

Abstract
Cerebral microbleeds (CMB) are a common MRI finding, representing underlying cerebral microhemorrhages (CMH). The etiology of CMB and microhemorrhages is obscure. We conducted a pathological investigation of CMH, combining standard and immunohistological analyses of postmortem human brains. We analyzed 5 brain regions (middle frontal gyrus, occipital pole, rostral cingulate cortex, caudal cingulate cortex, and basal ganglia) of 76 brain bank subjects (mean age ± SE 90 ± 1.4 years). Prussian blue positivity, used as an index of CMH, was subjected to quantitative analysis for all 5 brain regions. Brains from the top and bottom quartiles (n = 19 each) were compared for quantitative immunohistological findings of smooth muscle actin, claudin-5, and fibrinogen, and for Sclerosis Index (SI) (a measure of arteriolar remodeling). Brains in the top quartile (i.e. with most extensive CMH) had significantly higher SI in the 5 brain regions combined (0.379 ± 0.007 vs 0.355 ± 0.008; p < 0.05). These findings indicate significant coexistence of arteriolar remodeling with CMH. While these findings provide clues to mechanisms of microhemorrhage development, further studies of experimental neuropathology are needed to determine causal relationships.
Keywords: Arteriosclerosis, Autopsy, Blood-brain barrier, Cerebral microhemorrhage, Histology, Immunohistochemistry
INTRODUCTION
Cerebral microbleeds (CMB) are a common MRI finding in the brains of elderly individuals and mark the presence of cerebral microhemorrhage (CMH) (1–3). CMH can be identified by histochemical studies that detect hemosiderin using Prussian blue (PB) staining (2). CMB have been shown to be associated with poor functional outcomes in stroke patients (4), increased risk of ischemic strokes and intracerebral hemorrhage (5, 6), cognitive decline, and memory impairment (7). It is thus important to identify pathogenic mechanisms underlying CMB formation in order to devise proper prevention and treatment strategies (8).
CMB have well-established comorbidities of hypertension (9, 10), cerebral amyloid angiopathy (CAA) (10, 11), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (12, 13), Alzheimer disease (14), and chronic kidney disease (CKD) (15, 16). Other evidence suggests that CMB may share common mechanisms with small deep infarcts in the context of small vessel disease (17, 18). These associated risk factors and disease entities have been implicated in the development of arteriolar injury and capillary abnormalities involving the blood-brain barrier (BBB) (8). However, exact pathogenic mechanisms underlying the formation of microhemorrhages are still poorly understood. Thus, we devised a postmortem mechanistic study combining quantitative histopathological and immunohistological analyses of human brain specimens in an attempt to elucidate pathogenic mechanisms of CMH.
MATERIALS AND METHODS
Ethics and Data Availability Statements
This study was approved by the Institutional Review Board at University of California, Irvine. Requirements for patient consents were waived due to the retrospective postmortem design of the study. Data from this study are available upon request to the corresponding author from qualified investigators.
Study Population
This retrospective postmortem study examined brain bank specimens from participants of UCI Alzheimer’s Disease Research Center (ADRC) and The 90+ Study, who died between January 01, 2013 and September 30, 2015. A total of 124 participants were available. After applying exclusion criteria, 76 participants remained for analysis (Fig. 1). Excluded were participants with certain diagnoses (Trisomy 21, hereditary diffuse leukoencephalopathy with axonal spheroids, corticobasal degeneration, and frontotemporal lobar degeneration), participants with incomplete data, brains that were formalin fixed, or brains with tissue unavailable for regions of interest.
FIGURE 1.

Study population. *Excluded diagnoses: Trisomy 21, hereditary diffuse leukoencephalopathy with axonal spheroids, corticobasal degeneration, and frontotemporal lobar degeneration.
Brain Collection Procedures
Brain collection procedures were performed by the UCI ADRC neuropathology core and The 90+ Study. The brains were removed using a Stryker autopsy saw, and blood and cerebrospinal fluid were collected within an hour of death at the UCI Medical Center. The brain specimens were then taken to the UCI School of Medicine campus repository. For each specimen, the left hemisphere was fixed whole in 4% paraformaldehyde and the right hemisphere was divided into approximately twelve 2-cm coronal sections and frozen at –80°C. A few brain regions including the hippocampus and brainstem were dissected from the right hemisphere and frozen as well. Tissue fixation and freezing were done 1.5–2 hours after death.
