

Summary
A major obstacle to treating Alzheimer’s disease (AD) is our lack of understanding of the molecular mechanisms underlying selective neuronal vulnerability, which is a key characteristic of the disease. Here we present a framework integrating high-quality neuron type-specific molecular profiles across the lifetime of the healthy mouse, which we generated using bacTRAP, with postmortem human functional genomics and quantitative genetics data. We demonstrate human-mouse conservation of cellular taxonomy at the molecular level for neurons vulnerable and resistant in AD, identify specific genes and pathways associated with AD neuropathology, and pinpoint a specific functional gene module underlying selective vulnerability, enriched in processes associated with axonal remodeling, and affected by both amyloid accumulation and aging. We have made all cell-type specific profiles and functional networks available at alz.princeton.edu. Overall, our study provides a molecular framework for understanding the complex interplay between Aβ, aging, and neurodegeneration within the most vulnerable neurons in AD.
eTOC blurb :
Neurons display different levels of vulnerability to Alzheimer’s pathology. Roussarie, Yao et al. experimentally profile and computationally model several relevant neuron types. Using a mouse−human framework, they identify genes linking Aß, aging, and tau in vulnerable neurons. Finally, they show experimentally that PTB, regulator of tau splicing, contributes to vulnerability.
Introduction
Selective neuronal vulnerability is a shared property of most neurodegenerative diseases (Saxena and Caroni, 2011). In the early stages of Alzheimer’s disease (AD), the most common form of age-related dementia, clinical symptoms (such as memory loss) are caused by the selective degeneration of principal neurons of the entorhinal cortex layer II (ECII), followed by CA1 pyramidal cells in the hippocampus, and pyramidal neurons in neocortical association areas. In contrast, other brain regions, such as the primary sensory cortices, are relatively resistant to degeneration until later stages of the disease (Arnold et al., 1991; Bussière et al., 2003; Fukutani et al., 2000; Gómez-Isla et al., 1996; Hof and Morrison, 1990; Hyman et al., 1984; Morrison and Hof, 1997; West et al., 1994). The molecular basis for this selective vulnerability remains unknown.
AD is characterized by two major pathological hallmarks: accumulation of the Aβ peptide (the main constituent of amyloid plaques) and formation of neurofibrillary tangles (NFT, aggregates of hyperphosphorylated tau proteins which are thought to occur downstream of Aβ accumulation). Amyloid plaques do not accumulate in discrete brain areas. Rather, they are relatively widespread across most regions of the neocortex, followed by the entorhinal cortex and hippocampus of AD patients (Sepulcre et al., 2017; Thal et al., 2002). In contrast, NFTs exhibit the same regional pattern as neurodegeneration(Braak and Braak, 1991; Bussière et al., 2003; Lewis et al., 1987). The co-occurrence of NFTs and neurodegeneration, as well as the fact that the best pathological correlate for clinical symptoms to date is the extent of NFT formation (Arriagada et al., 1992; Brier et al., 2016; Giannakopoulos et al., 2003), highlight the importance of tau pathology. Genetic analyses have revealed the central role of microglial cells and of their crosstalk with neurons in the disease (Efthymiou and Goate, 2017). Selective neuronal vulnerability could be due to intrinsic properties of vulnerable neurons, or alternatively to the surrounding microglia. However, recent evidence suggests that the regional particularities of microglia are primarily driven by differences in the neighboring neurons (Ayata et al., 2018). Yet the molecular drivers for the neuronal component of the pathological cascade that leads from Aβ accumulation to NFT formation and neurodegeneration are still largely unknown.
To understand and model cell type-specific vulnerability in AD, we must thus gain insight into the molecular-level differences between healthy vulnerable and resistant neurons that predispose some neurons, before any pathological process becomes visible, to develop tau pathology earlier and faster than others. This requires high-quality cell type-specific profiles of both vulnerable and resistant neurons. While some neuron types of relevance to AD were profiled in a mouse hippocampal study (Cembrowski et al., 2016), the most vulnerable neuron type in early AD (ECII) has not previously been studied ex vivo. In humans, Small et al. profiled whole EC and dentate gyrus (DG) in control and AD patients (Small et al., 2005), and the Allen Brain Atlas (ABA) provides a large dataset for a number of human brain regions (Hawrylycz et al., 2015) , but neither of these studies are cell type-specific. A comprehensive dataset of neuron-specific AD-relevant profiles has been generated by Liang et al (Liang et al., 2007). However, while valuable, human samples, including those in the studies cited above, are inevitably subject to degradation and postmortem changes and, in the context of AD, do not allow for direct probing of the effect of aging and Aβ accumulation on gene expression.
Furthermore, a key challenge to achieving a molecular understanding of selective neuronal vulnerability in AD is that vulnerability and pathology are likely not simply the result of a few genes, or even pathways, acting in isolation. Deciphering the pathological cascade requires cell type-specific systems-level analysis and modeling of the complex molecular interactions that underpin the vulnerability of specific neurons to AD, going beyond differential gene expression and pathway enrichment analysis. Previous work examining whole brain lysates from AD patients and non-demented individuals (Miller et al., 2008; Mostafavi et al., 2018; Zhang et al., 2013) demonstrated the promise of network analyses in AD, but these studies were limited to larger brain regions and thus could not address cell type-specific vulnerability.
Here, we provide the first molecular framework to understand the interactions between age, Aβ, and tau within neurons. Our approach (Figure 1A) integrates the precision of cell-type specific profiling across age in the non-diseased mouse with computational modeling of human neuronal -omics (e.g., expression, interaction) data. Importantly, we show that the molecular identity of AD vulnerable and resistant neurons is largely conserved between mouse and human, justifying the use of the mouse to gain insight into specific vulnerability, which would be intractable in humans. All subsequent analyses use human data, generating human network models for each neuron type, and using disease signals from human quantitative genetics data, ensuring relevance for human AD neuropathology. The neuron-specific expression profiles and functional networks are available for download and exploration in an interactive web interface (alz.princeton.edu).
