

SUMMARY
Repurposing the broadly distributed native CRISPR-Cas systems in prokaryotes for genome editing is emerging as a new strategy for genetic manipulations. We recently reported the establishment of a single plasmid-mediated, one-step genome-editing technique in a multidrug-resistant genotype of the opportunistic pathogen Pseudomonas aeruginosa by harnessing its endogenous type I-F CRISPR-Cas system. The platform is readily applicable in additional type I-F CRISPR-containing clinical and environmental P. aeruginosa isolates. Herein, we provide the detailed protocol for the methodology.
For complete details on the establishment and exploitation of this protocol, please refer to Xu et al. (2019).
Graphical Abstract
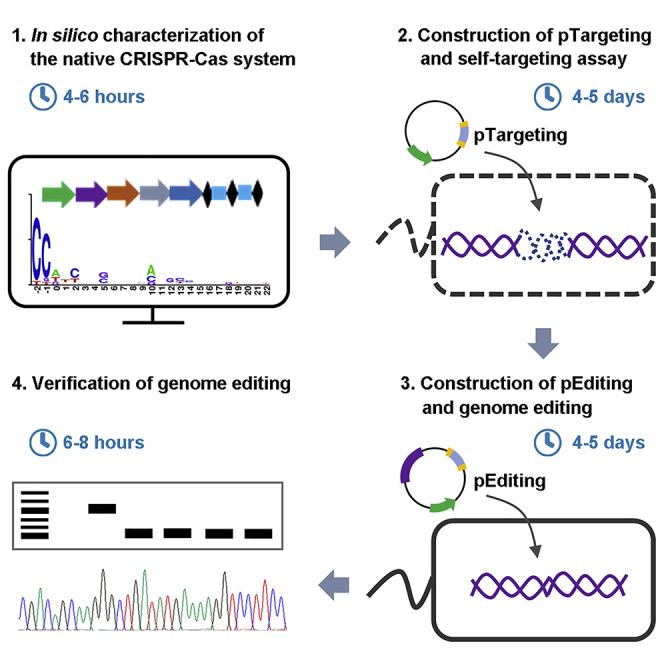
Highlights
-
•
Workflow to repurpose the native I-F CRISPR-Cas system for genome editing in PA154197
-
•
Procedures to characterize native CRISPR-Cas systems in the strains of interest
-
•
Potential solutions for troubleshooting and expanding the method to other strains
Repurposing the broadly distributed native CRISPR-Cas systems for genome editing is emerging as a new strategy for genetic manipulations in prokaryotes. We recently reported the establishment of a single plasmid-mediated, one-step genome-editing technique in a multidrug-resistant genotype of the opportunistic pathogen Pseudomonas aeruginosa by harnessing its endogenous type I-F CRISPR-Cas system. The platform is readily applicable in additional type I-F CRISPR-containing clinical and environmental P. aeruginosa isolates. Herein, we provide the detailed protocol for the methodology.
BEFORE YOU BEGIN
Timing: 4–6 h
-
1.
In silico analysis of the native CRISPR-Cas system in the P. aeruginosa isolate of interest
P. aeruginosa is a model organism for CRISPR-Cas research, especially the type I-F CRISPR-Cas system which constitutes ∼67% of all CRISPR-Cas systems present in the P. aeruginosa genomes sequenced so far (van Belkum et al., 2015). Belonging to the multi-effector class 1 CRISPR-Cas system, the subtype I-F is characterized by the signature Cas3 nuclease common in all type I systems and is distinguished from other type I systems by the presence of the cas8f gene (formerly csy1) and the unique cas2-cas3 fusion in its cas loci (Nethery and Barrangou, 2019). The one-step genome-editing technique we illustrate herein is applicable in P. aeruginosa isolates containing an active type I-F CRISPR-Cas system in their genomes. Hence, the first step towards the establishment of the technique is to search and identify the presence of a type I-F CRISPR-Cas locus in the genome of interest. We employed CRISPRCasFinder (Couvin et al., 2018) to search for the CRISPR-Cas system in PA154197, a multidrug-resistant (MDR) P. aeruginosa strain isolated from a case of blood stream infection in the Queen Mary Hospital, Hong Kong (Cao et al., 2019). Inputting the genome sequence of PA154197 as a query, CRISPRCasFinder identified a type I-F CRISPR-Cas system encompassing six cas genes with the signature cas8f gene and the unique cas2-3 fusion along with three CRISPR arrays PA154197_2, PA154197_5, and PA154197_6, each containing 10, 12, and 4 spacers, respectively, in the strain (Figure 1). Aligning these elements against the genome of PA154197 leads to the assembly of the complete type I-F CRISPR-Cas loci in this strain (Figure 1). The cas operon is sandwiched by the convergent CRISPR arrays PA154197_6 (4 spacers) and PA154197_5 (10 spacers), respectively, with the third CRISPR array PA154197_2 located 1,376,188-bp downstream of PA154197_5. The 28-bp consensus repeat in the three arrays differs by only one nucleotide and their spacers are identical in size (32 bp).
Figure 1.

