

Abstract
Atherosclerosis remains a leading cause of death and disability around the world and a major driver of health care spending. Nanomaterials have gained widespread attention due to their promising potential for clinical translation and use. We have developed a collagen‐targeted peptide amphiphile (PA)‐based nanofiber for the prevention of neointimal hyperplasia after arterial injury. Our goal was to characterize the pharmacokinetics and biodistribution of the collagen‐targeted PA to further its advancement into clinical trials. Collagen‐targeted PA was injected into the internal jugular vein of Sprague Dawley rats. PA concentrations in plasma collected at various times after injection (0 to 72 hours) were measured by liquid chromatography‐tandem mass spectrometry. Pharmacokinetics of the collagen‐targeted PA were characterized by a three‐compartment model, with an extremely rapid apparent elimination clearance resulting in a plasma concentration decrease of more than two orders of magnitude within the first hour after injection. This rapid initial decline in plasma concentration was not due to degradation by plasma components, as collagen‐targeted PA was stable in plasma ex vivo for up to 3 hours. Indeed, cellular blood components appear to be partly responsible, as only 15% of collagen‐targeted PA were recovered following incubation with whole blood. Nanofibers in whole blood also adhered to red blood cells (RBCs) and were engulfed by mononuclear cells. This work highlights the unique pharmacokinetics of our collagen‐targeted PA, which differ from pharmacokinetics of small molecules. Because of their targeted nature, these nanomaterials should not require sustained elevated plasma concentrations to achieve a therapeutic effect the way small molecules typically do.
Keywords: collagen‐targeted, nanofibers, neointimal hyperplasia, peptide amphiphiles, pharmacokinetics
HLPC‐MS/MS was used to assess plasma levels of I.V.‐injected peptide amphiphile (PA) molecules over time. PA PK were characterized by a 3‐compartment model, with extremely rapid apparent elimination clearance. PA were stable in plasma ex vivo, but adhered to red blood cells and were engulfed by mononuclear cells.
![]()
Abbreviations
- PA
peptide amphiphile
- PEG
polyethylene glycol
- Fmoc
fluorenylmethyloxycarbonyl
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilane
- HPLC
high‐performance liquid chromatography
- ACN
acetonitrile
- Q/TOF
quadrupole/time‐of‐flight
- MS/MS
tandem mass spectrometry
- HFIP
hexafluoroisopropanol
- EDTA
ethylenediaminetetraacetic acid
- PBS
phosphate‐buffered saline
- LC‐MS/MS
liquid chromatography‐tandem mass spectrometry
- TCEP
tris(2‐carboxyethyl)phosphine hydrochloride
- RBCs
red blood cells
- SEM
scanning electron microscopy
- PBMC
peripheral blood mononuclear cells
- DAPI
4′,6‐diamidino‐2‐phenylindole
- VC
central volume
- VSS
total volume of distribution
- VF
rapidly equilibrating tissue
- VS
slowly equilibrating tissue
- ClF
fast intercompartmental clearance
- ClE
elimination clearance
- ClS
slow intercompartmental clearance
- TEM
transmission electron microscopy
- SAXS
small‐angle X‐ray scattering
- H&E
hematoxylin and eosin; WBC, white blood cells
1. INTRODUCTION
Over the last decade, large strides have been made in the field of biomaterials. Developments in nanoscale engineering have occurred in a variety of medical disciplines, including biomaterials for tissue engineering, imaging, and drug delivery. Within drug delivery, peptide‐based nanomaterials allow for tissue‐specific binding and localization, variety in structure that can be tailored to specific needs, and diversity in therapy delivery and dosage. 1 , 2 Peptide amphiphiles (PAs) are a class of molecules consisting of a hydrophobic alkyl tail, a region containing amino acids with high propensities to form β‐sheets, a charged region composed of acidic or basic amino acids, and an epitope that can be modified for targeting purposes and/or specific biological activity. 3 Under aqueous conditions, PAs will self‐assemble into different tertiary nanostructures depending on various characteristics. For our application, these PAs form clearly defined “filamentous” assemblies, often with a high aspect ratio owing to the β‐sheet domain. 4 These assemblies can be structured as cylinders, ribbons, twisted structures, or aggregates of more than one fiber. The display of peptides and the ability to incorporate multi‐functionality through the co‐assembly of several different amphiphile monomers make PAs ideal delivery vehicles for targeting diseased tissues for both therapeutic and diagnostic applications.
Factors known to influence binding and in vivo biodistribution of PAs have been attributed to various physicochemical characteristics. Surface charge, shape, particle size, surface chemistry, and modifications made for specific applications have all been known to affect elimination clearance and biodistribution.
Generally speaking, smaller, spherical nanoparticles (3‐6 nm) are less likely to be taken up by the mononuclear phagocytic system than larger particles with higher surface charges. 5 Positively charged particles are readily taken up and cleared by the mononuclear phagocytic system due to their attraction to the negatively charged cell membrane of macrophages. 6 Depending on structure and modifications, smaller nanoparticles have the ability to cross the tight junctions between endothelial cells, leading to a wider distribution into various organs and renal excretion. 5 The effective pore size in normal intact endothelium is about 5 nm, leading larger particles to experience a longer circulation time due to the prolonged time needed for them to equilibrate with the extracellular fluid space. 5 The prolonged circulation time tends to favor elimination of larger nanoparticles by the mononuclear phagocytic system. Furthermore, larger particles tend to get sequestered in the sinusoids of the spleen and fenestra of the liver. 7 , 8
Surface modifications also play an integral role in biodistribution and elimination clearance. The molecular attachment of polyethylene glycol (PEG) of varying molecular weights to the particle surface (ie, PEGylation) cloaks certain parts of the surface, resulting in changes in tissue distribution and particle elimination. 9 Along with surface charge and modifications, shape can influence binding, biodistribution, and elimination clearance. Compared to spherical nanoparticles, gold nanorods are engulfed to a lesser extent by macrophages and have decreased accumulation in the liver. 8 , 10 In addition, the increased random motion experienced by nonspherical particles, such as tumbling and rolling, can lead to margination within the circulatory system. In our own work, we observed negligible binding of our spherical collagen‐binding nanoparticle to target sites in the vasculature compared to its nanofiber‐forming counterpart of similar diameter. 11
Given this unique set of characteristics that can be changed in various combinations for each nanoparticle system, and recognizing that it is difficult to predict the in vivo behavior of all nanoparticles, it is essential that the biodistribution and pharmacokinetics of each nanoparticle system that is designed, synthesized, and evaluated in vivo be assessed. In our earlier studies, we reported that fluorescently labeled PA nanofibers containing a collagen type IV‐binding sequence target the site of arterial injury after systemic intravenous injection in wild‐type Sprague Dawley rats. 11 Furthermore, S‐nitrosation of our collagen‐targeted PA nanofiber led to a 53% reduction in the development of neointimal hyperplasia 2 weeks following balloon angioplasty injury of the common carotid artery in rats and this effect was durable at 7 months. 12 Given the promising therapeutic potential of this collagen‐targeted PA nanofiber, the aim of the present study was to determine the pharmacokinetics and biodistribution of the collagen‐targeted PA nanofibers in Sprague Dawley rats. We hypothesized that the collagen‐targeted PA nanofibers will have a pharmacokinetic profile unlike that of other common small molecule therapeutics.
