

Abstract
Purpose:
To determine the dose limiting toxicities (DLTs), maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) of veliparib in combination with weekly topotecan in patients with solid tumors. Correlative studies were included to assess the impact of topotecan and veliparib on poly(ADP-ribose) levels in peripheral blood mononuclear cells, serum pharmacokinetics of both agents, and potential association of germline repair gene mutations with outcome.
Experimental Design:
Eligible patients had metastatic nonhematological malignancies with measurable disease. Using a 3+3 design, patients were treated with veliparib orally twice daily on days 1–3, 8–10 and 15–17 and topotecan intravenously on days 2, 9 and 16 every 28 days. Tumor responses were assessed by RECIST.
Results:
Of 58 patients enrolled, 51 were evaluable for the primary endpoint. The MTD and RP2D was veliparib 300 mg twice daily on days 1–3, 8–10 and 15–17 along with topotecan 3 mg/m2 on days 2, 9 and 16 of a 28-day cycle. DLTs were grade 4 neutropenia lasting >5 days. The median number of cycles was 2 (1–26). The objective response rate was 10%, with 1 complete and 4 partial responses. 22 patients (42%) had stable disease ranging from 4 to 26 cycles. Patients with germline BRCA1, BRCA2 or RAD51D mutations remained on study longer than those without homologous recombination repair (HRR) gene mutations (median 4 vs. 2 cycles).
Conclusions:
Weekly topotecan in combination with veliparib has a manageable safety profile and appears to warrant further investigation.
Keywords: Phase I, Veliparib, PARP inhibitors, Topotecan, Solid Tumors, Advanced Malignancies, NCT01012817
Introduction
Topoisomerase I (topo I) poisons are widely used for the treatment of various cancers (1–4). In particular, irinotecan has regulatory approval for colorectal cancer (5), and exhibits activity in non-small cell lung cancer and breast cancer (6). Nanoliposomal irinotecan used in combination with 5-fluorouracil and folinic acid has extensive activity against advanced pancreatic cancer (7). Topotecan (TPT) is approved for the treatment of ovarian, cervical and small cell lung cancers (8). Moreover, novel topo I poisons continue to be identified and tested in the clinical setting (9–12).
Despite their widespread clinical use, topo I poisons have limited efficacy. Topotecan, for example, exhibits a 17% overall response rate and a median overall survival of 57 weeks when administered as a daily 1-hour infusion for 5 consecutive days to patients with recurrent, platinum-resistant ovarian cancer (13). Because myelosuppression occurs in up to 75% of women receiving this therapy (14), alternative regimens have been investigated to improve or maintain efficacy while decreasing toxicity (15,16). A phase II study investigating topotecan administered on days 1, 8, and 15 of a 28-day cycle had a response rate of 18% in platinum refractory/resistant ovarian cancer (17). With this alternative schedule, myelosuppression was mild, with no grade 4 toxicities observed.
Preclinical studies have demonstrated that PARP inhibitors, which exhibit single-agent clinical activity in ovarian cancer, breast and prostate cancer (18–22), sensitize tumor cells to topo I poisons in vitro and in vivo (23–25). Multiple mechanisms appear to contribute to this sensitization. First, inhibition of PARP1 catalytic activity impairs the recruitment of tyrosyl-DNA phosphodiesterase 1 (TDP), an enzyme that participates in detoxification of trapped topo I-DNA covalent complexes (26). Second, topo I poisons are known to cause replication fork stalling and collapse (27–29); and PARP1 plays a central role in helping stabilize and restart these replication forks (30–32). Finally, genetic analysis has indicated that trapping of PARP1 at sites of DNA damage, which is an intrinsic feature of all PARP inhibitors, including veliparib (33), also contributes to topotecan/PARP inhibitor synergy (34).
In view of this synergy in preclinical models, there has been substantial interest in combining PARP inhibitors with topo I poisons in the clinic (18,35). However, all of the early phase trials of topo I poison/PARP inhibitor combinations reported to date have been limited in their ability to administer PARP inhibitor because of severe myelosuppression (35–37). In the case of topotecan, Kummar et al. administered the orally bioavailable PARP inhibitor veliparib (38) in combination with the 5-day topotecan regimen and found the combination very myelosuppressive, requiring reduction in doses of both agents (39). The maximum tolerated dose (MTD), veliparib 10 mg twice daily combined with topotecan 0.6 mg/m2/day × 5 days (39), represents 1/40th and less than 1/3 of the respective single-agent MTDs, respectively (40,41).
In view of these results, we sought to determine whether veliparib could be safely combined with the less myelosuppressive weekly topotecan regimen in patients with solid tumors. In addition to identifying the MTD and dose-limiting toxicities (DLTs) of this combination, the goals of the present study were to determine whether there was evidence of clinical activity, assess the impact of topotecan and veliparib on poly(ADP-ribose) polymer (PAR) levels in peripheral blood mononuclear cells, characterize the pharmacokinetics of both agents, and the evaluate the impact of germline DNA repair gene mutations on clinical activity of this combination.
Materials and Methods
Patients
Patients 18 years or older with any histologically confirmed solid tumor with measurable disease (longest diameter ≥2 cm with conventional CT) that was metastatic or unresectable who had relapsed less than a year from their prior platinum treatment were eligible. In addition patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, adequate bone marrow (as defined by an absolute neutrophil count ≥ 1500/mcL, hemoglobin ≥ 9.0 g/dL and platelets ≥ 100,00/mcL), renal (Cr ≤ 1.5 × the upper limit of normal (ULN)) and hepatic function (total bilirubin ≤ 1.5 × ULN, ALT and AST ≤ 2.5 × ULN in the absence of hepatic metastasis or ALT ≤ 3 × ULN and AST ≤ 5 × ULN in the presence of hepatic metastasis) as well as the ability to swallow and absorb oral medication. Initially there was no limit on the number of prior regimens, but this was amended at dose level 4 due to impact of prior therapies on marrow reserve. For women of child bearing potential, a negative serum or urine pregnancy test was required. Exclusion criteria included treatment with chemotherapy, radiation or immunotherapy within 4 weeks prior to receiving treatment (6 weeks if prior treatment was mitomycin C or a nitrosourea); prior treatment with a PARP inhibitor; prior radiation therapy to >25% of the bone marrow; uncontrolled intercurrent illness; immunocompromise (other than related to the use of glucocorticoids); known CNS metastases or seizure disorder; New York Heart Association class II or III heart disease or history of MI within 6 months; and other active malignancy other than non-melanoma skin cancer and carcinoma in situ of the cervix. All patients provided written informed consent.