Bioinformatic Analysis for the Presence of Native CRISPR-Cas Locus and Its Spacers, Repeat, and PAM Sequences
(A) Output results showing the presence of a type I-F CRISPR-Cas system by CRISPRCasFinder search following input of PA154197 genome sequence as the query. Blast of the identified CRISPR arrays and cas genes against the genome of PA154197 led to assembly of the complete native CRISPR-Cas loci in the strain. Diamonds and rectangles indicate the repeats and spacers in the CRISPR arrays, respectively. Number of spacers is indicated above each of the CRISPR arrays. Bended arrows (black) above the leader sequence (purple) indicate the orientation of CRISPR transcription.
(B) Alignment of 8-bp upstream sequences of protospacers identified from the bacteriophage and plasmid databases led to prediction of the PAM sequence 5′-CC-3′ (Blue). A frequency chart is plotted to visualize the PAM sequence by inputting all the available PAM-Protospacer sequences using WebLogo.
-
2.
PAM sequence characterization
Identification of protospacer adjacent motif (PAM) sequence is essential for assembling a guide mini-CRISPR array and repurposing the identified CRISPR-Cas systems for genome editing. To this end, all 26 spacers present in the three CRISPR arrays are manually extracted and aligned against the bacteriophage and plasmid databases using CRISPRTarget to acquire protospacer sequences (Biswas et al., 2013). Since PAM is located at the 5′-end of the protospacer in type I CRISPR-Cas systems, sequences 8-bp upstream of the identified protospacers are extracted and aligned to predict the PAM sequence (Figure 1). The PAM sequence of type I-F CRISPR-Cas system was identified as 5′-CC-3′ (Richter et al., 2014). A frequency chart is plotted to visualize the PAM sequence using WebLogo (Crooks et al., 2004). Authenticity of the PAM sequence is further validated by “Self-targeting activity assay” described in the Step-by-Step Method Details section.
-
3.
Design mini-CRISPR and primers to assemble pTargeting and pEditing
-
a)
mini-CRISPR and pTargeting: A mini-CRISPR guide is required to construct the targeting and editing plasmids for self-targeting activity assay and genome editing, respectively. A mini-CRISPR encompasses a 32-bp spacer sequence derived from a selected target gene in the chromosome of PA154197 immediately downstream of a selected 5′-CC-3′ PAM and two 28-bp repeat sequences flanking at both ends (Figure 2). In our study, the 88-bp mini-CRISPR array (28+32+28) is synthesized commercially and is provided in a plasmid termed as pUC57-mini-CRISPR in which the synthesized mini-CRISPR is cloned between the KpnI and BamHI sites of the pUC57 vector (BGI, China) (Figure 2). pTargeting is assembled by cloning the mini-CRISPR between the KpnI and BamHI sites in the platform plasmid pAY5211.
Figure 2.

Schematic Diagram of Constructing pTargeting and pEditing (Use Gene Deletion as an Example)
Step 1: An 88-bp mini-CRISPR which encompasses a 32-bp CC-preceding sequence (Dashed Box) within the target gene flanked by two 28-bp repeat sequences (Orange diamond) is submitted for commercial synthesis. Mini-CRISPR flanked by restriction sites KpnI and BamHI is supplied in the plasmid pUC57-mini-CRISPR.
Step 2: The mini-CRISPR is amplified by PCR using primers pUC57-F/R.
Step 3: After digestion with KpnI and BamHI, the purified mini-CRISPR is inserted into the plasmid pAY5211 which is treated with the same restriction enzymes, generating the targeting plasmid pTargeting. pTargeting is verified by sequencing using primers pMS-402-F/R.
Step 4: pTargeting is linearized by enzyme digestion with XhoI.
Step 5: 500-bp Up donor (Pink), containing a 15-bp 5′-end homology (Green) with the 3′ end of the linearized pTargeting and a 15-bp 3′-end homology (Magenta) with the Down donor, is amplified by PCR using primers Donor-UF/UR. 500-bp Down donor (Magenta), containing a 15-bp 5′-end homology with the 3′-end of the Up donor and a 15-bp 3′-end homology (Dark green) with the 5′ end of the linearized pTargeting, is amplified by PCR using primers Donor-DF/DR.
Step 6: Up Donor and Down donor are assembled into the linearized pTtargeting by Gibson assembly, generating the editing plasmid pEditing. pEditing is verified by sequencing using primers pMS-402-F/R. Detailed donor design for various types of genetic editing are presented in Figure 3.
-
b)
Editing Donor: In CRISPR-mediated genetic editing, various types of editing are achieved by the provision of desired donor sequences (Figure 3). For gene deletion, an editing donor typically consists of 500-bp upstream (Up donor) and 500-bp downstream (Down donor) of the desired deletion region. For gene insertion, an editing donor typically consists of 500-bp upstream (Up donor) and 500-bp downstream (Down donor) of the insertion site and the intended insertion fragment (Mid donor). For single nucleotide substitution, an editing donor is consisted of ∼1-kb DNA sequence spanning the editing site with desirable single nucleotide substitution located in the center. Depending on the types of editing, the corresponding primers are designed and synthesized commercially.
Figure 3.

