

Summary
Here, we describe a xeno-free, feeder-free, and chemically defined protocol for the generation of ventral midbrain dopaminergic (vmDA) progenitors from human pluripotent stem cells (hPSCs). This simple-to-follow protocol results in high yields of cryopreservable dopamine neurons across multiple hPSC lines. Wnt signaling is the critical component of the differentiation and can be finely adjusted in a line-dependent manner to enhance production of dopamine neurons for the purposes of transplantation, studying development and homeostasis, disease modeling, drug discovery, and drug development.
For complete details on the use and execution of this protocol, please refer to Gantner et al. (2020) and Niclis et al. (2017a).
Graphical Abstract
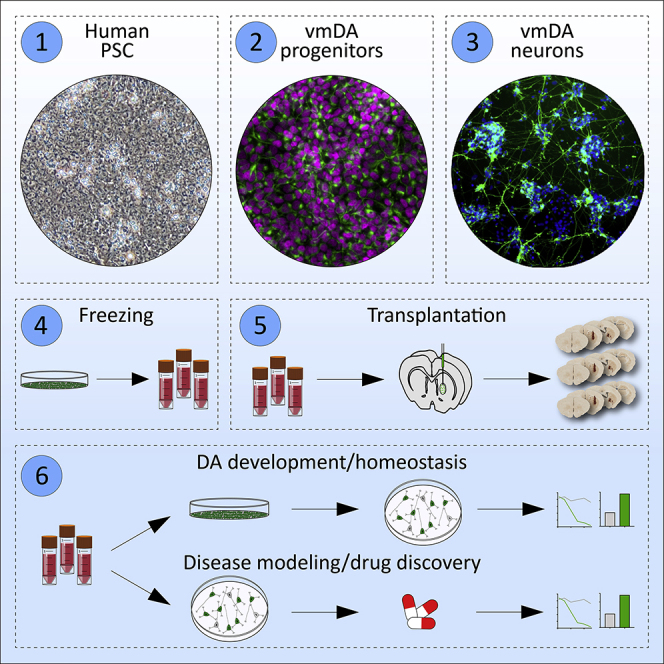
Highlights
-
•
Reproducible differentiation of human dopamine neurons from multiple hPSC lines
-
•
Dopamine progenitors can be cryopreserved for downstream applications
-
•
Dopamine neurons mature in vitro, enabling screening or developmental studies
-
•
Transplanted dopamine progenitors are capable of restoring motor function
Here, we describe a xeno-free, feeder-free, and chemically defined protocol for the generation of ventral midbrain dopaminergic (vmDA) progenitors from human pluripotent stem cells (hPSCs). This simple-to-follow protocol results in high yields of cryopreservable dopamine neurons across multiple hPSC lines. Wnt signaling is the critical component of the differentiation and can be finely adjusted in a line-dependent manner to enhance production of dopamine neurons for the purposes of transplantation, studying development and homeostasis, disease modeling, drug discovery, and drug development.
Before You Begin
Note: All steps should be carried out under sterile conditions and according to local regulatory requirements (e.g. Australian Std 2243 requiring all human tissues to be confined to Class II biosafety cabinets).
Note: Cells are cultured throughout at 37°C in 5% CO2.
Pluripotent Stem Cell Thawing
Timing: 2–3 h
-
1.
Coat a T25 flask with 5–10 μg/mL Laminin-521 and leave in the incubator for ≥2 h (see Tables 1 and 2 for laminin-521 dilution).
Note: 5 μg/mL of Laminin-521 is sufficient for cell lines that have been adapted to Laminin-521. For non-adapted lines (i.e. hPSC lines maintained on substrates other than laminin-521) 10 μg/mL may be required to ensure attachment during initial passages. Laminin-521 can also be added for 12–18 h at 4°C, although ≥2 h at 37°C is preferable.
CRITICAL: Laminin-521 must be diluted in PBS+/+. Diluting in PBS−/− will result in poor attachment and survival. Ensure that enough laminin solution is made to coat the entire flask throughout the coating period. Do not use flasks which have dried as cells will not attach in these areas.
-
2.
Equilibrate 15 mL pre-made mTeSR in a loosely closed 15 mL tube in the incubator for ≥15 min.
-
3.
Rapidly thaw a cryovial of hPSC in a 37°C water bath and transfer the contents to 10 mL prewarmed mTeSR solution using a P1000 pipette.
-
4.
Centrifuge cells at 260 × g (3 min at 21°C), aspirate supernatant, and resuspend the cell pellet in 5 mL mTeSR media, supplemented with 10 μM Y27632.
-
5.
Aspirate laminin from the T25 flask and immediately plate the cell suspension, gently rocking the flask multiple times up-and-down and then side-to-side to evenly distribute the cells.
Table 1.
Laminin-521 Dilution for 5 μg/mL
| Laminin-521 (μL) | PBS+/+ (μL) | Final Volume (μL) | |
|---|---|---|---|
| T25 | 150 | 2850 | 3000 |
| 48-well plate | 7.5 | 142.5 | 150 |
| 96-well plate | 3.5 | 66.5 | 70 |
Table 2.
