

 This article reviews the current progress in the development of SERDs as anti-breast cancer agents.
This article reviews the current progress in the development of SERDs as anti-breast cancer agents.
Abstract
Selective estrogen receptor downregulators (SERDs) are a novel class of compounds capable of reducing the ERα protein level and blocking ER activity. Therefore, SERDs are considered as a significant therapeutic approach to treat ER+ breast cancer in both early stage and more advanced drug-resistant cases. After the FDA approval of a steroidal drug, fulvestrant, as a SERD for the treatment of breast cancer in patients who have progressed on antihormonal agents, several molecules with diverse chemical structures have been rapidly developed, studied and evaluated for selective estrogen receptor downregulation activity. Here we compile the promising SERDs reported in recent years and discuss the chemical structure and pharmacological profile of the most potent compound of the considered series. Because of the availability of only a limited number of effective drugs for the treatment of breast cancer, the quest for a potent SERD with respectable activity and bioavailability is still ongoing. The goal of this article is to make available to the reader an overview of the current progress in SERDs and provide clues for the future discovery and development of novel pharmacological potent SERDs for the treatment of breast cancer.
1. Introduction
Breast cancer is the most frequently occurring cancer in women and is responsible for the maximum number of cancer-related deaths in women. Every year breast cancer is distressing the lives of around 1.2 million women and in 2018, it is estimated that 627 000 women died from breast cancer.1 Approximately 80% of post-menopausal women suffering from breast cancer have estrogen receptor α (ERα)-positive tumors.2,3 Endocrine therapy is the first-line treatment available for ER+ breast cancer patients and the available drugs are categorized into two major types: selective estrogen receptor modulators (SERMs) and aromatase inhibitors (AIs). Tamoxifen (1) (Fig. 1) is a very effective selective estrogen receptor modulator approved by the FDA and is currently in use for the treatment of ER+ breast cancer in pre- and post-menopausal women.4 Tamoxifen is marketed as a single Z (trans) isomer of p-β-dimethylaminoethoxy-1,2-diphenylbut-1-ene and exhibits both estrogenic and antiestrogenic activity depending upon the tissue. For example, it has mainly antiestrogenic effects on the breast but estrogenic effects on the uterus and liver. Tamoxifen is a lead compound that initiated SERM development for the treatment of various diseases, which include osteoporosis and rheumatoid arthritis, and for the application of the SERM concept for all the members of the nuclear receptor family.5 Aromatase is an enzyme that plays a significant role in the biosynthesis of important endogenous estrogens from androgens. Aromatase inhibitors such as anastrozole (3) (Fig. 1) are compounds that are competitive inhibitors of the enzyme aromatase and block the synthesis of estrogen by binding to the estrogen receptor.6,7 However, undesirably, 50% of patients either do not respond or acquire resistance within five years of treatment.8 Advanced-stage breast cancer is generally incurable. Treatment of reoccurring breast cancer is very challenging and long-term use of SERMs leads to the development of other cancers and drug resistance, thus making the situation fatal for the breast cancer patient.9–11 The development of ER+ and treatment-resistant phenotypes involve multiple mechanisms that mostly involve ERα activity.12–15 These include (a) ERα mutation that leads to ligand-independent constitutive activation of ER, (b) in the absence of estradiol, the activation of ERα because of signaling pathway activation and (c) variation in the expression of ERα cofactors responsible for the ERα modulator activity. Keeping in mind the significance of ERα as a potential target for breast cancer treatment, an enormous number of SERMs and aromatase inhibitors with diverse chemical structures were clinically tested in recent years.16–19 Tragically, none of them showed effective results in the treatment of advanced disease.20,21 The existence of only a limited number of effective drugs encourages scientists to develop new compounds that reduce the possibility of recurrence and drug resistance through complete inhibition of the pathway. For patients that have progressed on antihormonal therapy such as tamoxifen or aromatase inhibitors, the development of an ERα ligand that effectively antagonizes and degrades the ERα would be favorable. This results in the development of selective estrogen receptor downregulators (SERDs) that act as a pure antagonist by interfering with the binding of estradiol to estrogen receptors (ERs) and inducing the rapid downregulation of ER.22–26 SERDs effectively block endocrine-dependent and -independent ER signaling and are considered as a significant therapeutic approach to treat ER+ breast cancer in both early stage and more advanced drug-resistant cases.27,28
Fig. 1. Chemical structure of FDA-approved drugs (1–4) for the treatment of breast cancer.

Fulvestrant (4) (Fig. 1) is a first-generation SERD approved by the FDA in 2007 for the treatment of ER-positive, metastatic breast cancer in postmenopausal women following progression on prior endocrine therapy involving tamoxifen and aromatase inhibitors.29–35 Fulvestrant (4) is a steroidal molecule bearing a 4,4,5,5,5-pentafluoropentylsulfinyl alkyl group at the 7α position of estradiol. It is a high-affinity competitive antagonist that lacks an agonistic effect across all types of tests tissues and upon binding to an estrogen receptor it induces a denaturing structural change and accelerates the degradation of the estrogen receptor.36–38 However, the efficacy of fulvestrant (4) is limited in the clinic due to its low oral bioavailability and is administered monthly by intramuscular injection.39,40 The encouraging clinical effectiveness of fulvestrant (4) and the understanding of its mechanism of action motivated medicinal chemists to develop orally bioavailable SERDs through structural modification and optimization. Several attempts were made to design and synthesize fulvestrant-like compounds with increased polarity and solubility by modifying the long alkyl chain.41–43 However, such modifications failed to solve the problem of poor bioavailability and therefore in recent years continuous efforts towards the development of clinically effective oral SERDs resulted in the identification of several promising nonsteroidal SERDs.44–54 A Glaxo SmithKline (GSK) compound, GW-5638 (5) (Fig. 2), having a triphenylethylene skeleton and characteristic acrylic acid side chain was discovered as a second-generation nonsteroidal SERD and progressed to phase I clinical trials.55,56 Unfortunately, the development of GW 5638 (5) and its metabolite GW 7604 (6) (Fig. 2) was discontinued due to metabolism issues. GDC-0810 (ARN-810) (7) (Fig. 2) is a second-generation SERD effective in treating advanced metastatic breast cancer and underwent a clinical phase II trial, but tragically its clinical trial was recently discontinued.57,58 Other significant SERDs such as (RAD-1901) (8) (Fig. 2),59 AZD-9496 (Fig. 2) (9)60,61 and GDC-0927 (10) (Fig. 2)62,63 are currently under clinical trials.
Fig. 2. Chemical structure of compounds (5–10) in clinical trials for the treatment of breast cancer.

Indeed, in recent years with tremendous efforts, scientists delivered several molecules with promising SERD activity. However, increasing cases of drug-resistant breast cancer favor the requirement of potent SERDs that are not only capable of suppressing the tumor but also effectively complete clinical trials. One of the major objectives of our research group has been to support researchers around the world by giving them a current update on the recently reported anticancer compounds and their mechanism of action and applicability for cancer treatment.5,7,64–66 We have compiled this review on SERDs and recently reported steroidal and nonsteroidal novel SERDs are included for discussion with focus on the chemical structure and activity of the most active compound in the series. Readers will be acquainted with the current progress in SERD discovery, which will be advantageous in the designing of future novel SERDs.
2. Steroidal derivatives as SERDs
The studies conducted by the ICI Company (now Astra Zeneca) revealed that the introduction of a long alkyl side chain with selected residues at the end might generate compounds with strong antiestrogenic activity. This includes pure antiestrogens such as ICI 164384 (11) (Fig. 3) and ICI 182780 (4) that are completely devoid of estrogenic activity.67–69 Jean-Claude Blazejewski and coworkers decided to examine the steric hindrance effects associated with 7-α rigid side chains on the binding and endocrine properties of 7-α derivatives. Therefore, in 2003 the group synthesized two derivatives of estradiol, 7α-perfluorohexylestradiol (12) (Fig. 3) and 7α-trifluoromethylestradiol (13) (Fig. 3), and studied the endocrine properties on the MCF-7 breast cancer cell line.70 The result revealed that the perfluorination-induced enhancement of rigidity in compound 12 is not capable of affecting the binding affinity severely (IC50 = 1 μM). Compound 13 bearing a trifluoromethyl group at the 7α-position exhibited better binding affinity (IC50 = 20 nM) than compound 12. It was observed that fluorinated compound 13 was well tolerated in the ligand-binding pocket of estrogen receptors and activated ERs with slightly lower efficiency than estradiol (E2), that is, 1 : 10. Both compounds 13 and 12 worked as ER ligands and at 0.1 μM concentration downregulated the estrogen receptor (75% and 50%, respectively) less efficiently than E2.
Fig. 3. Chemical structure of ICI 164384 (11) and steroidal derivatives 12–14.

