perfused. This is important because in some cases the liver seems white only due to passive clearing of blood from the severed portal vein and not from active perfusion.
-
15.
After 30 s perform a second clamp, make sure you observe liver swelling and relaxation. If the liver is completely washed of blood, move to the next section.
Digestion
Here, Collagenase (Liberase) is perfused to the liver in order to digest collagen in the extracellular matrix, thereby facilitating cell dispersion (originally proposed by Berry and Friend, (1969)). Collagenase is a calcium-dependent enzyme and therefore calcium is included in the digestion buffer.
This section is shown in Methods Videos S3 and S4
-
16.
Stop the pump and quickly transfer the inlet tubing from perfusion buffer to the pre-warmed “DB-Liberase” tube. Quickly turn the pump back on.
Note: Make sure the tubing reaches the bottom of the 50 mL tube and make sure no bubbles got in the tubing (Troubleshooting 3).
-
17.
Clamp the liver one more time, prior to digestion buffer arriving to the liver. Make sure the liver swells and relaxes.
-
18.
While digestion buffer is perfused into the liver, clamp the portal vein every minute but no more than 3–4 times.
Note: When clamping while digestion buffer is perfused, the liver swells to maximal volume in the first clamp and does not relax. This is normal.
Note: Digestion buffer contains phenol red. This facilitates visualization of when the liver is perfused with digestion buffer rather than perfusion buffer (which does not contain phenol red).
-
19.
Remove the needle before air gets into the liver.
-
20.
Dissect out the liver gently (it is now very flimsy and frail): using forceps, grab the central connective tissue between the lobes and slightly lift upwards, using it as an anchor point. Cut all the connections of the liver to other organs, remove the gall bladder, place the liver in the tube labeled “DB-chilled” and put it back on ice.
Note: If needed, the liver can stay on ice for 30–40 min.
-
21.
Clean the pump with ethanol (see step 3).
Collagenase (Liberase) is perfused to the liver to facilitate hepatocyte dispersion. Steps 16–19.
The liver is dissected out gently by removing all connections to other organs. Step 20.
Hepatocyte Purification
Here, liver cells are released into suspension and viable hepatocytes are separated from dead hepatocytes and non-hepatocyte cells.
Steps 22–24 in this section are shown in Methods Video S5.
Note: These steps should be done in sterile conditions, in a biological hood (i.e., biosafety cabinet)
Note: Use only 25 mL serological pipettes. Smaller bore pipettes may reduce hepatocyte viability.
-
22.
Transfer liver and media to a 10 cm plate (not pre-coated with collagen).
-
23.
Rupture liver sack with fine tip forceps in a few locations along the liver surface and gently release cells using a cell lifter.
Note: Tilt the plate to have the liver submerged in the media to improve releasing of cells.
Note: The liver should tear apart easily. Do not cut the liver to pieces, leave it whole.
-
24.
Filter 5 mL of suspension through a 70 μm cell strainer into a 50 mL tube. Repeat with a new filter and a new tube for the remaining 5 mL.
-
25.
Add another 10 mL of cold plating media to rinse plate, add 5 mL of it to each filter.
-
26.
Spin at 50 × g for 2 min at 4°C (Figure 7A).
Figure 7.

Hepatocyte Purification
(A) Cells from the dissected liver are released into suspension and centrifuged. The pellet contains hepatocytes and the rest of the cells are left in the supernatant.
(B) The hepatocyte pellet (A) is re-suspended with Percoll solution and centrifuged again. The pellet contains viable hepatocytes while dead cells and debris are left in the supernatant.
The liver is removed to a 10 cm plate and is punctured repeatedly. Liver cells are released to the media and filtered. Steps 22–24.
Note: Centrifuge with low acceleration and low brake to minimize trauma to hepatocytes.
Note: Hepatocytes are denser than other liver cells. Due to low centrifugation force, only hepatocytes are pelleted while other cells are left in the supernatant.
-
27.
While the samples are spinning, prepare fresh Percoll solution from pre-chilled ingredients.
Note: Percoll is used for density separation of the viable hepatocytes from dead hepatocytes and cell debris.
-
28.
Aspirate most supernatant, leave ∼1 mL and resuspend the cells by swirling the tube.