Study Design
We sampled tissue from 5 brain regions for all 76 participants. The 5 sampled areas were taken from the following sites: Middle frontal gyrus at the level of the genu of the corpus callosum (MF), calcarine/pericalcarine cortex 2 cm rostral to the occipital pole (OCC), rostral cingulate cortex at the level of the genu of the corpus callosum (CGA), caudal cingulate cortex at the level of the splenium of the corpus callosum (CGP), and striatum at the level of the mammillary bodies (BG2). Tissue from these 5 brain regions were sectioned at 4.5 µm thickness and mounted onto plus-charged slides and stained for PB by the research service core at the UCI Medical Center Department of Pathology & Laboratory Medicine. PB-stained slides were screened by UCI neuropathologist (R.C.K.) for PB positivity and an area within each section was chosen for PB load quantification. Briefly, 2448 × 2048 and 1442 × 1206 pixels images for each selected area were photographed using Olympus BX51 microscope at 10× objective, Infinity 2 Camera, and INFINITY ANALYZE 6.5.0 software (Lumera Corporation, ON, Canada). PB load was measured by scanning each image by a Red Green Blue Area Analyzer program (RGB-AA program). The PB-positive foci were highlighted and a PB-positive area per image was generated based on the pixels per micron of the scale bar. Using the PB-positive area, the 76 participants were stratified based on PB positivity. The top and bottom quartiles (n = 19 each) were determined and used as the bases of the immunohistological comparisons. RGB-AA calculates area based on pixel threshold tracing. RGB color space works by combining various intensities of red, green, and blue ranging from 0 to 255, zero being the absence of the specific color and 255 being 100% of the color wave. The program works by using the selected color and calculating the number of pixels that fall within a specific threshold of said color. The pixels/micron determined from the scale bar of the image is then used by the program to calculate the total positive area. The specific RGB value for the PB stain and immunohistochemistry (IHC) stains were determined by opening respective sample images within the program and adjusting the RGB values until reaching a maximal coverage of the objects of interest (e.g. red chromogen or PB) and minimal coverage of the background.
To study the relationship between PB load and vascular smooth muscle cells and BBB structure and function, IHC was performed on the 5 brain regions (i.e. CGP, CGA, MF, OCC, and BG2) using red chromogen. IHC was done by the research service core at the UCI Medical Center Department of Pathology & Laboratory Medicine. The 5 brain regions of each case were stained for smooth muscle actin (SMA) (Ventana Medical Systems, Tucson, AZ), claudin-5 (Life Technologies, Carlsbad, CA), fibrinogen (Agilent Dako, Santa Clara, CA), and von Willebrand factor (VWF) (Abcam, Cambridge, UK). An area of interest for each IHC stain of each brain region was selected by UCI neuropathologist (R.C.K.). Using 4× and 10× microscope objectives, solitary, randomly selected microscopic fields were identified within the striatum and cerebral cortex and encircled with a marking pen. For the striatum, we created a 0.5-cm-diameter circle using 4× objective within putamen sampled at the level of the mammillary bodies. For the cerebral cortex, we created a 0.2-cm-diameter circle using 10× objective.
Analyses
Claudin-5, Fibrinogen, and VWF
Four adjacent images, 2448 × 2048 pixels, for the BG2 region and one image, 1442 × 1206 pixels, for CGP, CGA, MF, and OCC regions were photographed for each case using Olympus BX51 microscope at 10× objective, Infinity 2 Camera, and INFINITY ANALYZE 6.5.0 software. Total red chromogen-positive area (RCPA) was measured using RGB-AA program. The RCPA was standardized to the area analyzed and to the number of vessels. The number of vessels was determined by using VWF-stained sections. NIH Image J software cell counter tool was used to count the number of vessels within the area of interest.
Smooth Muscle Actin
SMA-stained arterioles were photographed at 60× objective; 10 vessels in BG2 and 5 vessels in CGP, CGA, MF, and OCC. Strict criteria were used in choosing the vessels: Vessels needed to be distended, in cross-section, shaped close to a circle or ellipse, and within size range of 10–125 µm in diameter. The RCPA was determined for each selected arteriole using the RGB-AA program. The RCPA for each vessel was standardized based on the diameter of the vessel. The diameter was calculated by measuring the length at the narrowest and widest points of the vessel and taking the average. Length measurements were done using NIH Image J software.