2. MATERIALS AND METHODS
2.1. Study approval
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85‐23, 1996) and approved by the Northwestern University Animal Care and Use Committee and the University of North Carolina Animal Care and Use Committee.
2.2. PA synthesis and labeling
The PAs and peptides were synthesized using standard fluorenylmethyloxycarbonyl (Fmoc) solid‐phase synthesis conditions as described previously. 11 Briefly, coupling reactions included Fmoc‐amino acids (4 equiv.), O‐(benzotriazol‐1‐yl)‐N,N,N’,N’tetramethyluronium hexafluorophosphate (3.95 equiv.), and diisopropylethylamine (6 equiv.) in dimethylformamide. For the aliphatic tail of the collagen‐targeted PA [sequence KLVWLPKCKKAAVVK(C12], lauric acid was attached to the ε‐amine of a lysine, which was deprotected by selective removal of the 4‐methyltrityl group using 2% trifluoroacetic acid (TFA) and 5% triisopropylsilane (TIPS) in CH2Cl2. Cleavage was performed using a TFA/TIPS/H2O/2,2’‐(ethylenedioxy)diethanethiol mixture (90:2.5:2.5:5). Purification by preparative‐scale high‐performance liquid chromatography (HPLC) was carried out on a Varian Prostar 210 HPLC system (Agilent Technologies; Santa Clara, CA, USA), eluting with a 2% acetonitrile (ACN) in water to 100% ACN gradient on a Jupiter Proteo C12 column (150 × 30 mm; Phenomenex; Torrance, CA, USA). TFA (0.1%) was added to both mobile phases to aid PA solubility during purification. Product‐containing fractions were confirmed by quadrupole/time‐of‐flight tandem mass spectrometry (6510 Q‐TOF MS/MS, Agilent Technologies), combined, and lyophilized after removing ACN by rotary evaporation.
Fluorescent labeling of the collagen‐targeted PA was achieved by reacting Alexa Fluor 546‐maleimide with four times excess of the collagen‐targeted PA in phosphate‐buffered saline (PBS) (pH 7.4). Any unreacted dye was removed by dialysis overnight in a 4k molecular weight cut‐off membrane. The labeled peptide was purified by HPLC, and only peptide conjugated with the fluorophore was collected, ensuring no free fluorophore and no unconjugated peptide. Both fluorescently labeled PA and unlabeled PA were dissolved in hexafluoroisopropanol (HFIP), an organic solvent known to disrupt hydrogen bonds, and mixed for at least 15 minutes. Samples were lyophilized to dryness to form a powder. After lyophilization in HFIP, samples were dissolved in water, aliquoted, and lyophilized again. The final product (Figure 1A) percentage of fluorescently labeled PA was 1.8 mol% relative to total PA concentration.
Figure 1.
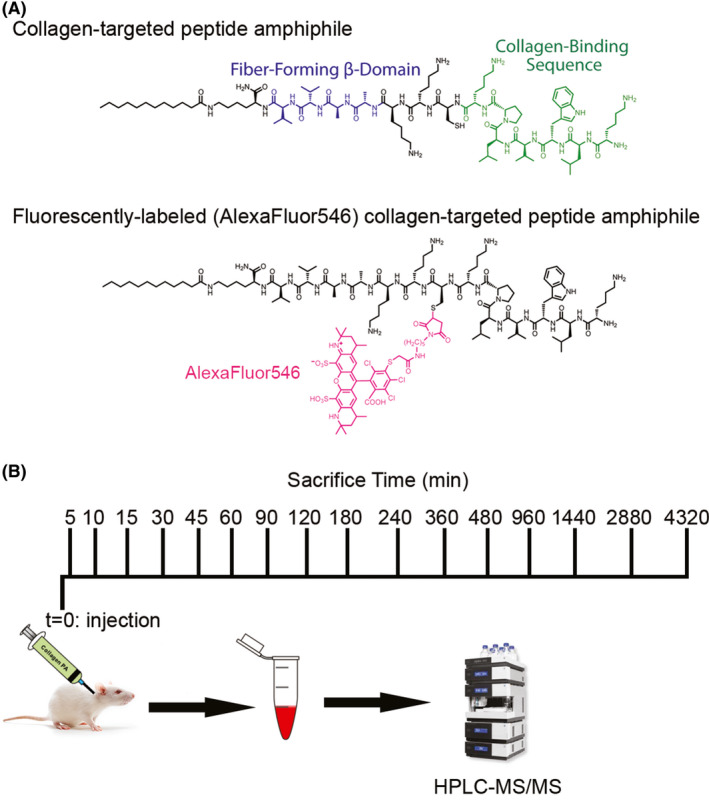
(A) Chemical structure of the fluorescently labeled (Alexa Fluor 546) collagen‐targeted peptide amphiphile (PA) and nonfluorescently labeled collagen‐targeted peptide amphiphile. (B) Schematic timeline of collagen‐targeted PA injection into the internal jugular vein at t = 0 minutes and subsequent times of blood collection by terminal cardiac puncture
2.3. Animal experiments
Anesthesia was induced in adult male Sprague Dawley rats, weighing 350‐400 g, with 5% isoflurane, and sedation of the animal was maintained by 1%‐3% isoflurane. The neck was shaved with an electric trimmer and prepped and draped in the standard surgical fashion. A midline neck incision was made and isolation of the left internal jugular vein was carried out. Once the internal jugular vein was isolated, 2.5 mg of PA dissolved in Hank's balanced salt solution was injected via the internal jugular vein using a 25 G needle. Pressure at the puncture site was maintained using a sterile Q‐tip until bleeding was no longer observed. The PA was allowed to circulate for 5, 10, 15, 30, 45, 60, 90, 120, 180, 240, 360, 480, 960, and 1440 minutes and 2, 3, and 6 days after administration, at which time the animal was euthanized and blood and urine were collected (Figure 1B; N = 3 per time point). Jugular vein injection was chosen over other intravenous routes for consistency with our previous report working with this nanostructure. 12 Skin was closed and the animal was allowed to wake up for all time points including and beyond 45 minutes.