Laminin-521 Dilution for 10 μg/mL
| Laminin-521 (μL) | PBS+/+ (μL) | Final Volume (μL) | |
|---|---|---|---|
| T25 | 300 | 2700 | 3000 |
| 48-well plate | 15 | 135 | 150 |
| 96-well plate | 7 | 63 | 70 |
Pluripotent Stem Cell Culture
-
6.
One day after thawing, wash once with PBS−/− to remove debris, and replace with fresh media.
-
7.
Continue to culture hPSC until they reach 70%–80% confluence, changing media every day.
Note: Generally, cells will recover quickly after thawing and should reach confluence within 5 days. However, if cells are growing slowly it is best to passage at a lower density (50%–60% confluence) or after 5–6 days.
-
8.
To passage hPSC, wash once in PBS−/− and add 2 mL of ReLeSR. Leave at 21°C and monitor cell morphology under the microscope until colonies are loosely attached (5–10 min).
-
9.
Remove ReLeSR carefully and rapidly tap the flask hard 3–4 times on each long edge to detach colonies.
-
10.
Resuspend by dissociating colonies in 2 mL equilibrated mTeSR using a P1000 pipette. Dilute cells (1:10–1:20) and transfer using a 5 mL serological pipette to a Laminin-521 coated T25 flask.
Note: Morphology during passage can vary between cell lines and the optimal timing of ReLeSR should be determined accordingly. The majority of cells should remain as 10–50 cell aggregates, removing the need for Y27632. Add Y27632 if passaged as single cells. Likewise, the growth rate of each line will differ, and passage ratios should be adjusted to compensate. We recommend passaging every 3–5 days.
Optional: Cryopreserve stem cells at low passage number by adding 10% DMSO, 30% KnockOut Serum Replacement and 60% mTeSR to isolated cell pellets (after ReLeSR dissociation) and slow-freeze at -80°C. 3–4 cryovials can be generated from a single, confluent T25 flask. Transfer to liquid nitrogen for long-term storage.
Pause Point: hPSC can be maintained by passaging every 3–5 days until the desired experimental start time. Thinking ahead and knowing the growth characteristics of the cell line are helpful in aligning cells at 70%–80% confluence with the desired start date.
Preparing Differentiation Media
Note: Always prepare media as close to use as possible and use within 2 weeks. It is best to make Induction Media the day of the experiment and Maturation Media at day 11. Where possible, add each factor (SHH, CHIR, etc.) to the base media on the day to minimize degradation.
Table 3.
Induction Media Composition
| Induction media (N2B27) | Volume (mL) |
|---|---|
| DMEM/F12 | 47.75 mL |
| Neurobasal media (NBM) | 47.75 mL |
| B27 -Vitamin A (1×) | 2 |
| N2 (1×) | 1 |
| Glutamax (1×) | 1 |
| Penicillin/Streptomycin (0.5×) | 0.5 |
| Total | 100 mL |
Table 4.
Maturation Media Composition
| Maturation Media (NBB27) | Volume (mL) |
|---|---|
| DMEM/F12 | 46.75 |
| Neurobasal media (NBM) | 46.75 |
| B27 +Vitamin A (1×) | 2 |
| N2 (1×) | 1 |
| Non-essential amino acids (1×) | 1 |
| ITS-A (1×) | 1 |
| Glutamax (1×) | 1 |
| Penicillin/Streptomycin (0.5×) | 0.5 |
| Total | 100 mL |
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BARHLl1 | Novus Biologicals | NBP1-86513, RRID:AB_11034569 |
| Mouse monoclonal anti-EN1 | DSHB | 4G11, RRID:AB_528219 |
| Goat polyclonal anti-FOXA2 | Santa Cruz Biotechnology | sc-6554, RRID:AB_2262810 |
| Chicken polyclonal anti-GFP | Abcam | ab13970, RRID:AB_300798 |
| Rabbit polyclonal anti-LMX1A | Millipore | AB10533, RRID:AB_10805970 |
| Rabbit polyclonal anti-NURR1/NUR77 | Santa Cruz Biotechnology | Sc-990, RRID:AB_2298676 |
| Goat polyclonal anti-OTX2 | R&D Systems | AF1979, RRID:AB_2157172 |
| Sheep polyclonal anti-PITX2 | R&D Systems | AF7388, RRID:AB_11128639 |
| Rabbit polyclonal anti-TH | Pel-Freez Biologicals | P40101-0, RRID:AB_461064 |
| Sheep polyclonal anti-TH | Pel-Freez Biologicals | P60101-0, RRID:AB_461070 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase® | Innovative Cell Technologies | AT104 |
| Ascorbic acid | Sigma-Aldrich | A4403 |
| B27™ Supplement or CTS™ B27™ Supplement | Gibco | 17504044 A1486701 |
| B27™ Supplement -vitA or CTS™ B27™ Supplement -vitA | Gibco | 12587010 A3353501 |
| Brain-derived neurotrophic factor | R&D Systems | 248-BDB |
| CHIR99021 | Miltenyi Biotech | 130-103-926 |
| Dibutyryl cAMP | Tocris Bioscience | 1141 |
| DMEM/F-12 or CTS™ DMEM/F-12 | Gibco | 11320033 A1370801 |
| Dimethyl sulfoxide | Sigma-Aldrich | D2650 |
| Glial cell line-derived neurotrophic factor | R&D Systems | 212-GD |
| GlutaMax™ Supplement | Gibco | 35050061 |
| Insulin-Transferrin-Selenium-Sodium Pyruvate (ITS-A) | Gibco | 51300044 |