Study Design
This phase I clinical trial was open at Mayo Clinic in Rochester MN, Jacksonville FL and Scottsdale, AZ. Patients were enrolled using a standard 3+3 study design in order to determine the MTD and recommended phase II dose (RP2D) of the combination of weekly topotecan with veliparib. A secondary objective was to identify any preliminary evidence of anti-tumor activity as assessed by objective response in patients with advanced solid tumors. Clinical correlates included identification of any pharmacokinetic interactions between veliparib and topotecan and assessment of the impact of topotecan and veliparib on poly(ADP-ribose) (PAR) levels in peripheral blood mononuclear cells (PBMCs). The study was conducted according to the Declaration of Helsinki. It was approved by the Mayo Clinic Institutional Review Board and registered at clinicaltrials.gov as NCT01012817.
Treatment Schedule (Supplementary Fig. S1)
Patients were treated with veliparib orally once (dose levels 1–6) or twice (dose levels 7–13) daily on Days 1–3, 8–10 and 15–17 and topotecan intravenously (IV) over 30 minutes on Days 2, 9 and 16 every 28 days. For Cycle 2 only, patients were treated with veliparib orally daily only on Days 8–10 and 15–17 and topotecan IV on Days 2, 9 and 16. This omission of veliparib on Days 1–3 of Cycle 2 allowed sampling of PBMCs and serum topotecan clearance in the absence of veliparib. Subsequent cycles were as described for Cycle 1. Premedications were not routinely used. Antiemetics were provided when needed as determined by the treating physician. Prophylactic use of 5-HT3 antagonists and other agents such as glucocorticoids, phenothiazines and benzodiazepines was permitted. Growth factor support was not recommended and not allowed during Cycle 1 of therapy, Prophylactic growth factor was allowed in subsequent cycles at physician discretion if a patient developed grade 3 or 4 neutropenia during a previous cycle. Patients were given a pill diary to record and remind patients of when to take the oral veliparib.
Efficacy Assessments
Response was assessed every 8 weeks. Modified response evaluation criteria in solid tumors (RECIST v.1.0) were used.
Pharmacokinetic (PK) assessments
PK studies were performed for veliparib and topotecan. For veliparib, blood samples were taken on Cycle 1 Day 1 and 2 prior to the dose of veliparib as well as 15 minutes, 30 minutes, 1, 2, 4, 6, and 9 hours after administration. For topotecan, samples were drawn on Cycle 1 Day 2 and Cycle 2 Day 2 prior to the topotecan infusion as well as 15 and 30 minutes after start of infusion and 15 and 30 minutes, 1, 1.5, 3.5, 5.5, and 8.5 hours after the end of the infusion. These samples were analyzed for both agents using validated high performance liquid chromatography assays for topotecan (42) and veliparib (39). Plasma concentration-time data were analyzed by standard non-compartmental methods using Phoenix® WinNonlin® 1.3 (Certara USA, Inc., Princeton, NJ).
PAR measurements
Whole blood samples for PBMC PAR assessment were obtained on Cycle 1 Day 1 (0, 2, 4 and 8 hours after veliparib), Cycle 1 Day 2 (0, 2, 4 and 8 hours after veliparib) and on Cycle 2 Day 2 (0, 2, 4 and 8 hours after topotecan). PAR levels were analyzed by sandwich ELISA in the National Clinical Target Validation Laboratory of the NCI as previously described (43).
Statistical and Analytical Methods
The phase I portion of this trial is a single arm phase I study utilizing 3+3 design to determine the MTD of the combination of veliparib and weekly topotecan in the treatment of solid tumors. If a patient failed to complete the initial cycle of therapy for reasons other than toxicity, the patient was regarded as unevaluable with respect to the goals of the study and an additional patient was treated at the current dose level; however, all toxicity information was utilized in the analysis.Objective responses, as defined per RECIST (v.1.0), were summarized by simple descriptive summary statistics delineating complete and partial responses (CR and PR, respectively) as well as stable and progressive disease (SD and PD, respectively).
Safety and DLT Definitions
All subjects receiving at least 1 dose of veliparib and topotecan were included in the safety analysis. All adverse events (AEs) were graded according to the CTCAE (version 4.0) grading system.
A DLT was defined as the occurrence of drug-related AEs during the first treatment cycle, including grade 4 anemia or thrombocytopenia at any time; grade 4 neutropenia persisting 5 days or longer; grade 3 nausea, diarrhea, or vomiting in spite of maximal supportive care and prophylaxis; clinically significant grade 3 or greater non-hematologic toxicity (not including alopecia, anorexia or fatigue); and serum creatinine ≥2 times baseline or ≥2 times ULN if baseline is less than the ULN.
The MTD was defined as the dose level below the one that caused DLTs in at least 2 of up to 6 patients. The RP2D was to be the MTD dose unless other safety considerations required lowering the dose.
Results
Patient characteristics
Fifty-eight patients were enrolled in the dose escalation phase of this trial of weekly topotecan and veliparib (Fig. S1) between November, 2009 and February, 2015. One patient enrolled but was never treated so was replaced. Six patients were replaced due to AEs unrelated to treatment or refusal of treatment, but toxicity information was included in the data analysis. Fifty-one patients completed at least one cycle of therapy. As shown in Table 1, the majority of evaluable patients enrolled in the study had ovarian, fallopian tube or primary peritoneal cancer (45 patients), all of whom had platinum resistant disease. Because of concern for severe myelosupression, which was seen when low doses of veliparib were combined with daily topotecan (39), the starting dose of veliparib in the present trial was significantly lower than single agent MTD of 400 mg bid. As a result, 13 dose levels were utilized in this trial (Table 2).
Table 1.
Patient demographics and baseline disease characteristics
| All evaluable patients (n=51) | |
|---|---|
| Median Age (range) | 56 (34–78) |
| %Male (n) | 1 (2%) |
| % Female (n) | 50 (98%) |
| ECOG PS | |
| 0 | 36 (70.6%) |
| 1 | 15 (29.4%) |
| 2 | 0 |
| Number of prior lines of therapy | |
| 1 | 13 (25.5%) |
| 2 | 27 (52.9%) |
| 3 | 6 (11.8%) |
| 4 | 1 (2.0%) |
| 5+ | 4 (8.0%) |
| Germline mutation status by BROCA | |
| No detected germline mutation detected | 33 (65.6%) |
| BRCA1 | 13 (25.4%) |
| BRCA2 | 1 (2.0%) |
| RAD51D | 1 (2.0%) |
| CHEK2 | 1 (2.0%) |
| FANCL | 1 (2.0%) |
| MSH6 | 1 (2.0%) |
| Primary diagnosis/histology | |
| Cervix/adenocarcinoma | 1 (2%) |
| Endometrial | 4 (8%) |
| • Endometrioid | 2 (4%) |
| • High grade serous | 2 (4%) |
| Small cell lung cancer | 1 (2%) |
| Ovarian, fallopian, primary peritoneal | 45 (88%) |
| • High grade serous | 38 (74%) |
| • Endometrioid | 3 (6%) |
| • Poorly differentiated adenocarcinoma | 1 (2%) |
| • Carcinosarcoma | 1 (2%) |
| • Mucinous (1) | 1 (2%) |
| • Clear cell (1) | 1 (2%) |
| Ovarian Cancer Cohort (n = 45) | |
| Number of prior regimens | |
| 1 | 7 (16%) |
| 2 | 11 (24%) |
| 3 | 9 (20%) |
| 4 | 5 (11%) |
| 5+ | 13 (29%) |
| Germline mutation status by BROCA | |
| No germline mutation detected | 29 (65%) |
| BRCA1 | 13 (29%) |
| BRCA2 | 1 (2%) |
| RAD51D | 1 (2%) |
| CHEK2 | 1 (2%) |
Table 2.