2.4. Blood and urine collection for pharmacokinetic analysis
Animals were euthanized with an isoflurane overdose and bilateral thoracotomies were performed. Prior to in situ perfusion, a 16 G needle attached to 5‐mL syringe was introduced via cardiac puncture into the left ventricle and placed under negative pressure. Whole blood was collected into commercially available anticoagulant‐treated tubes (BD Vacutainer, K2EDTA 4.0 mL, catalog # 367 862; Becton, Dickinson and Company; Franklin Lakes, NJ, USA). Cells were separated from plasma by centrifugation for 10 minutes at 1,000‐2,000 g using a refrigerated centrifuge (5804R; Eppendorf North America; Hauppauge, NY, USA). The resulting supernatant plasma was immediately transferred into a clean polypropylene tube using a Pasteur pipette. The samples were maintained at 2‐8°C while handling and stored at –80°C if analysis did not occur within 7 days. Urine samples were collected directly from the bladder using a 25 G needle attached to a 1‐mL syringe. Urine samples were transferred to a clean polypropylene tube and maintained at 2‐8°C while handling and stored at –80°C if analysis did not occur within 7 days. Blood and urine collection were done prior to in situ perfusion with PBS. Quantification of PA by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) was performed as described below. Fluorescence measurements were performed using 100 µL of sample in a Cytation 5 plate reader and imaging system (Biotek Instruments, Inc; Winooski, VT, USA; λex = 546 nm, λem = 573 nm).
2.5. Tissue processing for biodistribution analysis
Viscera harvested after in situ perfusion with PBS (250 mL) were fixed in 2% paraformaldehyde and processed as previously described for cross‐sectional fluorescence imaging and for hematoxylin and eosin (H&E) staining. 13
2.6. Collagen‐targeted peptide amphiphile concentration measurement and pharmacokinetic analysis
Plasma collagen‐targeted PA (C94H166N22O16S, [M + H]+4 = 473.82 mass‐to‐charge ratio (m/z)) concentrations were measured in duplicate by LC‐MS/MS after sample preparation by protein precipitation. For sample preparation, to a 0.1‐ml plasma sample aliquot was added 10 μL of a 10 μg/mL collagen‐targeted PA internal standard solution (an analog of the collagen‐targeted PA being studied; C78H138N18O12S. [M + H]+4 = 388.76 m /z) and 100 μL of aqueous 10‐mM tris (2‐carboxyethyl) phosphine (TCEP) hydrochloride, and the sample was mixed for 10 minutes at room temperature. 14 Four hundred microliters of formic acid, 0.1%, in ACN were then added to each sample, which was then mixed at room temperature for 10 minutes before being centrifuged at 10,000 rpm for 10 minutes. The supernatant of each sample was transferred to a well of a 96‐well plate, dried at 50°C under N2, and reconstituted with 100 µL of 70% formic acid, 0.1%, in water and 30% formic acid, 0.1%, in ACN.
A 20‐µL aliquot of the reconstituted sample was analyzed on an API 3000 LC‐MS/MS system (Thermo Fisher Scientific; Waltham, MA, USA) with an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA, USA). Samples were eluted from an Aeris Peptide XB‐C18, 3.6 µm, 2.1 × 100 mm column (Phenomenex; Torrance, CA, USA) at a mobile phase flow rate of 250 µL/min using a gradient beginning with a mobile phase consisting of 90% formic acid, 0.1%, in water and 10% formic acid, 0.1%, in ACN that changed to 5% formic acid, 0.1%, in water and 95% formic acid, 0.1%, in ACN over 7 minutes, after which it was maintained at that composition for another 3 minutes before being returned to the starting composition over 5 minutes and the column re‐equilibrated for 5 minutes. The tandem mass spectrometer was operated with its electrospray source in the positive ionization mode. The m/z of the precursor‐to‐product ion reactions monitored was 474.0 → 129.0 for the collagen‐targeted PA and 388.9 → 129.1 for the internal standard. The retention time of collagen‐targeted PA was approximately 6.6 minutes, while that of the internal standard was approximately 6.4 minutes. The linear range for plasma collagen‐targeted PA standard curves was 1.0 to 500 ng/mL, with coefficients of variation of 10% or less throughout the entire concentration range. The relative bias was less than 10% for concentrations from 5.0 to 500 ng/mL and less than 15% for concentrations less than 5 ng/mL. Fresh plasma standard curves were prepared in blank plasma and run on the day of analysis of plasma samples. Samples were diluted to fall within the calibration curve.
The plasma collagen‐targeted PA concentration vs time relationship after intravenous administration was modeled using the SAAM II software system (SAAM Institute; Seattle, WA, USA), implemented on a Windows‐based PC. Plasma concentration histories were modeled with a three‐compartment pharmacokinetics model using a naïve pooled data approach. 15 The SAAM II objective function used was the extended least‐squares maximum likelihood function using data weighted with the inverse of the model‐based variance of the data at the observation times. 16 Model misspecification was sought by visual inspection of the measured and predicted marker concentrations vs time relationships.
2.7. Collagen‐targeted peptide amphiphile stability in plasma and whole blood
To study the stability of collagen‐targeted PA in plasma or blood, a standard solution of 1.0 mg/mL collagen‐targeted PA with Alexa Fluor 546 in 1X PBS was diluted 1:100 with PBS to obtain 1 mL of a 10 µg/mL working solution. Fifty microliters of the 10 µg/mL working solution was added to 950 µL of blank rat plasma or whole blood to obtain 1.0 mL of 500 ng/mL collagen‐targeted PA solution and incubated at 37°C for 0, 5, 10, 15, 30, 60, 120, and 180 minutes. This was repeated for a total of three solutions for each of the time points. To obtain an equivalent plasma concentration in whole blood, 75 µL of 10 µg/mL working solution was added to 2,925 µL of whole blood to obtain 3.0 mL of 250 ng/mL collagen‐targeted PA solution and incubated at 37°C, for a total of three replicate solutions for the same time points. Once the incubation time had been reached, the samples were removed from the heat block, 300‐µL aliquots of whole blood from each replicate were transferred into appropriately labeled 1.5‐mL Eppendorf tubes, and the remaining whole blood samples were centrifuged at 2000 g for 5 minutes. Separated plasma was placed in a new 1.5‐mL Eppendorf tube. Internal standard (10 µL of 10 µg/mL solution) and 100 µL of 10‐mM TCEP were added. Samples were vortexed for 20 minutes at room temperature, then briefly centrifuged. Collagen‐targeted PA concentration was measured by LC‐MS/MS as described above.