| KnockOut™ serum replacement or CTS™ KnockOut™ serum replacement | Gibco | 10828010 12618012 |
| Laminin-521 | BioLamina | LN521 |
| Laminin - mouse | Sigma-Aldrich | L2020 |
| LDN193189 | Miltenyi Biotech | 130-103-925 |
| MEM Non-Essential Amino Acids | Gibco | 11140035 |
| mTeSR™1 or TeSR™2 | STEMCELL Technologies | 85880 05860 |
| N2 Supplement or CTS™ N2 Supplement | Gibco | 17502048 A1370701 |
| N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) | Sigma-Aldrich | D5942 |
| Neurobasal™ or CTS™ Neurobasal™ | Gibco | A3582901 A1371201 |
| PBS+/+ (with Calcium/Magnesium) | Gibco | 13492609 |
| PBS−/− | Gibco | 14190144 |
| Pierce™ 16% formaldehyde | Thermo Fisher | 28906 |
| Purmorphamine | Miltenyi Biotech | 130-104-465 |
| ReLeSR™ | STEMCELL Technologies | 05873 |
| SB431542 | R&D Systems | 1614 |
| SHH C25II | R&D Systems | 464-SH |
| TGFβ3 | Peprotech | 100-36E |
| Trypan blue | Sigma-Aldrich | T8154 |
| Y27632 (ROCK inhibitor) or Y27632-GMP | Tocris Bioscience | 1254 TB1254-GMP |
| Critical Commercial Assays | ||
| MycoAlert™ Mycoplasma Detection Kit | Lonza | LT07-218 |
| Experimental Models: Cell Lines | ||
| H9 PITX3-eGFP hESC | Colin Pouton Laboratory. (Watmuff et al., 2015) | N/A |
| RM3.5 PITX3-eGFP hiPSC | In house | N/A |
| Other | ||
| 48-well plate – Nunclon Delta surface | Thermo Fisher | 150687 |
| Bright-Line™ hemocytometer | Sigma-Aldrich | Z359629 |
| Cryovials – 1.5 mL | Thermo Fisher | 366656 |
| Mr. Frosty™ freezing container | Thermo Fisher | 5100-0001 |
| T25 Flask | Corning | 353108 |
Materials and Equipment
Laminin-521 Dilution (Tables 1 and 2)
Base Media Composition (Tables 3 and 4)
Step-By-Step Method Details
Plating hPSC for Differentiation
This step describes the passaging of hPSC cultures into monolayers appropriate for differentiation.
-
1.
Coat a 48-well plate with 5 μg/mL Laminin-521 and leave in the incubator for ≥2 h (see Tables 1 and 2 for laminin-521 dilution).
Note: Differentiation can proceed in any size multi-well plate or flask. Adjust the Laminin-521 dilution, seeding density, and media volume accordingly.
-
2.
Passage hPSC at 70%–80% confluency by washing once in PBS−/−, adding 2 mL Accutase, and placing in the incubator for 4–6 min. Monitor the morphology of the cells closely during this period. Once colonies show a characteristic bright halo and are close to detaching, gently pipette using a P1000 until ≥90% of the flask has detached.
-
3.
Rapidly dilute Accutase solution in ≥3 mL mTeSR and centrifuge at 260 × g for 3 min at 21°C.
-
4.
Aspirate supernatant, resuspend the cell pellet in fresh 21°C mTeSR supplemented with 10 μM Y27632 and count live cells using a hemocytometer.
Note: Resuspend hPSC at 1–2 × 106 cells/mL to enable rapid, accurate counting. Typically, we resuspend hPSC in 1 mL/5–10 cm2. Each cell line grows at a different density and therefore the correct volume to resuspend cells in should be determined individually.
-
5.
Seed 0.3 × 106 cells/cm2 (48 well surface area = ∼1 cm2) in ≥400 μL of mTeSR + Y27632. Rock the plate in a “figure eight” movement to evenly distribute cells and leave to settle for 10–15 min before placing in the incubator.
Note: It is crucial to start the differentiation with an even monolayer of cells. Carefully dissociating to a single cell solution is important as it allows for an accurate count and an even distribution upon seeding. It is equally important not to prolong exposure of the hPSCs to enzymatic dissociation as this will impair survival. The duration of enzymatic treatment and the balance between optimal dissociation and survival may vary between cell lines.
Note: Initial seeding density should be optimized for each cell line to ensure confluency 24 h after seeding. Typically, we seed 0.2–0.4 × 106 cells/cm2. Trypan blue exclusion dye can be utilized during cell counting to ensure that only viable cells are included in the final cell seeding calculation.
vmDA Progenitor Differentiation
This step describes the steps needed to differentiate hPSCs into vmDA progenitors.
See Figure 1A for a schematic of the differentiation.
-
6.
Day 0: 24 h after seeding check under the microscope that hPSC have survived and appear as a single, confluent (≥95%) monolayer with no obvious differentiation or uneven distribution.
Note: No (or very few small) gaps should be present the day after seeding the cells. If it is hard to see, wash twice with PBS−/− and check under the microscope. In addition, there should be very little death. If there is a large amount of death or cells are not confluent, it is best to discard the cells and start again.