Schematic Diagram of the Donor Sequence Assembly for Various Types of Genome Editing
(A) Donor for gene deletion consists of a 500-bp upstream sequence (Up donor, Pink) and a 500-bp downstream sequence (Down donor, Magenta) of the gene to be deleted. Up donor is amplified by PCR using primers Donor-UF/UR and contains a 15-bp 5′-end homology (Green) with the 3′ end of the linearized pTargeting (Figure 2) and a 15-bp 3′-end homology (Magenta) with the Down donor. Down donor is amplified by PCR using primers Donor-DF/DR and contains a 15-bp 3′-end homology (Dark green) with the 5′ end of the linearized pTargeting. Up Donor and Down donor are assembled into the linearized pTargeting by Gibson assembly (Figure 2).
(B) Donor for gene insertion consists of a 500-bp upstream sequence (Up donor, blue) and a 500-bp downstream sequence (Down donor, Magenta) of the insertion site and an n-bp inserted fragment (Mid donor, Orange) in the middle. Up donor is amplified by PCR using primers Donor-UF/UR and contains a 15-bp 5′-end homology (Green) with the 3′ end of the linearized pTargeting and a 15-bp 3′-end homology (Orange) with the 5′ end of Mid donor. Mid donor is amplified by PCR using primers Donor-MF/MR and contains a 15-bp 3′-end homology (Magenta) with the Down donor. Down donor is amplified by PCR using primers Donor-DF/DR and contains a 15-bp 3′-end homology (Dark green) with the 5′ end of the linearized pTargeting. Up Donor and Down donor are assembled into the linearized pTargeting by Gibson assembly.
(C) Donor for single nucleotide substitution consists of a 500-bp upstream sequence (Up donor, Blue) and a 500-bp downstream sequence (Down donor, Magenta) which span the editing site and contain the desirable single nucleotide substitution. Up donor is amplified by PCR using primers Donor-UF and Donor-UR with a point mutation (Red), and contains a desired point mutation (Red) and a 15-bp 5′-end homology (Green) with the 3′ end of the linearized pTargeting. Down donor is amplified by PCR using primers Donor-DF/DR and contains a 15-bp 5′-end homology (Blue) with the Up donor and a 15-bp 3′-end homology (Dark green) with the 5′ end of the linearized pTargeting. Up donor and Down donor are assembled into the linearized pTargeting by Gibson assembly.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| PA154197 | This study | N/A |
| PA150567 | This study | N/A |
| Ocean-100 | Kumagai et al., 2017 | N/A |
| DH5α | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kanamycin disulfate | Sigma-Aldrich | Cat#K1876 |
| KpnI | New England Biolabs | Cat#R3142 |
| BamHI | New England Biolabs | Cat#R3136 |
| XhoI | New England Biolabs | Cat#R0146 |
| Critical Commercial Assays | ||
| Quick Ligation™ Kit | New England Biolabs | Cat#M2200 |
| ClonExpress II One Step Cloning Kit | Vazyme | C112 |
| iProof™ High-Fidelity DNA Polymerase | Bio-Rad | Cat#1725301 |
| Taq DNA polymerase | Thermo Scientific | Cat#EP0406 |
| illustra GFX PCR DNA and Gel Band Purification Kit | GE Healthcare | 28-9034-70 |
| MiniBEST Agarose Gel DNA Extraction Kit | Takara | Cat#9761 |
| QIAprepTM Spin Miniprep Kit | Qiagen | Cat#27104 |
| Oligonucleotides | ||
| pUC57-F: 5′-AGGGTTTTCCCAGTCACGAC-3′ | This study | N/A |
| pUC57-R: 5′-AGCGGATAACAATTTCACAC-3′ | This study | N/A |
| pMS402-F: 5′-CCAGCTGGCAATTCCGA-3′ | This study | N/A |
| pMS402- R: 5′-AATCATCACTTTCGGGAAAG-3′ | This study | N/A |
| Mini-CRISPR: 5′-GTTCACTGCCGTATAG GCAGCTAAGAAANNNNNNNNNNNN NNNNNNNNNNNNNNNNNNNNGTTC ACTGCCGTATAGGCAGCTAAGAAA-3′ |
This study | N/A |
| Donor-UF: 5′-CCCTTTCGTCTTCACCT CGAGNNNNNNNNNNNNN NNNNNNN-3′ |
This study | N/A |
| Donor-DR: 5′-GATCAGGAACACCCCC TCGAGNNNNNNNNNNNNNNNNN NNN-3′ |
This study | N/A |
| Recombinant DNA | ||
| pAY5211 | This study | N/A |
RESOURCE AVAILABILITY
Requests for resources should be directed to and will be fulfilled by the Lead Contact, Aixin Yan (ayan8@hku.hk).