Dose Escalation and Response Table
| Dose Level | Veliparib (mg)* | Topotecan (mg/m2) | Number of patients treated | Mutation Status | Number of DLTs | Responses | |||
|---|---|---|---|---|---|---|---|---|---|
| CR | PR | SD | PD | ||||||
| 1 | 10 qd | 2 | 3 | BRCA1 | 0 | 0 | 0 | 2 | 1 |
| 2 | 20 qd | 2 | 3 | BRCA2 | 0 | 0 | 1 | 0 | 1 |
| 3 | 10 qd | 3 | 3 |
BRCA1 CHEK2 |
0 | 0 | 0 | 1 | 1 |
| 4 | 20 qd | 3 | 3 | BRCA1 (2) | 0 | 0 | 0 | 2 | 1 |
| 5 | 20 qd | 4 | 6 |
BRCA1 (2) RAD51D MSH6 |
3 | 1 | 0 | 4 | 1 |
| 6 | 30 qd | 4 | 6 | BRCA1 | 2 | 0 | 0 | 3 | 3 |
| 7** | 30 bid | 3 | 3 | BRCA1 | 0 | 0 | 0 | 3 | 0 |
| 8 | 50 bid | 3 | 3 | BRCA1 | 0 | 0 | 0 | 1 | 2 |
| 9 | 100 bid | 3 | 3 | BRCA1 | 0 | 0 | 0 | 1 | 2 |
| 10 | 150 bid | 3 | 6 | FANCL | 1 | 0 | 0 | 3 | 2 |
| 11 | 200 bid | 3 | 3 | 0 | 0 | 0 | 2 | 1 | |
| 12 (MTD) | 300 bid | 3 | 6 | BRCA1 | 1 | 0 | 3 | 0 | 2 |
| 13 | 400 bid | 3 | 3 | BRCA1 | 2 | 0 | 0 | 0 | 2 |
Abbreviations are:qd, one a day; bid, twice a day.
Dose level 7 and higher received twice daily veliparib
DLT and MTD determination
DLTs in the first cycle, consisting of myelosuppression (grade 4 neutropenia and grade 4 thrombocytopenia), were observed in 2 of 3 patients at dose level 6 and the study accrued 3 additional patients at dose level 5, all of whom experienced the DLT (grade 4 neutropenia, grade 4 thrombocytopenia). The study was temporarily closed while the dose schedule was reevaluated. Based on the premise that veliparib is being administered to enhance the efficacy of topotecan, the study was reopened at a reduced topotecan dose of 3 mg/m2 on days 1, 8 and 15 (3/4 the single-agent MTD) along with veliparib twice daily at dose level 7. At dose level 10, a DLT in one of the first three patients necessitated treating 6 patients with no further DLTs observed. Grade 4 neutropenia lasting >5 days was observed in 2 of 3 patients at dose level 13 and in 1 of 6 patients at dose level 12.Accordingly, the MTD and recommended phase 2 dose (RP2D) was dose level 12, i.e., veliparib 300 mg twice daily by mouth on days 1–3, 8–10 and 15–17 along with topotecan 3 mg/m2 intravenously on days 2, 9 and 16.
Safety
For cycle 1, the only grade 4 AEs at least possibly related to treatment in the study were cytopenias (neutropenia in 11 patients, thrombocytopenia in 3 patients and leukopenia in 2 patients) and one febrile neutropenia.For all cycles, the only grade 4 AEs at least possibly related to treatment involved bone marrow suppression, namely neutropenia (13), thrombocytopenia (3), leukopenia (3) and sepsis (1). The most common grade 3–4 AEs at least possibly related to treatment (Table 3) were again hematologic and included neutropenia (23), thrombocytopenia (12) and leukopenia (19) as well as grade 3 fatigue (1), grade 3 nausea (1) and electrolyte derangements (1). One patient received filgrastim while on therapy. Seventeen patients received red blood cell transfusions and one patient received a platelet transfusion for thrombocytopenia while on low molecular weight heparin.
Table 3.
Grade 3 and 4 Adverse Events* at Least Possibly Related to Therapy in the 51 Evaluable Patients (all cycles)
| Toxicity | Grade 3 | Grade 4 |
|---|---|---|
| Anemia | 16 (31.3%) | --- |
| Leukopenia | 16 (31.3%) | 3 (5.8%) |
| Neutropenia | 10 (19.6%) | 13 (22.8%) |
| Thrombocytopenia | 9 (17.6%) | 3 (5.8%) |
| Febrile Neutropenia | 1(2.0%) | --- |
| Sepsis | --- | 1 (2.0%) |
| Fatigue | 1 (2.0%) | --- |
| Nausea | 1 (2.0%) | --- |
| Hypokalemia | 1 (2.0%) | --- |
As per NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
Assessment of topotecan and veliparib pharmacokinetics
Fifty-four patients participated in pharmacokinetic (PK) studies for veliparib and topotecan, and data are available for forty-nine patients (Supplemental Table S1).Similar plasma concentration-time profiles were observed for topotecan and veliparib when administered alone or together as illustrated for a patient treated at the MTD in Fig. S2.The mean (± SD) half-life for veliparib was 6.1 ± 2.8 hr. Increasing the veliparib dose from 10 mg to 300 mg resulted in dose-proportional increases in Cmax and AUC0–9h values (Fig. 1A and Table S1).Veliparib AUC0–9h increased from 351 ± 69 ng*hr/ml (10 mg daily dose) to 15031 ± 3094 ng*hr/ml (300 mg twice daily dose). In order to determine the effect of topotecan on veliparib pharmacokinetics, we measured veliparib Cmax and AUC0–9h, on Day 1 when veliparib was given alone and on Day 2 when it was given with topotecan.The mean (± SD) ratio of (veliparib + topotecan)/veliparib alone was 1.10 ± 0.24 and 1.17 ± 0.19 for Cmax and AUC0–9h, respectively, consistent with little if any accumulation of veliparib. In order to determine the effect of veliparib on topotecan pharmacokinetics, we measured topotecan Cmax and AUC0–9h, on Cycle 1, Day 2 when topotecan was given with veliparib and on Cycle 2, Day 1 when it was given with alone.When given alone, there was substantial variability in topotecan clearance such that overlapping AUC values were observed for each dose level and a dose proportional increase in AUC was not observed as the dose was increased from 2 mg/m2 to 4 mg/m2 (Table S1).The mean (± SD) half-life and plasma clearance values for topotecan administered alone on Cycle 2 were 2.7 ± 0.6 hr and 10.1 ± 3.8 L/hr/m2, respectively.The mean (± SD) ratios of (veliparib + topotecan)/topotecan alone were 1.06 ± 0.20 for AUC0–9h and 0.97 ± 0.28 for CL, suggesting at most a small effect of veliparib on topotecan clearance (Fig. 1B and Table S1).