Sclerosis Index
Sclerosis Index (SI) is a measure of vascular wall thickness and arteriolar injury, with SI = 1 – (inner diameter/outer diameter). SMA-stained vessels were photographed under 60× objective, 10 vessels in BG2 and 5 vessels in CGP, CGA, MF, and OCC. Vessels were selected following the same criteria as the SMA RCPA analysis. SI for each selected vessel was calculated by measuring the inner and outer diameters using NIH Image J software. Two SI values were calculated per vessel, one at the widest axis and one at the narrowest axis of the arteriole. Final SI value per vessel was the average of the SI of the widest and narrowest axes (Fig. 2). All measurements within the study were done by observers blinded to the group designations of participants.
FIGURE 2.
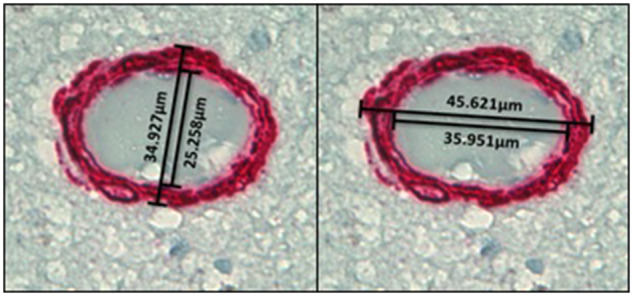
Illustrative images of Sclerosis Index (SI) measurement at 60× magnification. The final SI value per vessel is the average of the SI of the widest and narrowest axes of the arteriole.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 7 and Microsoft Excel 2010. Differences in the means of the variables between top and bottom PB load quartiles were analyzed using the Student t-test and in proportions using the Fisher exact test. Two-sided p < 0.05 was considered statistically significant.
RESULTS
Participant Characteristics
Of the 124 potential participants, 76 met all inclusion criteria detailed in Figure 1. The average age ± standard error of the 76 participants was 90 ± 10.4 years. The average PB positivity, used as an index of CMH, was 4384 ± 750 µm2 and ranged from 0 to 31 690 µm2. Age was not significantly related to PB load (correlation coefficient = 0.10).
Patients in the top quartile of PB positivity (i.e. with most extensive CMH) had on average significantly higher PB positivity than patients in the bottom quartile (12 839 ± 1930 vs 192 ± 47 µm2) (Table; Fig. 3). The mean age of participants in the top quartile (93 ± 2.6 years) was higher than that in the bottom quartile (85 ± 3.5 years). However, this difference in age was not significant. Most of the participants had hypertension (72% and 67% in the top and bottom quartiles). Although not statistically significant, histories of stroke or transient ischemic attack (TIA) were higher in the top quartile (22% vs 6%, p = 0.34) as was ApoE2 genotype (31% vs 8%, p = 0.20). Table highlights additional participant characteristics and shows a breakdown of the various diagnoses within each group.
TABLE.
