

Summary
Infections caused by drug-resistant Acinetobacter baumannii have posed a serious threat to global public health. However, genetic manipulation methods, the primary way to study pathogenesis and drug-resistance mechanisms, remain time consuming and inefficient. Here, we provide a detailed protocol for genetic manipulation, including gene deletion, insertion, and point mutation in A. baumannii using the platform.
For complete details on the use and execution of this protocol, please refer to Wang et al. (2019).
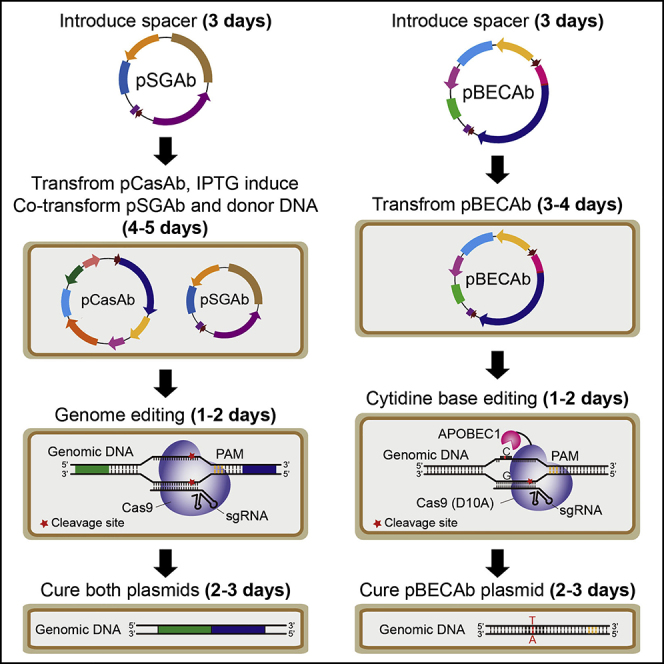
Graphical Abstract
Highlights
-
•
An optimized protocol for A. baumannii-competent cell preparation and electroporation
-
•
Achieving chromosomal gene deletion, insertion, and point mutation in A. baumannii
-
•
Inactivating the gene via cytidine deaminase-catalyzed C-to-T conversion in A. baumannii
-
•
The editing plasmids can be cured via sacB counter-selection
Infections caused by drug-resistant Acinetobacter baumannii have posed a serious threat to global public health. However, genetic manipulation methods, the primary way to study pathogenesis and drug-resistance mechanisms, remain time consuming and inefficient. Here, we provide a detailed protocol for genetic manipulation, including gene deletion, insertion, and point mutation in A. baumannii using the platform.
BEFORE YOU BEGIN
Examination of the Antibiotic Sensitivity of the Target Strain
TIMING: 1 day
To select the proper plasmids used for gene editing, the antibiotic sensitivity and the minimal inhibitory concentration (MIC) of the target A. baumannii strain needs to be examined beforehand.
-
1.
Streak the wild-type cells onto different lysogeny broth (LB) agar plates containing different antibiotics with serial 2-fold dilution concentrations. Use the antibiotic-free LB agar plate as a control.
Note: Several commonly used antibiotics are recommended at a final concentration of 100 μg/mL of apramycin, 50 μg/mL of kanamycin, and 100 μg/mL of spectinomycin.
-
2.