With the FDA approval of the steroidal molecule fulvestrant (4) as a first-generation SERD for the treatment of estrogen receptor positive (ER+) metastatic breast cancer in postmenopausal women, several attempts were made to design and synthesize potent steroidal SERDs with high efficacy and oral bioavailability.71,72 In particular, the long alkyl chain of fulvestrant (4) was modified to improve the polarity and solubility, although the efforts were unsuccessful with reference to bioavailability.42,43,73 Fulvestrant (4) is known to undergo rapid and extensive O-glucuronidation and O-sulfation to form inactive and water-soluble polar phase II metabolites.74–77 Guangdi Wang and coworkers observed that the introduction of a boronic acid group in antiestrogenic compounds such as 4-hydroxytamoxifen (2) (Fig. 1) and endoxifen (N-desmethyl-4-hydroxytamoxifen) reduced the first-pass metabolism of hydroxylated drug molecules and thus improved their bioavailability.78–89 Considering this information, in 2016 the group decided to synthesize a boronic acid derivative of fulvestrant (4) and expected that the modification will reduce glucuronidation and sulfation while maintaining the binding affinity and SERDs activity. The fulvestrant-3 boronic acid (14, ZB716) (Fig. 3) was prepared by introducing a boronic acid moiety on the C-3 position of 4 and evaluated for ER binding, downregulation and bioavailabilty.80 Compound 14 was as potent as 4 in its action as a SERD and exhibited good ERα binding affinity (IC50 = 4.1 nM) and ERα downregulating activity in both tamoxifen-sensitive (IC50 of 7.8 nM) and tamoxifen-resistant breast cancer cells (IC50 = 12.7 nM). Additionally, 14 exhibited enhanced oral bioavailability (AUC = 2547.1 ng h mL–1) than 4 (AUC = 158.4 ng h mL–1) in mice and therefore suggests its favorable clinical utility as an oral SERD.
3. Non-steroidal molecules as SERDs
3.1. Triaryl ethylene derivatives as SERDs
Two antiestrogens, GW 5638 (5) and its hydroxylated metabolite GW 7604 (6), bearing a triphenylethylene core similar to 4-hydroxytamoxifen (2) were discovered as SERDs and have the ability to inhibit the growth of tamoxifen-resistant breast tumors.56,81 The major structural difference between 6 and 2 is the presence of an acrylic acid side chain in place of a basic amine-containing side chain extending from the triphenylethylene core. To further explore the relative importance of the acrylic acid side chain on SERDs activity, R. V. Weatherman and coworkers in 2007 designed and synthesized two analogues of 6 by replacing the acrylic acid group with acrylamide or a methyl vinyl ketone.82 The estrogen receptor binding data suggest that the change of carboxylic acid (6) to either a carboxamide (15) (Fig. 4) or a methyl ketone (16) (Fig. 4) decreases the binding affinity of ligands for ERα (Ki values 27 ± 10 nM for 6, 240 ± 35 nM for 15 and 210 ± 30 nM for 16). Both the compounds exhibited similar activity to 6 for antagonism of estradiol-induced activation of ERα-mediated transcription and inhibition of estradiol-induced proliferation of the MCF-7 cell line. GW 7604 (6) was able to induce ERα degradation in a dose-dependent manner; however, compounds 15 and 16 were not as effective as 6 for ER degradation.
Fig. 4. Chemical structure of triaryl ethylene derivatives 15–17.

One of the major drawbacks associated with fulvestrant (4), a steroid-based selective estrogen receptor degrader (SERD), is its poor pharmaceutical properties. It is administered by intramuscular injections (500 mg total dose in 2 × 5 mL injections) that limits the total amount of drug administered and hence leads to potential incomplete receptor blockade.40,83,84 Moreover, GW 5638 (5), a nonsteroidal ER antagonist, emerged as a potent molecule for the treatment of tamoxifen-resistant cancer.55,85 The activity study of 4 and 5 in models of tamoxifen resistance suggests the critical role of ER in tamoxifen-resistant breast cancer. Therefore, in 2015, Nicholas D. Smith and coworkers explored scaffold 5 with the intention of identifying fully efficacious ERα degraders with good exposure and oral dosing.57 A series of small-molecule, that is triphenylalkene, analogues were synthesized and evaluated for antagonistic and ERα degrading activity. Compound 7 (GDC-0810/ARN-810) (Fig. 2) of the indazole series emerged as the most potent compound with high ERα binding affinity (IC50 = 6.1 nM) and works as a full transcriptional antagonist with no agonism (IC50 = 2 nM). It displays good potency and efficacy in ERα degradation (EC50 = 0.7 nM and 91% efficacy) and MCF-7 breast cancer cell viability (IC50 = 2.5 nM and 99% efficacy). Compound 7 exhibited robust activity in models of tamoxifen-sensitive and tamoxifen-resistant breast cancer and was subjected to clinical trials in women with locally advanced or metastatic estrogen receptor-positive breast cancer.
A potent SERD, GW 5638 (5), reached up to phase I clinical trial, but unfortunately, further development was discontinued because of the associated metabolism issue.57 The study of its mechanism of action revealed that the conversion of 5 into its metabolite 6 in human liver microsomes is low (around 16%) and that 6, having phenolic group, tends to be metabolized rapidly by phase II metabolizing enzymes through glucuronidation and sulfation. This reduces the bioavailability of 6 as an oral drug. Guangdi Wang and coworkers' study on boronic derivatives of antiestrogenic compounds such as 4-hydroxy tamoxifen (2) and endoxifen (N-desmethyl-4-hydroxytamoxifen) proposed that boronic acid or boronate is an oral bioisostere of a phenolic hydroxyl group.78,79 In view of this information, in 2016 the group developed an orally bioavailable steroidal SERD (fulvestrant-3 boronic acid, ZB716, 14).80 Further, the group decided to block the primary metabolizing site of GW 7604 (6) by introducing a boronic acid functional group in place of the phenolic hydroxyl group in anticipation that the boronic acid functional group would lock the primary site of phase II metabolism and maintain the pharmacology of the phenolic hydroxyl group in ER recognition and binding.54 A boron-modified GW 7604 (6) derivative GLL398 (17) (Fig. 4) was prepared and evaluated for SERD activity. Compound 17 strongly binds to ERα in a fluorescence resonance energy transfer binding assay (IC50 = 1.14 nM) and potently degrades ERα in MCF-7 breast cancer cells (IC50 = 0.21 μM). Additionally, compound 17 exhibited superior oral bioavailability (AUC = 36.9 μg h mL–1) in rats than 6 (AUC = 3.35 μg h mL–1). The strikingly favorable pharmacokinetic property of 17 makes it a promising oral SERD suitable for clinical evaluation.
3.2. Benzothiophene derivatives as SERDs
The eminent SERMs raloxifene (18) and arzoxifene (19) (Fig. 5) bear the benzothiophene core that is responsible for the potent modulation of ER-mediated biological activity and adequate information about their metabolic and pharmacokinetic properties is available in the literature.86–88 The acrylic acid side chain attached to a ligand core is important for effective SERD activity. Masaaki Kurihara prepared tamoxifen derivatives with long alkyl chains on the amine moiety as effective SERDs.89,90 Among the alkyl chain derivatives, the compounds bearing a decyl group were capable of inducing the downregulation of ERα. Next, to understand the relationship between the ligand core and the SERD activity the group selected a non-steroidal SERM raloxifene bearing a benzothiophene core.91,92 In 2016, the group designed and synthesized raloxifene derivatives with long alkyl chains at the amine moiety and evaluated their SERD activity.93 All the compounds of the series bind to ERα with IC50 values between 1.8 and 61 nM, but only two compounds having decyl (20) and dodecyl (21) groups (Fig. 5) exhibited SERD activity. Compounds 20 and 21 reduced the ER protein level in the cells at the concentration of 10 μM and exhibited good ER antagonistic effect (0.68 and 0.09 nM, respectively).
Fig. 5. Chemical structure of raloxifene (18), arzoxifene (19) and benzothiophene derivatives 20–24.

Gregory R. J. Thatcher and coworkers utilized the benzothiophene (BT) scaffold to design and synthesize novel ER ligands that exhibited substantial biological activity as SERMs, ER agonists (SEMs), and ShERPAs.86 Bearing in mind the effectiveness of the benzothiophene core, the group prepared a series of ER ligands by substituting an acrylate side chain at the benzothiophene 3-position and varying the substituents at the 2-position and evaluated them for orally bioavailable SERD activity.52 These BT derivatives exhibited promising activity both as degraders and as antagonists of ERα and inhibited the growth of endocrine-independent breast cancer cells with IC50 values ranging from 0.3 to 12.5 nM. In comparison to compound GDC-0810 (7), the novel BT-SERD potency and efficacy in in vitro and in vivo assays was equivalent or superior to that of compound 7. The most potent compound of the series was 22 (Fig. 5) which exhibited promising ER degradation (IC50 = 0.07 nM) and ER binding activity (Ki = 0.57 nM). Compound 22 was effective in inhibiting the growth of treatment-resistant (IC50 = 0.3 nM) and tamoxifen-sensitive (IC50 < 0.1 nM) breast cancer cells.
In 2018 George S. Tria et al. made efforts to develop nonsteroidal SERDs with an appropriate orally bioavailable profile and in this respect, the group thought to combine a nonsteroidal benzothiophene core of an arzoxifene (19) with an appropriately selected side chain, such as the carboxylic acid moiety found in GW 7604 (6). The group designed and synthesized a series of potent benzothiophene-containing compounds that exhibited oral bioavailability and preclinical activity as SERDs.44 The effect of various linkers such as ether and amine tethers, carbonyl connections and C–C bond tethers connected to the cinnamic acid side chain on the benzothiophene scaffold was examined. The substitution at the ortho position of the 2-aryl ring was favorable to achieve high potency and a substitution at the para position was helpful to attain the pharmacokinetic properties. Among all the benzothiophene-based compounds, compound 23 (LSZ102) (Fig. 5) bearing the difluoroethyl group with the para-fluoro group emerged as the most potent ERα degrader (IC50 = 0.2 nM) and ERα antagonist with high ERα transcription (IC50 = 6 nM) and ERα remaining % (17%). Compound 23 reached phase I/Ib clinical trial for the treatment of advanced or metastatic ERα+ breast cancer.