Characteristics of Brain Bank Participants and Those Available for Immunostaining
| All Potential Subjects | Paraformaldehyde-Fixed and All Brain Regions | Prussian Blue | Prussian Blue | |
|---|---|---|---|---|
| Top Quartile | Bottom Quartile | |||
| n = 113 | n = 76 | n = 19 | n = 19 | |
| Age, years | ||||
| Mean ± SE | 91 ± 1.2 | 90 ± 1.4 | 93 ± 2.6 | 85 ± 3.5 |
| Range | 46–110 | 58–110 | 68–110 | 58–101 |
| MMSE | ||||
| Mean ± SE | 17.4 ± 0.9 | 16.3 ± 1.1 | 13.8 ± 2.3 | 16.3± 2.2 |
| Range | 0–30 | 0–30 | 0–30 | 0–28 |
| Number missing data | 9 | 7 | 1 | 4 |
| Male | 40 (35%) | 29 (38%) | 9 (47%) | 7 (37%) |
| Smoker | 47 (44%) | 30 (43%) | 9 (50%) | 5 (29%) |
| Number missing data | 7 | 6 | 1 | 2 |
| Medical History | ||||
| Hypertension | 77 (72%) | 48 (69%) | 13 (72%) | 12 (67%) |
| Number missing data | 6 | 5 | 1 | 1 |
| Diabetes | 10 (9.3%) | 7 (9.9%) | 2 (11%) | 2 (11%) |
| Number missing data | 6 | 5 | 1 | 1 |
| Stroke | 18 (17%) | 9 (13%) | 4 (22%) | 1 (6%) |
| Number missing data | 7 | 7 | 1 | 1 |
| TIA | 24 (23%) | 12 (18%) | 4 (22%) | 1 (6%) |
| Number missing data | 10 | 8 | 1 | 1 |
| APOE genotype | ||||
| Number missing data | 24 | 17 | 3 | 7 |
| 23 | 10 (11%) | 6 (10%) | 3 (19%) | 1 (8%) |
| 24 | 2 (2%) | 2 (3%) | 2 (13%) | |
| 33 | 53 (60%) | 37 (63%) | 5 (31%) | 7 (58%) |
| 34 | 20 (22%) | 12 (20%) | 5 (31%) | 4 (33%) |
| 44 | 4 (4%) | 2 (3%) | 1 (6%) | |
| Diagnosis | ||||
| Normal | 54 (48%) | 31 (41%) | 8 (42%) | 6 (32%) |
| Alzheimer’s disease | 32 (28%) | 24 (32%) | 3 (16%) | 10 (53%) |
| Alzheimer’s disease with other pathology | 19 (17%) | 16 (21%) | 5 (26%) | 2 (11%) |
| With Lewy body | 2 | 2 | 1 | |
| With Lewy body, hippocampal sclerosis | 1 | 1 | ||
| With vascular dementia | 2 | 2 | 1 | 1 |
| With vascular dementia (amyloid angiography) | 3 | 3 | 2 | |
| With vascular dementia (amyloid angiography), hippocampal sclerosis | 2 | |||
| With hippocampal sclerosis | 8 | 7 | 2 | |
| With other | 1 | 1 | ||
| Vascular dementia | 2 (2%) | |||
| Hippocampal sclerosis | 3 (3%) | 2 (3%) | 2 (11%) | |
| Parkinson’s disease | 1 (1%) | 1 (1%) | ||
| Other | 2 (2%) | 2 (3%) | 1 (5%) | 1 (5%) |
| Prussian blue | ||||
| Mean ± SE | 4384 ± 750 | 12 839 ± 1930 | 192 ± 47* | |
| Range | 0–31 690 | 5435–31 690 | 0–663 | |
p < 0.05 for difference between top and bottom quartiles of Prussian blue load.
FIGURE 3.

Prussian blue stains of cerebral microhemorrhages. (A, B) From bottom quartile of Prussian blue load positivity. (C, D) From top quartile of Prussian blue load positivity.
Top and Bottom PB Positivity Quartile Differences in Claudin-5, Fibrinogen, and SMA Levels
Claudin-5 RCPA, an index for BBB tight-junction protein levels, was not significantly different between the top and bottom quartiles in any of the 5 brain regions (Fig. 4) or in all brain regions taken collectively. Fibrinogen RCPA, an index for BBB disruption, was also not significantly different between the top and bottom quartiles in the 5 brain regions (Fig. 4) or in all collective brain regions. SMA RCPA, a measure of SMA prevalence, was not significantly different for the top quartile versus bottom quartile, with or without adjustment for vessel diameter (data not shown).
FIGURE 4.