2.8. Collagen‐targeted peptide association with cellular components in whole blood
Whole blood from rats was collected into EDTA‐containing vacutainers (catalog # 367 861; Becton Dickinson). Whole blood was then incubated with 0.256 µg/mL of Alexa Fluor 546‐tagged collagen‐targeted PA nanofiber for 30 minutes at 37°C. To evaluate the possible association of collagen‐targeted PA nanofiber with red blood cells (RBCs), plasma was separated, and the cellular fraction was fixed in 3% glutaraldehyde for 20 minutes. Samples were dehydrated in a series of EtOH washes and then critically point dried, mounted, and coated with 5 nm of osmium before imaging using a Gemini 1525 sFEG scanning electron microscope (SEM; LEO Electron Microscopy Inc; Thornwood, NY, USA). To evaluate the collagen‐targeted PA nanofiber association with the mononuclear phagocytic system, peripheral blood mononuclear cell (PBMC) isolation was performed using a Histopaque‐1077 density gradient technique, according to the manufacturer's instruction (10 771; Sigma‐Aldrich; St. Louis, MO, USA). Further PBMC purification from RBC was performed using a homemade RBC lysis buffer (10 × buffer, 0.155M NH4Cl, 0.01M Tris‐HCl) to resuspend the cells, followed by centrifugation at 300 × g for 5 minutes. PBMCs were fixed to a coverslip using ice‐cold acetone. Cells were probed for CD45 (1 µg/mL, catalog # ab10558; Abcam; Cambridge, UK), diluted in IHC‐Tek diluent (1W‐1000; IHC World; Woodstock, MD, USA) followed by Alexa Fluor 488 goat anti‐rabbit IgG (2 µg/mL, A11008; Thermo Fisher Scientific). Nuclei were counterstained with 0.0012‐µM 4′,6‐diamidino‐2‐phenylindole (DAPI; catalog # D3571, Invitrogen; Waltham, MA, USA). ProLong Gold Antifade Reagent (catalog # P36930, Thermo Fisher Scientific) was used for mounting coverslips. Three‐dimensional image acquisition was performed using an LSM 780 laser scanning confocal microscope (Zeiss International; Germany). Three‐dimensional reconstructions were carried out using Imaris v9.5.0 (Bitplane AG; Zurich, Switzerland).
3. RESULTS
3.1. Collagen‐targeted peptide amphiphile pharmacokinetics
Plasma concentrations of the collagen‐targeted PA in plasma decreased in a multiexponential manner in the 72 hours after intravenous administration, with a decrease of two orders of magnitude within the first hour after intravenous administration (Figure 2A). These data were well characterized by a three‐compartment pharmacokinetic model (Figure 2B), as can be seen in Figure 2A. The model parameter values are reported in Table 1; note that estimates of parameter variability are not associated with naïve pooled data approach, hence they are not reported. Additionally, because of the unusual nature of the pharmacokinetics of this molecule, discussed later, elimination half‐life is not reported; however, the observed terminal half‐life was 42.6 hours.
Figure 2.
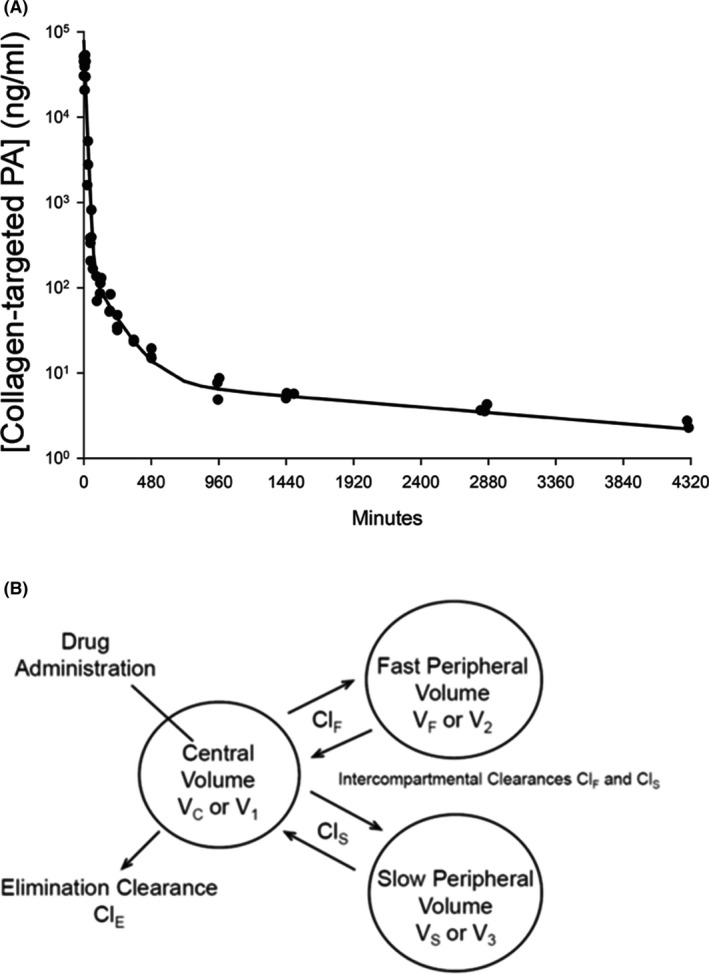
(A) Plasma PA concentration vs time relationship after intravenous administration to rats with the measured concentrations indicated by circles at each collection time (N = 3 at each time) and the fit of a three‐compartment pharmacokinetic model developed using a naïve pooled data approach indicated by the line. (B) The three‐compartment model. Three anesthetized rats were sacrificed at each time point after blood collection by cardiac puncture for plasma PA concentration measurement
Table 1.
Pharmacokinetic parameters derived from a three‐compartment model fit
| Pharmacokinetic Parameters | Values |
|---|---|
| Distribution Volumes (mL) | |
| Central, VC | 33 |
| Fast, VF | 12 |
| Slow, VS | 277 |
| Total, VSS | 322 |
| Clearance (mL/hour) | |
| Fast, ClF | 5 |
| Slow, ClS | 5 |
| Elimination, ClE | 172 |
The estimate of the central volume (VC) is consistent with the volume of plasma and interstitial fluid space in rats, and the relatively large total volume of distribution, VSS, is consistent with what might be expected for a molecule with both hydrophilic and lipophilic characteristics—that is, for an amphiphile. The first, fast exponential phase generally corresponds to distribution to rapidly equilibrating tissues by fast intercompartmental clearance, ClF. However, for the collagen‐targeted PAs in the present study, the extraordinary decrease in plasma collagen‐targeted PA concentrations during the first exponential phase corresponds to elimination clearance, ClE, the process that apparently removes the collagen‐targeted PAs from plasma irreversibly within the time frame of the study.