MATERIALS AND EQUIPMENT
-
•
Centrifuge (4°C)
-
•
Incubator (25°C and 37°C)
-
•
Shaking incubator (37°C)
-
•
Heat block (42°C)
-
•
NanoVue Plus Spectrophotometer (GE Healthcare, USA)
-
•
Molecular Imager® Gel Doc™ XR System (Bio-Rad, USA)
-
•
Synergy HTX Plate Reader (Bio Tek, USA)
-
•
ECM399 Electroporation System (BTX, USA)
-
•
S1000 Thermo cycler (Bio-Rad, USA)
-
•
2-mm Electroporation cuvette (ThermoFisher Scientific, USA)
-
•
96-well white plate (SPL, Korea)
CRITICAL: Use autoclaved Milli-Q H2O at all steps whenever Milli-Q H2O is needed.
Note: Any alternative commercial kits for PCR, enzymatic digestion, ligation etc. may be used as well, as long as the DNA products are obtained with sufficient quantity and quality as described in the steps below.
STEP-BY-STEP METHOD DETAILS
This protocol provides detailed procedures for one-step genome editing (Gene deletion, Gene insertion and Single nucleotide substitution) in the native type I-F CRISPR-Cas-containing MDR P. aeruginosa isolate PA154197 (Figure 2). A platform plasmid (pAY5211, Figure 4A) for constructing the customized targeting plasmid and editing plasmid is available upon request. pAY5211 contains a mini-CRISPR insertion site which is flanked by the KpnI and BamHI sites, a donor insertion site at the XhoI site, and a kanamycin-luminescence dual selection marker. Sequence information of pAY5211 is presented in Figure 4A.
Figure 4.

Sequence Information of the Platform Plasmid pAY5211 and the pEditing pAY5235 Constructed for mexB Deletion
(A) pAY5211 contains a kanamycin-resistant gene and a luciferase-encoding operon (luxCDABE) for the dual selection of transformants. Mini-CRISPR is inserted between KpnI and BamHI which is co-transcribed with the luxCDABE operon driven by the Ptat promoter. Donor fragments are assembled at the XhoI site. Primers pMS402-F/R are used to verify the constructs of pTargeting and pEditing.
(B) Sequence information of the mini-CRISPR and donor in pAY5235 employed to construct mexB deletion is shown.
Construction of the Targeting Plasmid (pTargeting)
-
1.
Select a region of ∼200 bp (236 bp in our case) in the pUC57-mini-CRISPR covering the mini-CRISPR and the flanking KpnI and BamHI sites. Amplify this region by Polymerase Chain Reaction (PCR) using iProof™ High-Fidelity DNA Polymerase (Bio-Rad, USA). Reaction mixture (a) and PCR cycles (b) are set up as below:
-
a)
iProof PCR Reaction setup:
| Component | Volume (μL) |
|---|---|
| 5×iProof GC Buffer | 10 |
| 100% DMSO solution | 1.5 |
| 10 mM dNTP mix | 1 |
| Primer 1 (pUC57-F, 10 μM) | 5 |
| Primer 2 (pUC57-R, 10 μM) | 5 |
| DNA template (pUC57-mini-CRISPR) | 1 |
| iProof DNA Polymerase | 0.5 |
| Milli-Q H2O | 26 |
| Total Volume | 50 |
-
b)
PCR cycles:
| Cycle Step | Temperature (°C) | Time | Number of Cycle |
|---|---|---|---|
| Initial Denaturation | 98 | 5 min | 1 |
| Denaturation | 98 | 30 s | ×35 |
| Annealing | 62 | 30 s | |
| Extension | 72 | 10 s | |
| Final Extension | 72 | 10 min | 1 |
-
2.
Purify the 236-bp PCR product using the MiniBEST Agarose Gel DNA Extraction Kit (TaKaRa, Japan) following the manufacture’s instruction.
Alternatives: If the KpnI-mini-CRISPR-BamHI fragment is synthesized and supplied in the linear double-stranded DNA form, it can be directly proceeded to Step 3.
-
3.
Purified PCR product from Step 2 and the plasmid pAY5211 are digested with KpnI and BamHI (NEB, USA). Digestion reaction is set up as below:
| Component | Volume (μL) |
|---|---|
| 10×CutSmart Buffer | 5 |
| pAY5211/ PCR product from Step 2 | X μL (12.5 μg/5 μg) |
| KpnI | 2 |
| BamHI | 2 |
| Milli-Q H2O | to final 50 |
| Total Volume | 50 |
-
4.
Incubate reactions at 37°C for 4 h.
-
5.
Purify the digested pAY5211 and 88-bp mini-CRISPR (with KpnI and BamHI digested ends) using the illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, USA) and the MiniBEST Agarose Gel DNA Extraction Kit, respectively.
Note: Use MiniBEST Agarose Gel DNA Extraction Kit or equivalent kits to purify the small-sized mini-CRISPR. The mini-CRISPR can be directly purified from the KpnI and BamHI digestion reaction. Thus, no gel purification is required. Use 20 μL Milli-Q H2O to elute the DNA fragments to ensure sufficient quantity for ligation in the next step.
-
6.