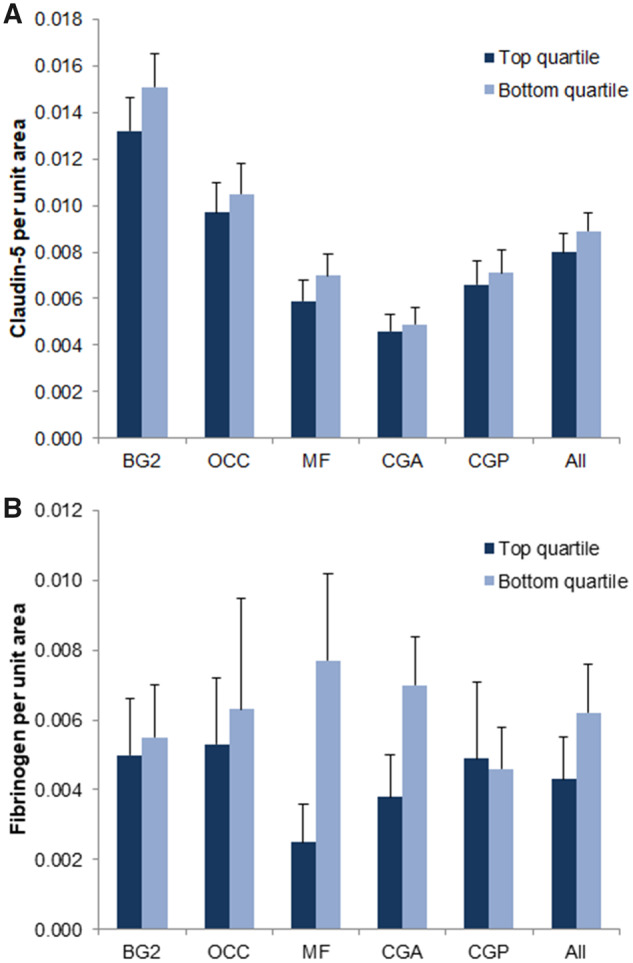
Top and bottom Prussian blue-positivity quartile differences in claudin-5 and fibrinogen immunostaining. Top and bottom Prussian blue-positivity quartile differences in claudin-5 (A) and fibrinogen (B) immunostaining in the following brain regions: Striatum at the level of the mammillary bodies (BG2), calcarine/pericalcarine cortex (OCC), middle frontal gyrus (MF), rostral cingulate cortex (CGA), caudal cingulate cortex (CGP). No significant differences in claudin-5 or fibrinogen immunoreactivity are noted between top and bottom quartiles.
Higher SI in Brains With More Extensive CMH Burden
SI was significantly higher in the top versus bottom quartile in the MF and CGA brain regions (0.410 ± 0.015 vs 0.358 ± 0.018; p = 0.03) and (0.376 ± 0.010 vs 0.341 ± 0.013; p = 0.04). We saw no significant difference in BG2, OCC, and CGP. However, patients with more extensive CMH burden (i.e. in the top PB positivity quartile) had significantly higher SI in all 5 brain regions collectively when compared with patients in the bottom quartile (0.379 ± 0.007 vs 0.355 ± 0.008; p = 0.04) (Fig. 5).
FIGURE 5.

Top and bottom Prussian blue-positivity quartile differences in Sclerosis Index (SI). Top and bottom Prussian blue positivity quartile differences in SI in the following brain regions taken separately and collectively: Striatum at the level of the mammillary bodies (BG2), calcarine/pericalcarine cortex (OCC), middle frontal gyrus (MF), rostral cingulate cortex (CGA), caudal cingulate cortex (CGP). (A) SI was significantly higher in the top quartile versus bottom quartile in the MF brain region (0.410 ± 0.015 vs 0.358 ± 0.018; p < 0.05), in the CGA region (0.376 ± 0.010 vs 0.341 ± 0.013; p < 0.05), and in all 5 brain regions taken collectively (0.379 ± 0.007 vs 0.355 ± 0.008; p < 0.05). (B, C) SI in the bottom and top quartiles, respectively, under 60× magnification. *p < 0.05.
DISCUSSION
We analyzed 76 brain bank specimens obtained from The 90+ Study and from the UCI Alzheimer’s Disease Research Center, with a focus on tissue derived from 5 brain regions that were subjected to quantitative analysis of PB staining for CMH detection. Our principal finding is significantly higher SI for top versus bottom quartile groups for PB positivity. We found no significant differences in demographic factors between the group of brains that had the most extensive CMH (i.e. top quartile for PB positivity) and the group with least extensive CMH (i.e. bottom quartile for PB positivity). There were trends for higher prevalence of clinical history of cerebrovascular disease (stroke or TIA) in the top quartile, as well as presence of the ApoE2 genotype, which has been linked to small vessel injury predisposing to hemorrhage (19); however, these differences were not statistically significant.