Ex vivo stability of the collagen‐targeted PAs in plasma revealed that the rapid decrease in plasma concentrations in the first hour after intravenous administration is not due to rapid degradation by plasma components, as PA was stable in plasma for 3 hours (Figure 3A). However, when incubated in whole blood, only 15% were recovered, suggesting that cellular blood components could be at least partially responsible for its rapid initial decline in plasma concentrations (Figure 3A). We proceeded to determine whether the PA was associated with the blood cell components. SEM revealed the PA fiber aggregates associated with RBCs (Figure 3B). Additionally, fluorescently labeled PA was also detected in white blood cells, suggesting that the mononuclear phagocytic system could account for some of the rapid clearance (Figure 3C).
Figure 3.
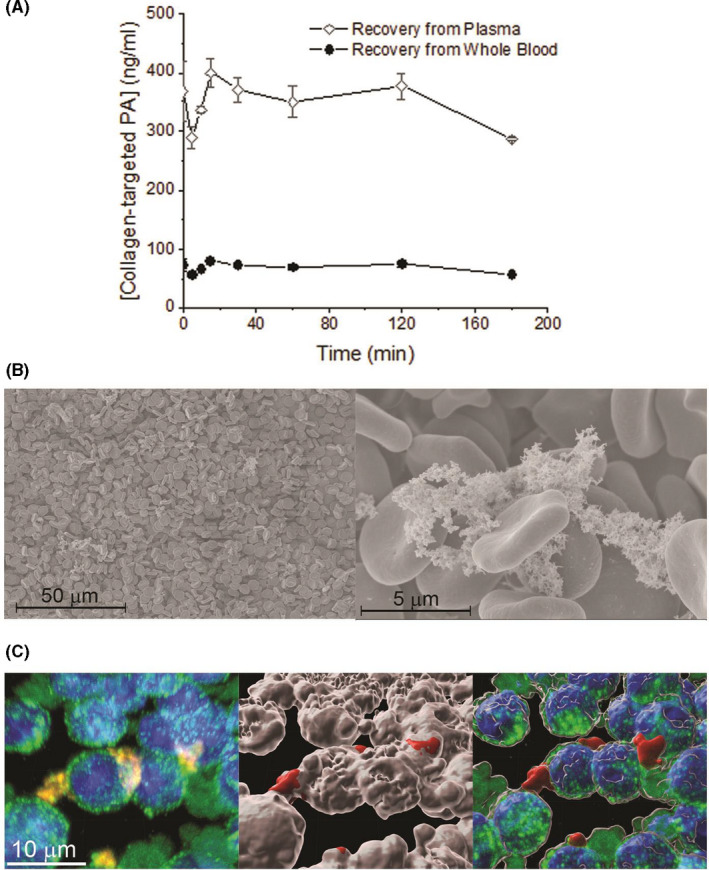
(A) Collagen‐targeted PA concentration measured in plasma and whole blood after incubation at 37°C. (B) SEM of red blood cells exhibiting some nanofibers on the surface of RBCs. (C) Confocal microscopy exhibiting nanofibers associated with white blood cells (left). Green = CD45, blue = DAPI, and red = Alexa Fluor 546‐tagged nanofiber. Surface 3‐D rendering showing the CD45‐positive surface in white, and the Alexa Fluor‐positive in red (middle); overlay of maximal projection and surface rendering suggesting the cells are taking up the collagen‐targeted PA (right)
The slow equilibration of the collagen‐targeted PA with the rapidly equilibrating tissue (VF) by fast intercompartmental clearance (ClF) and the even slower equilibration of the collagen‐targeted PA with the slowly equilibrating tissue (VS) by slow intercompartmental clearance (ClS) are additional unusual features of the pharmacokinetic model for this molecule in rats. A final unusual feature of the pharmacokinetic model is the apparent failure to reach a terminal, elimination phase even after sampling the blood for 3 days after administration of a single intravenous dose to rats. The half‐life of the terminal phase we observed was 42.6 hours. However, this should not be interpreted as the elimination half‐life because of the apparent failure to reach a terminal elimination phase in the classical drug elimination sense within the context of the study.
3.2. Biodistribution of collagen‐targeted peptide amphiphile
The collagen‐targeted PA with Alexa Fluor 546 has previously been characterized to form nanofibers at neutral pH by cryogenic transmission electron microscopy (TEM) and small‐angle X‐ray scattering (SAXS) analysis. 11 In this characterization, the collagen‐targeted PA nanofiber had a critical aggregation concentration (CAC) of < 300 nM. Therefore, in vivo experiments were carried out with 1.0 mL of a 1.3‐mM solution administered via internal jugular vein injection.
To investigate the in vivo biodistribution of fluorescently tagged collagen‐targeted PA, histologic cross‐sections of the liver, kidney, spleen, and lung were examined using fluorescence microscopy. Fluorescence was detected in the liver and spleen from day 1 to day 6 (Figure 4). In contrast, the kidney, heart, and lung did not show significant fluorescence at any of these time points. Even though it appeared that there was little presence of the collagen‐targeted PA in the kidney due to a lack of fluorescent signal, we examined the elimination of the collagen‐targeted PA by renal clearance. Urine was collected directly from the bladder and fluorescence measured. A significant amount of fluorescence was seen in the urine within the first 5 hours, followed by a rapid descent and a gradual increase at hours 20 to 60 and a continued presence at 80 hours (Figure S1). However, urine analysis for the presence of the collagen‐targeted PA by LC‐MS/MS did not show the presence of the target peptide. Hence, the fluorescence present in urine could be due to free fluorophore or labeled peptide fragments.
Figure 4.