Ligate the enzyme-digested pAY5211 (∼0.5 μg) and mini-CRISPR (∼0.1 μg) using the Quick Ligation Kit (NEB, USA) to generate pTargeting. Reaction mixture without addition of digested mini-CRISPR serves as the negative control. Ligation reaction is set up as below:
| Component | Volume (μL) |
|---|---|
| 2×Quick Ligation Reaction Buffer | 10 |
| Digested pAY5211 | X μL (0.5 μg) |
| Digested mini-CRISPR | X μL (0.1 μg) |
| Quick T4 DNA Ligase | 1 |
| Milli-Q H2O | to final 20 |
| Total Volume | 20 |
-
7.
Incubate the reaction at 25°C for 45 min.
-
8.
Mix the ligation mixture with 100 μL cold CaCl2 (0.1 mM) and 200 μL DH5α component cells gently and thoroughly. Chill on ice for 10 min.
-
9.
Heat shock the mixture at 42°C for 90 s and quickly chill it on ice for 5 min.
-
10.
Add 1 mL fresh LB broth into the mixture and recover the cells at 37°C with 220-rpm agitation for 1 h.
-
11.
Harvest the cells by centrifugation with 16,000 ×g for 1 min and resuspend them in 100 μL LB broth. Spread 100 μL cell suspension onto a LB agar plate containing 20 μg/mL kanamycin and incubate the plate at 37°C overnight (15–16 h) to obtain colonies. No colony should be recovered in the negative control (without digested mini-CRISPR).
Note: Overnight incubation in this protocol is approximately 15–16 h.
-
12.
Perform colony PCR to verify the construction of pTargeting (Figure 2, Step 3). Colony PCR reaction using the platform plasmid pAY5211 as the template serves as the negative control. PCR reaction (a) and cycles (b) are set up as below:
-
a)
Colony PCR Reaction setup:
| Component | Volume (μL) |
|---|---|
| 10×Taq buffer | 1 |
| 10 mM dNTP mix | 0.2 |
| Primer 1 (pMS402-F, 10 μM) | 1 |
| Primer 2 (pMS402-R, 10 μM) | 1 |
| DNA template (Colony suspension in 10 μL MilliQ H2O) | 0.2 |
| Taq DNA Polymerase | 1 |
| Milli-Q H2O | 5.6 |
| Total Volume | 10 |
-
b)
Colony PCR cycle:
| Cycle Step | Temperature (°C) | Time | Number of Cycle |
|---|---|---|---|
| Initial Denaturation | 95 | 5 min | 1 |
| Denaturation | 95 | 30 s | ×30 |
| Annealing | 62 | 30 s | |
| Extension | 72 | 1 min | |
| Final Extension | 72 | 10 min | 1 |
-
13.
Run an 1% agarose gel to examine the PCR product.
-
14.
Visualize bands with UV transillumination after the gel is stained by GelRed. PCR product of the expected pTargeting is 468 bp due to the replacement of 2-bp sequence flanked by KpnI and BamHI in pAY5211 by the 88-bp mini-CRISPR in pTargeting. PCR product of the negative control pAY5211 is 382 bp.
-
15.
Inoculate a validated colony into 5 mL LB broth supplemented with 20 μg/mL kanamycin and propagate the plasmid by growing the inoculum at 37°C with 220-rpm agitation overnight. Extract pTargeting from the overnight culture using the QIAprepTM Spin Miniprep Kit (Qiagen, Germany).
Pause Point: Plasmids can be stored at -20°C for ∼3 months
Self-Targeting Activity Assay
Note: This step is conducted to examine the self-targeting DNA interference activity of the native CRISPR-Cas system and to further validate the PAM sequence. This step only needs to be conducted once prior to the first genome-editing exploitation in the strain of interest.
pTargeting and the control plasmid pAY5211 are delivered into PA154197 cells, respectively, by electroporation following the steps from 24 to 31 (below). Since pTargeting contains a mini-CRISPR guide which targets a genomic locus in PA154197 and is expressed by a strong promoter Ptat, if the CRISPR-Cas system in PA154197 is active, the crRNA expressed from the mini-CRISPR will form a Cascade with the Cas complex and elicit site-specific DNA breakage in the genome, resulting in cell death. Hence, no colony should be recovered following pTargeting transformation relative to the high colony recovery of the pAY5211 transformation. As shown in Figure 5, expected colony recovery pattern was observed. The result showed that the native type I-F CRISPR-Cas system in PA154197 is active to interfere genomic DNA and confirmed the authenticity of the predicted PAM sequence. Moreover, the fact that no colony was recovered upon introduction of the pTargeting indicated that as in most bacteria, non-homologous end joining (NHEJ) does not occur in this strain. Once the self-targeting activity of the native CRISPR-Cas system is confirmed and the PAM sequence is validated, direct genome editing in this strain can be performed followingly (Steps 16 to 36).
Figure 5.

Representative Results of Genome Editing following Transformation of the Single pEditing Plasmid and Verification of the Desired Construct (mexB Deletion as an Example)
(A) Plates showing the colony recovery pattern following the transformation of the control plasmid pAY5211, the pTargeting, and the pEditing. Provision of the repair donor in pEditing resumed colony recovery, indicating the success of DNA repair through the donor-mediated homologous recombination (HR) and consequently the desired mexB deletion.