Figure 1.
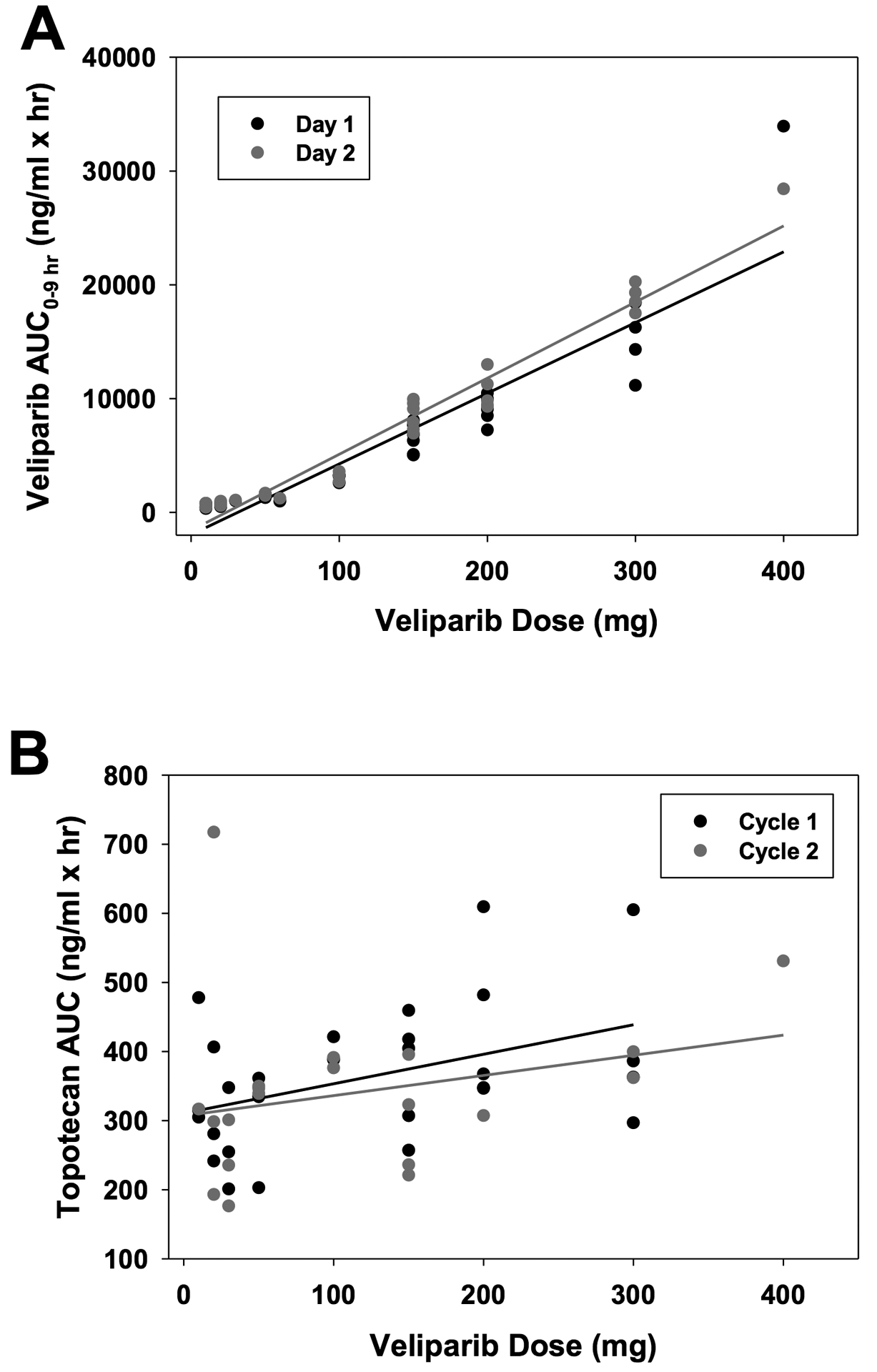
Graphs of veliparib AUC0–9h versus dose (A) and topotecan AUC0−∞ versus veliparib dose (B) following administration of escalating doses of 10 – 400 mg veliparib with 3 mg/m2 topotecan.
PAR measurements
Measurements of PAR after dosing on Day 1 of Cycle 1 (veliparib alone), Day 2 of Cycle 2 (topotecan alone) and Day 2 of Cycle 1 (topotecan + veliparib), allowed assessment of the effects of each drug alone as well as the combination. Of 47 patients who had PBMCs collected for evaluation of PAR levels, 33 had levels above the lower limit of quantitation (LLQ) at baseline (Day 1, prior to initiation of therapy).
PAR levels on Day 1 decreased in 32 of these 33 patients (Fig. S3A, top panel; p = 1.5 × 10−8 by sign test) at 2–8 hours after veliparib administration compared to baseline. The extent of PAR inhibition, which varied from 0 to at least 85%, increased with veliparib dose (Fig. S3A) and with veliparib exposure represented by AUC0–9h after the first dose (Fig. 2A).Prior to the Day 2 treatment (Day 2, 0 h), PAR levels were close to baseline at dose levels 1–6, reflecting poor target inhibition at the end of the 24-hour dosing interval (Fig. 2B; Fig. S3A, middle panel, closed circles). In contrast PAR levels were generally lower and, in many patients below the LLQ, at 12 hours after the last dose in the twice daily schedule of dose levels 8–13 (Fig. 2B and Fig. S3A, red circles).
Figure 2. Effects of treatment on PAR levels.