-
7.
Prepare Induction Media and supplement with 10 μM SB431542 and 200 nM LDN193189, SMAD inhibitors of the Transforming Growth Factor-β and Bone Morphogenetic Protein pathways, respectively.
-
8.
Gently wash each well twice with PBS−/− to remove residual mTeSR and replace media with 500 μL of supplemented Induction Media.
-
9.
Day 1: Change media to 500 μL Induction Media supplemented with 10 μM SB431542, 200 nM LDN193189, as well as ventralizing factors 100 ng/mL C25II SHH and 2 μM purmorphamine.
-
10.
Days 2 and 3: Change media to 500 μL (day 2) or 1 mL (day 3) Induction Media supplemented with 10 μM SB431542, 200 nM LDN193189, 100 ng/mL C25II SHH, 2 μM purmorphamine and 2.5 μM of the Wnt-agonist CHIR99021 to caudalize developing progenitors.
Note: Media is changed every 24 h and 500 μL/48-well is added at days 0–2. At day 3 and beyond media is changed every other day and the volume is increased to 1 mL/48-well.
-
11.
Day 5: Change media to 1 mL Induction Media supplemented with 200 nM LDN193189, 100 ng/mL C25II SHH, 2 μM purmorphamine and 2.5 μM CHIR99021.
-
12.
Days 7 and 9: Change media to 1 mL Induction Media supplemented with 200 nM LDN193189 and 2.5 μM CHIR99021.
-
13.
Day 11: Wash cultures twice in PBS−/− and switch base media to Maturation Media with 20 ng/mL rhBDNF, 20 ng/mL rhGDNF, 0.1 mM dibutyryl cAMP, 200 nM Ascorbic Acid, 1 ng/mL rhTGFβ3, and 10 μM DAPT (Maturation Media + BGCATD), supplemented with 2.5 μM CHIR99021.
Optional: Analyze cells at the end of progenitor patterning, day 13, for markers of vmDA progenitors by fixation and immunofluorescence or collection of cells for RT-qPCR. See below for the immunostaining protocol. By day 13 the majority (≥80%) of cells should co-express OTX2, FOXA2 and Nestin (Figure 2A; Table 5). Together, these markers indicate correct specification of ventral midbrain progenitors.
Figure 1.

Differentiation Overview and Morphological Changes during vmDA Differentiation
(A) Schematic representation of the differentiation protocol indicating when each factor is added to the base media to appropriately pattern hPSC toward the vmDA lineage.
(B) Morphological changes during differentiation. At day 3, the cells are a flat, uniform monolayer. By day 11, the density has increased, and the wells appear darker under the microscope. By days 18–20, fibers begin to appear as neurons accumulate and aggregate together and by day 25 vmDA neurons display extended axonal processes. Scale bar, 100 μm.
Figure 2.

Characterization of Developing vmDA Progenitors and Neurons with 2.5 μM CHIR99021
(A) Immunofluorescence analysis of developing vmDA progenitors differentiated from either H9:PITX3-eGFP or RM3.5:PITX3-eGFP using 2.5 μM CHIR99021. At day 13, vmDA progenitors should homogenously express OTX2, FOXA2 and Nestin, indicating correct midbrain floorplate lineage acquisition.
(B) At day 25, vmDA neurons expressing FOXA2, PITX3, and TH should be readily observed if differentiation has been successful. Scale bar, 100 μm.
Table 5.
Expression of vmDA Genes during Differentiation
| Nestin |
OTX2 |
FOXA2 |
LMX1A |
EN1 |
NURR1 |
PITX2 |
BARHL1 |
(PITX3)eGFP |
TH |
|
|---|---|---|---|---|---|---|---|---|---|---|
| 1:1000 | 1:1000 | 1:500 | 1:200 | 1:50 | 1:200 | 1:500 | 1:500 | 1:1000 | 1:1000 | |
| Day 13 | +++ | +++ | +++ | ++ | +/- | -/+ | +/- | +/- | - | - |
| Day 16 | +++ | +++ | +++ | ++ | ++ | ++ | +/- | +/- | +/- | +/- |
| Day 25 | ++ | +++ | +++ | ++ | +/- | + | +/- | +/- | ++ | ++ |
| Day 60 | +/- | ++ | ++ | + | +/- | +/- | +/- | +/- | +++ | +++ |
Expression level: - none; +/- low; + moderate; ++ high; +++ very high.
Note: If vmDA progenitors are too dense at day 13 for accurate quantification, it is possible to passage the cells with Accutase. If so, replate cells in Maturation Media + BGCATD, supplemented with 10 μM Y27632 at 0.3–0.45 × 106/cm2. Fix 24 h later, after cells have attached, for analysis or continued culture.
Terminal Differentiation and Long-Term Culture of vmDA Neurons
This step describes the long-term of culture of vmDA progenitors and the generation of vmDA neurons.
-
14.
Days 13–30. Change media every other day to Maturation Media + BGCATD.