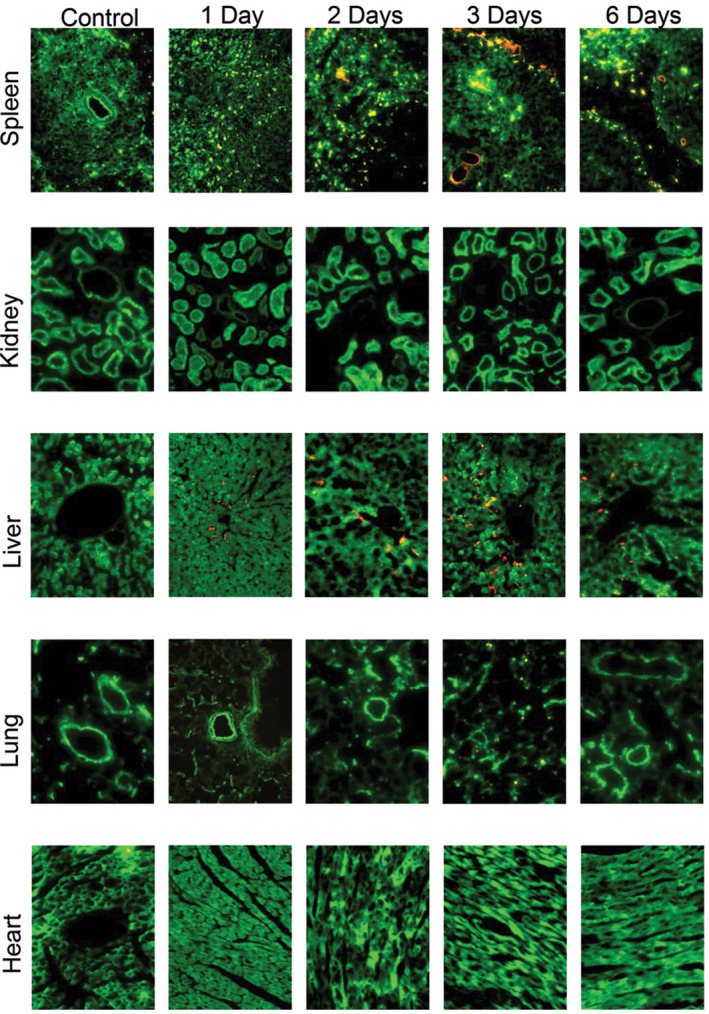
Fluorescent microscopy of spleen, kidney, liver, lung, and heart in control, non‐PA‐injected animals, and 1, 2, 3, 6 days after injection of collagen‐targeted PA nanofibers. A small amount of fluorescence was detected in the spleen and liver as early as 1 day after injection. Green = autofluorescence of viscera and red = Alexa Fluor 546‐tagged nanofiber; images taken using 20 × objective
Potential tissue damage of the heart, lung, liver, spleen, and kidney after collagen‐targeted PA administration was evaluated histologically. All tissue cross‐sections were stained with H&E, and then examined by light microscopy using the 5 × and 20 × objectives. As seen in the representative images in Figure 5, and as expected, viscera did not exhibit any architectural changes indicative of fibrosis compared to controls at 1, 2, 3, and 6 days after injection. As we have previously reported, no change in liver function profiles, coagulation panel, or chemistry panel was observed at 3 hours, or 1, 2, 3, and 7 days after injection. 12
Figure 5.
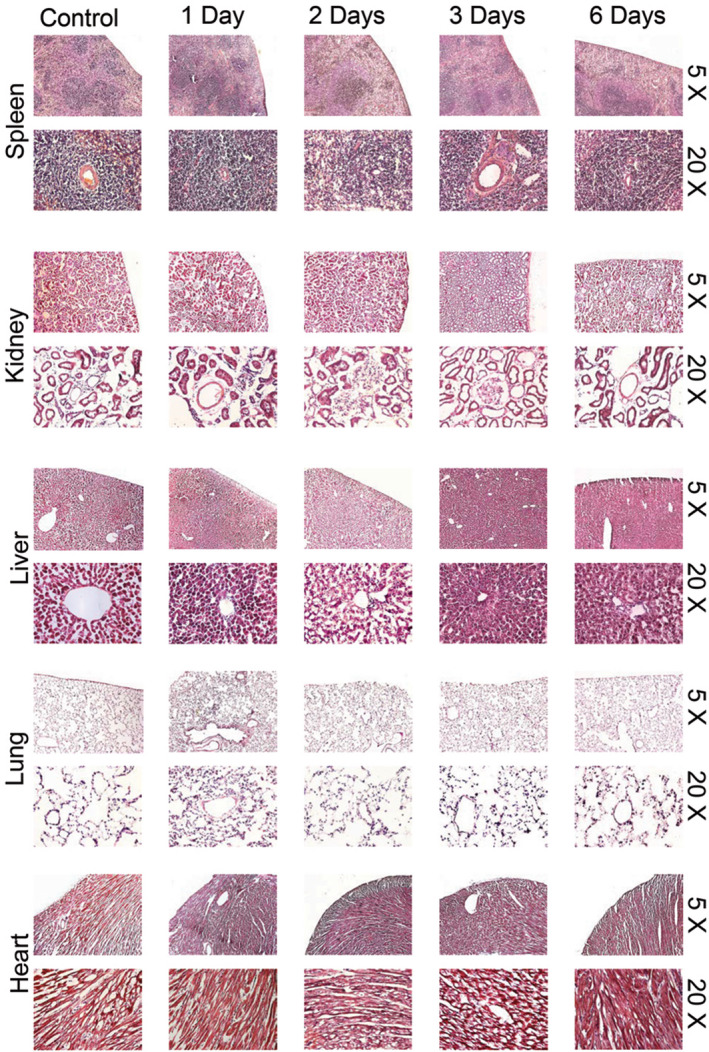
Hematoxylin and eosin‐stained spleen, kidney, liver, lung, and heart in control and collagen‐targeted PA nanofiber‐injected (0.5 mg/500 µL) animals at 1, 2, 3, and 6 days after injection. No significant fibrosis is observed in any viscera. Images taken using 5 × and 20 × objective
4. DISCUSSION
We have described the pharmacokinetics and biodistribution of our novel, biodegradable, collagen‐targeted PA nanofiber in rats. Detailed physicochemical characterizations of all materials used in this study, including histopathology, have already been reported. 11 , 12 In our first study using this targeted nanofiber, we tested different doses and determined that 2.5 mg achieved statistically significant binding at the site of carotid balloon injury. 11 The animals tolerated up to 10 mg. When testing the therapeutic potential of a nitrosated version of the nanofiber, we showed that 2.5 mg was sufficient to inhibit neointimal hyperplasia after balloon injury. 12 Therefore, we wanted to study the pharmacokinetics of the fiber using the same dose we had determined to be therapeutic. In the present study, we have found that the disposition of our PA after intravenous administration can be characterized by a three‐compartment pharmacokinetic model with an apparent elimination clearance that is responsible for the rapid and marked decrease in plasma PA concentrations within the first hour after injection. Further examination of the stability of the collagen‐targeted PA in plasma revealed that the rapid initial decline in plasma concentration is not due to rapid degradation by plasma components. However, upon incubation with whole blood, only 15% of the collagen‐targeted PA were recovered, suggesting that cellular blood components were at least partially responsible for the rapid clearance. Further analysis of whole blood found that the nanofiber adhered to RBCs and was engulfed by mononuclear cells. Taken together, these data suggest a pharmacokinetic profile of the collagen‐targeted PA nanofiber unlike that of most small molecule drugs.