(B) A representative gel image of colony PCR to verify ΔmexB in PA154197. PCR reaction using the wild-type (WT) strain as template yielded a band of 4,228 bp whereas that using recovered colonies from pEditing transformation as template yielded a band of 1,087 bp, indicating the deletion of 3,141-bp mexB gene.
Construction of the Editing Plasmid (pEditing)
-
16.
Amplify the donor DNA fragments using the genomic DNA of PA154197 as template and primer pairs (Donor-UF/UR and Donor-DF/DR) as indicated in Figure 2. Amplify and purify the donor DNA fragments following the procedures as described in Steps 1 and 2.
Note: Adjust annealing temperature for different primer pairs.
-
17.
Linearize pTargeting (∼12.5 μg) through XhoI digestion. XhoI digestion reaction is set up as below:
| Component | Volume (μL) |
|---|---|
| 10×CutSmart Buffer | 5 |
| pTargeting | X μL (12.5 μg) |
| XhoI | 2 |
| Milli-Q H2O | to final 50 |
| Total Volume | 50 |
-
18.
Incubate XhoI digestion reaction at 37°C for 4 h.
-
19.
Purify the linearized pTargeting using the illustra GFX PCR DNA and Gel Band Purification Kit.
-
20.
Use One Step Cloning Kit (Vazyme, China) to assemble the donor fragments into pTargeting (Figure 2). Assembly reaction without addition of donor fragments serves as the negative control. Assembly reaction is set up as below:
| Component | Volume (μL) |
|---|---|
| 5×CE MultiS Buffer | 4 |
| Linearized pTargeting | X μL (0.2 μg) |
| Donor fragments | X μL (0.02 μg for each fragment) |
| Exnase MultiS | 2 |
| Milli-Q H2O | to final 20 |
| Total Volume | 20 |
-
21.
Incubate reactions at 37°C for 30 min.
-
22.
Transform the reaction mixture into E. coli DH5α component cells as described in Steps 8 to 11. Following overnight incubation at 37°C on LB agar plates containing 20 μg/mL kanamycin, perform colony PCR using the primer pairs pMS402-F/R to verify the construct (pEditing) as described from Step 12 to 15. No colony should be recovered in the negative control (without addition of donor fragments).
Note: Expected size of the PCR product should be 1,468 bp if the upstream and downstream donors are 500 bp, respectively. The negative control colony PCR reaction using pTargeting as template will yield a product size of 468 bp.
-
23.
Sequence the plasmid (pEditing) using primer pairs (pMS402-F/R) to confirm the desired incorporation of mini-CRISPR and repair donor. An example of sequence information of the pEditing (pAY5235) to construct mexB-deletion was shown in Figure 4B.
Fast Preparation of Electro-component Cells and Electroporation of pEditing
-
24.
Overnight PA154197 cell culture is diluted 1:100 into 50 mL fresh LB broth and grow at 37°C with 220-rpm agitation for 4–5 h till OD600 is ∼0.6–0.8.
-
25.
Cell culture is chilled on ice for at least 30 min. Cells are collected by centrifugation at 4°C with 5,000 ×g for 15 min and washed three times with 25 mL cold Milli-Q H2O.
-
26.
Resuspend the resulting cell pellet (electrocompetent PA154197 cells) into 1 mL Milli-Q H2O.
-
27.
Mix 100 μL electrocompetent PA154197 cells with 1 μg pEditing, or pAY5211 (control plasmid) or pTargeting (negative control). Transfer the mixtures into electroporation cuvettes and apply electrical pulse at 2.3 kV, 6 ms (BTX, USA).
-
28.
Add 1 mL cold LB broth into each cuvette immediately after electroporation.
-
29.
Transfer the cells into 1.5 mL Eppendorf tubes and incubate at 37°C with 220-rpm agitation for 90 min.
Figure 6.

Prolonged Growth during Recovery of PA154197 Cells Electroporated with pEditing Resulted in Undesirable Cell Lysis
Suspension of cells electroporated with the control plasmid pAY5211 or pEditing following recovery for 120 min. Cell lysis and subsequent precipitation (red arrow) was observed in the cell suspension electroporated with pEditing but not with the control plasmid pAY5211.
-
30.
Harvest the cells by centrifugation and resuspend them in 100 μL LB broth. Spread the suspension onto LB agar plates containing 500 μg/mL kanamycin.
Note: Concentration of kanamycin for selection is dependent on the kanamycin susceptibility of the P. aeruginosa isolates subjected to editing. For example, 800 μg/mL is used for selection of another clinical isolate PA150567 and 500 μg/mL for an environmental isolate Ocean-100 (Kumagai et al., 2017).
-
31.
Incubate the plates at 37°C for 24 h.
Mutants Screening and Verification
-
32.
Inoculate the colonies recovered from Step 31 in 100 μL LB broth supplemented with 100 μg/mL kanamycin in a 96-well white plate (SPL, Korea).
-
33.
Incubate the 96-well white plate at 37°C with 220-rpm agitation for 3 h.
-
34.
Measure the luminescence intensity using the Synergy HTX Plate Reader (Bio Tek, USA).
-
35.
Select cell cultures with high luminescence value (>1000) for further colony PCR verification using primers located upstream and downstream of the donor regions (Veri-F/R, Figure 2).