Optional: Transplantation of vmDA progenitors is optimal between days 13–22. Wash cells twice in PBS−/−, add 400 μL/48-well of Accutase and incubate for 7–10 min at 37°C. Monitor the wells closely under a microscope and test detachment every few minutes by gently pipetting the Accutase directly onto the cells. Once cells begin to detach, gently pipette using a P1000 to collect ≥90% of the well and further dissociate vmDA progenitors. Accutase can be left on for up to 20 min for slowly detaching, more dense cultures. Arrest enzymatic digestion when cells have dissociated into small (5–20 cell) aggregates by adding Maturation Media + BGCATD, supplemented with 10 μM Y27632 and centrifuge at 260 × g (3 min at 21°C). Aspirate the supernatant, resuspend the cell pellet in (1 mL/cm2) Maturation Media + BGCATD + 10 μM Y27632 and count viable progenitors using a hemocytometer and Trypan blue exclusion. Centrifuge at 260 × g (3 min at 21°C), aspirate supernatant, and resuspend at 0.1–0.2 × 106/μL in Maturation Media + BGCATD + 10 μM Y27632 and store on ice prior to transplantation.
Note: From day 18–20, vmDA neurons will become increasingly abundant and can be indirectly observed by the formation of neuronal bunches and axonal projections. By day 25, these neuronal bundles become dense and highly arborized (Figure 1B), making dissociation challenging and resulting in poor survival after plating and following transplantation. Viability when counting cells for further culture or transplantation should be ≥90%. Cells should not be stored on ice for >5 h.
Optional: We routinely characterize our cultures at days 13, 16, and 25 to confirm the degree of vmDA specification and neuronal identity within the cultures. If desired, fix cultures or recover cells from wells at these stages for immunofluorescence and/or RT-qPCR. vmDA progenitors co-expressing LMX1A, EN1, NURR1 should be apparent by D16, and vmDA neurons co-expressing FOXA2 along with the postmitotic lineage markers PITX3 and TH should be readily apparent by D25 (Figure 2B; Table 5).
-
15.
From day 30 onwards, perform half-media changes, such that ∼400 μL is removed from each well and ∼500 μL of fresh, equilibrated media is added to maintain ∼1 mL/48-well. At these late stages of the differentiation, media changes can be done 2–3 times per week.
Note: For in vitro studies of DA development, homeostasis, disease modeling, or for drug screening approaches, vmDA neurons can be cultured until at least day 60, at which point electrophysiologically active vmDA neurons will be present.
Cryopreservation of vmDA Progenitors
This step describes the protocol for generating frozen stocks of vmDA progenitors for downstream applications.
Note: Cryopreserving vmDA progenitors between days 13 and 19 is ideal to balance lineage acquisition and survival.
-
16.
Label cryovials and cool freezing container to 4°C.
-
17.
Prepare freeze media containing 20% DMSO and 60% KnockOut serum replacement and 20% fresh Maturation Media + BGCATD + 10 μM Y27632.
Note: 0.5 mL of freeze media and 0.5 mL Maturation Media are needed per 1 mL cryovial. Typically, we cryopreserve vmDA progenitors at 3 × 106/mL.
-
18.
Wash wells twice in PBS−/− and add 400 μL/48-well of Accutase. Place in incubator for 7–10 min. Monitor cell morphology closely under the microscope.
-
19.
Dissociate cells by gentle pipetting using a P1000 and dilute Accutase in Maturation Media + BGCATD + Y27632.
-
20.
Centrifuge cells at 260 × g for 3 min, aspirate supernatant and resuspend in Maturation Media + BGCATD + 10 μM Y27632 (1 mL/cm2) at approximately 1 mL × 106/mL for accurate counting using a hemocytometer and Trypan blue exclusion.
-
21.
Centrifuge at 260 × g for 3 min, aspirate supernatant and resuspend in fresh Maturation Media + BGCATD + 10 μM Y27632 at 6 × 106/mL.
-
22.
Transfer 500 μL of the cell solution to each cryovial and add 500 μL of freeze media, pipetting twice to gently mix.
-
23.
Rapidly move vials to the freezing container and place at -80°C.
-
24.
After ≥48 h, transfer cryovials to LN2 for long-term storage.
Recovery of Cryopreserved vmDA Progenitors
See Figure 3 for an overview of vmDA progenitor maturation following thawing.
Figure 3.

vmDA Progenitors Can Be Cryopreserved and Thawed with High Viability
(A) Differentiation schematic highlighting two time points where vmDA progenitors can be frozen and/or transplanted with high viability.
(B) Live/dead analysis of DNA content using FACS highlights that cells are viable after cryopreservation at days 13 or 19.
(C) By day 25, thawed and replated vmDA progenitors mature into FOXA2+/PITX3-eGFP+ vmDA neurons in vitro. Scale bar, 100 μm.
This step describes how to thaw cryopreserved vmDA progenitors for use in downstream applications.
-
25.
Coat 48-well plates with 10 μg/mL Laminin-521 and place in incubator for ≥2 h.
-
26.
Equilibrate 15 mL Maturation Media + BGCATD + 10 μM Y27632 at 37°C for ≥15 min.
-
27.
Rapidly thaw a cryovial of vmDA progenitors in a 37°C water bath and transfer the contents to 10 mL of prewarmed media using a P1000 pipette.
-
28.
Centrifuge at 260 × g for 3 min, aspirate supernatant and resuspend in Maturation Media + BGCATD + 10 μM Y2732.