While the pharmacokinetics of this PA molecule seem rather extraordinary, a careful review of the literature reveals that its pharmacokinetics are not unique. Much of what one expects of the pharmacokinetics of a novel molecule is based on the vast experience of both preclinical pharmacologists and clinical pharmacologists with traditional small molecular entities. While the pharmacokinetic profile of the collagen‐targeted PA in the present study is very dissimilar from that of traditional small molecular entities, it is similar to that of another molecule with similar physicochemical properties, amiodarone, a cationic amphiphilic antiarrhythmic agent. 17 , 18 Indeed, these authors describe a rapid and profound decrease in plasma amiodarone concentrations immediately after dosing due to trapping in tissue depots with very long release times. Similar disposition patterns have also been reported for metallic nanoparticles. 19 , 20 A novel approach to characterizing both the short‐ and the long‐term pharmacokinetic behavior of amiodarone has been described by Weiss, who has noted that the concept of steady‐state and elimination half‐life is not meaningful for a drug that has a pharmacokinetic profile like this. 18 Given the similar disposition of amiodarone and the collagen‐targeted PA in the present study, the approach proposed by Weiss 18 or approaches used by others such as Pollak 21 may be useful in guiding the designs of studies aimed at characterizing the disposition of this and similar drugs and the development of therapeutic approaches for their safe and effective use. It is important to point out that the nanostructure used in this study exists in dynamic equilibrium between the fibers and the free PA in solution. The CAC for this fiber is below 1 µM. 11 , 12 However, by 1 hour, the plasma concentration falls below the CAC and the majority of the peptide should be in solution. Thus, we infer that, soon after administration, the PAs that had not bound to either circulating blood cells or tissue to which they had been exposed quickly oxidize to disulfides, and the unbound PA spent much of the remaining time in the body as such.
Despite the rapid clearance of the collagen‐targeted PA from plasma, we have previously reported that when administered after balloon angioplasty, the targeted PA nanofiber remains at the site of arterial injury for 2‐3 days. 11 , 12 This suggests that unlike small drugs, the therapeutic effect of targeted nanomaterials might not be as dependent on their pharmacokinetics. The rapid clearance of the collagen‐targeted PA from plasma may be due to processes including organ sequestration, uptake by the mononuclear phagocytic system, metabolism, and renal excretion. Analysis of viscera showed that only the liver and, to a lesser extent, the spleen had significant fluorescence 1 day after intravenous injection. Although this suggests that organ sequestration may be at least partially responsible for the rapid plasma clearance, we explored other possible mechanisms. Despite the presence of fluorescence in the urine 1 hour after injection, urine analysis by LC‐MS/MS did not detect the presence of intact collagen‐targeted PA, suggesting urinary excretion of PA degradation products. Nonetheless, the kinetic profile of urinary fluorescence excretion does not match the rapid clearance from plasma. Although plasma proteases could potentially rapidly degrade the PA, in vitro studies found that plasma concentrations of the collagen‐targeted PA were stable over time, suggesting that plasma proteases are not responsible for the rapid clearance. There was rapid sequestration of the PA by cellular blood components in in vitro studies and SEM images provided evidence of the presence of the collagen‐targeted PA on the surface of RBCs. In addition, there was evidence of white blood cell uptake of the PA. These data suggest that association with the RBCs and uptake by the mononuclear phagocytic system might also contribute to the rapid clearance from plasma determined by our pharmacokinetic analysis.
Peptide amphiphiles have a great deal of promise due to their potential role in a variety of disciplines, including targeting diseased tissues for both therapeutic and diagnostic applications, as well as tissue regeneration. Previous work from our laboratories has shown the continued inhibition of neointimal hyperplasia by our collagen‐targeted PA 7 months after carotid artery balloon angioplasty in Sprague Dawley rats. 12 Furthermore, PAs from our laboratories have been used to decrease hemorrhage after liver injury and induce plaque regression at the aortic root. 22 , 23 Moreover, other groups have shown the utility of PAs as scaffolds for tissue regeneration, tumor regression, and improved MRI resolution. 1
There are some limitations of our current study. First, as our aim was to determine the pharmacokinetics of a nanostructure with potential therapeutic applications, we did not assess the collagen‐binding sequence alone. The binding sequence alone is not of interest as a therapeutic and did not exhibit evidence of assembly. 11 Instead, we compared our PA to the substantial extant data on prototypical small molecules in the literature. Second, the presence of fluorescence in liver and spleen, as well as RBCs and white blood cells, suggests that these organs might be partially responsible for removing PA from plasma. However, because we detected the fluorophore and not the native peptide, it is possible that the fluorescence present in the organs and blood cells (and urine) was due to PA fragments. Thus, we do not have an unequivocal explanation for the rapid plasma clearance of our collagen‐targeted PA and have not elucidated the mechanism of rapid plasma concentration decline. The blood and plasma stability studies in vitro pointed to some possible role of blood cells in the decline of PA concentration in vivo. Even though we detected PA associated with RBC and WBC, there was not a quantitative assessment of the PA fraction associated with these cells. Therefore, we cannot conclude that the blood cells are accountable for the rapid decline in plasma concentration. Additionally, the literature shows that the phagocytic system is a preferred mechanism of removing nanostructures from plasma. We performed the pharmacokinetic studies in animals that did not undergo balloon injury. Hence, we did not study if the arterial injury or surgical intervention affects the pharmacokinetic profile of the targeted nanofiber. However, we reported that 0.3 nmoles (5.7 µg ‐ less than 1%) of the initial dose binds and remains bound for up to 4 days. 11 This is in stark contrast with the rapid decrease of more than two orders of magnitude from plasma we observe in the present study. Therefore, we believe that the amount bound to the site of vascular injury does not affect the drug disposition.
Finally, it is important to address the possibility that the fluorescently labeled PA behaves differently from the nonlabeled peptide. However, fluorescently labeled PA and nonlabeled PA were co‐assembled with only 1.8 molar% fluorescent peptide. Even if the labeled PA behaved differently when dissociated, which is a distinct possibility, the majority of the building blocks of the fiber are always unlabeled. Importantly, even though we acknowledge that modifications to the peptide backbone can possibly change the drug disposition, it is impractical to perform a full pharmacokinetic study for each version of PA we try for a new application.