We used SI as a measure of the severity of arteriolar injury in this study (20). The higher mean arteriolar SI for the top PB positivity quartile group suggests a relationship between arteriolar injury and microhemorrhage development. We have proposed that development of CMB may be mediated by arteriolar injury with resulting dysfunctional cerebral blood flow regulation (8); these findings are consistent with that model. Arteriolar stiffness can also result in high pulsatile pressures in cerebral arterioles, with resulting endothelial and smooth muscle cell injury (21). Janaway et al have also reported association between basal ganglia hemosiderin and indices of microvascular disease (including infarcts and arteriolar injury), and suggested that basal ganglia hemosiderin may reflect the consequence of local ischemic injury (22). Our findings are not inconsistent with that hypothesis. However, our findings of widespread arteriolar injury extending well beyond the basal ganglia is consistent with a model of microhemorrhage formation in which the arteriole functions as both source (via local extravasation) and mediator of microhemorrhage formation.
Several important factors complicate analysis of CMH formation using PB staining. First, the timing of any microhemorrhage cannot be ascertained by this stain, as its presence simply indicates a nonacute process (23). This may help explain why we found no indication that BBB injury corresponded with microhemorrhage formation; neither changes in BBB structure (i.e. claudin-5) nor functional elements (i.e. fibrinogen) were associated with microhemorrhage load. Any acute BBB injury predisposing to microhemorrhage formation may have resolved by the time of this autopsy sample. Alternatively, microhemorrhages may develop at a site other than the BBB (22) or at the BBB via mechanisms independent of capillary injury (24). Second, PB staining does not distinguish between hemoglobin-derived and nonhemoglobin-derived iron. Systemic iron uptake is complex (25), and brain iron accumulates in a manner that is largely age-dependent (26, 27). The basal ganglia, especially globus pallidus, is the site of greatest iron accumulation (26, 27). This occurs most rapidly up to age 30, but continues to increase up to age 50–60 and perhaps beyond (28). It is unclear what proportion of brain iron is incorporated into hemosiderin versus nonhemosiderin ferritin, both of which may be identified by PB staining. These complexities argue for a cautionary note in the interpretation of our findings.
This study has several strengths. We have utilized a strictly quantitative approach for analysis of CMH. The postmortem design and access to a human brain bank provided us with a detailed examination of specific brain regions of interest and allowed us to make direct associations between microhemorrhage histopathology, arteriolar SI, and immunohistological findings. The sampling of 5 brain regions also shed light on the possibility of having different mechanisms of microvascular injury and microhemorrhage formation depending on the brain region. This study is also subject to some limitations. The participants who were included for analysis belonged to the oldest-old age group with a mean age of 90, and so our findings may not be generalizable to a younger population. With respect to the comparative analysis of markers of microvascular injury and SI, we cannot make causal associations between arteriolar injury and microhemorrhage formation due to the cross-sectional study design. Finally, we did not attempt to identify the vascular source of the microhemorrhages, which has been attributed to both capillaries and arterioles (2, 22).
In conclusion, we demonstrated significant coexistence of arteriolar injury with CMH. The exact relationship between CMH and arteriolar injury remains to be determined. Arteriolar injury may be mechanistically linked to CMB development, or may simply reflect common origin.
ACKNOWLEDGMENT
We thank Natalie Chan and Sean Rafferty for development of the quantitative histology program.