-
29.
Add 10 mL plating media and resuspend by gentle swirling.
-
30.
Add 10 mL Percoll solution and mix thoroughly by inverting the tube several times.
-
31.
Spin at 200 × g for 10 min at 4°C. (Figure 7B)
Note: Centrifuge with low acceleration and low brake to minimize trauma to hepatocytes.
-
32.
Aspirate most supernatant, leave ∼1 mL and resuspend the cells by swirling the tube
Note: The pellet contains live purified hepatocytes.
-
33.
Add 20 mL plating media.
-
34.
Spin at 50 × g for 2 min at 4°C.
Note: Centrifuge with low acceleration and low brake to minimize trauma to hepatocytes.
-
35.
Aspirate supernatant, add 20 mL plating media.
Hepatocyte Plating
Here hepatocytes are counted and plated on collagen-coated cell culture plates/wells.
-
36.
Count viability with 1:1 dilution trypan blue.
-
37.
Plate cells according to Table 1. Be sure to plate cells evenly across the plate/well.
Table 1.
Desired Plating Density of Hepatocytes
| Cell Culture Plate | Number of Cells per Plate/Well |
|---|---|
| 12-well plate | 2 x 105 per well |
| 6-well plate | 4 x 105 per well |
| 10cm plate | 4 x 106 |
| 15cm plate | 10 x 106 |
-
38.
Place cells in a humidified CO2 incubator.
-
39.
Pre-warm maintenance media to 37°C.
-
40.
After 3 h change medium to warm maintenance media.
-
41.
Keep cells in a humidified CO2 incubator for 16–20 h.
Expected Outcomes
A typical hepatocyte yield is 30–50 × 106 viable cells per mouse and the viability is ∼95%. When hepatocytes adhere to the surface (3 h after plating) they have a spherical shape and they do not take up much space. Thus, they seem very sparsely plated (Figure 8A). Only 16–20 h following plating, hepatocytes acquire their typical hexagonal “chicken-wire” shape (Figure 8B). Hepatocytes do not survive in culture for prolonged periods and cell passaging is unattainable. Moreover, hepatocytes lose hepatic features with every day they stay in culture (Azimifar et al., 2014). It is therefore advised to perform the experiment no later than 1 day following plating. Some experiments may require longer periods and therefore can be initiated a few hours after plating. In some cases, starting the experiment 3 h after plating can lead to better outcomes (e.g., viral infection or siRNA transfection).
Figure 8.

Hepatocyte Plating
(A) Hepatocytes are plated and after 3 h have adhered to the surface, acquiring a spherical shape.
(B) Twenty-four hours following plating hepatocytes acquire their typical hexagonal shape.
Limitations
In most cases, if hepatocyte yield is above 15 X 106, viability is ∼95% and cells were plated at the indicated density (Table 1), the cells will be in good condition (Figure 8). When hepatocyte yield is low or cells are plated too densely or too sparsely, downstream experiments may be compromised.
Troubleshooting
Problem 1
The vena cava was cannulated but the liver does not blanch.
Potential Solution
This is one of the most common reasons for failure of the protocol. In many cases, the needle punctures the vena cava twice, therefore both entering and exiting the vein. In some cases, you’ll observe a bulb of fluid forming next to the vein as a result. Try to gently pull the needle back into the vena cava without removing the needle completely. If you see liver blanching, continue to next step. If this does not work, remove the needle completely and re-cannulate the vein in a location closer to the liver.
Alternative cannulation approaches: some researchers prefer cannulating the vena cava using a catheter followed by suture ligation which significantly reduces the chances of an exit puncture. However, we found this approach is lengthy and causes other technical issues further compromising the procedure.
Problem 2
The liver does not swell and relax in clamp/release cycles.
Potential Solution
This means that the liver is not perfused, see Troubleshooting 1. In most cases if swell/release is not observed, hepatocyte yield will be very low.
Problem 3
Bubbles appear in the tubing.
