

The range of clinical responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is extremely broad. Although most patients with coronavirus disease 2019 (COVID-19) present with a mild upper respiratory tract infection and then recover, some infected patients develop pneumonia, acute respiratory distress syndrome, multi-organ failure, and death. Clues to the pathogenesis of severe COVID-19 may lie in the systemic inflammation and thrombosis observed in infected patients. We propose that severe COVID-19 is a microvascular disease in which coronavirus infection activates endothelial cells, triggering exocytosis, a rapid vascular response that drives microvascular inflammation and thrombosis.
Both arterial and venous thromboembolism are common in patients with severe COVID-19. The incidence of venous thromboembolic events in patients with COVID-19 admitted to intensive care units ranges from 20% to 35%, and deep venous thrombosis has been identified in 70% to 100% of patient who died from COVD-19.1 Furthermore, arterial thrombosis resulting in stroke or myocardial infarction occurs in up to 4% of patients with COVID-19 hospitalized in intensive care units. Patients with severe COVID-19 often have laboratory findings consistent with a hypercoagulable state, suggesting widespread thrombosis and fibrinolysis, as well as elevated levels of D-dimer, von Willebrand factor (VWF), and factor VIII. These patients also manifest a hyperinflammatory state, or “cytokine storm,” characterized by elevated levels of inflammatory markers such as C-reactive protein and interleukin-6, which have been linked to severity of pneumonia and mortality.
Endothelial injury is an underlying mechanism that might link inflammation and thrombosis in severe COVID-19. Autopsy studies have suggested that both endothelial inflammation and microvascular thrombosis are prominent, with inflammatory cells attached to the endothelium of small vessels in lung, kidney, heart, and liver.2 Moreover, VWF, which is released from endothelial cells after vascular injury, and P-selectin, which is also released from activated endothelial cells, are both markedly elevated in several series of patients with COVID-19.3,4 The combination of high VWF levels, elevated P-selectin levels, microvascular thrombosis, and microvascular inflammation all suggest that microvascular injury may be a common trigger for both the inflammatory and thrombotic complications of COVID-19.5
We propose that the coronavirus SARS-CoV-2 triggers a unique endothelial response, endothelial exocytosis, which simultaneously activates 2 parallel pathways, microvascular inflammation and microvascular thrombosis, ultimately leading to hyperinflammation and diffuse thrombosis characteristic of severe COVID-19. Exocytosis is a rapid secretory response to injury in which diverse agonists bind to endothelial cell surface receptors, triggering endothelial granules to fuse with the endothelial membrane and release granule contents into the blood stream.
Endothelial granules contain VWF, P-selectin, and other proinflammatory cytokines. During exocytosis, endothelial cells release VWF, which mediates platelet adherence and aggregation. Additionally during exocytosis, endothelial cells also externalize P-selectin where it mediates leukocyte adherence to the vessel wall (Figure).
Figure.
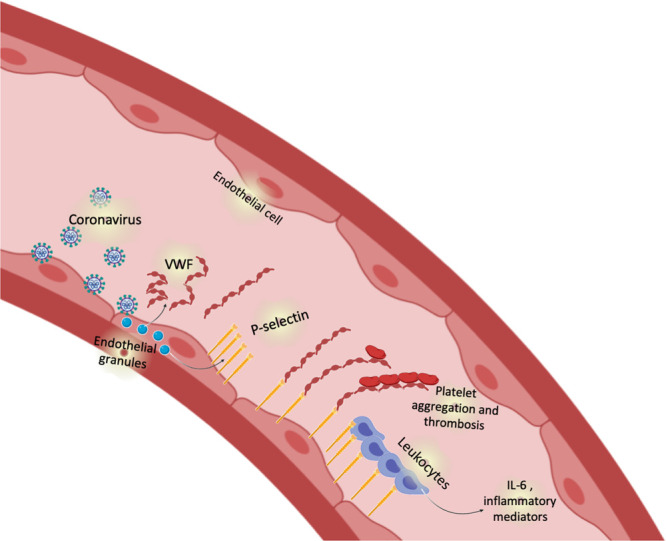
Endothelial exocytosis in coronavirus disease 2019 (COVID-19). We propose that the coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) injures endothelial cells. Endothelial cells respond to viral injury by exocytosis, mobilizing granules that release von Willebrand factor and P-selectin. P-selectin plays a dual role, accelerating thrombosis by binding to platelets, and mediating vascular inflammation by interacting with leukocytes.
P-selectin and VWF are not only stored together in the same granule but also interact to promote thrombosis. After exocytosis, P-selectin is displayed on the outer surface of the endothelial cells where it interacts with ultralong multimers of VWF, anchoring one end of the VWF multimer to the endothelial surface. Long strings of VWF unfurl from endothelial cells and platelets bind to these VWF strands, resembling pearls on a string. The synergistic relationship between P-selectin and VWF may lead to parallel activation of inflammatory and thrombotic pathways characteristic of severe COVID-19.
There are several mechanisms by which SARS-CoV-2 coronavirus might activate endothelial exocytosis. The first possibility is that SARS-CoV-2 directly activates exocytosis by binding to an endothelial surface receptor such as angiotensin-converting enzyme 2. The second possibility is that SARS-CoV-2 indirectly activates exocytosis by activating host responses and a plethora of cytokines which in turn activate exocytosis. A third possibility is that SARS-CoV-2 directly infects endothelial cells and viral polypeptides activate the exocytic machinery.