Incubate the plates for approximately 16 hours at 37°C, and then determine the antibiotic sensitivity and the MIC of the target strain.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli DH5α | WEIDI | CAT# DL1001 |
| A. baumannii ATCC 17978 | ATCC | ATCC® 17978 |
| A. baumannii XH386 | (Fang et al., 2016) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Apramycin sulfate | Sangon | CAT# A600090 |
| Kanamycin sulfate | Sangon | CAT# A600286 |
| Spectinomycin hydrochloride | Meilunbio | CAT# MB1497 |
| Glycerol | Hushi | CAT# 10010618 |
| Sucrose | Sangon | CAT# A502792-0500 |
| LB Broth Powder | Sangon | CAT# A507002-0250 |
| LB Agar Powder | Sangon | CAT# A507003-0250 |
| 2×Es Taq MasterMix (Dye) | CoWin Biosciences | CAT# CW0690M |
| T4 Polynucleotide Kinase | TAKARA | CAT# 2021S |
| T4 DNA Ligase | NEB | CAT# M0202S |
| BsaI-HFv2 | NEB | CAT# R3733S |
| HiPure Plasmid Micro Kit | Magen | CAT# P1001-03C |
| Ezup Column Bacteria Genomic DNA Purification Kit | Sangon | CAT# B518255-0050 |
| Recombinant DNA | ||
| pAT04 | (Tucker et al., 2014) | N/A |
| pCasAb-apr | This Paper | Addgene 121998 |
| pSGAb-km | This Paper | Addgene 121999 |
| PSGAb-spe | This Paper | Addgene 122000 |
| PBECAb-apr | This Paper | Addgene 122001 |
| Other | ||
| Gene Pulser Xcell™ Electroporation System | Bio-rad | CAT# 165-2660; CAT# 165-2662 |
| 2 mm electroporation cuvette | Bio-Rad | CAT# 1652082 |
STEP-BY-STEP METHOD DETAILS
Spacer Cloning
-
1.Pick a 20 bp spacer ahead of the PAM sequence (5’-NGG-3’) in the target locus, and then synthesize the two oligonucleotides as follows:
- spacer-F: 5’-tagtNNNNNNNNNNNNNNNNNNNN-3’
- spacer-R: 5’-aaacN’N’N’N’N’N’N’N’N’N’N’N’N’N’N’N’N’N’N’N’-3’
For example, the selected spacer and PAM sequence for the oxyR gene is 5’-ATTGTAGAGCGCCTAGAACA-TGG-3’. The sequences of oxyR_spacer oligonucleotides are as follows:
oxyR-spacer-F: 5’-tagtATTGTAGAGCGCCTAGAACA-3’
oxyR-spacer-R: 5’-aaacTGTTCTAGGCGCTCTACAAT-3’
Note: A spacer design tool (e.g. sgRNAcas9 software) can be used for selecting a suitable spacer sequence (Xie et al., 2014). N’ denotes reverse complement of N in forward primer.
-
2.Phosphorylate the oligonucleotides as indicated below and incubate them at 37°C for 1 hour.
- 5 μL 10 × T4 DNA Ligase buffer (TAKARA)
- 1 μL Spacer-F (100 μM)
- 1 μL Spacer-R (100 μM)
- 1 μL T4 Polynucleotide Kinase (TAKARA)
- 42 μL ddH2O
Note: Gently mix all the reagents and collect them by a quick spin. The order of addition of the reagents can be random.
-
3.
Add 0.5 μL of 5 M NaCl into the phosphorylated product. Incubate the reaction solution at 95 °C for 5 min and then slowly cool it down to room temperature (20-30°C) using a thermocycler, during the process of which the temperature decreases by 1°C per 10s.
Alternatives: You can anneal the oligonucleotides using hot water instead of a thermocycler. Incubate the tube containing the aforementioned reaction solution in a hot water bath of boiling water for 5 min. Then get the tube out of the water bath and let the solution slowly cool down to room temperature (20-30°C).
-
4.Dilute the annealed oligonucleotides 20 folds with ddH2O. Clone the annealed oligonucleotide into the sgRNA expression plasmid as indicated below.
- 1 μL sgRNA expression plasmid (50 ng/μL)
- 1 μL Diluted annealed oligonucleotides
- 1 μL 10 × T4 DNA Ligase buffer (NEB)
- 0.5 μL T4 DNA Ligase
- 0.5 μL BsaI-HFv2
- 6 μL ddH2O
Note: The sgRNA expression plasmids in this protocol contain pSGAb-km, pSGAb-spe and pBECAb-apr.