Figure 1:
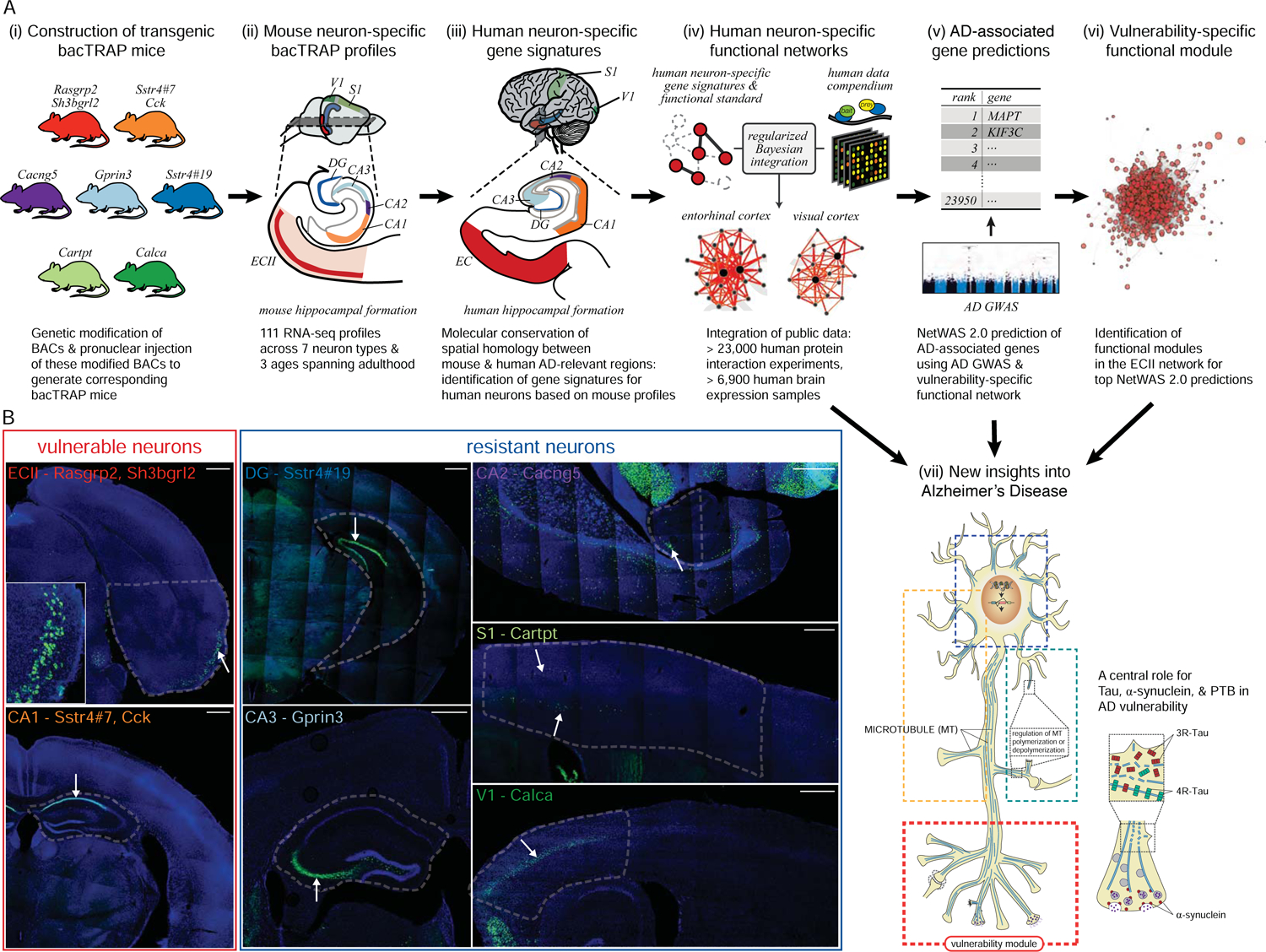
An integrative experimental genomics and bioinformatics framework, combining mouse and human data, to identify genes and pathways involved in Alzheimer’s Disease. A. Overview of our framework. (i) To obtain molecular profiles of 7 types of neurons most vulnerable and resistant to AD using the bacTRAP technology, we constructed bacTRAP mice for each of them (see b). (ii) 111 neuron-specific high-quality ex vivo expression profiles were obtained for each neuron type at three different ages (5, 12 and 24 months), using bacTRAP followed by deep sequencing: bacTRAP allows for fast isolation of actively translated RNA with minimal alterations of mRNA content after the death of the animal, and quantitative assessment of gene expression over a large range of expression levels. (iii) Using these data, we generated neuron-specific molecular signatures in mouse and human and created a spatial homology map between the two organisms. (iv) We used these neuron-specific signatures to construct 7 neuron-specific functional networks, through Bayesian integration of a compendium of over 30,000 human experiments. (v) We identified genes functionally associated with AD pathology by combining the network for the most vulnerable neuron (ECII) with an AD tau pathology GWAS (Beecham et al., 2014) using our NetWAS 2.0 machine learning approach. (vi) These genes form distinct functional modules in the vulnerable neuron-specific network, with one module in particular capturing vulnerability-specific signal. (vii) Our analyses point to the involvement of neurotransmitter release and axonogenesis in AD vulnerability, as well as a central role for the regulation of tau and α-synuclein by the RNA-binding protein PTB. Overall, we map AD-associated processes and their potential regulation by aging and Aβ in ECII neurons, providing the first molecular dissection of the AD pathological cascade within vulnerable neurons. B. bacTRAP transgenic mice generated for the molecular profiling of vulnerable and resistant neurons. For each line, brain sections were stained with an anti-eGFP antibody (green) and counterstained with DAPI (blue). Genes whose regulatory regions we used for driving eGFP-L10a expression are indicated in each frame. For ECII and CA1, we show a section from the Rasgrp2- and from the Sstr4#7-bacTRAP line respectively, but also used Sh3bgrl2- and Cck-bacTRAP lines for subsequent analyses. The dashed line delineates the brain region dissected out for bacTRAP. Arrows point to the neurons of interest, which overexpress eGFP-L10a. Scale bar: 500 μm (ECII: principal neurons of the layer II of the entorhinal cortex, CA1, CA2, CA3: pyramidal neurons of hippocampus CA1, CA2, CA3 respectively, DG: granule neurons of dentate gyrus, S1, V1: pyramidal neurons of primary somatosensory and visual cortex respectively.)
Results:
Cell type-specific profiling of mouse neurons with differential vulnerability to AD
To investigate the selective vulnerability of neurons in AD, we generated cell-type specific expression profiles spanning the entirety of adulthood for vulnerable and resistant neurons using the bacTRAP (Bacterial Artificial Chromosome – Translating Ribosome Affinity Purification) technology in wild-type mice (Doyle et al., 2008; Heiman et al., 2008). The bacTRAP technology enabled us to assay AD-relevant neuronal cell types with genome-wide coverage, measure transcripts ex vivo (as opposed to postmortem), and specifically capture actively translated (rather than all transcribed) genes.
We focused on the vulnerable principal neurons of ECII and pyramidal neurons of CA1, and on five types of resistant neurons, namely pyramidal neurons of CA2, CA3, primary visual cortex (V1), primary somatosensory cortex (S1), and granule cells of DG. Specifically, we constructed different transgenic mouse lines for each type of neuron, overexpressing the ribosomal protein L10a fused to the green fluorescent protein (GFP) under the transcriptional control of a driver specific to that type of neuron (Figure 1B). The bacTRAP procedure then consists of immunoprecipitation of GFP-tagged polysomes from GFP-L10a-expressing cells, thus isolating actively translated neuron-specific mRNAs for RNA-sequencing. Previous work using bacTRAP or similar technologies (e.g., RiboTag) has demonstrated the strong enrichment for cell type-specific signal from cells expressing the tagged ribosomal protein (Brichta et al., 2015; Clarke et al., 2018; Doyle et al., 2008; Heiman et al., 2008; Sanz et al., 2009; Sun et al., 2015).