The proposed mechanism for the action of traditional SERDs suggests that the induction of misfolding of the ER protein ultimately leads to proteasome-dependent ERα protein degradation.25 Most of the potent SERD molecules are capable of inducing degradation of the ER protein in ER+ breast cancer cells, but they are not able to completely degrade the ER protein.37,94 In recent years, the proteolysis targeting chimeras (PROTACs) have been rationally designed and developed as degrader molecules against target proteins. PROTACs are heterobifunctional small molecules containing two ligands that are tethered together by a chemical linker. A PROTAC works by inducing selective intracellular proteolysis and one ligand binds to the target protein of interest and another ligand to an E3 ligase system. Recruitment of the E3 ligase to the target protein induces polyubiquitylation and protesomal degradation of the target proteins in the cells. PROTACs have gained much attention because of the availability of potent and small drug-like molecules for a number of E3 ligase systems and degraders for a number of proteins.95–99 Naito et al. reported some PROTAC-like ERα degraders, named as specific and nongenetic IAP-dependent protein erasers (SNIPERs). The ER SNIPER molecules not only effectively induce partial degradation of the ER protein but also induce autoubiquitylation and proteasomal degradation of the E3 ligase, the cIAP1 protein, therefore limiting degradation and therapeutic efficacy.100,101
Shaomeng Wang and coworkers proposed that the molecules, which are able to fully degrade the ER protein, could be more effective for the treatment of ER+ metastatic breast cancer. In 2019, the group designed and synthesized small-molecule ERα degraders based on the proteolysis targeting chimera (PROTAC) concept and further performed extensive structure–activity relationship (SAR) studies.102 In this strategy, the ER ligand raloxifene (18) linked to the E3 ligase ligands CRBN (cereblon) and VHL (von Hippel–Lindau) via various long-chain linkers. This study provided compound 24 (ERD-308) (Fig. 5) as a potent and effective PROTAC ER degrader that exhibited DC50 (concentration causing 50% of protein degradation) values of 0.17 and 0.43 nM in MCF-7 and T47D ER+ breast cancer cell lines, respectively. In addition, 24 induces >95% of ER degradation at concentrations as low as 5 nM in both cell lines. Compound 24 effectively induces complete ER degradation and very effectively inhibits cell proliferation and therefore was selected for lead optimization for advanced ER+ breast cancer.
3.3. Coumarin and chromene derivatives as SERDs
It is apparent that most of the available SERMs and SERDs bear phenolic groups, which mimic the estradiol skeleton and are responsible for estrogen receptor related activity.103,104 However, the glucuronidation and sulfation of the phenols generates poor oral bioavailability and therefore supports the requirement of oral SERDs. Recently, 7-hydroxy-chomarin 25 (SS5020),50 tamoxifen derivative 26 (SS1020)51 and 7-hydroxy quinoline derivatives 27 and 28 (Fig. 6)47 emerged as selective ER downregulators with the belief that the phenyl acrylic acid moiety is responsible for the potency. Greene et al. in 2005 proposed the mechanism for the downregulation by phenylacrylic acid, which involves a shift in helix 12 that leads to degradation of the receptor.85 Considering the strong binding (IC50 = 0.0081 μM), downregulating ER activity (IC50 = 1.18 μM) and low lipophilicity of 25, James S. Scott et al. in 2015 synthesized a series of 3-aryl-7-hydroxycoumarins and evaluated them for selective estrogen receptor downregulator activity.45 The effect of various C4-linker, C3-aryl and core substitutions on the selected scaffold was examined for the ER binding, downregulation, lipophilicity and in vitro properties. Compound 29 (Fig. 6) bearing a 7-hydroxy group emerged as the most potent compound in terms of ER binding (IC50 = 0.0005 μM), ER MCF7 downregulation (IC50 = 0.0027 μM) and PR MCF7 antagonism (IC50 = 0.0039 μM). However, reduction in ligand-lipophilicity efficiency was observed for 29 with LLE = 6.5 in comparison to the initial hit compound 25 (LLE = 4.4). Further optimization and structural modifications to increase bioavailability led to the discovery of compound 30 (Fig. 6) with improved pharmacokinetic profiles and reasonable potency, i.e. ER binding (IC50 = 0.015 μM), ER MCF7 downregulation (IC50 = 0.100 μM), PR MCF7 antagonism (IC50 = 0.18 μM) and LLE = 4.5. Compound 30 bearing no hydroxyl group at the 7-position of the coumarin ring was considered as an attractive in vivo tool compound to evaluate the potential of an oral SERD.
Fig. 6. Chemical structure of SS5020 (25), SS1020 (26), 7-hydroxy quinoline derivatives (27 and 28), coumarin derivatives (29 and 30), EM 652 (31) and chromene derivatives (32–34).

In the literature, necessary information is available regarding the structural elements required to obtain compounds with high affinity for ER binding; however, certainly very little information is available to maximize ERα degradation activity while maintaining antagonistic activity. Therefore, Nicolous D. Smith and his group committed towards optimization of ERα degradation efficacy and identification of novel SERDs with strong oral bioavailability. The group utilized the in-cell Western assay that monitored the levels of ERα in MCF-7 breast cancer cells to identify a promising SERD, ARN-810/GDC-0810 (7). Further attempts were made to preliminarily optimize the chromene scaffold found in EM-652 (31) (Fig. 6) to obtain a novel SERD with high efficacy, oral bioavailability and ERα degrader activity in a model of tamoxifen-resistant breast cancer.105 The estradiol, EM-652 (31) and the most potent ER ligands bear two hydroxyls on the core structure responsible for key interactions in the ligand-binding domain (LBD) of ERα.106 Moreover, the attached side chain to this core mostly modulates the position of helix 12 of ER and therefore plays a major role in determining the antagonist/agonist properties of the ligand. The free phenolic groups of these molecules are susceptible towards in vivo glucuronidation.107 This is the reason why 31 had low exposure following oral dosing in mice (AUC = 0.09 μg h mL–1; 10 mg kg–1 orally). Some of the recent reports suggest that the positioning of the phenol OH group on the bicyclic core has a dramatic effect on the PK properties. The dihydrobenzoxathiin and chromane scaffolds with 6-OH phenol isomers exhibited higher rat oral bioavailability than the corresponding 7-OH phenol isomers.104,108 Bearing in mind all this information, the group decided to optimize the chromene core with a 6-OH group for higher ERα degradation efficacy.62 A series of chromene derivatives were synthesized to study the effect of the ring size and alkyl substitution of the side chain and to maximize ERα degradation efficacy as monitored by the MCF-7 in-cell Western assay. Most of the derivatives exhibited promising ERα degradation efficacy (IC50 < 1.0 nM) and antagonistic activity (IC50 < 0.5 nM).
The SAR study confirms that the difference in the ERα degradation efficacy was reliant on the ring size and substitution pattern of the side chain. Among all the chromene derivatives, compound 32 (Fig. 6) was identified as a leading compound with promising ERα binding (IC50 = 5.1 nM), ERα degradation (IC50 = 0.2 nM) and efficacy (91%), and MCF-7 antiproliferation (IC50 = 0.2 nM). The diastereoisomer of 32 which includes 33 (Fig. 6) (ERα binding IC50 = 2.8 nM; ERα degradation IC50 = 0.1 nM and efficacy 91%, and MCF-7 antiproliferation IC50 = 0.1 nM) exhibited a more promising profile in comparison to the diastereoisomer 34 (Fig. 6) (ERα binding IC50 = 136 nM, ERα degradation IC50 = 9.7 nM and efficacy 90%, and MCF-7 antiproliferation IC50 = 59 nM).
3.4. Benzoxepin derivatives as SERDs
Irene Barrett and coworkers utilized the benzoxepin scaffold in place of the triarylethylene core to synthesize a novel SERM 35 (Fig. 7) having potential as an ER binding ligand.109 Next, in 2018 the group developed a benzoxepin template with structural modifications to afford nonsteroidal SERDs and made efforts to optimize its ER degradation activity.49 The effect of the conformationally restricted benzoxepin ligand core structure and the amido, phenylacrylic acid, phenoxyacetic acid, phenoxybutyric acid, and penta-2,4-dienoic acid substituents on the activity of the ER was studied. The data suggest that these compounds have a high ER binding affinity with IC50 mostly in the nanomolar range and acrylic acid ligands were found to be ERα selective. Compound 36 (Fig. 7) bearing a phenylpenta-2,4-dienoic acid substituent on the benzoxepin core exhibited promising antiproliferative activity (IC50 = 1.6 μM) and was found to downregulate ERα and ERβ expression in MCF-7 breast cancer cells at 10 μM dosage. On the other hand, compound 37 (Fig. 7) having a phenoxybutyric acid derivative was not antiproliferative despite possessing antiestrogenic activity (IC50 = 0.43 μM), but selectively downregulated ERβ at 10 μM dosage. Therefore, compound 36 was chosen as a promising lead for the development of a clinically relevant SERD, whereas compound 37 was considered applicable to study the role of ERβ in cancer cells.
Fig. 7. Chemical structure of benzoxepin derivatives 35–37.

3.5. Tetrahydroisoquinoline derivatives as SERDs
James S. Scott and coworkers, after the successful investigation of a series of coumarin-based compounds as potent SERDs, decided to explore the phenolic tetrahydroisoquinoline (THIQ) scaffold with the purpose of identifying a potent, orally bioavailable SERD.45,46 The group selected phenolic THIQs because compounds 38 and 39 (Fig. 8) bearing phenolic THIQs with basic side chains act as antagonists at the receptors and belong to SERMs.110–112 The optimization around the core skeleton, alkyl side chain, and pendant aryl ring provided compounds with high potency mostly in the subnanomolar range. The most potent compound of the series was 40 (Fig. 8), obtained by replacement of the basic side chain with acrylic acid, which exhibited encouraging ER binding and ER degradation activities, i.e. IC50 = 19 and 1.3 nM, respectively, and bioavailability in rats (86%).