-
5.Perform the Golden Gate assembly reaction inside a thermocycler using the parameter indicated below (approximately 4 hours) (Figure 1).
- 25 cycles at 37°C for 3 min, and 16°C for 4 min;
- 50°C for 5 min;
- 80°C for 10 min;
- Hold at 16°C.
-
6.
Transform 10 μL assembled product into 100 μL E. coli DH5α competent cells according to the manufacturer's protocol. The cells are plated onto an LB agar plate supplemented with the corresponding antibiotic and the plate is incubated at 37°C for 14-16 hours.
-
7.Randomly pick several colonies to verify the successful cloning of the spacer by colony PCR with the primers of Spacer-F/M13R. The M13R primer sequence is 5’-CAGGAAACAGCTATGACC-3’). Make a colony PCR mix as indicated below, and keep on ice.
- 10 μL 2×Es Taq MasterMix (Dye)
- 0.5 μL Spacer-F (10 μM)
- 0.5 μL M13R (10 μM)
- 0.5 μL bacteria solution
- 8.5 μL ddH2O
Note: Do not add more than 1 μL of bacteria solution into a 20 μL of colony PCR reaction.
-
8.Perform the PCR protocol inside a thermocycler using the parameter indicated below (approximately 1.5 hours). 5-10 colonies are recommended for the PCR screening.
- 95°C for 10 min;
- 28 cycles at 95°C for 30 s, 54°C for 30 s, and 72°C for 20 s;
- 72°C for 5 min;
- Hold at 16°C.
CRITICAL: It is crucial that an initial denaturation of 10 min at 95°C for colony PCR.
-
9.
Run an agarose gel to check for the size of the PCR product, of which the theoretical size is 224 bp. Select 1∼2 positive colonies confirmed by colony PCR. Inoculate the colonies into 5 mL LB broth containing the corresponding antibiotic and incubate the cultures at 37°C for 16 hours with shaking at 250 rpm.
-
10.
Extract the spacer-introduced plasmids by plasmid miniprep kit according to the manufacturer's protocol. Determine the concentration of the extracted plasmid using NanoDrop. Confirm the correct spacer insertion by DNA sequencing using the M13R primer.
PAUSE POINT: The plasmids can be stored at −20°C for several months.
Figure 1.

Inserting a 20 bp Spacer Fragment into the sgRNA Expression Plasmid via Golden Gate Assembly
Preparation of Plasmid-free Electrocompetent Cells
-
11.
Streak a wild-type A. baumannii strain onto an antibiotic-free LB agar, and incubate at 37 °C until colonies are visible (approximately 16 hours).
-
12.
Pick a fresh single colony, and inoculate it into 5 mL of LB broth and incubate the culture at 37 °C for 16 hours with shaking at 250 rpm.
-
13.
Dilute 500 μL overnight culture into 50 mL of LB broth, and incubate the culture at 37°C with shaking at 250 rpm (approximately 3 hours).
-
14.
Chill the culture on ice immediately for 15 min, when the optical density at 600 nm (OD600) of the cell culture reached to 0.5-0.7.
-
15.
Collect the cells by centrifugation at 4000 × g for 5 min at 4°C and discard the supernatant.
-
16.
Resuspend the cell pellet with 15 mL of sterile ice-cold ddH2O, and repeat step 15.
-
17.
Resuspend the cell pellet with 15 mL of sterile ice-cold 10% v/v glycerol, and repeat step 15.
Alternatives: The 10% v/v glycerol can be replaced by ddH2O when the freshly prepared electrocompetent cells are used immediately without freezing.
-
18.
Resuspend the cell pellet with 500 μL of sterile ice-cold 10% v/v glycerol.
-
19.
Dispense the cell suspension into 50 μL aliquots in sterile 1.5 mL microfuge tubes. Freeze the electrocompetent cells in liquid nitrogen and store them at -80°C.
Note: The sterile microfuge tubes at 0-25°C can meet the experimental requirement.