Note: Luminescence reading of cultures containing pEditing is usually more than 1,000 after incubation, while the false positive colonies have undetectable luminescence intensity. For colony PCR verification, include a negative control reaction using the genomic DNA of PA154197 as template.
-
36.
Verify the desired genome editing by DNA sequencing of the PCR product.
Curing of pEditing for the Next Round of Editing
-
37.
Streak the edited colony from Step 35 on a LB agar plate (without kanamycin supplement).
-
38.
Incubate the plate at 37°C overnight. Plasmid curing is confirmed by inoculating the colony into LB broth supplemented with 100 μg/mL kanamycin. No growth after overnight culture indicates the loss of the editing plasmid.
-
39.
Single colony with desired mutation can be used for next round of editing or other physiological studies.
Note: In some cases, multiple (2 to 3) rounds of curing are required for complete loss of the plasmid.
EXPECTED OUTCOMES
Representative plates resulting from the electroporation of pAY5211 (control plasmid), pTargeting (negative control), or pEditing are shown in Figure 5A. Compared with the transformation of pTargeting which resulted in no colony recovery due to self-targeting, provision of a repair donor in the pEditing resumed transformants recovery, suggesting the occurrence of DNA repair at the self-targeted site through homologous recombination (HR) and consequently the desired genome editing. An example of PCR verification of the desired gene deletion is shown in Figure 5B. The fact that the PCR products of these colonies displayed either the size of the WT or the desired full-length gene deletion suggested that microhomology-mediated end joining (MMEJ) did not occur or was not frequent.
LIMITATIONS
This native type I-F CRISPR-Cas-mediated genome-editing protocol is limited to P. aeruginosa isolates that express active type I-F CRISPR-Cas system. Currently, strains that do not contain such a native CRISPR-Cas system or those containing a degenerated CRISPR-Cas system are not editable by this method.
Similar to the limitations encountered in other CRISPR-based genetic applications, constructing gene insertion and single nucleotide substitution is dependent on the presence of a PAM sequence within a strict range of the desired editing site. For gene insertion, presence of a PAM sequence within the 32-bp upstream of the desired insertion site is required. For single nucleotide substitution, presence of a PAM sequence at the desired editing site or within 2-bp upstream of the site is required. The editing efficiency is strictly dependent on the distance between the PAM and the desired editing site in these applications (Figure 7).
Figure 7.

Dependence on the Presence of a PAM at Proximity of the Desired Editing Site in the Native Type I-F CRISPR-Cas-Mediated Single Nucleotide Substitution (A Fragment in the mexB Gene as an Example)
Success of single nucleotide substitution (N to A) was examined in the PAM and PAM proximal region in a selected region in mexB. Editing plasmids containing a repair donor (U and D) for site specific point mutation were introduced into PA154197. For each of these mutagenesis, five randomly selected transformants with high luminescence intensity were subjected to colony PCR and DNA sequencing using primers Seq-F/Seq-R. Sequencing results demonstrated that mutations only in the PAM and PAM proximal 2 nucleotides can be efficiently achieved.
TROUBLESHOOTING
Problem
PAM prediction fails using CRISPRTarget
Potential Solution
If the online alignment using CRISPRTarget fails to predict PAM sequences, an alternative experimental PAM screening strategy, PAM depletion assay, can be considered. PAM depletion assay employs a plasmid library of all possible 3–6-nt PAM sequences to screen for the effective PAM. Detailed description for PAM depletion assay was reported by Walker et al. (Walker et al., 2020).
Problem
Native type I-F CRISPR-Cas system is inactivated by anti-CRISPR elements in the strains of interest.
Potential Solution
To exploit the native CRISPR-Cas-mediated genome editing in these strains, one potential solution is to delete the anti-CRISPR element first using other genetic approaches. Alternatively, overexpression of the common anti-CRISPR repressor Aca can be considered to inhibit the expression of anti-CRISPR (Stanley et al., 2019).
Problem
Non-effective targeting
Potential Solution
-
1.
For deleting a gene which contains multiple available targets (PAM-Protospacer) to select, try an alternative target and assemble a corresponding new mini-CRISPR to achieve targeting. Alternatively, we recommend designing a mini-CRISPR cassette simultaneously incorporating 2–3 spacers (e.g. Repeat+Spacer 1 + Repeat + Spacer 2 + Repeat) to avoid potentially non-effective targeting by one spacer.
-
2.
For other editing which requires the presence of a PAM sequence within a strict range of the desired editing site, we recommend a two-step In-Del method as described in our Cell Reports Resource article (Xu et al., 2019).
Problem
Low editing efficiency to delete genes with large sizes (i.e. >3 kb)
Potential Solution
Increase the donor size can help to increase the editing efficiency.
Problem
Low yield of electroporation
Potential Solution
pAY5211 contains an origin of transfer (oriT). Try to use conjugation to deliver the editing plasmid into the isolate of interest. Detailed procedures for conjugation were reported by Withers et al. (Withers et al., 2014).