Potential Solution
Quickly break apart bubbles by flicking the tubing with your finger. Smaller bubbles tend to attach to the tubing rather than travel through it.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ido Goldstein (ido.goldstein@mail.huji.ac.il).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
Acknowledgments
This work was carried out with the aid of grants from the Canadian Institutes of Health Research (CIHR), the International Development Research Centre (IDRC), the Israel Science Foundation (ISF, grant nos. 1469/19 and 3533/19), the Azrieli Foundation and the Abisch-Frenkel Foundation. M.C. is supported by the Golda Meir Fellowship. We thank Dr. Yaarit Adamovich-Tamam and Dr. Keren Bahar-Halpern for help in establishing the protocol.
Author Contributions
M.C. helped in protocol optimization and wrote the manuscript. I.G. developed the protocol and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100086.
References
- Azimifar S.B., Nagaraj N., Cox J., Mann M. Cell-type-resolved quantitative proteomics of murine liver. Cell Metab. 2014;20:1076–1087. doi: 10.1016/j.cmet.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Batista T.M., Garcia-Martin R., Cai W., Konishi M., O'Neill B.T., Sakaguchi M., Kim J.H., Jung D.Y., Kim J.K., Kahn C.R. Multi-dimensional transcriptional remodeling by physiological insulin in vivo. Cell Rep. 2019;26:3429–3443.e3. doi: 10.1016/j.celrep.2019.02.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M.N., Friend D.S. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciano D.A. Development and utilization of primary hepatocyte culture systems to evaluate metabolism, DNA binding, and DNA repair of xenobiotics. Drug Metab. Rev. 2000;32:1–13. doi: 10.1081/dmr-100100561. [DOI] [PubMed] [Google Scholar]
- Goldstein I., Baek S., Presman D.M., Paakinaho V., Swinstead E.E., Hager G.L. Transcription factor assisted loading and enhancer dynamics dictate the hepatic fasting response. Genome Res. 2017;27:427–439. doi: 10.1101/gr.212175.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I., Paakinaho V., Baek S., Sung M.H., Hager G.L. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat. Commun. 2017;8:1849. doi: 10.1038/s41467-017-02055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I., Yizhak K., Madar S., Goldfinger N., Ruppin E., Rotter V. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab. 2013;1:9. doi: 10.1186/2049-3002-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig J.E., Goldblatt P.J., Hinton D.E., Lipsky M.M., Chacko J., Trump B.F. Mouse liver cell culture. I. Hepatocyte isolation. In Vitro. 1981;17:913–925. doi: 10.1007/BF02618288. [DOI] [PubMed] [Google Scholar]
- Li W.C., Ralphs K.L., Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- Lin L., Valore E.V., Nemeth E., Goodnough J.B., Gabayan V., Ganz T. Iron transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4. Blood. 2007;110:2182–2189. doi: 10.1182/blood-2007-04-087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton K.W., Deloria L.B., Mannering G.J. Effects of polyribonoinosinic acid polyribocytidylic acid and a mouse interferon preparation on cytochrome P-450-dependent monooxygenase systems in cultures of primary mouse hepatocytes. Mol. Pharmacol. 1978;14:672–681. [PubMed] [Google Scholar]
- Seglen P.O. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- Severgnini M., Sherman J., Sehgal A., Jayaprakash N.K., Aubin J., Wang G., Zhang L., Peng C.G., Yucius K., Butler J. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 2012;64:187–195. doi: 10.1007/s10616-011-9407-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiller M., Latta M., Kresse M., Lucas R., Wendel A. Toxicity of nutritionally available selenium compounds in primary and transformed hepatocytes. Toxicology. 2004;201:21–30. doi: 10.1016/j.tox.2004.03.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The anesthetized mouse is positioned on the dissection tray. The mouse fur and skin are cut in a “U” shape. Mouse intestine and the rest of viscera are moved to the right and both the portal vein and vena cava are revealed. Steps: 7–10.
The inferior vena cava is cannulated and the liver is perfused to wash out blood and circulating cells as well as to eliminate calcium via EDTA. Steps: 11–15.
Collagenase (Liberase) is perfused to the liver to facilitate hepatocyte dispersion. Steps 16–19.
The liver is dissected out gently by removing all connections to other organs. Step 20.
The liver is removed to a 10 cm plate and is punctured repeatedly. Liver cells are released to the media and filtered. Steps 22–24.
Data Availability Statement
This study did not generate any unique datasets or code.