-
29.
Plate progenitors at 0.3–0.45 × 106/cm2.
Optional: Count vmDA progenitors using a hemocytometer to ascertain an accurate count of surviving cells. Live/dead analysis using FACS can also be used with a subfraction of the cells, in addition to Trypan blue exclusion. Viability immediately post-thaw is typically 60%–70%, although a further proportion may undergo apoptosis after re-plating.
Optional: Transplant vmDA progenitors directly after thawing. Wash once in PBS−/− after counting, centrifuge, aspirate supernatant and resuspend in Maturation Media + BGCATD + 10 μM Y27632 at 0.1–0.2 × 106/μL for transplantation.
Optional: For long-term culture of vmDA neurons for studying development, drug screening, and/or disease modeling, wash cells once in PBS−/− the day after thawing and replace media with 1 mL fresh Maturation Media + BGCATD. Proceed with steps 14–15 to continue the differentiation.
Note: The concentration of rhGDNF and Y27632 can be doubled to 40 ng/mL and 20 μM, respectively, if survival upon thawing is poor.
Immunostaining of vmDA Progenitors and Neurons
This step outlines how to undertake immunocytochemical profiling of generated vmDA progenitors.
-
30.
Wash wells once with PBS−/− to remove debris and add 200 μL 4% paraformaldehyde. Leave at 21°C for 10 min.
-
31.
Aspirate paraformaldehyde and wash 3 times with PBS−/−, leaving wells in PBS−/− for at least 2 min between washes.
-
32.
Prepare blocking buffer by adding 1 mL of species-specific serum (same as secondary antibodies) and 0.6 mL 10% Triton X-100 to 18.4 mL PBS-Azide 0.05%.
-
33.
Add 200 μL/48-well of blocking buffer to each well for 30–60 min.
-
34.
Dilute the primary antibodies in blocking buffer. Aspirate blocking buffer and add 200 μL/48-well of primary antibody solution. Incubate at 21°C for 12–18 h.
-
35.
Wash wells 3 times in PBS−/− to remove primary antibodies and block cells again for 30–60 min by adding 200 μL blocking buffer.
-
36.
Dilute appropriate secondary antibodies in blocking buffer and add 200 μL of secondary antibody solution to each well. Incubate cultures in the dark at 21°C for 1.5–2 h.
-
37.
Wash wells in PBS−/− and add 1 μg/mL DAPI (diluted in PBS−/−) for 10 min.
-
38.
Wash wells twice in PBS−/− and image.
Note: It is optimal to perform immunofluorescence immediately after fixation of the cells. If long-term storage is necessary, ensure sufficient PBS-Azide is added to each well (1 mL), to account for evaporation during refrigeration and minimize exposure to light to prevent fluorochrome fading.
Expected Outcomes
One day after seeding, undifferentiated hPSC should tessellate the culture surface such that there are no (or very small) gaps between cells. As differentiation proceeds cells will actively proliferate and appear denser, until individual cells can no longer be resolved under the microscope (typically day 5). At the early stages of differentiation there should be very little cell death. By day 13 the majority of the culture should appear homogenous in distribution and density across the culture well, co-express FOXA2 and OTX2 (≥80%) and will begin to appear darker under the microscope as cell density increases (Figures 1B and 2; Table 5). Note, some death may occur following the transition from Induction to Maturation Media. Addition of the γ-secretase inhibitor DAPT, which drives cell cycle exit, often leads to clear neuronal processes by day 20, although this is cell line-dependent and does not necessarily reflect the final TH+ vmDA neuron yield.
By day 20 TH+ (or PITX3-eGFP+) neurons are readily observable and are increasingly observed until peaking at day 40 (D20: ∼5%; D25: ∼25%; D40: ∼50%) (Figure 2; Table 5; See Niclis et al. (2017a) for more detailed quantification across numerous hPSC lines). vmDA neurons should also co-express FOXA2, OTX2, NURR1, EN1, LMX1A (Figure 2; Table 5). vmDA progenitors and neurons will increasingly clump together after day 30 as they continue to mature. At day 40 vmDA neurons in culture are capable of synthesizing and metabolizing dopamine, and by day 60 vmDA neurons display characteristic vmDA electrophysiological properties (Niclis et al., 2017a).
For the purpose of transplantation, we typically obtain total cell yields of 1–2 × 106/cm2 at time points amenable to implantation (days 13–22). Demonstrative of the functionality and integration of these cells, we observe correction in the unilateral 6-hydroxydopamine rat model of DA depletion/Parkinson’s disease in amphetamine-induced rotational asymmetry after 12–16 weeks.
Limitations
We have successfully utilized this protocol to generate vmDA neurons from multiple hESC and hiPSC cell lines (including H9, HES3, RUES2, 409B2 parental lines and several subclones). However, differentiation efficiency does vary between hPSC lines. We have found that the majority of this variation is Wnt-dependent. As such, we recommend optimization of the CHIR99021 concentration for each cell line. Typically, titration between 2–3 μM is sufficient to optimize vmDA yield (Figure 4). This is particularly important for iPSC-based disease modeling studies, that typically derive numerous cell lines.
Figure 4.

CHIR99021 Concentration Determines the Rostrocaudal Positioning of Developing Neural Progenitors
Titrating CHIR99021 is advisable for each individual cell line.