The cytokine storm in severe COVID-19 is likely caused by an auto-amplifying loop centered on injured endothelial cells. Endothelial exocytosis initiates leukocyte and platelet adherence to capillaries, which leads to microvascular obstruction, microthrombosis, and vascular inflammation. Extensive vascular inflammation can injure tissue which releases cytokines such as interleukin 6, interleukin 1β, and tumor necrosis factor α, which in turn can trigger endothelial exocytosis. In this construct, coronavirus serves as the initiator of vascular injury, with subsequent endothelial exocytosis that perpetuates a cycle of vascular injury and tissue inflammation contributing to the prolonged hyperinflammatory state in severe COVID-19.
This pathophysiologic construct might inform several potential novel host-directed therapies for COVID-19. Inhibitors of endothelial exocytosis may represent a novel therapeutic approach to the vasculopathy of COVID-19. Inhibition of P-selectin would block part of the pathway downstream of endothelial injury, including leukocyte rolling and platelet adherence to the vessel wall. Compounds that target the interaction of P-selectin with its ligand PSGL-1 (P-selectin glycoprotein ligand 1) include small molecules, aptamers, ligand decoys, and antibodies. For example, small molecules like quinoline salicylate derivatives such as PSI-697 (2-[4-chlorobenzyl]-3-hydroxy-7,8,9,10-tetrahydrobenzo[h] quinoline-4-carboxylic acid) inhibit P-selectin and decrease leukocyte rolling in rodent models of inflammation. Oligonucleotide aptamers such as ARC5690 inhibit leukocyte binding to endothelial cells in vitro. Decoy ligands such as recombinant PSGL-1 decrease thrombosis in animals. Finally, monoclonal antibodies have been developed against P-selectin, including inclacumab and crizanlizumab. Crizanlizumab is currently approved for preventing vaso-occlusive crisis in sickle cell disease, which shares a number of pathophysiologic mechanisms with COVID-19 vasculopathy. The CRITICAL trial (Crizanlizumab in COVID-19 Vasculopathy; NCT04435184) is currently testing the hypothesis that crizanlizumab can reduce mediators of thrombosis and inflammation in patients with moderate COVID-19.
Inhibition of VWF would interfere with another part of the endothelial secretory pathway. Agents such as abciximab or eptifibatide block one of the cellular receptors of VWF, the glycoprotein IIb/IIIa receptor on platelets. Compounds targeting VWF or VWF receptors could decrease the risk of thrombosis in patients with COVID-19.
In summary, accumulating clinical, laboratory, and autopsy evidence suggests that endothelial exocytosis plays a central role in the pathogenesis of severe COVID-19. Endothelial release of P-selectin and VWF activate 2 parallel pathways, leukocyte adherence and platelet aggregation, that lead to the cytokine storm and massive thrombosis that are characteristic of severe COVID-19. Clinical trials are needed that test the efficacy of drugs that target the pathway of endothelial exocytosis in patients with COVID-19.
Sources of Funding
This work was supported by National Institutes of Health/National Heart Lung and Blood Institute grants R01 HL134894 and R61 HL141791 and the Michel Mirowski, MD Professorship in Cardiology (C.J.L.).
Disclosures
Dr Lowenstein reports research grants from Novartis. In addition, Johns Hopkins University plans to file a use patent for crizanlizumab for COVID-19. Dr Lowenstein is the principal investigator of the CRITICAL trial which tests the safety and efficacy of a P-selectin inhibitor in patients with COVID-19. Dr Solomon reports grants and personal fees from Alnylam, grants and personal fees from Amgen, grants and personal fees from AstraZeneca, grants from Bellerophon, grants and personal fees from Bristol Myers Squibb, grants from Celladon, grants and personal fees from Gilead, grants and personal fees from GlaxoSmithKline, grants from Ionis, grants from Lone Star Heart, grants from Mesoblast, grants and personal fees from MyoKardia, grants from National Institutes of Health/National Heart, Lung, and Blood Institute, grants and personal fees from Novartis, grants from Sanofi Pasteur, grants and personal fees from Theracos, personal fees from Akros, grants and personal fees from Bayer, personal fees from Corvia, personal fees from Ironwood, personal fees from Merck, personal fees from Roche, personal fees from Takeda, personal fees from Quantum Genomics, personal fees from AoBiome, personal fees from Janssen, personal fees from Cardiac Dimensions, grants from Eidos, grants and personal fees from Cytokinetics, personal fees from Tenaya, personal fees from Daichi-Sankyo, personal fees from Arena, personal fees from Dinaqor, personal fees from Cardurion, personal fees from Tremeau, personal fees from Moderna, outside the submitted work; and Dr Solomon is a co-investigator of the CRITICAL trial which tests the safety and efficacy of a P-selectin inhibitor in patients with COVID-19.
Footnotes
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
References
- 1.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res. 2020;191:148–150. doi: 10.1016/j.thromres.2020.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020;190:62 doi: 10.1016/j.thromres.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–e582. doi: 10.1016/S2352-3026(20)30216-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Sullivan JM, Gonagle DM, Ward SE, Preston RJS, O’Donnell JS. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020;7:e553–e555. doi: 10.1016/S2352-3026(20)30215-5 [DOI] [PMC free article] [PubMed] [Google Scholar]