We first performed multidimensional scaling analysis of the resulting bacTRAP data and found that the samples (3–12 biological replicates per neuron type per age) clustered primarily by tissue location, as expected, with a clear separation between the ECII, hippocampal regions (CA1, CA2, CA3, DG), and neocortical regions (S1 and V1) (Figure 2A). We further verified the expression patterns of known neuron type-specific markers (Figure 2B) and identified the top enriched genes for each neuron type in our data (Figure 2C, Table S1). Comparisons with the semiquantitative in situ hybridization (ISH) data in the ABA (Figure S1) show that our data includes the cell-type specific signals in these datasets, while providing substantially higher regional and quantitative, genome-scale coverage. Thus, our approach provides a high quality, genome-wide assay of ex vivo neuron type-specific expression in AD-vulnerable and AD-resistant regions of the brain. We provide an interactive web interface (alz.princeton.edu) to explore these expression data.
Figure 2:
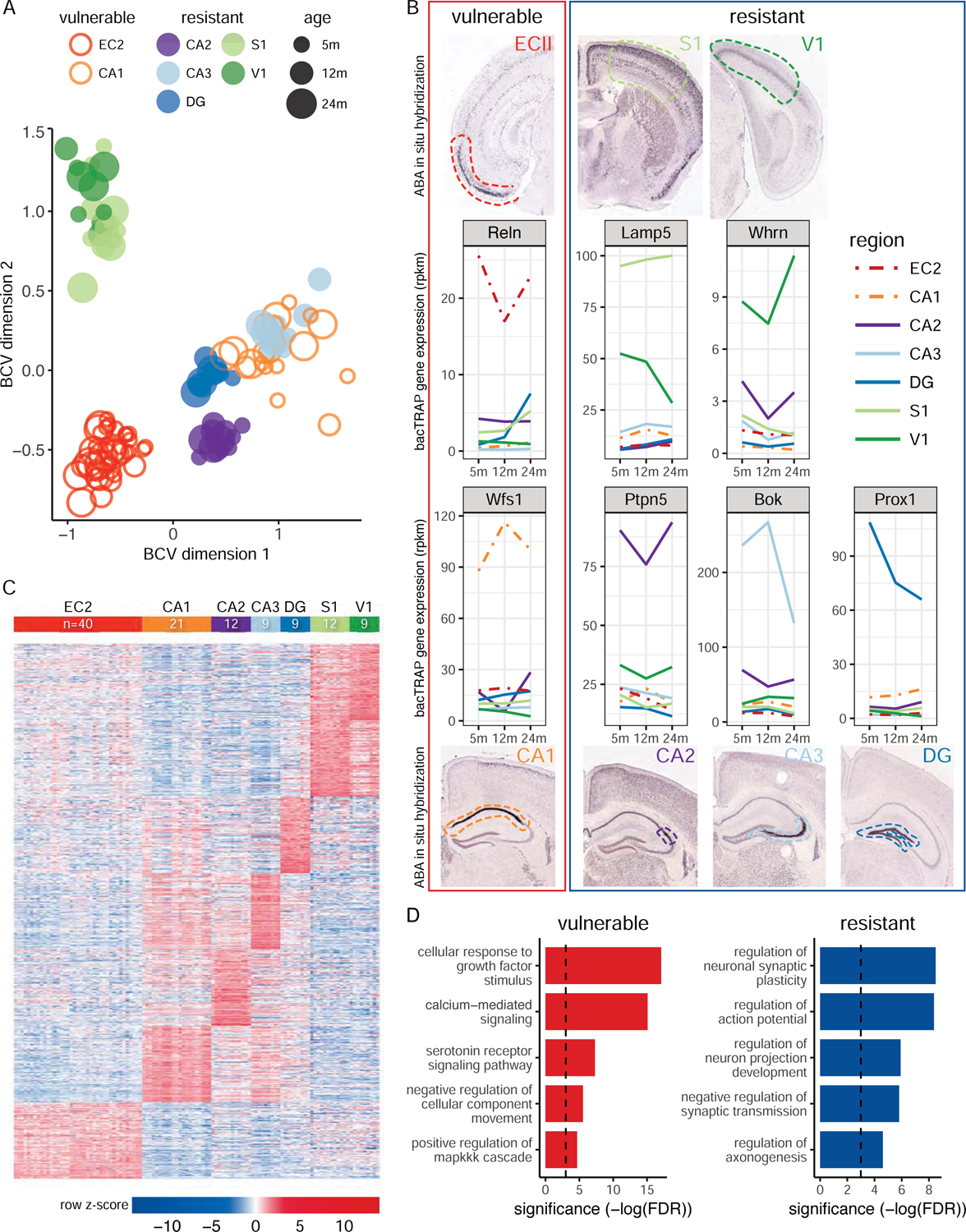
Molecular characterization of vulnerable and resistant neurons. A. Multidimensional scaling analysis of all samples demonstrating clustering of samples by region of origin. Each dot represents one sample (two mice pooled). Red (ECII) and orange (CA1) dots correspond to AD vulnerable neurons. Purple (CA2), light blue (CA3), dark blue (DG), light green (S1), dark green (V1), dots correspond to resistant neurons. Increasing dot sizes represent increasing mouse ages (5, 12 and 24 months). B. Verification of our quantitative, neuron-specific RNAseq profiles for known markers by ISH (ABA). Expression of previously described cell type-specific markers across the seven types of neurons, Reln for ECII neurons (Pesold et al., 1998), Wfs1 for CA1 pyramidal neurons (Lein et al., 2004), Ptpn5 (or Step) for CA2 pyramidal neurons (Kohara et al., 2014), Bok for CA3 pyramidal neurons (Lein et al., 2004), Prox1 for DG granule neurons (Liu et al., 2000), Whrn for V1 layer IV (Dugas-Ford et al., 2012), and Lamp5 (or C20orf103) for S1 layers II and III (Zeng et al., 2012). For each marker, we show the expression at 5, 12, and 24 months of age; each color represents a different type of neuron; we also show for each gene an ISH image from the ABA that shows expression in the corresponding neurons. Image credit: Allen Institute. C. Heatmap of gene expression for the top 500 genes enriched in each neuron type. For each gene (rows, grouped by neuron type in which they are enriched) and sample (columns, grouped by cell type, including all three different ages), the row-normalized log2 (RPKM) is displayed, showing that hundreds of genes are enriched in each type of neuron. D. Pathways enriched in vulnerable (red) and resistant (blue) neurons, with their significance (−log(FDR)) of enrichment. See also Table S1, Figure S1.