-
20.
Perform the steps 21-26 to determine the transformation efficiency of the prepared competent cells.
Electroporation of pCasAb-apr Plasmid
-
21.
Take a tube of the plasmid-free A. baumannii electrocompetent cells out from the -80°C freezer (see step 19), and thaw it on ice for several minutes.
-
22.
Add approximately 50 ng pCasAb-apr plasmid into the thawed A. baumannii electrocompetent cells, and then gently transfer the mixture into an ice-cold 2 mm electroporation cuvette.
Note: The pAT04 plasmid can serve as positive transformation control; water can serve as a negative transformation control.
-
23.
Perform the electroporation using a Gene Pulser Xcell™ Electroporation System with the following parameters: 2.5 kV, 200Ω and 25 μF.
Note: Please adjust the appropriate parameters when using different electroporation systems.
-
24.
After being pulsed, immediately add 1 mL of prechilled antibiotic-free LB broth into the cuvette to resuspend the cells.
-
25.
Transfer the culture into a sterile 1.5 mL microfuge tube and incubate it for 1 h at 37°C with shaking.
-
26.
Plate 50 μL of the transformed cells on an LB agar plate (90 mm in diameter) supplemented with 100 μg/mL apramycin, and incubate the plate at 37°C until the colonies are visible (14-16 hours).
Preparation of pCasAb-harboring Electrocompetent Cells
-
27.
Pick a single colony of the pCasAb-harboring A. baumannii strain from step 26, and inoculate it into 5 mL of LB broth containing 100 μg/mL apramycin and incubate the culture at 37°C for 16 hours with shaking at 250 rpm.
-
28.
Dilute 500 μL overnight culture into 50 mL of LB broth containing 100 μg/mL apramycin, and incubate the culture with shaking (250 rpm) for 1-1.5 hours at 37°C.
-
29.
Add 50 μL of 1M IPTG into the culture to induce the expression of the RecAb recombinases and the Cas9 nuclease, when the OD600 value of the culture reaches to 0.1-0.15.
Note: IPTG at a final concentration of 1 mM is recommended.
-
30.
Incubate the culture with shaking (250 rpm) at 37°C for another 2 h.
-
31.
Chill the culture on ice for 15 min.
-
32.
Collect the cells by centrifugation at 4000 × g for 5 min at 4°C, and discard the supernatant.
-
33.
Resuspend the cell pellet with 15mL of sterile ice-cold ddH2O, and repeat step 32.
-
34.
Resuspend the cell pellet with 15mL of sterile ice-cold 10% v/v glycerol, and repeat step 32.
-
35.
Resuspend the cell pellet with 500 μL of sterile ice-cold 10% v/v glycerol.
-
36.
Dispense the cell suspension into 50 μL aliquots in sterile 1.5 mL microfuge tubes.
Note: The sterile microfuge tubes at 0-25°C can meet the experimental requirement.
The Two-Plasmid pCasAb/pSGAb-Mediated Genome Editing
-
37.
Add approximately 200 ng spacer-introduced pSGAb-km plasmid and 3 μL of 100 μM ssDNA or 300 ng dsDNA donor repair template into a tube of freshly prepared pCasAb-harboring A. baumannii electrocompetent cells, and then gently transfer the mixture into an ice-cold 2 mm electroporation cuvette (Figure 2). For example, the sequence of 80-nt ssDNA donor DNA for oxyR gene deletion is 5’- TTGATGAAGTGCATCAACAATTACCGAAGATTCAGTTGCAATGATCTCGATTTATCACGTCTCATTTTGCTTGAAGAAGG-3’.
Note: The empty pSGAb-km plasmid can be used as positive transformation control. The spacer-introduced pSGAb-km plasmid without the insertion of a donor DNA can be used as negative transformation control. ssDNA donor template can be used for gene deletion and point mutation. dsDNA donor template can be used for point mutation and gene insertion. Long homologous template (>200 bp each) can improve the efficiency of gene insertion. The deletion length flanking Cas9 cleavage sites should be similar and no more than 1 kb each.