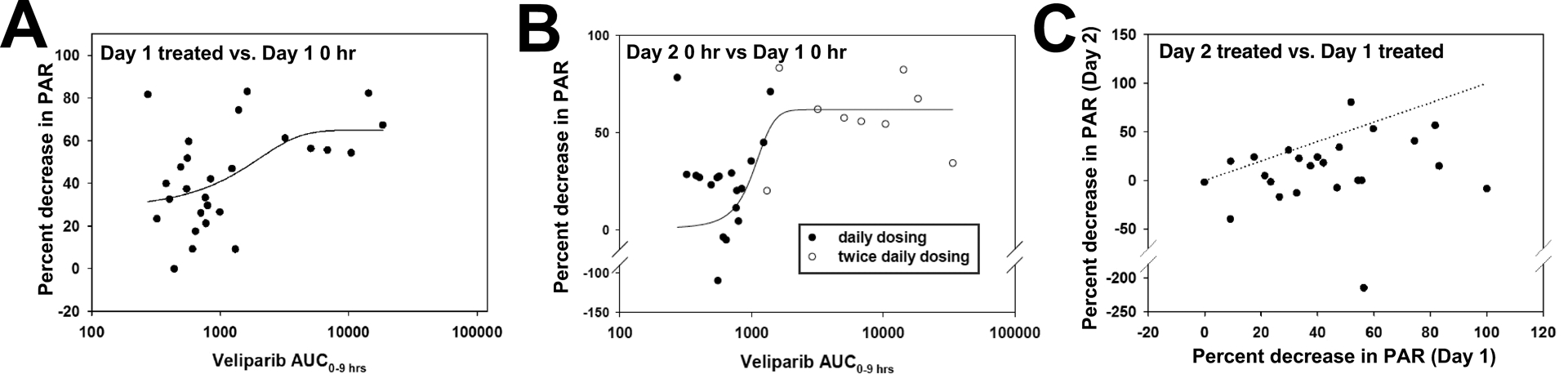
A, percent decrease in PAR levels at 2–8 hours on Day 1 versus veliparib AUC0–9h. Shown is the mean decrease at 2, 4 and 8 hours after veliparib treatment. Values at or below the LLQ were assumed to be at the LLQ. Line shows fit of data to a 3-parameter sigmoidal equation by non-linear regression using SigmaPlot (version 12.0). R2 = 0.25, p = 0.031. B, percent decrease in PAR levels at 0 hours on Day 2 versus veliparib AUC0–9h on Day 1. Samples with values below the LLQ at 0 h on Day 2 were omitted. Line shows fit of data to a 3-parameter sigmoidal equation by non-linear regression using SigmaPlot (version 12.0). R2 = 0.33, p = 0.007. Closed circles, patients receiving once daily dosing; open circles, twice daily dosing. C, relationship between mean decrease in PAR levels at hours 2–8 on Day 1 relative to Day 1 baseline vs. decrease in PAR levels at hours 2–8 on Day 2 relative to Day 2 baseline.Dashed line indicates equal % inhibition on Days 1 and 2.
Baseline PAR levels on Day 30 were 130 ± 100% (range 44–540%) of baseline PAR levels on Day 1 (Fig. S3A, lower panel, open circles).After treatment with topotecan alone, PAR levels on Day 30 increased in 11 of 20 quantifiable pairs of samples, decreased in 7 pairs and remained unchanged in 2 (Figs. S3A and S3B). While this weak trend (p = 0.12 by sign test) is suggestive of earlier reports that topo I poisons activate PAR synthesis in tissue culture lines in vitro (30,34), topo I levels are known to be low in resting PBCs (44), which also have few advancing replication forks to collide with topo I-DNA complexes to produce additional damage that would activate PARP1. Accordingly, changes in PAR did not correlate with topotecan peak concentrations or AUC (Fig. S3B).
Finally, after treatment with the combination on Day 2, PBMC PAR levels decreased relative to the Day 2 baseline in 14 patients, were unchanged in 4 and increased in 6 (p = 0.04 by sign test; Fig. 2C and Fig. S3A, lower panel). The percentage change from baseline on Day 2 was generally smaller than on Day 1 (Fig. 2C, p = 3.7 × 10−4 by sign test) reflecting diminished ability to suppress PAR below the lower baseline levels on Day 2 and/or the impact of TPT on polymer synthesis.
Antitumor activity
Evaluable patients (n=51) received 1 to 26 cycles of therapy. The mean number of cycles for all patients enrolled was 4.5 cycles.The median number of cycles was two. One patient with primary peritoneal cancer had a CR and remained on study for 14 cycles before disease progression.Four patients had PRs as their best response. An additional 22 patients had stable disease for at least four months as their best response, with nine patients having stable disease for six or more cycles and three for nine or more cycles.
Because the vast majority of patients on the study had ovarian carcinoma, most responses were seen within this group. Of the six patients who did not have ovarian carcinoma, one woman with endometrial carcinoma remained on study for 26 cycles with PR as her best response and the other five did not have objective responses. Of the ovarian carcinoma patients, 1 had a CR and 3 had a PR (n=45; 9%), with an additional 21 maintaining stable disease. Ten women with ovarian cancer remained on study for 6–18 cycles.
Overall, the objective response rate for the trial was 10%, with an additional 42% achieving SD. The correlation between dose level and response was not significant. Instead clinical responses (CR, PR, SD) were seen across multiple dose levels (Table 2). The CR was seen at dose level 5 (20 mg veliparib and 4 mg/m2 topotecan). This dose of topotecan was subsequently reduced due to DLTs at this dose level. However, three of the four PRs were seen at dose level 12, the MTD.
Relationship between germline HRR gene mutations and time on treatment
56 of the 58 patients underwent germline BROCA mutation analysis (45). Within the group, there were 14 patients with deleterious germline BRCA1 mutations, two with BRCA2 mutations, one with a RAD51D mutation, and one with a CHEK2 mutation as well as one patient with an MSH6 mutation (endometrial cancer). Among the 45 evaluable ovarian cancer patients with BROCA testing, those with germline BRCA mutations (n=14) received a mean of 5.8 cycles and median of 4 cycles, whereas those without germline BRCA mutation (n=31) received a mean and median of 4.4 and 2.0 cycles, respectively. Within the BRCA mutated ovarian cancers (n = 14), there were 0 CR, 3 PR, and 7 SD, whereas in the germline BRCA wildtype ovarian cancers (n = 31), there were 1 CR, 1 PR, and 14 SD. One ovarian cancer patient had a Rad51D mutation and maintained stable disease for 14 cycles. Accordingly, in the 15evaluable patients with BRCA or RAD51D mutation, the mean number of cycles was 6 and the median number of cycles was 4. Interestingly, the endometrial cancer patient with a germline MSH6 mutation had stable disease as her best response and also remained on study for 26 cycles before disease progression.