To characterize molecular signatures for AD vulnerable cells in the non-disease state, we compared gene expression profiles of ECII and CA1 neurons against the five AD-resistant neuron types in wild-type mice (Table S1). Among the significantly enriched processes we found many AD-relevant pathways (Figure 2D). Furthermore, one of the gene sets most enriched in vulnerable neurons was annotated Alzheimer’s disease-associated genes (KEGG AD genes, one-sided Wilcoxon rank-sum test, p-value < 9.41×10−7). These results support the hypothesis that there are intrinsic differences between ECII and CA1 neurons, present even in healthy individuals. We aim to leverage these differences in our framework to understand why these neurons are preferential substrates for the development of AD pathogenesis.
Neuron-specific spatial homology between mouse and human
An important question for interpreting model organism studies of AD is whether the molecular identity of neurons is conserved between mouse and human. Previous comparisons using spatially resolved semiquantitative ISH (Zeng et al., 2012) or transcriptomics and proteomics without cellular resolution (Carlyle et al., 2017; Strand et al., 2007; Walker and Jucker, 2017) have suggested that mouse and human regional expression patterns are correlated, but the conservation of expression across neuronal subtypes requires further exploration.
In humans, fully quantitative data at cell-type specific resolution is lacking across the regions most relevant for AD. However, the discrete brain structure microarray data from the ABA (Hawrylycz et al., 2015) captures enough regional specificity for an expression-based comparison between the seven mouse neuronal subtypes and 205 human brain regions. We calculated a spatial homology score between molecular signatures for each mouse neuron type and each human brain region, generating 1,435 pairwise spatial homology measurements. Remarkably, of all these possible mappings, we found a near perfect match between each mouse profile and its corresponding relevant human brain region (Figure 3, Table S2, Figure S2A, p-value < 0.0001, permutation test, n=10,000). This confirms the validity of leveraging the power of ex vivo neuron-specific molecular profiles in the mouse to gain relevant insight into the molecular characteristics of the most vulnerable neurons in human AD. While there are differences in lifespan and other factors relevant to AD that may facilitate the degeneration of human neurons (Walker and Jucker, 2017), our comparison supports the notion that physiological differences between vulnerable and resistant neurons are conserved. This study provides, to our knowledge, the first systematic evidence that the molecular identity of AD-relevant neuron types is conserved between the mouse and human brain. This supports our approach of combining the cell type-specific signals in healthy mouse neurons with the AD-relevant signals in large collections of human data.
Figure 3:
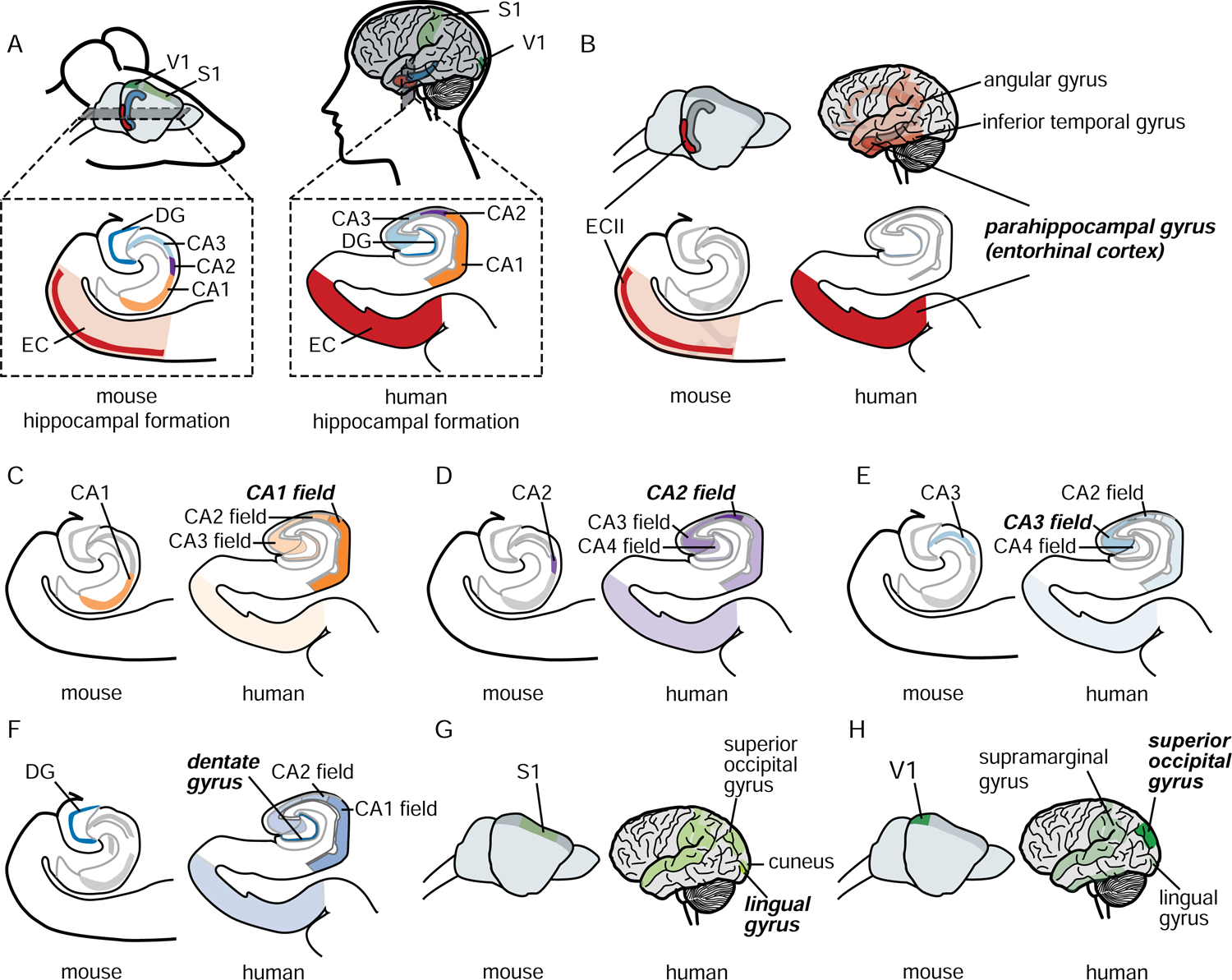
Conservation of molecular identity of seven AD-vulnerable and -resistant neuronal types between mouse and human. A. Location in the mouse and human brain of the seven brain regions included in this study (lateral view of the whole brain and close-up of the hippocampal formation). To validate the use of mouse profiles for the study of the human disease, we compared the molecular signatures of the mouse neurons derived from 111 mouse bacTRAP samples with 205 human brain region-specific expression profiles from the Allen Institute (B – H). For each mouse neuron type (B: ECII, C: CA1, D: CA2, E: CA3, F: DG, G: S1, H: V1), the human brain regions with molecular signatures closest to each mouse neuron type are highlighted (the more opaque the color of a brain region, the higher the similarity with the mouse neuron type). Note that we find a near perfect correspondence between mouse neurons and human brain regions. See also Table S2 and Figure S2A.