-
38.
Perform the electroporation using a Gene Pulser Xcell™ Electroporation System with the following parameters: 2.5 kV, 200Ω and 25 μF.
Note: Adjust the appropriate parameters when using different electroporation systems.
-
39.
After being pulsed, immediately add 1 mL of prechilled antibiotic-free LB broth into the cuvette to resuspend the cells.
-
40.
Transfer the culture into a sterile 1.5 mL microfuge tube and incubate it for 1.5 h at 37°C with shaking for recovery and genome editing.
-
41.
Plate 100 μL of the transformed cells on an LB agar plate (90 mm in diameter) supplemented with 100 μg/mL apramycin and 50 μg/mL kanamycin, and incubate the plate at 37°C until colonies are visible (approximately 16 hours).
Note: Plating 100 μL of the transformed cells is sufficient to obtain enough colonies.
-
42.Randomly pick several colonies to verify the success of the genome editing by colony PCR. For example, the sequences of two primers for verifying the oxyR gene deletion are as follows.
- oxyR-seq-F:5’-ACTTTATCGGGCGGCATTAT-3’
- oxyR-seq-R:5’-GTACGGCTAGGTGCGTCTTC-3’
Note: Screening of 10 colonies is recommended. In general, the genome editing efficiency is more than fifty percent.
-
43.
Perform DNA sequencing using one of the specific primers employed above (for example, oxyR-seq-F or oxyR-seq-R) to confirm the desired genome editing.
Figure 2.

Scheme for pCasAb/pSGAb-Mediated Genome Editing in A. baumannii
Cure Both the pCasAb-apr and pSGAb-km Plasmids
-
44.
Inoculate the desired A. baumannii mutant in 5 mL of antibiotic-free LB broth and shake it at 250 rpm for 14-16 hours at 37°C.
-
45.
Streak a fraction of culture onto an LB agar plate supplemented with 5% w/v sucrose, and incubate it at 37°C until colonies are visible (approximately 16 hours).
Note: The LB agar supplemented with 5% w/v sucrose is sterilized at 115°C for 15-20 min.
-
46.
Pick and streak several colonies onto LB agar plates supplemented with or without 100 μg/mL apramycin or 50 μg/mL kanamycin, respectively. Incubate the plates at 37°C overnight. If the colonies can only grow on the antibiotic-free LB agar plate, it indicates that both the pCasAb-apr plasmid and the pSGAb-km plasmid are cured successfully in the colonies (Figure 3).
Note: Screening of 2–4 colonies is recommended.
Figure 3.

Curing of Plasmids pCasAb and pSGAb in A. baumannii
The strain can only grow on the antibiotic-free LB agar plate, rather than the plate supplemented with apramycin or kanamycin, confirming that the pCasAb-apr and the pSGAb-km plasmids can be cured simultaneously. Apr, apramycin; Km, kanamycin.
The Single-Plasmid pBECAb-Mediated Cytidine Base Editing
-
47.
Take a tube of the plasmid-free A. baumannii electrocompetent cells out from the -80°C freezer (see step 19), and thaw it on ice for several minutes.
-
48.
Add 50-200 ng spacer-introduced pBECAb-apr plasmid into the thawed A. baumannii electrocompetent cells, and then gently transfer the mixture into an ice-cold 2 mm electroporation cuvette (Figure 4).
Note: The spacer-free pBECAb-apr plasmid can serve as positive transformation control; water can serve as negative transformation control.
-
49.
Perform the electroporation using a Gene Pulser Xcell™ Electroporation System with the following parameters: 2.5 kV, 200Ω and 25 μF.
Note: Adjust the appropriate parameters when using different electroporation systems.
-
50.
After being pulsed, immediately add 1 mL of prechilled antibiotic-free LB broth into the cuvette to resuspend the cells.
-
51.