Discussion
There has been significant interest in combining PARP inhibitors with other agents such as DNA damaging agents, anti-angiogenics and immune checkpoint inhibitors such as anti-PD-1 antibodies and IDO inhibitors. Trials examining the latter combinations are currently underway. PARP inhibitors are currently FDA approved for the 3rd or 4th line therapy of ovarian cancers with somatic or germline BRCA1 or BRCA2 mutations as well as a maintenance therapy for platinum responsive recurrent ovarian cancer regardless of BRCA status. Although these are broad indications, the combination of topotecan and veliparib could potentially be used for women with platinum resistant disease and no BRCA mutation.
Starting with the less myelosuppressive weekly schedule of topotecan, the present study identified the MTD and RP2D of the topotecan/veliparib combination. In addition, as discussed in greater detail below, this study also established that inhibition of poly(ADP-ribose) polymer formation in PBMCs was detectable but modest at PARP inhibitor doses administered. Moreover, the regimen was associated with objective responses and disease stabilization in patients with platinum-resistant ovarian cancer whether germline BRCA mutations were present or not. All of these observations have potential implications for the future development of topo I poison/PARP inhibitor combinations.
The combination of weekly topotecan with veliparib administered for 3 days around the time of each topotecan dose was generally well tolerated, with a manageable safety profile in patients with advanced solid malignancies. The MTD of the combination was veliparib 300 mg twice daily by mouth on Days 1–3, 8–10 and 15–17 along with topotecan 3 mg/m2 intravenously on Days 2, 9 and 16. This represents ¾ of topotecan MTD in single-agent studies as well as ¾ of daily dose of veliparib administered at the MTD, albeit on a more contracted schedule.
Several prior clinical trials have evaluated combinations of camptothecins and PARP inhibitors (36,39,46,47). In a trial of veliparib with topotecan administered daily for 5 days (39), the MTDs of veliparib and topotecan were much lower than in the present trial (10 mg BID and 0.6 mg/m2/dose, respectively) and hematological toxicity precluded dose escalation. When olaparib was combined with the daily x 3 topotecan regimen (36), the MTDs were olaparib 100 bid and topotecan 1 mg/m2– both far below the individual single-agent MTDs– and neutropenia precluded dose escalation. Examination of the olaparib/irinotecan combination on two different schedules (47) established that administration of irinotecan every two weeks along with olaparib twice daily on days 1–5 was the better tolerated regimen, although only 50 mg of olaparib/dose (1/6 the daily dose of olaparib at the single-agent MTD) could be administered at the RP2D of the combination. Finally, a recently published phase I of the veliparib/irinotecan combination established the MTD as irinotecan 100 mg/m2 on days 1 and 8 with 40 mg of veliparib twice daily for 14 days of every 21-day cycle. In each of these cases, a PARP inhibitor was added to a regimen that already had myelosuppression as one of its prominent toxicities; and the result was dose-limiting marrow toxicity at relatively modest PARP inhibitor concentrations. In contrast, the weekly regimen of topotecan is less myelosuppressive. Although dose limiting toxicity was observed at dose level 5 (20 mg veliparib twice daily on days 1–3, 8–10 and 15–17) when we attempted to administer topotecan at its single-agent MTD of 4 mg/m2 on days 2, 9 and 16, we were able to easily escalate the PARP inhibitor 10-fold higher by fixing the topotecan at ¾ of its single-agent MTD. The PK of topotecan and veliparib in this study were similar to those found in a previous study of the combination (39).The PK of veliparib were not altered by coadministration with topotecan over the broad range of veliparib doses studied in this trial.While there might be a slight effect of veliparib on topotecan clearance, it is very modest and does not appear to be clinically significant, as the 3 mg/m2 topotecan dose was tolerable over a 10 – 400 mg dose range for veliparib.
In the prior studies of veliparib combined with topotecan or irinotecan (39,46), reductions in PBMC PAR levels were seen despite the fact that the doses of veliparib were far below the single-agent MTD. In the phase 1 trial that evaluated veliparib with the 5 day topotecan regimen, paired tumor biopsies from three patients, all obtained at doses above the MTD, also demonstrated a >75% decrease in PAR levels at 4–6 hours post therapy. In the current study we likewise observed a reduction in PAR levels in PBMCs after administration of veliparib. In our study there was also a trend toward an overall decrease in PAR levels at higher veliparib exposures (Fig. 2A).Moreover, as observed in the Day 2 pretreatment samples (Fig. 2B), PAR levels had substantially rebounded from their nadir before the end of the 24-hour dosing interval at dose levels 1–6.At dose levels 8–13, which involved twice daily dosing, suppression at 12 hours after the last dose was somewhat greater, but polymer was readily detected at 20–70% of baseline in many samples (Fig. 2B, open circles), suggesting that bulk PAR synthesis was not being effectively suppressed throughout the interval between doses. Whether this would also have been observed in tumor tissue is currently unknown. Nonetheless, the observation that clinical responses (Table 2) as well as dose-limiting marrow suppression (Table 3) occur at veliparib doses that give incomplete suppression of PAR synthesis might be explained by earlier preclinical studies showing that veliparib traps PARP1 and PARP2 on damaged DNA (33) and PARP trapping contributes to topo I poison/PARP inhibitor synergy (34).
The present results also need to be placed in the context of recent PARP inhibitor combinations. Bang et al. have reported that olaparib does not increase the response rate to paclitaxel in relapsed gastric cancer (http://oncologypro.esmo.org/Meeting-Resources/ESMO-2016/Olaparib-in-combination-with-paclitaxel-in-patients-with-advanced-gastric-cancer-who-have-progressed-following-first-line-therapy-Phase-III-GOLD-study). It is important to emphasize that olaparib was administered at doses far below the MTD in that study and there was little evidence to suggest that olaparib would synergize with a microtubule inhibitor. Unpublished results also suggest that veliparib fails to enhance the response rate to carboplatin and paclitaxel in non-small cell lung cancer and triple negative breast cancer (https://news.abbvie.com/news/abbvie-announces-topline-results-from-two-phase-3-studies-investigating-veliparib-in-combination-with-chemotherapy-for-treatment-patients-with-advanced-or-metastatic-squamous-non-small-cell-lung-cancer-and-early-stage-triple-negative-breast-cancer.htm), again mirroring preclinical results that failed to observe synergy between PARP inhibitors and platinum (34,48). In contrast, a variety of studies in tissue culture and xenograft models have demonstrated synergy between topo I poisons and PARP inhibitors, including veliparib (23–26,34). These observations, coupled with the safety profile described in the present study, suggest that further study of the topotecan/ veliparib combination might be warranted. On the other hand, PARP inhibitors have also been shown to increase death receptor expression, leading to enhanced sensitivity to cytokines such as FAS ligand and TRAIL that are produced by cytotoxic T cells and activated monocytes (49). These latter observations, along with promising results in immunocompetent mouse models and a variety of additional studies, provide a preclinical basis for PARP inhibitor/immunotherapy trials.