In silico modeling of gene networks in AD-relevant neuronal cell types
AD neurodegeneration is the result of multiple molecular-level changes to the system of interacting genes and pathways within vulnerable neurons. We model this system with cell-type specific functional networks, i.e., maps of functional relationships between proteins in the specific cellular contexts of the different types of neurons. Specifically, a functional relationship represents the common involvement of two proteins, either directly or indirectly, in a biological pathway in the cell type of interest. We recently developed a regularized Bayesian network integration method to construct tissue-specific functional networks (Greene et al., 2015). These network-level models are an effective first approximation of the functional landscape of a cell and have been successfully applied to the study of diseases (Greene et al., 2015; Krishnan et al., 2016; Song et al., 2016) . It was, however, previously impossible to apply this method to construct networks at neuron-specific resolution because of limitations in high-quality cell type-specific gene expression annotations in human. Given the strong concordance between our mouse neuron-specific molecular signatures and their corresponding human brain regions, we used these signatures as positive examples to extract cell-specific signal from a large human data compendium including thousands of gene expression, protein-protein interaction, and shared regulatory profile datasets to construct human neuron type-specific functional networks. We have made the resulting seven in silico human genome-wide network models, each representing one AD-vulnerable or resistant neuron type in the non-disease state, available both for download and dynamic, query-based exploration at http://alz.princeton.edu.
To identify functional characteristics and differences specific to neuron types vulnerable or resistant to AD, we examined the functional cohesiveness of biological processes (i.e., a measure of network connectivity among genes known to be part of that process) in each corresponding functional network model (Table S1). We found that pathways neuroprotective in AD (Caraci et al., 2008; Liu et al., 2014; Tesseur et al., 2006)appeared more cohesive in AD resistant neurons than in vulnerable neurons, namely the transforming growth factor beta receptor signaling pathway (in DG) and the canonical Wnt signaling pathway (in DG, S1 and V1). On the other hand, mitochondrial processes like apoptotic mitochondrial changes and mitochondrial fission were more cohesive in CA1 and ECII respectively, which is consistent with the saliency of mitochondrial dysfunction at early stages of the disease (Du et al., 2010). Strikingly, we found that the processes with largest functional cohesiveness in vulnerable compared to resistant neurons were all related to microtubule organization. This is the first evidence that these tau-regulated processes may intrinsically differ between healthy vulnerable and resistant neurons.
Identifying AD-associated genes through integration of AD GWAS and the ECII functional network
To identify potential genes involved with AD neuropathology, we then combined these network models of vulnerable neuron function with unbiased disease signal from human quantitative genetics data. Specifically, we developed an approach, Network-Wide Association Study 2.0 (NetWAS 2.0), that extends our previously described (Greene et al., 2015) framework with a probabilistic subsampling method to take into account gene-level confidence from quantitative genetics studies. This machine learning approach leverages genome-wide association studies (GWAS) in conjunction with a functional network specific to the region of interest to identify cell type-specific network patterns predictive of a disease, reranking all genes based on disease relevance significantly better than the original GWAS (Greene et al., 2015).
Because many cortical neurons eventually develop pathology at late stages of AD, we expect to find disease-relevant signal with all network models, with the network model for the most vulnerable neuron type capturing the bulk of vulnerability signal. To specifically focus on the neuronal genes leading to degeneration as opposed to the varied other disease components that eventually lead to cognitive decline, we used a neuropathologically rather than clinically defined AD GWAS, namely a GWAS for Braak stage (NFT pathology-based staging) (Beecham et al., 2014). We thus applied NetWAS 2.0 using the network model for the most vulnerable neuron (ECII) to reprioritize genes based on this Braak-stage GWAS (Table S3). Remarkably, MAPT (microtubule-associated protein tau, the gene that encodes tau, the primary component of NFTs) was ranked first among all 23,950 reprioritized genes. MAPT was not even nominally significant in the initial GWAS (initial GWAS tau p-value = 0.269). Notably, a rare variant of Tau has been shown to increase AD risk (Coppola et al., 2012) as well as neuronal vulnerability (Silva et al., 2016), and a tau haplotype was associated with EC atrophy (Desikan et al., 2015). This illustrates the power of NetWAS 2.0 to extract important disease-relevant signals that may be hidden in the original GWAS. The identification of genes associated with Tau pathology using the Braak-stage GWAS was further corroborated by significantly overlapping results obtained by applying NetWAS 2.0 to two recent independent neuropathologically-associated AD GWAS (one-sided Fisher’s exact test comparing overlap of top 10% genes: NFT-GWAS based NetWAS, p-value < 2.2×10−16; CSF pTau-GWAS based NetWAS, p-value < 2.2×10−16) (Chibnik et al., 2018; Deming et al., 2017).
Overall, while the original GWAS for Braak stages was somewhat enriched for known AD-annotated genes (KEGG AD genes, one-sided Wilcoxon rank-sum test, p-value < 0.199), the reprioritized gene ranking was much more significantly enriched for these genes (one-sided Wilcoxon rank sum test, p-value < 4.55×10−8). We also observed strong enrichment of genes involved in regulation of Aβ accumulation and NFT formation (one-sided Wilcoxon rank-sum test, Amyloid: p-value < 1.29×10−10, NFT-1: p-value < 2.2×10−16, NFT-2: p-value < 2.2×10−16, respectively for gene sets curated by curators independent from the analyses, Table S4) (Figure 4A, B). Known AD neuroprotective pathways, like neurotrophin signaling (Nagahara et al., 2009) and Wnt signaling pathway (Liu et al., 2014; Tesseur et al., 2006) were also predicted to be strongly associated (Table S3). Lastly, we highlight the association of neurotransmitter secretion with AD (FDR < 2.57×10−24). Dysregulation of this pathway is one of the most prominent effects of Aβ accumulation (Abramov et al., 2009), and the resulting hippocampal network hyperactivity was suggested to be a crucial contributor to AD pathogenesis (Bakker et al., 2012; Palop and Mucke, 2010; Vossel et al., 2013). As the AD signal in NetWAS 2.0 comes only from unbiased GWAS data (i.e., no prior AD disease knowledge was incorporated), the NetWAS 2.0 results thus provide a data-driven, unbiased prioritization of AD-associated processes out of the many pathways that, over time, have been associated with tau pathology.