(A) Neural progenitors at day 13 exposed to increasing levels of [CHIR99021] (0.5–3.0 μM) demonstrating a loss of fore/midbrain marker OTX2 and increased EN1 at higher concentrations. PITX2 and BARHL1 are expressed in the rostral midbrain and are helpful to fine-tune CHIR99021 concentration. We have found that 2.5 μM consistently generated vmDA progenitors from multiple hPSC cell lines.
(B) PITX3-eGFP+/TH+ vmDA neurons are born only after appropriate CHIR99021 addition and are progressively lost when [CHIR99021] is too high and cultures are overcaudalized. Scale bar, 100 μm.
Even under optimal conditions, where OTX2+/FOXA2+, putative vmDA progenitors make up ≥90% of the culture, only ∼50% of these will mature into TH+ vmDA neurons. This is likely due to the aberrant maintenance of an immature state in vitro or the generation of adjacent midbrain lineages. Typically, we do not observe significant 5-HT, GABA or Glutamate (≥2%) contamination within vmDA cultures. If specific analysis of the vmDA population is desired (e.g. for in vitro disease modeling of vmDA phenotypes, vmDA-centric drug development, or grafting in vivo), we suggest the use of an appropriate fluorescent reporter (such as PITX3-GFP or TH-GFP) to allow for isolation (e.g., for multi-omic analyses) and/or tracking in vitro or after transplantation. See Niclis et al. (2017b) for an example of how a fluorescent reporter can augment analysis.
Finally, the use of fully defined media, utilizing xenogeneic-free reagents (amenable to clinical translation), results in less variability and better efficiency between differentiations and cell lines (Niclis et al., 2017a). However, the use of animal product-free reagents can add unnecessary expense when such stringency may not be required. For other applications we recommend using the more traditional media formulations, which are typically less expensive and work equally well. We regularly interchange xenogeneic and xenogeneic-free reagents (e.g. mTeSR2/mTeSR1) without altering the protocol, although this should be determined on a case-by-case basis by the end user.
Troubleshooting
Problem
hPSC do not survive or appear non-uniform at the beginning of the differentiation.
Potential Solution
The quality of the starting stem cells is crucial. Only commence differentiations when the hPSC are ∼70%–80% confluent and display a uniform, circular morphology in mTeSR (or comparable conditions, such as E8 or MEF co-culture). OCT4 immunofluorescence can aid in verifying the degree of stemness of a given cell line, prior to, or at the beginning of a differentiation. Likewise, careful monitoring during Accutase passaging is essential. Cells should appear round and display a characteristic bright halo but not be detached from the flask. Triturate cells using a P1000 to detach and rapidly place into warmed mTeSR. If cells are not confluent and/or overconfluent after 24 h alter the seeding density (typically 1.5–4.0 × 106/cm) and check that the ROCK inhibitor, Y27632, has been added. Lastly, we recommend routine mycoplasma testing. However, even with these troubleshooting tips, we can observe inter-personnel and inter-laboratory variability, highlighting the essential need to conduct multiple differentiations.
Problem
D13 vmDA progenitors do not express OTX2, FOXA2, or EN1.
Potential Solution
Successful early specification of the cultures should yield ≥80% OTX2+/FOXA2+ progenitors by day 13. Lack of FOXA2 expression is typically observed with inadequate SHH signaling (i.e. insufficient ventralization of the culture). Increasing SHH or purmorphamine concentration is normally sufficient to rescue FOXA2. Most importantly, the balance of Wnt signaling is crucial to restrict developing progenitors to an appropriate vmDA lineage (Figure 4A). If the concentration of CHIR99021 is too low, progenitors will co-express OTX2+/FOXA2+ but fail to express the more caudally restrictive protein Engrailed-1 (EN1). Added to this, the presence of PITX2+ and/or BARHL1+ progenitors within the cultures indicates a rostral midbrain identity of the cells – a problem that can be resolved by subtle increases in CHIR99021 concentration (Kirkeby et al., 2017). Conversely, high CHIR99021 doses will lead to reduced OTX2 expression, as progenitors are caudalized beyond the midbrain and instead adopt a more hindbrain-like fate (Figure 4A). Evidently, fine-tuning of the CHIR99021 concentration is crucial, with doses of 2–3 μM typical. Of note, subtle gradations, as small as 0.2 μM, can notably alter cell fate. For any given hPSC line, the highest possible concentration of CHIR99021, without jeopardizing the proportion of OTX2 and FOXA2 co-expressing cells, will maximize EN1 expression and ensure vmDA identity (Figure 4). We do not routinely use FGF8 in our cultures as we find adjusting the concentration of CHIR99021 is sufficient to caudalize progenitors appropriately. In our hands, FGF8 can also introduce significant heterogeneity into cultures. However, if FGF8 is added, the concentration of CHIR99021 should be lowered, noting that both factors promote caudalization of developing progenitors.
Problem
Immunofluorescence analysis is not clear or difficult to interpret.