The vast majority of the patients enrolled in the present trial had ovarian cancer. At the time the trial started, cells lacking BRCA1, BRCA2 and other homologous recombination (HR) repair proteins were known to be hypersensitive to veliparib (50); and deleterious BRCA1 or BRCA2 mutations were known to be frequent in ovarian cancer. Additional data indicated that cells lacking BRCA2 are also hypersensitive to topo I poisons and are sensitized even further by veliparib (34). Although responses were noted in patients without BRCA mutations, the above observations prompted us to examine the relationship between germline mutations in DNA repair genes and response.
In ovarian cancer patients with deleterious germline DNA repair pathway mutations, the objective response rate was 25% on this veliparib/topotecan regimen even though all patients had platinum-resistant disease. In contrast, there were no responses, only disease stability, in the phase 1 study combining veliparib with the 5-day schedule of topotecan (39). In the combination of veliparib with irinotecan (46), 9 patients with ovarian cancer enrolled, all with documented BRCA germline mutations. Among these 9 patients, 1 (with platinum sensitive disease) achieved a PR and 6 achieved SD (1 with platinum sensitive and 5 with platinum resistant cancer).
In the present study, ovarian cancer patients with germline BRCA1, BRCA2 or RAD51 mutations were also on study somewhat longer (mean 6 cycles, median 4 cycles) than patients without germline mutations (mean 4.4 cycles, median 2). This trend, however, was not statistically significant and was also confounded by the fact that patients received different doses of veliparib and topotecan across 13 dose levels. Likewise, among the 16 patients with germline homologous recombination defects, there was a trend toward increased clinical benefit (3 PR, 8 SD among 15 patients vs. 1 CR + 1 PR + 14 SD among 30 patients). This also did not reach statistical significance (p = 0.21 by Fisher’s exact test) and must interpreted in light of the wide variation in doses administered.
In summary, when veliparib is added to the weekly topotecan regimen, hematological toxicity is dose-limiting, but both agents can be administered at ¾ of their individual agent MTDs. This regimen has a manageable safety profile and exhibits signs suggestive of potential activity, particularly in ovarian cancer patients with deleterious germline repair pathway mutations. Accordingly, a phase 2 clinical trial is currently underway in platinum resistant ovarian carcinoma.
Supplementary Material
Translational Relevance:
There is significant interest in combining PARP inhibitors with DNA damaging agents, especially in cancers with frequent defects in the HRR pathway such as ovarian cancer. Preclinical observations indicate that addition of PARP inhibitors to topoisomerase I-directed agents such as topotecan results in increased antitumor efficacy in vitro and in vivo. When veliparib or olaparib was combined with daily topotecan for 5 days every 3 weeks, however, the regimen was quite myelosuppressive. This phase I study investigated the addition of veliparib to weekly topotecan, a less myelosuppressive but routinely used regimen, in patients with advanced solid malignancies. The results demonstrated a manageable safety profile and early signs of activity as manifested by responses or disease stability for ≥ 4 months in 37% of patients. A phase 2 clinical trial in advanced platinum resistant ovarian carcinoma is underway.
Acknowledgements
The authors would like to thank all patients and oncology providers for their participation in this study. Special thanks go to the research assistants who helped collect data for this study and Sarah A. Buhrow for assistance with preparation of figures.
Acknowledgments
Supported in part by UM1 CA186686 (C. Erlichman, P. Haluska), 5K12 CA090628 (A. Wahner Hendrickson) and P50 CA136393 (L. Hartmann, H. Long, K. Flatten, P. Haluska, S. Kaufmann) from the National Cancer Institute as well as support to A. Wahner Hendrickson, E. Swisher and S. Kaufmann from Stand Up To Cancer - Ovarian Cancer Research Fund Alliance - National Ovarian Cancer Coalition Dream Team Translational Research Grant (Grant Number:SU2C-AACR-DT16-15).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed by the authors.
Clinical trial information: NCT01012817
References
- 1.Khadka DB, Cho WJ. Topoisomerase inhibitors as anticancer agents: a patent update. Expert Opin Ther Pat 2013;23(8):1033–56. [DOI] [PubMed] [Google Scholar]
- 2.Kumler I, Brunner N, Stenvang J, Balslev E, Nielsen DL. A systematic review on topoisomerase 1 inhibition in the treatment of metastatic breast cancer. Breast Cancer Res Treat 2013;138(2):347–58. [DOI] [PubMed] [Google Scholar]
- 3.Beretta GL, Gatti L, Perego P, Zaffaroni N. Camptothecin resistance in cancer: insights into the molecular mechanisms of a DNA-damaging drug. Curr Med Chem 2013;20(12):1541–65. [DOI] [PubMed] [Google Scholar]
- 4.Moukharskaya J, Verschraegen C. Topoisomerase 1 inhibitors and cancer therapy. Hematol Oncol Clin North Am 2012;26(3):507–25. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert DC, Chalmers AJ, El-Khamisy SF. Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets. Br J Cancer 2012;106(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sparreboom A, Zamboni WC. Topoisomerase I-Targeting Drugs In: Chabner BA, Longo DL, editors. Cancer Chemotherapy and Biotherapy. Fourth ed: Lippincott Williams & Wilkins; 2006. p 371–413. [Google Scholar]
- 7.Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387(10018):545–57. [DOI] [PubMed] [Google Scholar]
- 8.Riemsma R, Simons JP, Bashir Z, Gooch CL, Kleijnen J. Systematic Review of topotecan (Hycamtin) in relapsed small cell lung cancer. BMC Cancer 2010;10:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munster PN, Daud AI. Preclinical and clinical activity of the topoisomerase I inhibitor, karenitecin, in melanoma. Expert Opin Investig Drugs 2011;20(11):1565–74. [DOI] [PubMed] [Google Scholar]
- 10.Jeong W, Park SR, Rapisarda A, Fer N, Kinders RJ, Chen A, et al. Weekly EZN-2208 (PEGylated SN-38) in combination with bevacizumab in patients with refractory solid tumors. Invest New Drugs 2014;32(2):340–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santi DV, Schneider EL, Ashley GW. Macromolecular prodrug that provides the irinotecan (CPT-11) active-metabolite SN-38 with ultralong half-life, low C(max), and low glucuronide formation. J Med Chem 2014;57(6):2303–14. [DOI] [PubMed] [Google Scholar]