Figure 4:
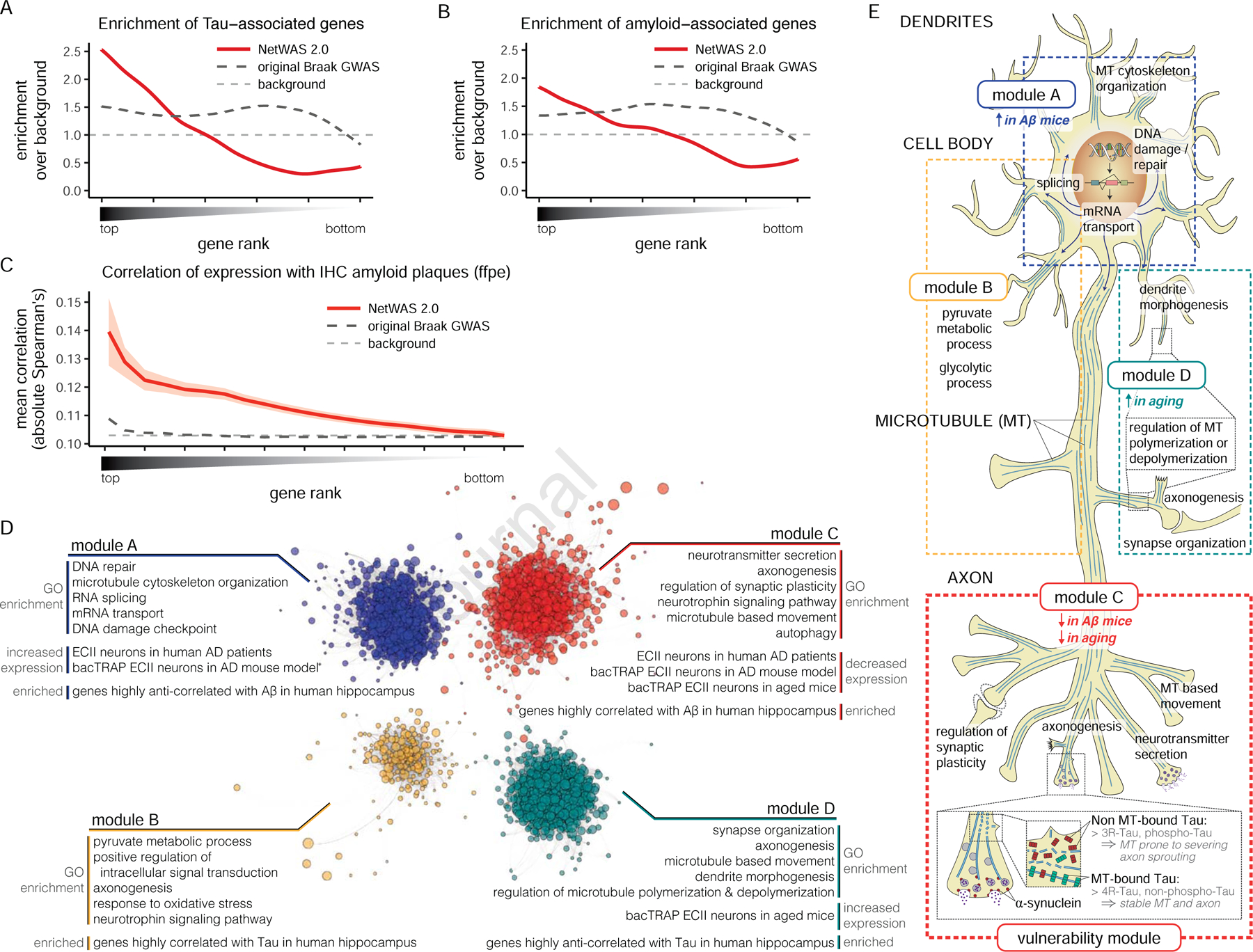
Prediction, validation, and functional analysis of genes associated with AD pathology. A, B. Top NetWAS 2.0 gene predictions show significantly higher enrichment of NFT- (A) and Aβ-associated genes (B) (curated by an independent expert, Table S4) than the original GWAS used as input to NetWAS 2.0. C. Human hippocampal expression levels of top AD-associated gene predictions are highly correlated with amyloid plaque amounts in the ACT cohort (Miller et al., 2017). The x-axis represents the proportion of top NetWAS 2.0 genes obtained. The average absolute values of correlations between gene expression level and amyloid plaques across the subset of genes are plotted (NetWAS 2.0 predictions in red with 95% confidence interval; Braak GWAS in black; background genes in grey). D. Clustering of the top 10% NetWAS genes using a shared-nearest-neighbor-based community-finding algorithm identifies functional modules corresponding to distinct AD-associated processes. We indicate pathways enriched in each module, as well as the association of each module with aging and AD pathology in both our data (independent from our functional network analysis) and external datasets. Each dot represents a gene (where size inversely correlates with the NetWAS 2.0 ranking, i.e. larger dots represent top ranked genes). Network layout (ForceAtlas) by gephi (Bastian et al., n.d.) of ECII-specific network posterior probabilities above prior are shown (comembership score ≥ 0.75 based on 1000 subsamples for visual clarity). E. Representation of pathways enriched in each module (d) in ECII neurons. Microtubules (MT) are represented in blue. Enrichment for genes modulated by Aβ and aging is indicated for each module. Module A is enriched in neuronal cell body processes, while module C includes many axonal processes. Modules A, B, and D may be generally associated with tau pathology in many types of projection neurons, while module C may capture the surplus of vulnerability from ECII neurons. The module includes both structural and functional axonal remodeling pathways, suggesting that axonal plasticity is key to the degeneration process in AD. Concomitant actions of Aβ and aging on module C genes might perturb crosstalk between axon remodeling processes and eventually impinge on SNCA and MAPT function. Inset: magnified view of an axon terminal. β-synuclein, a regulator of neurotransmitter release, binds to synaptic vesicles (grey circles), to the membrane of the presynaptic active zone, and to MTs. Both forms of tau (3R in red, and 4R in green) are present along MT in the axons, with 4R (as well as non-phosphorylated tau) having higher affinity to MT than 3R (as well as hyperphosphorylated tau). Tau-bound MTs are less stable and more prone to severing, a requirement for axon sprouting and axonal plasticity. PTBP1 regulates both tau isoform usage and α-synuclein levels. See also Table S3–6 and Figure S2B.
Beyond these well-characterized associations, one of the most significantly enriched pathways in the NetWAS 2.0 results was a microtubule-related process: regulation of microtubule cytoskeleton organization (FDR < 1.38×10−27) (Table S3). This is consistent with our connectivity analysis, where we discovered that microtubule-regulating pathways were particularly cohesive in vulnerable neurons. Together, these results support the hypothesis that microtubule-regulating pathway genes may cooperate with MAPT for the formation of NFTs in vulnerable neurons. Our data also strongly support a role for mRNA splicing and transport in AD pathogenesis (RNA splicing, FDR = 4.48×10−10; RNA transport, FDR = 3.32×10−16). RNA binding proteins in these processes have recently emerged as major players in various non-AD neurodegenerative diseases (Ramaswami et al., 2013), and recent studies suggested a possible involvement in AD (TIA1 protects against tau-mediated degeneration (Apicco et al., 2018), CELF1 is one of the main GWAS hits (Lambert et al., 2013), and the activity of ELAVL proteins is altered in AD brains(Scheckel et al., 2016)).