In conclusion, while our collagen‐targeted PA nanofiber exhibits great therapeutic and diagnostic benefits in the clinical realm, the present study highlights the uniqueness of the pharmacokinetics of this nanoparticle. Indeed, pharmacokinetics of certain targeted nanoparticles may not matter in the same way the pharmacokinetics of small molecules matter because their therapeutic effect appears to be unrelated to the maintenance of effective plasma concentrations. These unique characteristics should be taken into consideration when designing studies to test the efficacy of these nanomaterials.
5. DATA SHARING AND DATA ACCESSIBILITY
All data generated during the course of the work in this manuscript will be made available upon request.
DISCLOSURES
The authors report no financial conflict of interest as it pertains to the work in this manuscript.
AUTHOR CONTRIBUTIONS
Participated in research design: Avram, Bahnson, Kassam, Kibbe, and Stupp. Conducted experiments: Bahnson, Cartaya, Jiang, and Kassam. Performed data analysis: Avram, Bahnson, and Kassam. Wrote or contributed to the writing of the manuscript: Avram, Bahnson, Cartaya, Jiang, Kassam, Kibbe, Stupp, and Tsihlis.
Supporting information
Fig S1
ACKNOWLEDGMENTS
The authors acknowledge the Peptide Synthesis Core Facility of the Simpson Querrey Institute at Northwestern University.
Kassam HA, Bahnson EM, Cartaya A, et al. Pharmacokinetics and biodistribution of a collagen‐targeted peptide amphiphile for cardiovascular applications. Pharmacol Res Perspect. 2020;8:e00672 10.1002/prp2.672
These authors (HAK and EMB) contributed equally and share first authorship.
Funding information
This work was supported by the National Institutes of Health, National Heart Lung and Blood Institute [Grants R01HL116577, K01HL145354]; and the Office of the Director [Grant S10OD021786]. Confocal imaging was performed at the UNC Neuroscience Microscopy Core supported, in part, by the NIH‐NINDS P30 NS045892 and the NIH‐NICHD U54 HD079124. AC was supported by the National Institutes of Health, National Institute of General Medicine [Grant T32GM007040]. This work was not presented at a national meeting.
REFERENCES
- 1. Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22:969‐976. [DOI] [PubMed] [Google Scholar]
- 2. Peters D, Kastantin M, Kotamraju VR, et al. Targeting atherosclerosis by using modular, multifunctional micelles. Proc Natl Acad Sci U S A. 2009;106:9815‐9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chung EJ, Mlinar LB, Sugimoto MJ, Nord K, Roman BB, Tirrell M. In vivo biodistribution and clearance of peptide amphiphile micelles. Nanomedicine. 2015;11:479‐487. [DOI] [PubMed] [Google Scholar]
- 4. Hendricks MP, Sato K, Palmer LC, Stupp SI. Supramolecular assembly of peptide amphiphiles. Acc Chem Res. 2017;50:2440‐2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano‐sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond). 2008;3:703‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rattan R, Bhattacharjee S, Zong H, et al. Nanoparticle‐macrophage interactions: A balance between clearance and cell‐specific targeting. Bioorg Med Chem. 2017;25:4487‐4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burns AA, Vider J, Ow H, et al. Fluorescent silica nanoparticles with efficient urinary excretion for nanomedicine. Nano Lett. 2009;9:442‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9:1521‐1532. [DOI] [PubMed] [Google Scholar]
- 9. Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13:399‐409. [DOI] [PubMed] [Google Scholar]
- 10. Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009;26:235‐243. [DOI] [PubMed] [Google Scholar]
- 11. Moyer TJ, Kassam HA, Bahnson ES, et al. Shape‐dependent targeting of injured blood vessels by peptide amphiphile supramolecular nanostructures. Small. 2015;11:2750‐2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bahnson ES, Kassam HA, Moyer TJ, et al. Targeted Nitric Oxide Delivery by Supramolecular Nanofibers for the Prevention of Restenosis After Arterial Injury. Antioxid Redox Signal. 2016;24:401‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vavra AK, Havelka GE, Martinez J, et al. Insights into the effect of nitric oxide and its metabolites nitrite and nitrate at inhibiting neointimal hyperplasia. Nitric Oxide. 2011;25:22‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toyo'oka T. Recent advances in separation and detection methods for thiol compounds in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3318‐3330. [DOI] [PubMed] [Google Scholar]
- 15. Kataria BK, Ved SA, Nicodemus HF, et al. The pharmacokinetics of propofol in children using three different data analysis approaches. Anesthesiology. 1994;80:104‐122. [DOI] [PubMed] [Google Scholar]
- 16. Barrett PH, Bell BM, Cobelli C, et al. SAAM II: Simulation, Analysis, and Modeling Software for tracer and pharmacokinetic studies. Metabolism. 1998;47:484‐492. [DOI] [PubMed] [Google Scholar]
- 17. Campos Moreno E, Merino Sanjuan M, Merino V, Nacher A, Martin Algarra RV, Casabo VG. Population modelling to describe pharmacokinetics of amiodarone in rats: relevance of plasma protein and tissue depot binding. Eur J Pharm Sci. 2007;30:190‐197. [DOI] [PubMed] [Google Scholar]
- 18. Weiss M. The anomalous pharmacokinetics of amiodarone explained by nonexponential tissue trapping. J Pharmacokinet Biopharm. 1999;27:383‐396. [DOI] [PubMed] [Google Scholar]
- 19. Lin Z, Monteiro‐Riviere NA, Riviere JE. Pharmacokinetics of metallic nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7:189‐217. [DOI] [PubMed] [Google Scholar]
- 20. Mager DE, Mody V, Xu C, et al. Physiologically based pharmacokinetic model for composite nanodevices: effect of charge and size on in vivo disposition. Pharm Res. 2012;29:2534‐2542. [DOI] [PubMed] [Google Scholar]
- 21. Pollak PT, Bouillon T, Shafer SL. Population pharmacokinetics of long‐term oral amiodarone therapy. Clin Pharmacol Ther. 2000;67:642‐652. [DOI] [PubMed] [Google Scholar]
- 22. Morgan CE, Dombrowski AW, Rubert Perez CM, et al. Tissue‐Factor Targeted Peptide Amphiphile Nanofibers as an Injectable Therapy To Control Hemorrhage. ACS Nano. 2016;10:899‐909. [DOI] [PubMed] [Google Scholar]
- 23. So MM, Mansukhani NA, Peters EB, et al. Peptide amphiphile nanostructures for targeting of atherosclerotic plaque and drug delivery. Adv Biosyst. 2018;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Data Availability Statement
All data generated during the course of the work in this manuscript will be made available upon request.