Fig. 8. Chemical structure of tetrahydroisoquinoline derivatives 38–42.

Considering the SERM properties of bioavailable tetrahydroisoquinoline (THIQ) 41 (Fig. 8),111,112 Heather E. Burks and coworkers in 2017 selected 41 for further modification and substituted the amino ethoxy side chain with the acrylic acid moiety in a view to obtain an ERα degradation phenotype.48 The tetrahydroisoquinoline derivative 42 (Fig. 8) emerged as the most potent ERα antagonist and SERD, exhibiting good oral bioavailability, antitumor efficacy, and SERD activity in vivo. The introduction of the isopropyl substituent at the p-position of the N-aryl ring improved ERα affinity (ERα binding IC50 of 17 nM) and activity on ERα transcription assay (ERα transcription IC50 of 30 nM) and ERα degradation assays (15% ERα remaining, ERα degradation IC50 of 0.9 nM). The good pharmacokinetic properties such as oral bioavailability, strong antagonistic and ERα degradation properties of THIQ 42 make it a promising SERD molecule for further study.
3.6. Tricyclic indazole derivatives as SERDs
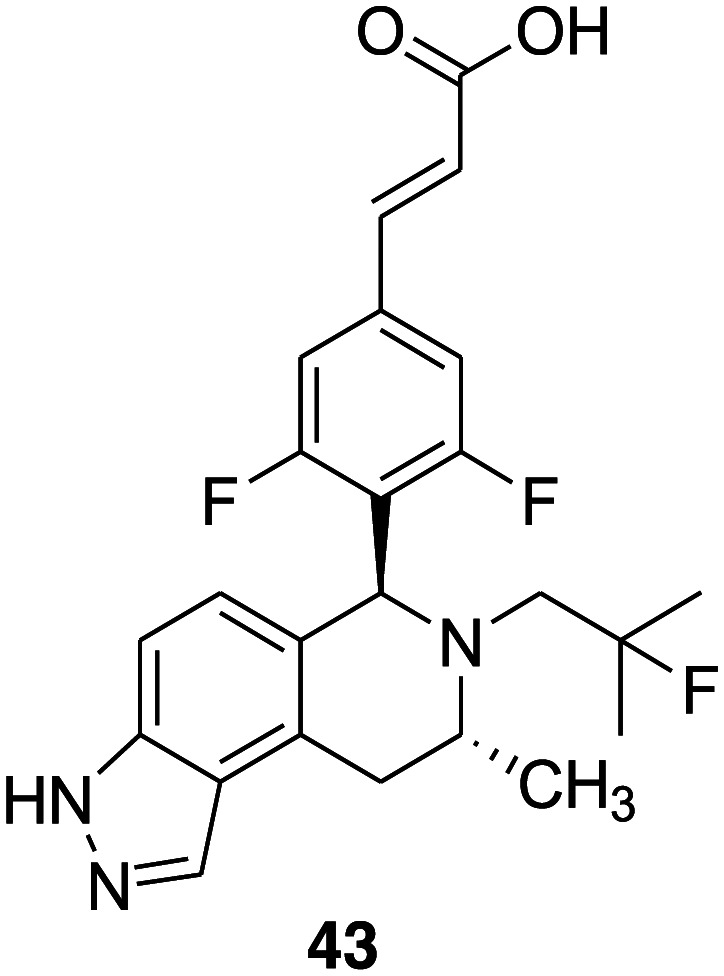
James S. Scott et al. reported a series of THIQ phenols that exhibited encouraging ER degrader–antagonist profile, including substantial ER binding and ER degradation activity.46 It was observed that the phenol functionality is responsible for glutathione (GSH) trapping in an in vitro microsome based-trapping assay as well as critical for potency. Therefore, the group decided to examine a potential phenol replacement further and employ tricyclic indole and indazole cores, which are present in some of the recently reported SERDs such as AZD 9496 (9).60 A series of tricyclic indazole derivatives were synthesized as selective estrogen receptor degrader antagonists and the introduction of an indazole group in the tetrahydroisoquinoline scaffold in place of phenol led to the removal of a reactive metabolite signal in an in vitro glutathione-trapping assay.113 Compound 43 (Fig. 9) emerged as a representative compound of the series with desirable estrogen receptor α degrader–antagonist profile (ER binding pIC50 = 9.5 and ER DR pIC50 = 9.6) and demonstrated tumor growth inhibition (80%) activity in a xenograft model of breast cancer at a dose of 20 mg kg–1 administered orally once daily.
Fig. 9. Chemical structure of tricyclic indazole derivative 43.
3.7. Diphenyl alkane derivatives as SERDs
Masaaki Kurihara and coworkers in 2015 reported 4-OH tamoxifen (2) derivatives with long alkyl chains similar to fulvestrant as an inducer of ER degradation.90 These compounds were synthesized and assayed as a mixture of geometric isoforms, i.e. E and Z isomers. Later, observation revealed that the triphenylethylene skeleton 2 is easily isomerized from the active Z form to an inactive E form in solution and is possibly responsible for a decrease in ER antagonistic and ER-downregulating activities.114 Moreover, the diphenyl skeleton acts as a steroidal skeleton mimic, having good chemical stability, easy to synthesize in comparison to the triphenylethylene skeleton of 2 and above all diphenylheptane skeleton based compounds acts as a potent anti-estrogen with an IC50 value of 4.5 nM from a reporter gene assay.115,116 Taking all this information in consideration, the group in 2017 decided to design and synthesize a series of diphenylalkane derivatives bearing several long alkyl chains on the hydroxy group and evaluated their biological properties such as ER degradation, binding affinity, transcriptional activity and anti-proliferation activity.117 Among all the compounds, 44 (Fig. 10) emerged as a novel selective estrogen degradation inducer with an ERα downregulation ratio at 10 μM = 32%, ERα binding affinity IC50 = 4.9 nM and ERα antagonistic activity IC50 = 45 nM. Computational docking analysis was performed to predict the binding mode of 44 to ERα. The study suggested that the interaction between the hydrogen atom on the amino group of 44 and the carboxylic acid of Glu351 of ERα leads to the binding of the long alkyl chain to the hydrophobic groove of ERα. The amino group and the optimal length of the long alkyl chain in the diphenylheptane skeleton are considered important for ERα downregulating activity.
Fig. 10. Chemical structure of diphenyl alkane derivatives 44 and 45.

ERs are mainly analyzed using Western blotting or other antibody-based technologies, which are associated with several limitations such as low throughput and not being quantitative. Therefore, in 2019 Tomomi Noguchi-Yachide and his coworkers developed a quantitative, high-throughput, luciferase-based SERD assay for the evaluation of SERD activity. Further, to evaluate its utility, the group also designed and synthesized a series of diphenylmethane (DPH) derivatives.118 The compounds were screened using a luciferase-based SERD assay and the structure–activity relationship (SAR) was established. The results revealed that the SERD activity of DPH derivatives was dependent on the structure of the ligand core and on the substituent of the phenolic oxygen. The most potent compound of the series was 45 (Fig. 10) with promising ERα antagonistic (IC50 = 44 nM) and down-regulatory activities (EC50 = 93 nM and Emax = 59%). Additionally, a docking study was performed on compound 45 and the crystal structure of the human ERα ligand-binding domain. The docking results revealed that the ethoxycarbonyl group of 45 does not form an ionic or hydrogen bonding interaction with Asp351 or the h11–h12 loop on ERα. Therefore, it confirms that the SERD efficiency depends upon the nature of the substituent on phenolic oxygen; its lack of stabilizing interactions with ERα may be important for effective SERD activity.
3.8. Cyclofenil, bicyclononane and adamantyl analogues as SERDs
The compound GW-7604 (6), a hydroxylated analogue of GW 5638, shows high estrogen receptor binding affinity.5,56,119 The X-ray crystallographic structure of 6 with ERα shows the interaction of the carboxylic acid group of the ligand with the positive end of the helix-12 dipole that results in the tilting of helix-12 and therefore suggests the significance of the carboxylic acid functional group in the acrylic acid side chain.85 Additionally, the reports suggest that the hydroxytriarylethylene (TAE) core of 6 similar to hydroxytamoxifen and diethylstilbestrol might be subjected to cis–trans isomerization in solution.120,121 However, Karen J. Kieser et al. realized that no substantial work was done to explore the importance of the side chain position, alternative groups and triarylethylene core structure on SERDs activity. Therefore, in 2010, a new series of ligands synthesized with acidic side chains of varying lengths and linkages built on different ligand core structures such as cyclofenil and bicyclononane were examined for SERDs activity.25 These new series of ER ligands bearing a cyclofenil and bicyclononane core skeleton were symmetrical and therefore devoid of stereoisomers. The results revealed that the alteration in the ligand core structure and the attached acrylic acid substituent of 6 affects the SERD activity. Most of the structural variants exhibited binding affinities for ERα and ERβ higher than that of 6. The compounds maintaining the acrylic acid side chain on the core skeleton, cyclofenil 46 and bicyclononane 47 (Fig. 11), effectively moderated the activity of ER. These two compounds redistributed and downregulated the ER in a similar manner to the SERD 6; however, the cyclofenil analogue (46) was less effective. The relative binding affinity for ERα and ERβ of the cyclofenil analogue (46) (2.1 ± 5 and 11.3 ± 3, respectively) is considerably lower than that of the GW compound (6) (20.3 ± 6 and 28.3 ± 7, respectively). However, the bicyclononane analogue (47) exhibited comparable affinity (17.7 ± 4 and 25.9 ± 5, respectively). Most of the compounds exhibited insignificant selectivity for either ERα or ERβ; however, the compounds of the cyclofenil series were found to be more ERβ selective. The cyclofenil acrylic acid compound (46) exhibited ∼5-fold selectivity in favor of ERβ.