Potential Solution
By day 13, cultures can be extremely dense, making analysis of individual cells difficult. Passaging cells for analysis can often be extremely helpful for accurate quantification. For this purpose, it is best to passage day 13 progenitors (after CHIR99021 is withdrawn), using Accutase, and re-seeding cells onto 10 μg/mL Laminin-521 coated plates at a density of 0.3–0.45 × 106/cm2. Cultures should then be fixed 24 h later, proceeding with immunofluorescence as normal. Ensure that Y27632 is added to minimize death, as this may obscure the yield of vmDA progenitors. Of note, EN1 can be difficult to detect by immunofluorescence. It is best to use a fresh batch of the antibody and/or utilize RT-qPCR to assess EN1 expression.
Problem
vmDA progenitors do not survive cryopreservation and/or transplantation.
Potential Solution
Identifying the optimal maturation stage of vmDA progenitors is critical to their survival during cryopreservation or transplantation. Typically, we transplant vmDA progenitors at days 13–23, shortly after TH-expressing vmDA neurons first begin to be evident in culture. Of note, transplantation of immature/insufficiently specified progenitors or overly mature/postmitotic neurons from cultures will yield low proportions of surviving vmDA neurons within grafts. Likewise, we routinely cryopreserve vmDA progenitors between days 13–19 with excellent survival (>75%) upon thawing of both day 13 and day 19 progenitors (Figure 3). Ensuring that the thaw media is made fresh and prewarmed prior to thawing a vial can impact cell survival. If survival remains poor, the concentration of GDNF and Y27632 in the thaw media can be doubled.
Problem
vmDA neurons do not survive long-term culturing.
Potential Solution
vmDA neurons in culture can deteriorate after day 40. However, vmDA neurons in our culture system do mature into electrophysiologically active cells, capable of metabolizing dopamine and expressing mature vmDA subtype specific markers (GIRK2+ and Calbindin+) over extended time frames (>50 Days) (Niclis et al., 2017a). To support long-term culturing, we recommend seeding differentiations onto a high concentration of laminin-521 (10 μg/mL), and only changing half of the media every 3 days from day 30, rather than every 2. Mouse laminin (1–2 μg/mL) can also be supplemented into the Maturation Media from day 30 to support attachment and enhance viability.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Clare Parish (clare.parish@florey.edu.au).
Materials Availability
This study did not generate new unique reagents, cell or mouse lines.
Data and Code Availability
This study did not generate a new dataset.
Acknowledgments
The authors thank current and past members of the Thompson and Parish laboratories for continuous discussion and refinement of the protocol. In particular, we thank Jonathan Niclis, Isabelle de Luzy, and Jennifer Durnall for their contribution. C.W.G. was supported by an Australian Postgraduate Award. C.L.P. was supported by a National Health and Medical Research Council (NHMRC) Australia, Senior Research Fellowship. This work was supported by an NHMRC Australia project grant (APP1102704 to C.L.P.) and Stem Cells Australia (to C.L.P.). The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
Author Contributions
Conceptualization, C.W.G., A.C.-C., L.H.T., and C.L.P.; Investigation, C.W.G.; Writing – Original Draft, C.W.G., A.C.-C., L.H.T., and C.L.P.; Writing – Review & Editing, C.W.G., A.C.-C., L.H.T., and C.L.P.; Funding Acquisition, L.H.T. and C.L.P.; Supervision, L.H.T. and C.L.P.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Carlos W. Gantner, Email: cg731@cam.ac.uk.
Clare L. Parish, Email: clare.parish@florey.edu.au.
References
- Gantner C.W., de Luzy I.R., Kauhausen J.A., Moriarty N., Niclis J.C., Bye C.R., Penna V., Hunt C.P.J., Ermine C.M., Pouton C.W. Viral delivery of GDNF promotes functional integration of human stem cell grafts in Parkinson’s disease. Cell Stem Cell. 2020;26:511–526.e5. doi: 10.1016/j.stem.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Kirkeby A., Nolbrant S., Tiklova K., Heuer A., Kee N., Cardoso T., Ottosson D.R., Lelos M.J., Rifes P., Dunnett S.B. Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-based therapy for Parkinson’s disease. Cell Stem Cell. 2017;20:135–148. doi: 10.1016/j.stem.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclis J.C., Gantner C.W., Alsanie W.F., McDougall S.J., Bye C.R., Elefanty A.G., Stanley E.G., Haynes J.M., Pouton C.W., Thompson L.H. Efficiently specified ventral midbrain dopamine neurons from human pluripotent stem cells under xeno-free conditions restore motor deficits in Parkinsonian rodents. Stem Cells Transl. Med. 2017;6:937–948. doi: 10.5966/sctm.2016-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclis J.C., Gantner C.W., Hunt C.P.J., Kauhausen J.A., Durnall J.C., Haynes J.M., Pouton C.W., Thompson L.H., Parish C.L. A PITX3-EGFP reporter line reveals connectivity of dopamine and non-dopamine neuronal subtypes in grafts generated from human embryonic stem cells. Stem Cell Rep. 2017;9:868–882. doi: 10.1016/j.stemcr.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watmuff B., Hartley B.J., Hunt C.P.J., Fabb S.A., Pouton C.W., Haynes J.M. Human pluripotent stem cell derived midbrain PITX3(eGFP/w) neurons: a versatile tool for pharmacological screening and neurodegenerative modeling. Front. Cell. Neurosci. 2015;9:104. doi: 10.3389/fncel.2015.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate a new dataset.