

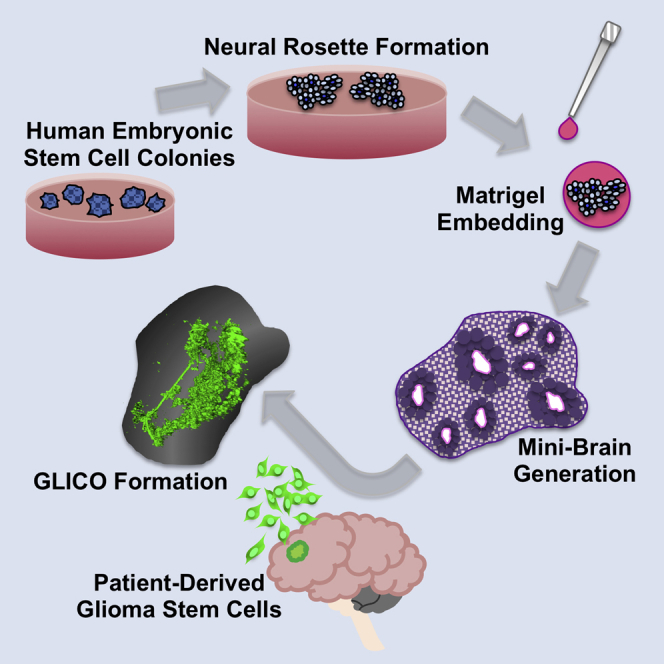
Summary
Glioblastoma (GBM) remains a devastating disease with a median survival of less than two years. Current preclinical models are unable to accurately reflect the complexity of human GBM. We recently established a cerebral organoid glioma (GLICO) model to study the invasion and biology of patient-derived glioma stem cells in miniature replicas of the human brain. Through the dissemination of our detailed methodology, we aim to encourage other scientists to further build upon our existing model for studying these destructive tumors.
For complete details on the use and execution of this protocol, please refer to Linkous et al. (2019).
Graphical Abstract
Highlights
-
•
Cerebral organoids can be generated from hESCs as well as iPSCs
-
•
Cerebral organoid gliomas (GLICOs) enhance the study of GBM biology and invasion
-
•
Patient-derived tumor cells migrate toward, invade and proliferate in human mini-brains
-
•
The GLICO model allows for human GBM tumor formation in less than 2 weeks
Glioblastoma (GBM) remains a devastating disease with a median survival of less than two years. Current preclinical models are unable to accurately reflect the complexity of human GBM. We recently established a cerebral organoid glioma (GLICO) model to study the invasion and biology of patient-derived glioma stem cells in miniature replicas of the human brain. Through the dissemination of our detailed methodology, we aim to encourage other scientists to further build upon our existing model for studying these destructive tumors.
Before You Begin
TIMING: 1 h for tumor cell isolation; 2-4 weeks for expansion of culture
Prepare Glioblastoma Tumor Samples
-
1.
Following informed consent, tumor samples classified as glioblastoma, based on the World Health Organization (WHO) criteria, were obtained from patients undergoing surgical treatment at the National Institutes of Health (NIH) or from Weill Cornell Medicine/New York Presbyterian Hospital in accordance with the appropriate Institutional Review Boards.
-
2.
Within 1–3 h after surgical removal, tumors were washed in PBS and enzymatically dissociated into single cells. Tumor cells were cultured in NBE medium.
-
3.
Regular mycoplasma screening was performed using the MycoAlert Detection Kit (Lonza Inc.).