Fig. 11. Chemical structure of cyclofenil (46), bicyclononane (47) and adamantyl analogues 48 and 49.

John A. Katzenellenbogen and his coworkers have explored the adamantane-based core, which is structurally simple, having planar symmetry and devoid of geometrical isomerism, as an ER ligand.25,120,122 Further, to search for new and more effective antiestrogens, the group designed and synthesized a series of compounds having an adamantyl ligand core with various acrylic acid side chains.53 The simple adamantyl ligand core with no stereochemical issues allowed the rapid synthesis of acrylate ketone, ester, and amide analogues. All compounds displayed high binding affinity for estrogen receptor α (ERα) and nearly all proved to have Ki values in the single-digit nanomolar range or below. All the compounds were evaluated for antiproliferative and ERα-downregulating (SERD) efficacies and the experimental data suggest that the efficacies and potencies of these molecules are dependent on the chemical structure of the side chain. The extension of the acrylic acid side chain as a secondary amide improved the affinity, efficacy and potency in surpassing the E2-driven proliferation of MCF-7 cells and in downregulating ERα levels, whereas the tertiary amine side chain was found to be less effective. The secondary carboxamides were found to give the best cellular activities, and 3-hydroxypropylamide was as efficacious as fulvestrant (4) in suppressing cell proliferation and gene expression. 3-Hydroxypropyl amide 49 and its parent compound 48 (Fig. 11) with a carboxylic acid side chain was selected as a promising candidate for further study in breast cancer cell lines having activated mutation in ERα and in vivo with a xenograft model. Compound 49 was found to be more potent than its parent carboxylic acid compound 48 in terms of antiproliferative activity (IC50 = 1.9 nM and 31 nM, respectively) and ERα-downregulating efficacy (IC50 = 0.53 nM and 17 nM, respectively), whereas their RBA ERα % (47% and 51%, respectively) and Ki values (0.43 and 0.40 nM, respectively) were comparable.
3.9. Oxabicycloheptene sulfonamides as SERDs
Hai-Bing Zhou and coworkers in 2012 reported a series of oxabicycloheptene sulfonamides (OBHSAs) as estrogen receptor antagonists.123 Crystal structure studies and phenotypic assays suggested that these compounds are SERDs with a new mechanism of action.124 Later, in 2019, to improve the potency of OBHSA as SERDs, the group designed and synthesized a new class of compounds bearing an OBHSA core structure and different side chains, e.g., basic side chains, long alkyl acid side chains, and glycerol ether side chains.125 The goal was to mimic the degrons of proteolysis targeting chimera (PROTAC) and then examine the structure–activity relationships of these PROTAC-like hybrid compounds. Most of the compounds of the series were effective in inhibiting MCF-7 cell proliferation and exhibited ERα degradation activity and a detailed structure–activity relationship was established. Among all, the most promising compounds were 50, 51 and 52 (Fig. 12) bearing an N-trifluoroethyl side chain and a para- methoxy group at the phenyl group of the sulfonamide. Compounds 50, 51, and 52 at the concentration of 10 μM efficiently degraded ERα with 99%, 99% and 81% degradation efficacies, respectively, and exhibited good antiproliferative activity (IC50 = 8.4, 6.1 and 2.8 μM, respectively). The docking study of these compounds with ERα suggested the significance of the basic side chain, N-trifluoroethyl group and para-methoxy group for ERα degradation efficacy. This study provides new options for discovering novel scaffolds in the development of novel PROTACs targeting ER for breast cancer therapy.
Fig. 12. Chemical structure of oxabicycloheptene sulfonamides 50–52.

4. Conclusion
In conclusion, we can undoubtedly mention that in recent years SERDs emerged as one of the possible approaches to treat drug-resistant breast cancer. During the past decade, researchers around the world significantly contributed to the discovery and development of novel SERDs. Fulvestrant, a steroidal SERD approved by the FDA as a drug for the treatment of breast cancer, and several non-steroidal molecules such as RAD-1901, AZD-9496 and GDC-0927 etc. are in clinical trials. Considerable progress has been made in developing effective SERDs with good bioavailability. Here we discussed the recently reported potent SERDs with an emphasis on their chemical structure and pharmacological profile. Considering the diverse chemical structure of available SERDs, we have grouped them into two categories, steroidal and non-steroidal, followed by the subcategory of the latter one. The positive outcome of SERDs in breast cancer treatment specifies that SERDs have tremendous scope in the pharmacotherapy of breast cancer and further research in this area could provide encouraging results for the breast cancer patient.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
The authors, Dr. Shagufta and Dr. Irshad Ahmad are thankful to the School of Graduate Studies and Research, American University of Ras Al Khaimah, UAE for its support.
References
- World Health Organisation, Cancer: Diagnosis and Treatment. https://www.who.int/cancer/treatment/en/, 2017.
- Katzenellenbogen J. A., Mayne C. G., Katzenellenbogen B. S., Greene G. L., Chandarlapaty S. Nat. Rev. Cancer. 2018:377–388. doi: 10.1038/s41568-018-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryfonidis K., Zardavas D., Katzenellenbogen B. S., Piccart M. Cancer Treat. Rev. 2016;50:68e81. doi: 10.1016/j.ctrv.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Jordan V. C. Br. J. Pharmacol. 1993;110(2):507–517. doi: 10.1111/j.1476-5381.1993.tb13840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagufta, Ahmad I. Eur. J. Med. Chem. 2018;143:515–531. doi: 10.1016/j.ejmech.2017.11.056. [DOI] [PubMed] [Google Scholar]
- Benson J. R., Ravisekar O. Curr. Cancer Ther. Rev. 2007;3:67–79. [Google Scholar]
- Ahmad I., Shagufta Eur. J. Med. Chem. 2015;102:375–386. doi: 10.1016/j.ejmech.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Dixon J. M. New J. Sci. 2014:1–27. [Google Scholar]
- Mills J. N., Rutkovsky A. C., Giordano A. Curr. Opin. Pharmacol. 2018;41:59–65. doi: 10.1016/j.coph.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone A., De Angelis C., Trivedi M. V., Osborne C. K., Schiff R. Breast. 2015;24(Suppl 2):S60–S66. doi: 10.1016/j.breast.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenbakh-Lamin K., Ben-Baruch N., Yeheskel A., Dvir A., Soussan-Gutman L., Jeselsohn R., Yelensky R., Brown M., Miller V. A., Sarid D., Rizel S., Klein B., Rubinek T., Wolf I. Cancer Res. 2013;73:6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- Toy W., Weir H., Razavi P., Lawson M., Goeppert A. U., Mazzola A. M., Smith A., Wilson J., Morrow C., Wong W. L., De Stanchina E., Carlson K. E., Martin T. S., Uddin S., Li Z., Fanning S., Katzenellenbogen J. A., Greene G., Baselga J., Chandarlapaty S. Cancer Discovery. 2016;7:277–287. doi: 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell D. P., Wardell S. E. Curr. Opin. Pharmacol. 2010;10:620–628. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove E. A., Sutherland R. L. Nat. Rev. Cancer. 2009;9(9):631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Herynk M. H., Fuqua S. A. Adv. Exp. Med. Biol. 2007;608:130–143. doi: 10.1007/978-0-387-74039-3_10. [DOI] [PubMed] [Google Scholar]
- Brueggemeier R. W., Hackett J. C., Diaz-Cruz E. S. Endocr. Rev. 2005;26(3):331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- Chumsri S., Howes T., Bao T., Sabnis G., Brodie A. J. Steroid Biochem. Mol. Biol. 2011;125(1–2):13–22. doi: 10.1016/j.jsbmb.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin S., Pickar J. H. Maturitas. 2014;80(1):52–57. doi: 10.1016/j.maturitas.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Patel H. K., Bihani T. Pharmacol. Ther. 2018;186:1–24. doi: 10.1016/j.pharmthera.2017.12.012. [DOI] [PubMed] [Google Scholar]