Association of NetWAS 2.0 genes with AD pathology
We next investigated the link between key drivers of the AD pathological cascade (Aβ accumulation and age) and AD vulnerability-associated genes identified by NetWAS 2.0 analysis. Recall that we leveraged a tau pathology-based GWAS (Beecham et al., 2014) for the NetWAS 2.0 predictions, thus prioritizing genes that may modulate NFT formation. To enable the direct analysis of the ECII-specific effects of Aβ accumulation in AD, we crossed our ECII-bacTRAP mice with an AD mouse model (APP/PS1 mice). These mice overexpress mutant APP and PSEN1 and have increased levels of Aβ in the cortex and hippocampus (Borchelt et al., 1997). We profiled ECII neurons at 6 months of age, when the first plaques are starting to form (Table S4). Genes significantly downregulated in APP/PS1 mice were strongly enriched in our top NetWAS 2.0 gene predictions (one-sided Wilcoxon rank-sum test, p-value < 9.21×10−14). Additionally, genes modified by aging in ECII of wild-type mice (24- vs. 5-month-old mice) (Table S4) were also strongly enriched at the top of our ranking (one-sided Wilcoxon rank-sum test, p-value < 4.19×10−13). Our finding that Aβ and aging modulate the expression of genes predicted by NetWAS 2.0 to be associated with tau pathology indicates that these genes might connect Aβ accumulation and NFT formation in the age-dependent AD pathological cascade.
To examine the possible relationship between top NetWAS 2.0 genes and human AD pathology directly, we then used data from two independent human datasets. The Adult Changes in Thought study (ACT) (Miller et al., 2017) provides paired gene expression data and pathology measurements from hippocampal samples of elderly individuals at risk for dementia. For each gene, we calculated the correlation between expression level and amount of amyloid plaques. We found that expression of our top gene predictions was significantly more correlated with amyloid burden than either background or genes implicated in the original Braak-stage GWAS (bootstrap p-value < 0.0001, Figure 4C). Top NetWAS 2.0 predictions obtained using the ECII functional networks were also more significantly correlated with amyloid plaque amount than top predictions obtained using the functional networks for resistant neurons (Figure S2B). Furthermore, our predictions were very significantly enriched in genes differentially downregulated in ECII neurons of sporadic AD patients measured in a different study (relative to control patients, one-sided Wilcoxon rank-sum test, p-value < 2.2×10−16) (Liang et al., 2008). Together, this consensus of results indicates that the top NetWAS 2.0 gene predictions highlight genes that participate in the AD pathological cascade within neurons.
Identification of AD-associated functional modules
To better understand the processes and pathways through which these genes are associated with NFT formation and AD, we clustered the genes with top NetWAS 2.0 ranks into functional modules within the ECII network (Figure 4D, Table S5), using a shared-nearest-neighbor-based community-finding algorithm (Blondel et al., 2008) . We identified four modules, each enriched in distinct AD-associated processes, including RNA splicing (module A), metabolism (module B), neurotransmitter release (module C), and neuron differentiation (module D). Interestingly, several pathways were shared across multiple modules, including microtubule organization (A, C, D) and axonogenesis (B, C, D), supporting a central role for these processes in AD pathogenesis (Table S5).
We then further characterized these functional modules by examining their relationship to aging as well as Aβ accumulation and NFT formation in vulnerable neurons (Table S5). We found that the neurotransmitter secretion-related module C genes showed decreased expression in the context of Aβ accumulation in the mouse (our APP/PS1 mouse profiling) and have significantly lower expression in aged wild-type mice. Thus, module C is a good candidate for linking Aβ accumulation with aging in the AD pathological cascade. Furthermore, module C was the only module with ECII-specific signal for tau pathology (i.e., significantly enriched in genes downregulated in ECII neurons of AD patients, but not strongly correlated with tau in non-ECII regions of the human hippocampal formation [ACT study]). Additionally, only module C demonstrated significantly tighter cohesiveness in ECII versus resistant neurons (Student’s t-test, intersection-union test, p-value < 0.0135). Thus, while modules A, B, and D may represent pathways common to general AD progression in any neuron type, module C may confer the surplus of susceptibility specific to ECII neurons. As this vulnerability-specific module represents processes related to both axon structural remodeling and presynaptic excitability, it is tempting to speculate that specific AD vulnerability of ECII neurons may be linked to their lifelong maintenance of a state of high axonal plasticity (Figure 4E).
Functional association of α-synuclein, tau, and PTB in ECII neurons
To identify genes in this vulnerability-specific module that underlie ECII susceptibility in early stage AD, we examined the connectivity and centrality of the module members across all seven neuron-specific networks. Intuitively, two genes are tightly connected in a specific neuronal context if they have a high confidence link in the functional network for that neuronal type; this suggests involvement of these genes in shared processes. A highly central gene is one that has many high confidence links with other genes across the network, indicating involvement of this gene in a wide array of processes. Within module C, MAPT (tau) was the most centrally connected out of all 668 module C genes, and our analysis pointed to SNCA (encoding α-synuclein) as potentially driving the ECII specificity of this vulnerability-specific module. This is based on the finding that not only are MAPT and SNCA tightly connected to each other in the ECII network, but α-synuclein also has the highest differential network centrality between ECII and the resistant neurons (Table S6). This suggests that α-synuclein is associated with many more processes in ECII neurons than in other types of neurons, that tau cooperates with α-synuclein in many of these processes, and that α-synuclein may contribute to NFT formation upon dysregulation of these processes. This association between MAPT and SNCA in the context of AD neuronal vulnerability is supported by previous work demonstrating physical as well as functional interaction between these two proteins in other neurodegenerative disorders (reviewed in (Moussaud et al., 2014)). For example, tau and α-synuclein have been previously described to influence each other’s aggregation into pathological lesions in Parkinson’s disease as well as in mice overexpressing these genes (Emmer et al., 2011; Giasson et al., 2003; Khandelwal et al., 2010, 2012). However, a role for endogenous α-synuclein in the formation of NFT has not been previously described, although a large proportion of AD patients present α-synuclein pathology (Hamilton, 2000).
PTB, a regulator of alternative splicing (Llorian et al., 2010), was the most highly connected protein to both α-synuclein and tau in the ECII network. It has not been previously linked to adult neural function and has often been thought to be expressed during development and downregulated in the adult (Boutz et al., 2007; Zheng et al., 2012). PTB has not been previously associated with AD, although a mini gene screen in cancer cell lines identified it as capable of regulating the splicing of tau exon 10 (Wang et al., 2004). Here, we found that PTB was actively translated in all adult mouse neurons profiled, and we detected significant amounts of the protein by Western blot on adult mouse EC lysates (Figure S3A). Furthermore, we analyzed transcriptomic data from laser capture microdissected ECII neurons in humans (Liang et al., 2008) and from diverse brain regions in GTEx (GTEx consortium) and found adult PTB expression in both (Figure S3B). We also examined publicly available AD expression datasets and found that in AD compared to control patients, PTB is specifically upregulated in the parahippocampal gyrus, where the EC is located (Figure S3C, logFC = 0.22, p-value = 0.0004, Amp-AD - https://agora.ampadportal.org/genes) and in laser capture microdissected ECII neurons (logFC = 0.39, p-value = 0.00099, AD vs. control, GSE5281) (Dunckley et al., 2006).