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| ROCK inhibitor Y-27632 2HCl (50 μM final concentration; Dissolve 5 mg in 2.96 mL water for a 5 mM stock. Dilute stock 1:100 in neural induction medium for final of 50 μM) | Selleck Chemicals | Cat# S1049; N/A |
| DMEM/F12 Medium | Hyclone | Cat# SH30126.01; N/A |
| L-Glutamine-200 mM (100X) | Life Technologies | Cat# 25030-149; N/A |
| Recombinant human EGF 1mg | R&D Systems | Cat# 3718-FB-01M; N/A |
| Recombinant human bFGF 1mg | R&D Systems | Cat# 236-EG-01M; N/A |
| N2 supplement | Life Technologies/GIBCO | Cat# 17502-048; N/A |
| Glutamax | Thermo Fisher Scientific | Cat# 35050-061; N/A |
| Minimum Essential Medium-Nonessential Amino Acids (MEM-NEAA) | Thermo Fisher Scientific | Cat# 11140-050; N/A |
| Neurobasal Medium | Life Technologies | Cat# 21103-049; N/A |
| Neurobasal-A Medium | Life Technologies | Cat# 10888-022; N/A |
| B27 supplement WITHOUT vitamin A | Life Technologies/GIBCO | Cat# 12587-010; N/A |
| 2-mercaptoethanol | Thermo Fisher Scientific | Cat# 21985-023; N/A |
| Insulin | Sigma | Cat# I9278-5ml; N/A |
| Matrigel™ Membrane Matrix for Organoid Droplets | Corning | Cat# 356234; N/A |
| B27 supplement WITH vitamin A | Life Technologies/GIBCO | Cat# 17504-044; N/A |
| hESC-Qualified Matrigel™ | Corning | Cat# 354277; N/A |
| Normocin | Invivogen | Cat# Ant-nr-2; N/A |
| 0.5M EDTA pH 8.0 | Invitrogen | Cat# 15575-038; N/A |
| mTeSR1 | STEM CELL TECHNOLOGIES | Cat# 05850; N/A |
| STEMdiff™ Neural Induction Medium | STEM CELL TECHNOLOGIES | Cat# 05835; N/A |
| STEMdiff™ Neural Rosette Selection Reagent | STEM CELL TECHNOLOGIES | Cat# 05832; N/A |
| Penicilin-Streptomycin solution (20mL) | Life Technologies/GIBCO | Cat# 15140148; N/A |
| Experimental Models: Cell Lines | ||
| Patient-Derived Glioma Stem Cells: 827, 1228, 211, 923, 308, 0728, 0320, 0517, 0607, 0810, and 1206 | National Institutes of Health and Weill Cornell Medicine/NYU Presbyterian Hospital | N/A |
| NIH-registered human H1 (WA01) embryonic stem cells | WiCell Research Institute | Cat# WA01; RRID: CVCL_9771 |
| NIH-registered human H9 (WA09) embryonic stem cells | WiCell Research Institute | Cat# WA09; RRID: CVCL_9773 |
| Other | ||
| Ultra low-attachment 96-well round bottom plates | Corning | Cat# 7007; N/A |
Materials and Equipment
Note: The media and components listed in this protocol have been specifically selected to eliminate mesoderm and endoderm formation within the cerebral organoids.
Alternatives: Human induced pluripotent stem cells (iPSCs) can also be used to generate cerebral organoids using this protocol.
STEMdiff Neural Induction Medium
-
•
From STEMCELL TECHNOLOGIES INC
-
•
Catalog #05835
Preparation of hESC Medium
-
•
To prepare stem cell medium, add 5x mTeSR1 supplement to mTeSR1 basal medium. Then add 1:1000 Normocin.
-
•
Prior to culturing cells, coat 6-well plates with hESC-Qualified Matrigel (1 aliquot in 12mL cold DMEM F12; check aliquot volume for each lot number by using the Corning web site— (https://www.corning.com/worldwide/en/products/life-sciences/resource-library.html).
-
•
To coat a 6-well plate, you will need 1mL of diluted matrigel per well.
-
•
Place plates at 37°C for at least 1 h prior to use.
Note: Unused, coated plates can be wrapped in parafilm and stored at 4°C.
Differentiation Medium (250 ml): Store at 4C for Up to 2 Weeks
| 125 mL DMEM/F12 |
| 125 mL Neurobasal |
| 1.25 mL N2 supplement |
| 2.5 mL B27 ± vitamin A supplement (Type I is without Vitamin A; Type II is WITH Vitamin A) |
| 62.5 μL insulin |
| 87.5 μL 2-ME solution (1:100 dilution in DMEM/F12) |
| 2.5 mL Glutamax supplement |
| 1.25 mL MEM-NEAA |
| 2.5 mL P/S |
NBE Medium for Glioma Stem Cells
| 1000 mL Neurobasal-A Medium |
| 5 mL L-Glutamine, 200 mM |
| 5 mL N2 supplement, 100X |
| 10 mL B27 supplement without vitamin A, 50X |
| 1 mL Heparin sodium, 2 mg/mL |
| 1 mL EGF, 25 μg/mL |
| 1 mL bFGF, 25 μg/mL |
| 10 mL P/S |
Step-by-Step Method Details
Thawing hESCs
This step provides a detailed procedure for thawing hESCs.
Note: Due to the sensitivity of hESCs and hESC-derived tissue, all steps in this protocol should be considered mandatory and should be followed exactly to ensure success.
-
1.
Warm mTeSR1 in 37°C water bath (∼20 min).
-
2.
Remove Matrigel-coated plates from the incubator and place in TC hood.