- Deshmane V. L., Krishnamurthy S., Melemed A. S., Peterson P., Buzdar A. U. J. Clin. Oncol. 2007;25:4967–4973. doi: 10.1200/JCO.2006.09.5992. [DOI] [PubMed] [Google Scholar]
- Johnston S. R. D. Clin. Cancer Res. 2001;7:4376–4387. [Google Scholar]
- Dauvois S., Danielian P. S., White R., Parker M. G. Proc. Natl. Acad. Sci. U. S. A. 1992;89(9):4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell S. J., Johnston S. R. D., Howell A. Best Pract. Res., Clin. Endocrinol. Metab. 2004;18:47–66. doi: 10.1016/j.beem.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Carlson R. W. Clin. Breast Cancer. 2005;6:S5–S8. doi: 10.3816/cbc.2005.s.008. [DOI] [PubMed] [Google Scholar]
- Kieser K. J., Kim D. W., Carlson K. E., Katzenellenbogen B. S., Katzenellenbogen J. A. J. Med. Chem. 2010;53:3320–3329. doi: 10.1021/jm100047k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell D. P., Wardell S. E., Norris J. D. J. Med. Chem. 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis M. J., Llombart-Cussac A., Feltl D., Dewar J. A., Jasiówka M., Hewson N., Rukazenkov Y., Robertson J. F. R. J. Clin. Oncol. 2015;33:3781–3786. doi: 10.1200/JCO.2015.61.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofanilli M., Turner N. C., Bondarenko I., Ro J., Im S. A., Masuda N., Colleoni M., DeMichele A., Loi S., Verma S., Iwata H., Harbeck N., Zhang K., Theall K. P., Jiang Y., Bartlett C. H., Koehler M., Slamon D. Lancet Oncol. 2016;17:425–439. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- Di Leo A., Jerusalem G., Petruzelka L., Torres R., Bondarenko I. N., Khasanov R., Verhoeven D., Pedrini J. L., Smirnova I., Lichinitser M. R., Pendergrass K., Garnett S., Lindemann J. P. O., Sapunar F., Martin M. J. Clin. Oncol. 2010;28:4594–4600. doi: 10.1200/JCO.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- Howell A. Cancer Treat. Rev. 2005;31:S26. doi: 10.1016/j.ctrv.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Howell A., Abram P. Cancer Treat. Rev. 2005;31:S3. doi: 10.1016/j.ctrv.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Wakeling A. E., Dukes M., Bowler J. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- Vergote I., Robertson J. F. R. Br. J. Cancer. 2004;90:S11–S14. doi: 10.1038/sj.bjc.6601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. F. R., Lindemann J., Garnett S., Anderson E., Nicholson R. I., Kuter I., Gee J. M. W. Clin. Breast Cancer. 2014;14:381–389. doi: 10.1016/j.clbc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Pritchard K. I., Rolski J., Papai Z., Mauriac L., Cardoso F., Chang J., Panasci L., Ianuli C., Kahan Z., Fukase K., Lindemann J. P. O., MacPherson M. P., Neven P. Breast Cancer Res. Treat. 2010;123:453–461. doi: 10.1007/s10549-010-1022-9. [DOI] [PubMed] [Google Scholar]
- Chia S., Gradishar W., Mauriac L., Bines J., Amant F., Federico M., Fein L., Romieu G., Buzdar A., Robertson J. F. R., Brufsky A., Possinger K., Rennie P., Sapunar F., Lowe E., Piccart M. J. Clin. Oncol. 2008;26:1664–1670. doi: 10.1200/JCO.2007.13.5822. [DOI] [PubMed] [Google Scholar]
- Wittmann B. M., Sherk A., McDonnell D. P. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- Lai A. C., Crews C. M. Nat. Rev. Drug Discovery. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. F. R. Oncologist. 2007;12:774–784. doi: 10.1634/theoncologist.12-7-774. [DOI] [PubMed] [Google Scholar]
- van Kruchten M., de Vries E. G., Glaudemans A. W., van Lanschot M. C., van Faassen M., Kema I. P., Brown M., Schröder C. P., de Vries E. F., Hospers G. A. Cancer Discovery. 2015;5:72–81. doi: 10.1158/2159-8290.CD-14-0697. [DOI] [PubMed] [Google Scholar]
- Yoneya T., Tsunenari T., Taniguchi K., Kanbe Y., Morikawa K., Yamada-Okabe H., Lee Y. H., Lee M. H., Kwon L. S. Oncol. Rep. 2009;21:747–755. [PubMed] [Google Scholar]
- Kanbe Y., Kim M. H., Nishimoto M., Ohtake Y., Yoneya T., Ohizumi I., Tsunenari T., Taniguchi K., Ichi Kaiho S., Nabuchi Y., Araya H., Kawata S., Morikawa K., Jo J. C., Kwon H. A., Lim H. S., Kim H. Y. Bioorg. Med. Chem. Lett. 2006;16:4959–4964. doi: 10.1016/j.bmcl.2006.06.047. [DOI] [PubMed] [Google Scholar]
- Jiang Q., Zheng S., Wang G. Future Med. Chem. 2013;5:1023–1035. doi: 10.4155/fmc.13.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tria G. S., Abrams T., Baird J., Burks H. E., Firestone B., Gaither L. A., Hamann L. G., He G., Kirby C. A., Kim S., Lombardo F., Macchi K. J., McDonnell D. P., Mishina Y., Norris J. D., Nunez J., Springer C., Sun Y., Thomsen N. M., Wang C., Wang J., Yu B., Tiong-Yip C. L., Peukert S. J. Med. Chem. 2018;61:2837–2864. doi: 10.1021/acs.jmedchem.7b01682. [DOI] [PubMed] [Google Scholar]
- Degorce S. L., Bailey A., Callis R., De Savi C., Ducray R., Lamont G., MacFaul P., Maudet M., Martin S., Morgentin R., Norman R. A., Peru A., Pink J. H., Plé P. A., Roberts B., Scott J. S. J. Med. Chem. 2015;58:3522–3533. doi: 10.1021/acs.jmedchem.5b00066. [DOI] [PubMed] [Google Scholar]
- Scott J. S., Bailey A., Davies R. D. M., Degorce S. L., Macfaul P. A., Gingell H., Moss T., Norman R. A., Pink J. H., Rabow A. A., Roberts B., Smith P. D. ACS Med. Chem. Lett. 2016;7:94–99. doi: 10.1021/acsmedchemlett.5b00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra W. J., Patel H. S., Liang X., Blanc J. B. E., Heyer D. O., Willson T. M., Iannone M. A., Kadwell S. H., Miller L. A., Pearce K. H., Simmons C. A., Shearin J. J. Med. Chem. 2005;48:2243–2247. doi: 10.1021/jm040154f. [DOI] [PubMed] [Google Scholar]
- Burks H. E., Abrams T., Kirby C. A., Baird J., Fekete A., Hamann L. G., Kim S., Lombardo F., Loo A., Lubicka D., Macchi K., McDonnell D. P., Mishina Y., Norris J. D., Nunez J., Saran C., Sun Y., Thomsen N. M., Wang C., Wang J., Peukert S. J. Med. Chem. 2017;60:2790–2818. doi: 10.1021/acs.jmedchem.6b01468. [DOI] [PubMed] [Google Scholar]
- O'Boyle N. M., Barrett I., Greene L. M., Carr M., Fayne D., Twamley B., Knox A. J. S., Keely N. O., Zisterer D. M., Meegan M. J. J. Med. Chem. 2018;61:514–534. doi: 10.1021/acs.jmedchem.6b01917. [DOI] [PubMed] [Google Scholar]
- Suzuki N., Liu X., Laxmi Y. R. S., Okamoto K., Kim H. J., Zhang G., Chen J. J., Okamoto Y., Shibutani S. Int. J. Cancer. 2011;128:974–982. doi: 10.1002/ijc.25659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxmi Y. R. S., Liu X., Suzuki N., Kim S. Y., Okamoto K., Kim H. J., Zhang G., Chen J. J., Okamoto Y., Shibutani S. Int. J. Cancer. 2010;127:1718–1726. doi: 10.1002/ijc.25167. [DOI] [PubMed] [Google Scholar]
- Xiong R., Zhao J., Gutgesell L. M., Wang Y., Lee S., Karumudi B., Zhao H., Lu Y., Tonetti D. A., Thatcher G. R. J. J. Med. Chem. 2017;60:1325–1342. doi: 10.1021/acs.jmedchem.6b01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J., Guillen V. S., Sharma A., Zhao Y., Ziegler Y., Gong P., Mayne C. G., Srinivasan S., Kim S. H., Carlson K. E., Nettles K. W., Katzenellenbogen B. S., Katzenellenbogen J. A. J. Med. Chem. 2017;60:6321–6336. doi: 10.1021/acs.jmedchem.7b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zheng S., Guo S., Zhang C., Zhong Q., Zhang Q., Ma P., Skripnikova E. V., Bratton M. R., Wiese T. E., Wang G. ACS Med. Chem. Lett. 2017;8:102–106. doi: 10.1021/acsmedchemlett.6b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson T. M., Henke B. R., Momtahen T. M., Charifson P. S., Batchelor K. W., Lubahn D. B., Moore L. B., Oliver B. B., Sauls H. R., Triantafillou J. A., Wolfe S. G., Baer P. G. J. Med. Chem. 1994;37:1550–1552. doi: 10.1021/jm00037a002. [DOI] [PubMed] [Google Scholar]