Transfer the culture into a sterile 1.5 mL microfuge tube and incubate it at 37°C for 1.5 hours with shaking (250 rpm) for recovery and base editing.
Note: The sterile microfuge tubes at 0°C–25°C can meet the experimental requirement.
-
52.
Plate 25 μL of the transformed cells on an LB agar plate (90 mm in diameter) supplemented with 100 μg/mL apramycin, and incubate the plate at 37°C until colonies are visible (approximately 16 hours).
-
53.
Randomly pick several colonies to amplify the target region covering the editable sites by colony PCR.
Figure 4.

Scheme for pBECAb-Mediated Cytidine Base Editing in A. baumannii
For example, to convert the Q37(CAG) of the blaTEM-1D gene to a premature stop codon (TAG) in the A. baumannii XH386 strain, the spacer and PAM sequence 5’-AGATCAGTTGGGTGCACGAG-3’ is selected. The sequences of blaTEM-1D_spacer oligonucleotides are as follows:
blaTEM-1D-spacer-F: 5’-tagtAGATCAGTTGGGTGCACGAG-3’
blaTEM-1D-spacer-R: 5’-aaacCTCGTGCACCCAACTGATCT-3’
The sequences of the primers for amplifying the blaTEM-1D_spacer region are as follows:
blaTEM-1D-seq-F: 5’- TGGCACTTTTCGGGGAAATG -3’
blaTEM-1D-seq-R: 5’- CACGCTCGTCGTTTGGTATG -3’
-
54.
Examine the desired C-to-T conversion by DNA sequencing using one of the specific primers employed above (for example, blaTEM-1D-seq-F or blaTEM-1D-seq-R).
Note: Screening of 3 colonies is recommended.
Cure the pBECAb-apr Plasmid
-
55.
Inoculate the desired A. baumannii mutant into 5 mL of antibiotic-free LB broth and shake it at 250 rpm for 14-16 hours at 37°C.
-
56.
Streak a fraction of culture onto an LB agar plate supplemented with 5% w/v sucrose, and incubate it at 37°C until colonies are visible (approximately 16 hours).
Note: The LB agar supplemented with 5% w/v sucrose is sterilized at 115°C for 15–20 min.
-
57.
Pick and streak several colonies onto LB agar plates supplemented with or without 100 μg/mL apramycin, respectively. Incubate the plates at 37°C overnight. If the colonies can only grow on the antibiotic-free LB agar plate, it indicates that the pBECAb-apr plasmid is cured successfully in the colonies (Figure 5).
Note: Screening of 2–4 colonies is recommended.
Figure 5.

Curing of Plasmid pBECAb in A. baumannii
The strain can only grow on the antibiotic-free LB agar plate, rather than the plate supplemented with apramycin, confirming that the pBECAb-apr plasmid can be cured easily cured in A. baumannii. Apr, apramycin.
EXPECTED OUTCOMES
Rapid and precise chromosomal gene deletion, insertion, and point mutation were achieved in various A. baumannii strains with the utilization of the two-plasmid pCasAb/pSGAb system. Highly efficient gene inactivation was observed via cytidine deaminase-catalyzed programmed C-to-T conversion. After desired editing, all these editing plasmids were successfully cured using the sacB counter-selection. Details of genome editing and base editing can be found in our recent Cell Chemical Biology publication (Wang et al., 2019).
LIMITATIONS
It is difficult to use the two-plasmid pCasAb/pSGAb system for gene editing in the sites located on mobile genetic elements, such as plasmids and transposons. The double-strand DNA streak (DSB) on the plasmids produced by Cas9 nuclease can lead to plasmid removal without repair. The DSB on the transposons may cause undesired recombination between repetitive sequences. Alternatively, the cytidine base editing system can be used for gene inactivation by converting four codons (CAA, CAG, CGA, and TGG) into premature stop codons (TAA, TAG, and TGA).