-
3.
Remove vial of hESCs from liquid nitrogen and thaw 1-2mins in 37°C water bath.
-
4.
Transfer contents of vial to 15mL tube, and then add 5-10mL mTeSR1 dropwise to cells. Do NOT create single-cell suspension!
-
5.
Spin @200 rcf for 3 min, pour off/aspirate supernatant, and resuspend cell pellet in 2 mL mTeSR1. Again, do NOT create single-cell suspension.
-
6.
Aspirate hESC-Qualified Matrigel from one well of a 6-well plate and add the 2 mL hESC suspension to the well.
-
7.
Incubate at 37°C for 12-24 h.
-
8.
The next day, aspirate the media and rinse the well with 1 mL of warm DMEM F12. Aspirate the DMEM F12 and replace with 2 mL of fresh, warmed mTeSR1. Examine cell survival/morphology.
Feeding hESCs
This step provides a detailed protocol for the daily maintenance of hESC cultures.
-
1.
On the day following either thawing or passaging, if you see a lot of dead cells, wash once with 1mL warm DMEM F12 before feeding.
Note: It is normal for cells to resemble differentiated cells on the first day post-thaw, but colonies should adopt normal morphology after a few days of routine feeding.
-
2.
Aspirate media from well, and feed the cells with 2mL fresh, warm mTeSR1 daily, monitoring cells for appropriate morphology (See Figure 1).
Figure 1.

Human Embryonic Stem Cell (hESC) Colony Formation
Normal morphology of healthy, hESC colonies 48 h after passaging; scale bar, 400 μm.
Passaging hESCs
This step describes how to passage hESCs to ensure their health throughout the experiment.
-
1.
hESCs usually need to be passaged every 5-7 days. It is best to passage them when they are no more than 80% confluent to prevent spontaneous differentiation.
-
2.
When ready to passage, aspirate the media and wash once with PBS. Aspirate PBS and add 1 mL of PBS containing EDTA (0.5M EDTA diluted 1:1000 in PBS). Incubate at 37°C for approximately 5-7 min to dissociate the cells. Check the morphology of cells at 5 min. If colonies are rounded up, proceed to Step 3.
-
3.
Carefully aspirate the EDTA and wash gently with 1 mL sterile PBS. Allotted wash time should be less than 10 s.
-
4.
Aspirate the Matrigel from a new, coated well(s).
-
5.
Carefully aspirate the PBS wash and gently remove the cells using a 5 mL serological pipette that contains 2 mL of warm mTeSR1 medium.
CRITICAL: Do not over-pipet; avoid creating a single-cell suspension!
-
6.
Transfer the cells to the new well.
Note: The passaging ratio differs based on the experiment and the cell line in hand, but generally is 1:2 depending on confluency.
-
7.
Examine morphology day by day. Colonies should be well circumscribed, i.e., differentiated cells appear fibroblastic or dense over-confluent colony cores.
Making Embryoid Bodies (EBs)
This step describes how to dissociate confluent hESCs into a single-cell suspension that will ultimately promote the formation of embryoid bodies.
-
1.
When hESC colonies are ready for splitting (80% confluent), passage according to protocol (Steps 2, 3, and 5 above), but resuspend cell pellet in warm STEMdiff Neural Induction Medium containing 1:100 Rock Inhibitor. When resuspending, create a single-cell suspension.
-
2.
Plate cells into individual wells of a low-attachment, round-bottom 96-well plate (150 μL per well). Plate approximately 7,000-9,000 cells per well.
-
3.
Perform partial medium change (∼100 μL) every other day for 4-5 days, with Rock inhibitor for the first 2 days. Monitor morphology of EBs (Figure 2).
Figure 2.

Embryoid Body Formation after 5 Days
Morphology of healthy embryoid body, left; morphology of unhealthy embryoid body, right; scale bars, 400 μm.
Forming Neural Rosettes
This step describes how to promote neural induction and generation of neural rosettes.
-
1.
The inclusion of a neural rosette formation step can be used to enrich for neural progenitors and improve efficiency of neuronal differentiation in 3D organoid cultures (Mariani et al., 2015). When EBs are about 500-600 μm in diameter and begin to brighten and have smooth edges (Figure 2, left), transfer ≤ 10 EBs to one hESC-Qualified Matrigel-coated well of a 6-well plate. (Coating procedure is the same as the one you use when culturing hESCs). Note: Use a p1000 tip to transfer embryoid bodies to coated well. Make sure you have at least 2 mL of warm STEMdiff Neural Induction Medium covering the embryoid bodies in each coated well. Transfer of embryoid bodies to coated well should be completed within 10 min to avoid any issues with EB survival or detachment from coated well surface.