- 12.Perez EA, Awada A, O’Shaughnessy J, Rugo HS, Twelves C, Im SA, et al. Etirinotecan pegol (NKTR-102) versus treatment of physician’s choice in women with advanced breast cancer previously treated with an anthracycline, a taxane, and capecitabine (BEACON): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol 2015;16(15):1556–68. [DOI] [PubMed] [Google Scholar]
- 13.Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 2001;19(14):3312–22. [DOI] [PubMed] [Google Scholar]
- 14.ten Bokkel Huinink W, Gore M, Carmichael J, Gordon A, Malfetano J, Hudson I, et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J Clin Oncol 1997;15:2183–93. [DOI] [PubMed] [Google Scholar]
- 15.Morris R, Munkarah A. Alternate dosing schedules for topotecan in the treatment of recurrent ovarian cancer. Oncologist 2002;7 Suppl 5:29–35. [DOI] [PubMed] [Google Scholar]
- 16.Rowinsky EK. Weekly topotecan: an alternative to topotecan’s standard daily x 5 schedule? Oncologist 2002;7(4):324–30. [DOI] [PubMed] [Google Scholar]
- 17.O’Malley DM, Azodi M, Makkenchery A, Tangir J, McAlpine J, Kelly M, et al. Weekly topotecan in heavily pretreated patients with recurrent epithelial ovarian carcinoma. Gynecol Oncol 2005;98(2):242–8. [DOI] [PubMed] [Google Scholar]
- 18.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012;12(12):801–17. [DOI] [PubMed] [Google Scholar]
- 19.Plummer R PARP inhibitors. Clin Adv Hematol Oncol 2012;10(5):322–3. [PubMed] [Google Scholar]
- 20.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet 2014;384(9951):1376–88. [DOI] [PubMed] [Google Scholar]
- 21.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J Clin Oncol 2015;33:1397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castro E, Mateo J, Olmos D, de Bono JS. Targeting DNA Repair: The Role of PARP Inhibition in the Treatment of Castration-Resistant Prostate Cancer. Cancer J 2016;22(5):353–6. [DOI] [PubMed] [Google Scholar]
- 23.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res 2000;6(7):2860–7. [PubMed] [Google Scholar]
- 24.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst 2004;96(1):56–67. [DOI] [PubMed] [Google Scholar]
- 25.Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther 2007;6(3):945–56. [DOI] [PubMed] [Google Scholar]
- 26.Das BB, Huang SY, Murai J, Rehman I, Ame JC, Sengupta S, et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res 2014;42(7):4435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Avemann K, Knipper R, Koller T, Sogo JM. Camptothecin, A Specific Inhibitor of Type I DNA Topoisomerase, Induced DNA Breakage at Replication Forks. Mol Cell Biol 1988;8:3026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin CG, Snapka RM. Exposure to Camptothecin Breaks Leading and Lagging Strand Simian Virus 40 DNA Replication Forks. Biochem Biophys Res Commun 1990;168(1):135–40. [DOI] [PubMed] [Google Scholar]
- 29.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 2013;20(3):347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimura K, Takebayashi S, Taguchi H, Takeda S, Okumura K. PARP-1 ensures regulation of replication fork progression by homolgous recombination on damaged DNA. J Cell Biol 2008;183:1203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryant HE, Petermann E, Schultz N, Jemth A-S, Loseva O, Issaeva N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009;28(17):2601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016;535(7612):382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol Cancer Res 2015;13(11):1465–77. [DOI] [PubMed] [Google Scholar]
- 34.Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, et al. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase inhibitors reflects poisoning of both enzymes. J Biol Chem 2012;287:4198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8(362):362ps17. [DOI] [PubMed] [Google Scholar]
- 36.Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs 2012;30(4):1493–500. [DOI] [PubMed] [Google Scholar]
- 37.ten Berge RL, Meijer CJLM, Dukers DF, Kummer JA, Bladergroen BA, Vos W, et al. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood 2002:4540–6. [DOI] [PubMed] [Google Scholar]
- 38.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 2007;13(9):2728–37. [DOI] [PubMed] [Google Scholar]
- 39.Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res 2011;71(17):5626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowinsky EK, Grochow LB, Hendricks CB, Ettinger DS, Forastiere AA, Hurowitz LA, et al. Phase I and Pharmacologic Study of Topotecan:A Novel Topoisomerase I Inhibitor. J Clin Oncol 1992;10(4):647–56. [DOI] [PubMed] [Google Scholar]
- 41.Huggins-Puhalla SL, Beumer JH, Appleman LJ, Tawbi HA. A Phase I Study of Chronically Dosed, Single-Agent Veliparib (ABT-888) in Patients with Either BRCA 1/2-mutated cancer (BRCA+), platinum-refractory ovarian cancer, or basal-like breast cancer (BRCA-wt). J Clin Oncol 2012;30:Abstr 3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufmann SH, Karp JE, Letendre L, Kottke TJ, Safgren S, Greer J, et al. Phase I and Pharmacological Study of Infusional Topotecan and Carboplatin in Relapsed and Refractory Leukemia. Clin Cancer Res 2005;11:6641–9. [DOI] [PubMed] [Google Scholar]
- 43.Ji J, Kinders RJ, Zhang Y, Rubinstein L, Kummar S, Parchment RE, et al. Modeling pharmacodynamic response to the poly(ADP-Ribose) polymerase inhibitor ABT-888 in human peripheral blood mononuclear cells. PLoS One 2011;6(10):e26152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaufmann SH, Charron M, Burke PJ, Karp JE. Changes in Topoisomerase I Levels and Localization During Myeloid Maturation In Vitroand In Vivo. Cancer Res 1995;55:1255–60. [PubMed] [Google Scholar]
- 45.Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol 2016;2(4):482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, et al. Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor Veliparib (ABT-888) in Combination with Irinotecan in Patients with Advanced Solid Tumors. Clin Cancer Res 2016;22(13):3227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen EX, Jonker DJ, Siu LL, McKeever K, Keller D, Wells J, et al. A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Invest New Drugs 2016;34(4):450–7. [DOI] [PubMed] [Google Scholar]
- 48.Huehls AM, Wagner JM, Huntoon CJ, Geng L, Erlichman C, Patel AG, et al. Poly(ADP-Ribose) polymerase inhibition synergizes with 5-fluorodeoxyuridine but not 5-fluorouracil in ovarian cancer cells. Cancer Res 2011;71(14):4944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng XW, Koh B, Zhang J-S, Flatten KS, Schneider PA, Billadeau DD, et al. Poly(ADP-Ribose) Polymerase Inhibitors Sensitize Cancer Cells to Death Receptor-Mediated Apoptosis by Enhancing Death Receptor Expression. J Biol Chem 2014;289:20543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashworth A A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J Clin Oncol 2008;26(22):3785–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.