- Willson T. M., Norris J. D., Wagner B. L., Asplin I., Baer P., Brown H. R., Jones S. A., Henke B., Sauls H., Wolfe S., Morris D. C., Mcdonnell D. P. Endocrinology. 1997;138:3901–3911. doi: 10.1210/endo.138.9.5358. [DOI] [PubMed] [Google Scholar]
- Lai A., Kahraman M., Govek S., Nagasawa J., Bonnefous C., Julien J., Douglas K., Sensintaffar J., Lu N., Lee K. J., Aparicio A., Kaufman J., Qian J., Shao G., Prudente R., Moon M. J., Joseph J. D., Darimont B., Brigham D., Grillot K., Heyman R., Rix P. J., Hager J. H., Smith N. D. J. Med. Chem. 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- Joseph J. D., Darimont B., Zhou W., Arrazate A., Young A., Ingalla E., Walter K., Blake R. A., Nonomiya J., Guan Z., Kategaya L., Govek S. P., Lai A. G., Kahraman M., Brigham D., Sensintaffar J., Lu N., Shao G., Qian J., Grillot K., Moon M., Prudente R., Bischoff E., Lee K. J., Bonnefous C., Douglas K. L., Julien J. D., Nagasawa J. Y., Aparicio A., Kaufman J., Haley B., Giltnane J. M., Wertz I. E., Lackner M. R., Nannini M. A., Sampath D., Schwarz L., Manning H. C., Tantawy M. N., Arteaga C. L., Heyman R. A., Rix P. J., Friedman L., Smith N. D., Metcalfe C., Hager J. H. eLife. 2016;5:e15828. doi: 10.7554/eLife.15828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner F., Shomali M., Paquin D., Lyttle C. R., Hattersley G. Anti-Cancer Drugs. 2015;26:948–956. doi: 10.1097/CAD.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Savi C., Bradbury R. H., Rabow A. A., Norman R. A., De Almeida C., Andrews D. M., Ballard P., Buttar D., Callis R. J., Currie G. S., Curwen J. O., Davies C. D., Donald C. S., Feron L. J. L., Gingell H., Glossop S. C., Hayter B. R., Hussain S., Karoutchi G., Lamont S. G., MacFaul P., Moss T. A., Pearson S. E., Tonge M., Walker G. E., Weir H. M., Wilson Z. J. Med. Chem. 2015;58:8128–8140. doi: 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- Weir H. M., Bradbury R. H., Rabow A. A., Buttar D., Callis R. J., Curwen J. O., De Almeida C., Ballard P., Hulse M., Donald C. S., Feron L. J. L., Karoutchi G., MacFaul P., Moss T., Norman R. A., Pearson S. E., Tonge M., Davies G., Walker G. E., Wilson Z., Rowlinson R., Powell S., Sadler C., Richmond G., Ladd B., Pazolli E., Mazzola A. M., D'Cruz C., De Savi C. Cancer Res. 2016;76:3307–3318. doi: 10.1158/0008-5472.CAN-15-2357. [DOI] [PubMed] [Google Scholar]
- Nagasawa J., Govek S., Kahraman M., Lai A., Bonnefous C., Douglas K., Sensintaffar J., Lu N., Lee K., Aparicio A., Kaufman J., Qian J., Shao G., Prudente R., Joseph J. D., Darimont B., Brigham D., Maheu K., Heyman R., Rix P. J., Hager J. H., Smith N. D. J. Med. Chem. 2018;61:7917–7928. doi: 10.1021/acs.jmedchem.8b00921. [DOI] [PubMed] [Google Scholar]
- Kahraman M., Govek S. P., Nagasawa J. Y., Lai A., Bonnefous C., Douglas K., Sensintaffar J., Liu N., Lee K., Aparicio A., Kaufman J., Qian J., Shao G., Prudente R., Joseph J. D., Darimont B., Brigham D., Heyman R., Rix P. J., Hager J. H., Smith N. D. ACS Med. Chem. Lett. 2019;10:50–55. doi: 10.1021/acsmedchemlett.8b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagufta, Ahmad I. MedChemComm. 2017;8:871–885. doi: 10.1039/c7md00097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagufta, Ahmad I., Panda G. Eur. J. Med. Chem. 2017;133:139–151. doi: 10.1016/j.ejmech.2017.03.054. [DOI] [PubMed] [Google Scholar]
- Shagufta, Ahmad I. Eur. J. Med. Chem. 2016;116:267–280. doi: 10.1016/j.ejmech.2016.03.058. [DOI] [PubMed] [Google Scholar]
- Bowler J., Lilley T. J., Pittam J. D., Wakeling A. E. Steroids. 1989;54:71–99. doi: 10.1016/0039-128x(89)90076-7. [DOI] [PubMed] [Google Scholar]
- Wakeling A. E., Dukes M., Bowler J. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- Wakeling A. E., Bowler J. J. Steroid Biochem. Mol. Biol. 1998;30:141–147. [Google Scholar]
- Blazejewski J. C., Wilmshurst M. P., Popkin M. D., Wakselman C., Laurent G., Nonclercq D., Cleeren A., Ma Y., Seo H. S., Leclercq G. Bioorg. Med. Chem. 2003;11:335–345. doi: 10.1016/s0968-0896(02)00457-1. [DOI] [PubMed] [Google Scholar]
- Robertson J. F. R. Oncologist. 2007;12:774–784. doi: 10.1634/theoncologist.12-7-774. [DOI] [PubMed] [Google Scholar]
- Ohno S., Rai Y., Iwata H., Yamamoto N., Yoshida M., Iwase H., Masuda N., Nakamura S., Taniguchi H., Kamigaki S., Noguchi S. Ann. Oncol. 2010;21:2342–2347. doi: 10.1093/annonc/mdq249. [DOI] [PubMed] [Google Scholar]
- Yoneya T., Tsunenari T., Taniguchi K., Kanbe Y., Morikawa K., Yamada-Okabe H., Lee Y. H., Lee M. H., Kwon L. S. Oncol. Rep. 2009;21:747–755. [PubMed] [Google Scholar]
- Yoneya T., Taniguchi K., Tsunenari T., Saito H., Kanbe Y., Morikawa K., Yamada-Okabe H. Anti-Cancer Drugs. 2005;16:751–756. doi: 10.1097/01.cad.0000171515.27439.de. [DOI] [PubMed] [Google Scholar]
- Hoffmann J., Bohlamann R., Heinrich N., Hofmeister H., Kroll J., Künzer H., Lichtner R. B., Nishino Y., Parczyk K., Sauer G., Gieschen H., Ulbrich H. F., Schneider M. R. J. Natl. Cancer Inst. 2004;96:210–218. doi: 10.1093/jnci/djh022. [DOI] [PubMed] [Google Scholar]
- Chouinard S., Tessier M., Vernouillet G., Gauthier S., Labrie F., Barbier O., Belanger A. Mol. Pharmacol. 2006;69:908–920. doi: 10.1124/mol.105.015891. [DOI] [PubMed] [Google Scholar]
- Wu B., Kulkarni K., Basu S., Zhang S., Hu M. J. Pharm. Sci. 2011;100:3655–3681. doi: 10.1002/jps.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Zhong Q., Zhang Q., Zheng S., Miele L., Wang G. Breast Cancer Res. Treat. 2015;152:283–291. doi: 10.1007/s10549-015-3461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q., Zhang C., Zhang Q., Miele L., Zheng S., Wang G. BMC Cancer. 2015;15:665. doi: 10.1186/s12885-015-1621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zheng S., Akerstrom V. L., Yuan C., Ma Y., Zhong Q., Zhang C., Zhang Q., Guo S., Ma P., Skripnikova E. V., Bratton M. R., Pannuti A., Miele L., Wiese T. E., Wang G. J. Med. Chem. 2016;59:8134–8140. doi: 10.1021/acs.jmedchem.6b00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayaratne A. L., Nagel S. C., Paige L. A., Christensen D. J., Norris J. D., Fowlkes D. M., McDonnell D. P. Endocrinology. 1999;140:5828–5840. doi: 10.1210/endo.140.12.7164. [DOI] [PubMed] [Google Scholar]
- Fan M., Rickert E. L., Chen L., Aftab S. A., Nephew K. P., Weatherman R. V. Breast Cancer Res. Treat. 2007;103:37–44. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- Johnston S., Cheung K. Curr. Med. Chem. 2010;17:902–914. doi: 10.2174/092986710790820633. [DOI] [PubMed] [Google Scholar]
- Gradishar W. J., Sahmoud T. Clin. Breast Cancer. 2005;6:S23–S29. doi: 10.3816/cbc.2005.s.011. [DOI] [PubMed] [Google Scholar]
- Wu Y. L., Yang X., Ren Z., McDonnell D. P., Norris J. D., Willson T. M., Greene G. L. Mol. Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Xiong R., Patel H. K., Gutgesell L. M., Zhao J., Delgado-Rivera L., Pham T. N. D., Zhao H., Carlson K., Martin T., Katzenellenbogen J. A., Moore T. W., Tonetti D. A., Thatcher G. R. J. J. Med. Chem. 2016;59:219–237. doi: 10.1021/acs.jmedchem.5b01276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelhamid R., Luo J., VandeVrede L., Kundu I., Michalsen B., Litosh V. A., Schiefer I. T., Gherezghiher T., Yao P., Qin Z., Thatcher G. R. J. ACS Chem. Neurosci. 2011;2:256–268. doi: 10.1021/cn100106a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Kastrati I., Ashgodom R. T., Lantvit D. D., Overk C. R., Choi Y., Van Breemen R. B., Bolton J. L., Thatcher G. R. J. Drug Metab. Dispos. 2009;37:161–169. doi: 10.1124/dmd.108.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoda T., Okuhira K., Kato M., Demizu Y., Inoue H., Naito M., Kurihara M. Bioorg. Med. Chem. Lett. 2014;24:87–89. doi: 10.1016/j.bmcl.2013.11.078. [DOI] [PubMed] [Google Scholar]
- Shoda T., Kato M., Harada R., Fujisato T., Okuhira K., Demizu Y., Inoue H., Naito M., Kurihara M. Bioorg. Med. Chem. 2015;23:3091–3096. doi: 10.1016/j.bmc.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Schmid C. R., Sluka J. P., Duke K. M. Tetrahedron Lett. 1999;40:675–678. [Google Scholar]