This study was supported by NIH NS20989 and NIA R01AG021055 and P50AG016573.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Charidimou A, Krishnan A, Werring DJ, et al. Cerebral microbleeds: A guide to detection and clinical relevance in different disease settings. Neuroradiology 2013;55:655–74 [DOI] [PubMed] [Google Scholar]
- 2. Fisher M, French S, Ji P, et al. Cerebral microbleeds in the elderly: A pathological analysis. Stroke 2010;41:2782–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fazekas F, Kleinert R, Roob G, et al. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: Evidence of microangiopathy-related microbleeds. Am J Neuroradiol 1999;20:637–42 [PMC free article] [PubMed] [Google Scholar]
- 4. Kim TW, Lee SJ, Koo J, et al. Cerebral microbleeds and functional outcomes after ischemic stroke. Can J Neurol Sci 2014;41:577–82 [DOI] [PubMed] [Google Scholar]
- 5. Bokura H, Saika R, Yamaguchi T, et al. Microbleeds are associated with subsequent hemorrhagic and ischemic stroke in healthy elderly individuals. Stroke 2011;42:1867–71 [DOI] [PubMed] [Google Scholar]
- 6. Lee SH, Bae HJ, Kwon SJ, et al. Cerebral microbleeds are regionally associated with intracerebral hemorrhage. Neurology 2004;62:72–6 [DOI] [PubMed] [Google Scholar]
- 7. Cordonnier C, Van der Flier WM, Sluimer JD, et al. Prevalence and severity of microbleeds in a memory clinic setting. Neurology 2006;66:1356–60 [DOI] [PubMed] [Google Scholar]
- 8. Fisher M. Cerebral microbleeds and thrombolysis: Clinical consequences and mechanistic implications. JAMA Neurol 2016;73:632–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vernooij MW, van der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: The Rotterdam Scan Study. Neurology 2008;70:1208–14 [DOI] [PubMed] [Google Scholar]
- 10. Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol 2009;8:165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Graff-Radford J, Botha H, Rabinstein AA, et al. Cerebral microbleeds: Prevalence and relationship to amyloid burden. Neurology 2019;92:e253–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dichgans M, Holtmannspötter M, Herzog J, et al. Cerebral microbleeds in CADASIL: A gradient-echo magnetic resonance imaging and autopsy study. Stroke 2002;33:67–71 [DOI] [PubMed] [Google Scholar]
- 13. Lee JS, Kang C, Park SQ, et al. Clinical significance of cerebral microbleeds locations in CADASIL with R544C NOTCH3 mutation. PLoS One 2015;10:e0118163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noguchi-Shinohara M, Komatsu J, Samuraki M, et al. Cerebral amyloid angiopathy-related microbleeds and cerebrospinal fluid biomarkers in Alzheimer’s disease. J Alzheimers Dis 2016;55:905–13 [DOI] [PubMed] [Google Scholar]
- 15. Kim SH, Shin DW, Yun JM, et al. Kidney dysfunction and cerebral microbleeds in neurologically healthy adults. PLoS One 2017;12:e0172210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oh MY, Lee H, Kim JS, et al. Cystatin C, a novel indicator of renal function, reflects severity of cerebral microbleeds. BMC Neurol 2014;14:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauer A, van Veluw SJ, William CM, et al. Microbleeds on MRI are associated with microinfarcts on autopsy in cerebral amyloid angiopathy. Neurology 2016;87:1488–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fisher M. The challenge of mixed cerebrovascular disease. Ann N Y Acad Sci 2010;1207:18–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charidimou A, Boulouis G, Greenberg SM, et al. Cortical superficial siderosis and bleeding risk in cerebral amyloid angiopathy. Neurology 2019;93:e2192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ighodaro ET, Abner EL, Fardo DW, et al. Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab 2017;37:201–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo WK, Lee JM, Park MH, et al. Cerebral microbleeds are independently associated with arterial stiffness in stroke patients. Cerebrovasc Dis 2008;26:618–23 [DOI] [PubMed] [Google Scholar]
- 22. Janaway BM, Simpson JE, Hoggard N, et al. Brain haemosiderin in older people: Pathological evidence for an ischaemic origin of magnetic resonance imaging (MRI) microbleeds. Neuropathol Appl Neurobiol 2014;40:258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu S, Grigoryan MM, Vasilevko V, et al. Comparative analysis of H&E and Prussian blue staining in a mouse model of cerebral microbleeds. J Histochem Cytochem 2014;62:767–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang R, Castillo J, Zambon AC, et al. Brain endothelial erythrophagocytosis and hemoglobin transmigration across brain endothelium: Implications for pathogenesis of cerebral microbleeds. Front Cell Neurosci 2018;12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ganz T. Systemic iron homeostasis. Physiol Rev 2013;93:1721–41 [DOI] [PubMed] [Google Scholar]
- 26. Hallgren B, Sourander P. The effect of age on the non‐haemin iron in the human brain. J Neurochem 1958;3:41–51 [DOI] [PubMed] [Google Scholar]
- 27. Aquino D, Bizzi A, Grisoli M, et al. Age-related iron deposition in the basal ganglia: Quantitative analysis in healthy subjects. Radiology 2009;252:165–72 [DOI] [PubMed] [Google Scholar]
- 28. Milton WJ, Atlas SW, Lexa FJ, et al. Deep gray matter hypointensity patterns with aging in healthy adults: MR imaging at 1.5 T. Radiology 1991;181:715–9 [DOI] [PubMed] [Google Scholar]