-
2.
Change medium every day for 5-6 days. Examine the morphology daily. You should clearly see the formation of neural rosettes within 3-4 days of plating the embyroid bodies (Figure 3A).
Figure 3.

Neural Rosette Clusters
(A) Normal morphology of primary neural rosette clusters at Day 4.
(B) Suboptimal neural rosette cluster with necrotic center (arrow).
(C) Normal morphology of rosettes during secondary rosette formation; scale bars, 500 μm.
-
3.
Once rosettes are readily apparent with radial arrangements of columnar cells and lumen-like centers (Day 5 or Day 6), aspirate the medium and wash with PBS for 1 min. Apply STEMdiff Neural Rosette Selection Reagent to the well and incubate at 37°C for one h. Please note that rosette clusters may develop necrotic centers if not detached at Day 5 or Day 6 (Figure 3B); clusters with necrotic cores should be discarded.
-
4.
Aspirate the STEMdiff Neural Rosette Selection and wash with PBS for 1 min. Using a p1000 tip containing 1 mL of STEMdiff Neural Induction Medium, propel the medium quickly to dislodge the rosettes clusters from the coated well and—using a 5 mL serological pipette, transfer the rosettes clusters to a new Matrigel-coated well for a second round of rosette formation. Use an additional 1 mL of STEMdiff Neural Induction Medium to dislodge any remaining rosettes. Rosettes should lift off, leaving only more differentiated neural cells behind (Figure 4).
Figure 4.

Detachment of Neural Rosette Clusters
Successful removal of neural rosette clusters from Matrigel-coated well; scale bar, 500 μm.
-
5.
Change medium daily for 5-6 days (monitoring for healthy, rosette formation each day). Morphology for secondary rosettes should closely-resemble that of primary rosettes (Figure 3C). Once rosettes exhibit radial arrangements of columnar cells with lumen-like centers, apply the neural rosette selection reagent as before, followed by a one-h incubation, and a PBS wash.
Development of Cerebral Tissue
This step describes how to embed the neural rosette clusters in Matrigel and promote differentiation into a mature, cerebral organoid.
-
1.
After the PBS wash, use a p1000 tip containing Differentiation Type I Medium to remove the rosette clusters; rosette clusters are approximately 1-2 mm in diameter. Using the p1000 tip, transfer the clusters one by one to sterile Parafilm in a 10 cm dish. The goal is to embed one cluster in one droplet of Matrigel.
-
2.
Remove excess media from each drop and add ∼75 μL of Matrigel (#356234, NOT hESC-qualified) to each rosette cluster. Position each cluster in the center of the droplet using a p200 pipet tip.
-
3.
Place Parafilm (should be in a 10 cm Petri dish) at 37°C for 30 min to allow Matrigel to polymerize.
-
4.
Using a sterile p200 tip, gently roll off Matrigel droplets into a 10 cm dish containing 10 mL Differentiation Type I Medium. Place plate in incubator without shaking for 72 h.
-
5.
After 3 days, transfer plate to an orbital shaker (85 rpm) in 10 mL Differentiation Type II Medium in a standard TC incubator.
-
6.
Change media every 3-4 days and monitor for morphology.
-
7.
Cerebral organoids are ready for analysis when tissue completely fills matrigel droplet and have been growing on the shaker for at least 2 weeks (Figure 5). Histopathological evaluation of resultant organoids should be performed by a board-certified pathologist using the neuronal and glial markers as originally described (Linkous et al., 2019).
Figure 5.

Stage-Specific Morphology of Cerebral Organoids
(A) Bright-field microscopy of cerebral organoid maturation.
(B) Hematoxylin and Eosin staining of cerebral organoids at 4 weeks and 8 weeks post-Matrigel embedding; scale bars, 400 μm.
Generation of Cerebral Organoid Glioma (GLICO) Tumors
This step describes how to initiate patient-derived brain tumors within cerebral organoids.
-
1.
For co-culture experiments, transfer individual organoids to a 24-well plate (one organoid per well).
-
2.
Remove excess media from organoids and gently wash organoids twice with PBS. Allotted wash time is 1 min per wash. Do not physically disturb the organoids, as they can shear easily.
-
3.
Seed 10,000 fluorescently-labeled glioma stem cells (GSCs) into each organoid-containing well (10,000 GSCs/2 mL of NBE per well).
-
4.
Incubate plates at 37°C for 24 h (no shaking).
-
5.