- Grese T. A., Cho S., Finley D. R., Godfrey A. G., Jones C. D., Lugar C. W., Martin M. J., Matsumoto K., Pennington L. D., Winter M. A., Adrian M. D., Cole H. W., Magee D. E., Phillips D. L., Rowley E. R., Short L. L., Glasebrook A. L., Bryant H. U. J. Med. Chem. 1997;40:146–167. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]
- Shoda T., Kato M., Fujisato T., Misawa T., Demizu Y., Inoue H., Naito M., Kurihara M. Bioorg. Med. Chem. 2016;24:2914–2919. doi: 10.1016/j.bmc.2016.04.068. [DOI] [PubMed] [Google Scholar]
- Marsaud V., Gougelet A., Maillard S., Renoir J. M. Mol. Endocrinol. 2003;17:2013–2027. doi: 10.1210/me.2002-0269. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Deng Q., Zhao H., Xie M., Chen L., Yin F., Qin X., Zheng W., Zhao Y., Li Z. ACS Chem. Biol. 2018;13:628–635. doi: 10.1021/acschembio.7b00985. [DOI] [PubMed] [Google Scholar]
- Zhang C., Han X. R., Yang X., Jiang B., Liu J., Xiong Y., Jin J. Eur. J. Med. Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Liu T., Jiao Q., Ji J., Tao M., Liu Y., You Q., Jiang Z. Eur. J. Med. Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- Schiedel M., Herp D., Hammelmann S., Swyter S., Lehotzky A., Robaa D., Oláh J., Ovádi J., Sippl W., Jung M. J. Med. Chem. 2018;61:482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Crew A. P., Raina K., Dong H., Qian Y., Wang J., Vigil D., Serebrenik Y. V., Hamman B. D., Morgan A., Ferraro C., Siu K., Neklesa T. K., Winkler J. D., Coleman K. G., Crews C. M. J. Med. Chem. 2018;61:583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Ohoka N., Okuhira K., Ito M., Nagai K., Shibata N., Hattori T., Ujikawa O., Shimokawa K., Sano O., Koyama R., Fujita H., Teratani M., Matsumoto H., Imaeda Y., Nara H., Cho N., Naito M. J. Biol. Chem. 2017;292:4556–4570. doi: 10.1074/jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N., Morita Y., Nagai K., Shimokawa K., Ujikawa O., Fujimori I., Ito M., Hayase Y., Okuhira K., Shibata N., Hattori T., Sameshima T., Sano O., Koyama R., Imaeda Y., Nara H., Cho N., Naito M. J. Biol. Chem. 2018;293:6776–6790. doi: 10.1074/jbc.RA117.001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Hu B., Wang M., Xu F., Miao B., Yang C. Y., Wang M., Liu Z., Hayes D. F., Chinnaswamy K., Delproposto J., Stuckey J., Wang S. J. Med. Chem. 2019;62:1420–1442. doi: 10.1021/acs.jmedchem.8b01572. [DOI] [PubMed] [Google Scholar]
- Musa M., Cooperwood J., Khan M. O. Curr. Med. Chem. 2008;15:2664–2679. doi: 10.2174/092986708786242877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q., Blizzard T. A., Morgan J. D., Birzin E. T., Chan W., Yang Y. T., Pai L. Y., Hayes E. C., Dasilva C. A., Warrier S., Yudkovitz J., Wilkinson H. A., Sharma N., Fitzgerald P. M. D., Li S., Colwell L., Fisher J. E., Adamski S., Reszka A. A., Kimmel D., Dininno F., Rohrer S. P., Freedman L. P., Schaeffer J. M., Hammond M. L. Bioorg. Med. Chem. Lett. 2005;15:1675–1681. doi: 10.1016/j.bmcl.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Gauthier S., Caron B., Cloutier J., Dory Y. L., Favre A., Larouche D., Mailhot J., Ouellet C., Schwerdtfeger A., Leblanc G., Martel C., Simard J., Merand Y., Belanger A., Labrie C., Labrie F. J. Med. Chem. 1997;40:2117–2122. doi: 10.1021/jm970095o. [DOI] [PubMed] [Google Scholar]
- Veeneman G. H. Curr. Med. Chem. 2005;12(9):1077–1136. doi: 10.2174/0929867053764662. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran A., McKeand W. E., Sullivan P., DeMaio W., Stoltz R., Scatina J. A. Drug Metab. Dispos. 2009;37:1219–1225. doi: 10.1124/dmd.108.023861. [DOI] [PubMed] [Google Scholar]
- Kim S., Wu J., Chen H. Y., Birzin E. T., Chan W., Yang Y. T., Colwell L., Li S., Dahllund J., DiNinno F., Rohrer S. P., Schaeffer J. M., Hammond M. L. Bioorg. Med. Chem. Lett. 2004;14:2741–2745. doi: 10.1016/j.bmcl.2004.03.074. [DOI] [PubMed] [Google Scholar]
- Barrett I., Meegan M. J., Hughes R. B., Carr M., Knox A. J. S., Artemenko N., Golfis G., Zisterer D. M., Lloyd D. G. Bioorg. Med. Chem. 2008;16:9554–9573. doi: 10.1016/j.bmc.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Chesworth R., Zawistoski M. P., Lefker B. A., Cameron K. O., Day R. F., Mangano F. M., Rosati R. L., Colella S., Petersen D. N., Brault A., Lu B., Pan L. C., Perry P., Ng O., Castleberry T. A., Owen T. A., Brown T. A., Thompson D. D., DaSilva-Jardine P. Bioorg. Med. Chem. Lett. 2004;14:2729–2733. doi: 10.1016/j.bmcl.2004.03.077. [DOI] [PubMed] [Google Scholar]
- Renaud J., Bischoff S. F., Buhl T., Floersheim P., Fournier B., Halleux C., Kallen J., Keller H., Schlaeppi J. M., Stark W. J. Med. Chem. 2003;46:2945–2957. doi: 10.1021/jm030086h. [DOI] [PubMed] [Google Scholar]
- Renaud J., Bischoff S. F., Buhl T., Floersheim P., Fournier B., Geiser M., Halleux C., Kallen J., Keller H., Ramage P. J. Med. Chem. 2005;48:364–379. doi: 10.1021/jm040858p. [DOI] [PubMed] [Google Scholar]
- Scott J. S., Bailey A., Buttar D., Carbajo R. J., Curwen J., Davey P. R. J., Davies R. D. M., Degorce S. L., Donald C., Gangl E., Greenwood R., Groombridge S. D., Johnson T., Lamont S., Lawson M., Lister A., Morrow C. J., Moss T. A., Pink J. H., Polanski R. J. Med. Chem. 2019;62:1593–1608. doi: 10.1021/acs.jmedchem.8b01837. [DOI] [PubMed] [Google Scholar]
- McCague R., Jarman M., Leung O. T., Foster A. B., Leclercq G., Stoessel S. J. Steroid Biochem. Mol. Biol. 1998;31:545–547. doi: 10.1016/0022-4731(88)90005-2. [DOI] [PubMed] [Google Scholar]
- Maruyama K., Noguchi-Yachide T., Sugita K., Hashimoto Y., Ishikawa M. Bioorg. Med. Chem. Lett. 2010;20:6661–6666. doi: 10.1016/j.bmcl.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Maruyama K., Nakamura M., Tomoshige S., Sugita K., Makishima M., Hashimoto Y., Ishikawa M. Bioorg. Med. Chem. Lett. 2013;23:4031–4036. doi: 10.1016/j.bmcl.2013.05.067. [DOI] [PubMed] [Google Scholar]
- Misawa T., Fujisato T., Kanda Y., Ohoka N., Shoda T., Yorioka M., Makishima M., Sekino Y., Naito M., Demizu Y., Kurihara M. MedChemComm. 2017;8:239–246. doi: 10.1039/c6md00553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanjyo S., Ohgane K., Yoshioka H., Makishima M., Hashimoto Y., Noguchi-Yachide T. Bioorg. Med. Chem. 2019;27:1952–1961. doi: 10.1016/j.bmc.2019.03.042. [DOI] [PubMed] [Google Scholar]
- Robertson D. W., Katzenellenbogen J. A., Long D. J., Rorke E. A., Katzenellenbogen B. S. J. Steroid Biochem. 1982;16:1–13. doi: 10.1016/0022-4731(82)90137-6. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen J. A., Carlson K. E., Katzenellenbogen B. S. J. Steroid Biochem. 1985;22:589–596. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- Airy S. C., Sinsheimer J. E. Steroids. 1981;38:593–603. doi: 10.1016/0039-128x(81)90057-x. [DOI] [PubMed] [Google Scholar]
- Seo J. W., Comninos J. S., Chi D. Y., Kim D. W., Carlson K. E., Katzenellenbogen J. A. J. Med. Chem. 2006;49:2496–2511. doi: 10.1021/jm0512037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Zhang C., Nwachukwu J. C., Srinivasan S., Cavett V., Zheng Y., Carlson K. E., Dong C., Katzenellenbogen J. A., Nettles K. W., Zhou H. B. Org. Biomol. Chem. 2012;10:8692–8700. doi: 10.1039/c2ob26531a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S., Nwachukwu J. C., Bruno N. E., Dharmarajan V., Goswami D., Kastrati I., Novick S., Nowak J., Cavett V., Zhou H. B., Boonmuen N., Zhao Y., Min J., Frasor J., Katzenellenbogen B. S., Griffin P. R., Katzenellenbogen J. A., Nettles K. W. Nat. Chem. Biol. 2017;13:111–118. doi: 10.1038/nchembio.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhang S., Zhang J., Hu Z., Xiao Y., Huang J., Dong C., Huang S., Zhou H. B. Eur. J. Med. Chem. 2019:48–61. doi: 10.1016/j.ejmech.2019.03.058. [DOI] [PubMed] [Google Scholar]