We have comprehensively investigated the activity window of the single plasmid pBECAb system, which is from positions 3 to 8 (counting the base distal from the PAM as position 1) (Figure 6). The Cs within the activity window can be converted into Ts efficiently. However, the conversion frequency of the Cs outside the activity window reduces dramatically. In addition, the editing efficiency is affected by the kind of base immediately at 5′ of the target C, which follows the order: TC ≥ CC ≥ AC > GC.
Figure 6.

Schematic Diagram Illustrating the Activity Window, which is from Positions 3 to 8 in the 20 bp Spacer
TROUBLESHOOTING
Problem
My A. baumannii strain is not sensitive to apramycin, kanamycin, or spectinomycin.
Potential Solution
Some of clinically isolated A. baumannii strains are usually resistant to multiple antibiotics. To modify the genome of MDR A. baumannii strains, you can first perform the drug sensitivity test for your strain, and then replace the antibiotic selection marker in pCasAb-apr, pSGAb-km, pSGAb-spe, and pBECAb-apr plasmids with any other proper selection markers by Gibson assembly.
Problem
Only a few colonies are obtained using the pCasAb/pSGAb system, and no colony is successfully edited.
Potential Solution
Does the transformation of the spacer-free pSGAb-km plasmid produce a lawn of colonies? If so, the result indicates that the CRISPR-Cas9 have cleaved the genomic DNA, but no recombination event occurs. Please confirm the correctness of the IPTG solution in your lab and the gene sequence of RecAb on the pCasAb plasmid you used. If all these are correct, the result indicates that the electroporation efficiency of the pCasAb-harboring cells is low. Please re-prepare the electrocompetent competent cells according to the preparation method described above.
Problem
For cytidine base editing, the results of DNA sequencing show that only overlapping peaks appearing in the target editing site.
Potential Solution
It indicates that the cells in a single colony are not 100% edited. You can obtain a pure mutant by streaking the partially edited colonies onto an LB agar containing the corresponding antibiotic and incubating it plate at 37°C overnight.
Problem
Only low efficiency of C-to-T conversion can be detected.
Potential Solution
Make sure the target C is located at the positions 3 to 8 (activity window) of the 20 bp spacer, and avoid use the spacer containing the GC target site.
Acknowledgments
We thank the support from Analytical Instrumentation Center (#SPST-AIC10112914), SPST, ShanghaiTech University.
Author Contributions
Y.W. and Z.W. wrote the protocol. Q.J. edited and revised the protocol.
Declaration of Interests
Two patents have been submitted for the two-plasmid pCasAb/pSGAb genome editing system and the single-plasmid pBECAb base editing system.
Contributor Information
Yu Wang, Email: wangyu@shanghaitech.edu.cn.
Quanjiang Ji, Email: quanjiangji@shanghaitech.edu.cn.
References
- Fang Y., Quan J., Hua X., Ye F., Li X., Wang J., Zhi R., Shang S., Yu Y. Complete genome sequence of Acinetobacter baumannii XH386 (ST208), a multi-drug resistant bacteria isolated from pediatric hospital in China. Genomics Data. 2016;7:269–274. doi: 10.1016/j.gdata.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker A.T., Nowicki E.M., Boll J.M., Knauf G.A., Burdis N.C., M Stephen T., Davies B.W. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. Mbio. 2014;5:e01313–e01314. doi: 10.1128/mBio.01313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang Z., Chen Y., Hua X., Yu Y., Ji Q. A Highly Efficient CRISPR-Cas9-Based Genome Engineering Platform in Acinetobacter baumannii to Understand the H2O2-Sensing Mechanism of OxyR. Cell Chem. Biol. 2019;26:1732–1742. doi: 10.1016/j.chembiol.2019.09.003. [DOI] [PubMed] [Google Scholar]
- Xie S., Shen B., Zhang C., Huang X., Zhang Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS one. 2014;9:e100448. doi: 10.1371/journal.pone.0100448. [DOI] [PMC free article] [PubMed] [Google Scholar]