Wash organoids once with PBS (1 min) and transfer to 6-well plates containing 2 mL of Differentiation Type II medium per well.
-
6.
Place the plate on the orbital shaker at 37°C and monitor daily for tumor growth (Figure 6).
Figure 6.
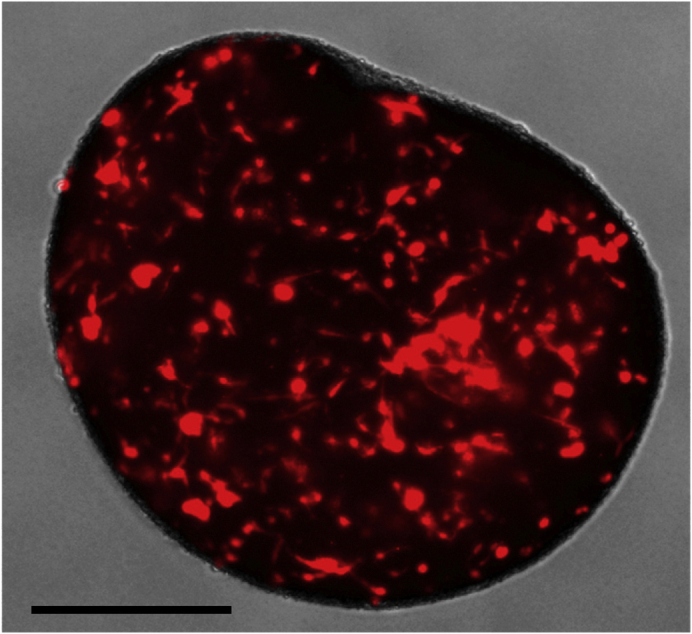
GLICO Formation
Tumor cell infiltration of cerebral organoid one week after co-culture; scale bar, 500 μm.
Note: The number of GSC-derived GLICO tumors that form can vary depending on the cell line, however, tumor invasion into the organoid should be evident within 48 h of co-culture for the majority of cell lines.
Note: Depending on the invasive and proliferative properties of the glioma stem cell line, tumor cells can invade the entire organoid within 2 weeks of co-culture (Linkous et al., 2019, Ogawa et al., 2018).
-
7.
If the desired endpoint requires that some normal cerebral organoid tissue should remain, then GLICOs should be harvested before tumor invasion has reached capacity.
Expected Outcomes
GLICO tumors should be evident within 48 h of co-culture initiation. Quality and batch control of all samples, including verification of organoid histopathology, was performed by a board-certified neuropathologist. GLICOs are formed using fluorescently-labeled glioma stem cells so that tumor growth can be monitored daily by fluorescence microscopy. Details of cell labeling can be found in our recent Cell Reports publication (Linkous et al., 2019). Histopathological evaluation of resultant organoids should be performed by a board-certified pathologist using the neuronal and glial markers as originally described (Linkous et al., 2019).
Limitations
Successful cerebral organoid generation is dependent upon the health of the hESC cultures. Optimal hESC cultures contain less than 10% of differentiated colonies; if more than 10% of colonies exhibit differentiation, one must either 1) Remove differentiated cells using stereomicroscopic techniques or 2) Thaw a new vial of cells.
Troubleshooting
Problem
Extensive cell death of hESCs after passaging
Possible Solution
Do not allow EDTA incubation to exceed 5-7 min; use gentle pipette motion to resuspend cells in fresh media
Problem
Matrigel droplets will not solidify
Possible Solution
Aliquot Matrigel upon arrival; avoid freezing/thawing
Problem
Cerebral organoids are shearing upon transfer/manipulation
Possible Solution
Use a 25 mL serological pipette to gently collect and transfer organoids
Acknowledgments
Work in H.F.’s laboratory is supported by an NIH Director Pioneer Award (1DP1CA228040-01).
Author Contributions
A.L. and H.F. conceived the project and wrote the protocol. A.L. designed, performed, and analyzed the experimental protocol.
Declaration of Interests
The authors declare no competing financial interests.
References
- Linkous A., Balamatsias D., Snuderl M., Edwards L., Miyaguchi K., Milner T., Reich B., Cohen-Gould L., Storaska A., Nakayama Y. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019;26:3203–3211.e5. doi: 10.1016/j.celrep.2019.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J., Coppola G., Zhang P., Abyzov A., Provini L., Tomasini L., Amenduni M., Szekely A., Palejev D., Wilson M. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa J., Pao G.M., Shokhirev M.N., Verma I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018;23:1220–1229. doi: 10.1016/j.celrep.2018.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]