Problem
Genome editing in P. aeruginosa isolates with other types of CRISPR-Cas systems or in other prokaryotic/eukaryotic cells lacking a native CRISPR-Cas system.
Potential Solution
For genome editing based on the native type I-F CRISPR-Cas system in other species, consider using species-compatible plasmid and promoters to express the mini-CRISPR. For species containing native CRISPR-Cas system belonging to other subtypes, the similar workflow described here is still applicable except that plasmid vector, PAM sequence, repeat sequence implemented should be compatible with the corresponding CRISPR-Cas subtypes and the host strains. For example, type I-B and I-E systems have been harnessed for genome editing in Clostridium and Lactobacillus, respectively, by applying plasmids pCParray-delcpaAIR and pTRK1183 and PAM sequences of 5′-AATTG-3′ and 5′-AAA-3′, respectively (Pyne et al., 2016, Hidalgo-Cantabrana et al., 2019).
ACKNOWLEDGMENTS
We acknowledge Dr. Ming Li and Prof. Hua Xiang (Institute of Microbiology, Chinese Academy of Sciences) for their advice and stimulating discussion of the protocol with us. This work was supported by the Hong Kong University Grants Council General Research Fund (HKU17142316 and HKU19180422 to A.Y.) and Seed Funding for Strategic Interdisciplinary Research Scheme (HKU 2017 to A.Y.).
AUTHOR CONTRIBUTIONS
Writing, Z.X., Y.L., and A.Y.; Funding Acquisition, A.Y.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Contributor Information
Zeling Xu, Email: zelingxu@connect.hku.hk.
Aixin Yan, Email: ayan8@hku.hk.
References
- Biswas A., Gagnon J.N., Brouns S.J., Fineran P.C., Brown C.M. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H., Xia T., Li Y., Xu Z., Bougouffa S., Lo Y.K., Bajic V.B., Luo H., Woo P.C., Yan A. Uncoupled quorum sensing modulates the interplay of virulence and resistance in a multidrug-resistant clinical Pseudomonas aeruginosa isolate belonging to the MLST550 clonal complex. Antimicrob. Agents Chemother. 2019;63:e01944-18. doi: 10.1128/AAC.01944-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., Rocha E.P., Vergnaud G., Gautheret D., Pourcel C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–W251. doi: 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Cantabrana C., Goh Y.J., Pan M., Sanozky-Dawes R., Barrangou R. Genome editing using the endogenous type I CRISPR-Cas system in Lactobacillus crispatus. Proc. Natl. Acad. Sci. U S A. 2019;116:15774–15783. doi: 10.1073/pnas.1905421116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y., Yoshizawa S., Nakamura K., Ogura Y., Hayashi T., Kogure K. Complete and draft genome sequences of eight oceanic Pseudomonas aeruginosa strains. Genome Announc. 2017;5:e01255-17. doi: 10.1128/genomeA.01255-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nethery M.A., Barrangou R. Predicting and visualizing features of CRISPR-Cas systems. Methods Enzymol. 2019;616:1–25. doi: 10.1016/bs.mie.2018.10.016. [DOI] [PubMed] [Google Scholar]
- Pyne M.E., Bruder M.R., Moo-Young M., Chung D.A., Chou C.P. Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium. Sci. Rep. 2016;6:1–15. doi: 10.1038/srep25666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C., Dy R.L., McKenzie R.E., Watson B.N., Taylor C., Chang J.T., McNeil M.B., Staals R.H., Fineran P.C. Priming in the Type IF CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014;42:8516–8526. doi: 10.1093/nar/gku527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S.Y., Borges A.L., Chen K.-H., Swaney D.L., Krogan N.J., Bondy-Denomy J., Davidson A.R. Anti-CRISPR-associated proteins are crucial repressors of anti-CRISPR transcription. Cell. 2019;178:1452–1464.e13. doi: 10.1016/j.cell.2019.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A., Soriaga L.B., LaFave M.C., Akella S., Veyrieras J.-B., Barbu E.M., Shortridge D., Blanc B., Hannum G., Zambardi G. Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio. 2015;6:e01796-15. doi: 10.1128/mBio.01796-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Lanahan A.A., Zheng T., Toruno C., Lynd L.R., Cameron J.C., Olson D.G., Eckert C.A. Development of both type I-B and type II CRISPR/Cas genome editing systems in the cellulolytic bacterium Clostridium thermocellum. Metab. Eng. Commun. 2020;10:e00116. doi: 10.1016/j.mec.2019.e00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers T.R., Yin Y., Hongwei D.Y. Identification of novel genes associated with alginate production in Pseudomonas aeruginosa using mini-himar1 mariner transposon-mediated mutagenesis. J. Vis. Exp. 2014:e51346. doi: 10.3791/51346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Li M., Li Y., Cao H., Miao L., Xu Z., Higuchi Y., Yamasaki S., Nishino K., Woo P.C. Native CRISPR-Cas-mediated genome editing enables dissecting and sensitizing clinical multidrug-resistant P. aeruginosa. Cell Rep. 2019;29:1707–1717.e3. doi: 10.1016/j.celrep.2019.10.006. [DOI] [PubMed] [Google Scholar]