We tested experimentally whether PTB can regulate SNCA and MAPT mRNA, by measuring gene expression and differential exon usage by RNAseq after PTB knockdown in human neuroblastoma cells in vitro. Silencing PTB increased both total SNCA levels and the inclusion of MAPT exon 10, while there was no effect on any other MAPT exon (Figure 5A); we confirmed these results using quantitative PCR (Figure 5B, Figure 5C, Figure S3D). Regulation of exon 10 is of high relevance for tau pathology, as its inclusion gives rise to four- rather than three-microtubule binding repeat tau (4R- and 3R-tau respectively). An imbalance between 4R- and 3R-tau has been repeatedly shown to give rise to tau pathology in different tauopathies as well as in AD (reviewed in (Liu and Gong, 2008)).
Figure 5: Modulation of MAPT splicing and SNCA levels by PTB.
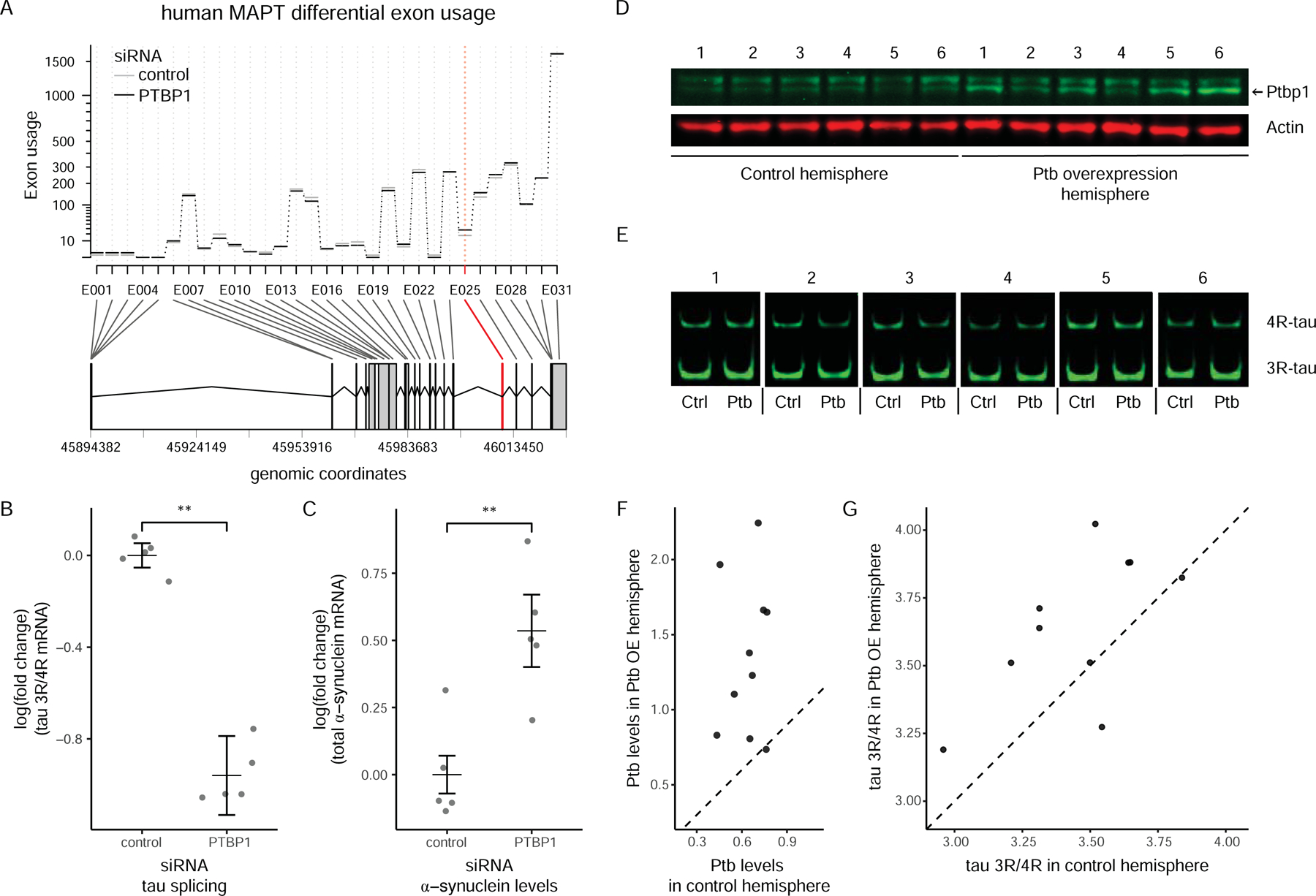
(A–C) Silencing of PTB in SH-SY5Y neuroblastoma cells. mRNA was purified after 3 days and analyzed mRNA by RNAseq.
A. DEXSeq diagram showing which exonic parts of MAPT are changed. Only exon 10 is significantly changed (DEXseq adjusted p-value = 0.028).
B. qPCR measurement of 3R / 4R tau ratio. The proportion of 3R tau significantly decreases, showing that PTB promotes exon 10 exclusion (t-test, p < 0.002).
C. qPCR measurement of SNCA levels. α-synuclein is significantly increased (t-test, p < 0.006).
(D–G) Overexpression of Ptb in the EC of htau phage artificial chromosome (PAC) mice, which transgenically overexpress the entire wild-type human tau gene. Empty AAV1 particles or particles containing a cDNA for mouse Ptb (∆exon 9 – predominant isoform in adult neurons) were injected stereotaxically in the EC, in opposite sides of the brain. qPCR and Western blots were run on bulk lysates of the EC, one week after injection. Ptb overexpression and control sides were paired.
D. western blot detection of Ptb in the EC in vivo. Numbers over the gel pictures represent individual mice.
E. Fluorescent PCR assay to test differential usage of human tau exon 10 on reverse-transcribed RNA from EC lysates. 3R-tau and 4R-tau in the lysate yield amplicons of different lengths. Numbers over the gel pictures represent individual mice.
F. Quantification of Ptb protein overexpression in the EC on the Ptb OE (overexpression) side compared to the control side, by quantitative western-blot as in D (2 experiments, n = 10 mice total, paired one-tail Student t-test p-value = 8.3e-4).
G. Quantification of human tau exon 10 splicing using fluorescent PCR as in E on EC lysates of injected mice. There is a significant decrease in human exon 10 usage (increase in human 3R-tau) upon Ptbp1 overexpression (2 experiments, n = 10 mice total, paired one-tailed Student t-test = 0.012). Of note, tau splicing is measured on bulk lysates of EC, potentially yielding a modest effect because of ECII-specific signal dilution. See also Figure S3 and S4.
To investigate directly the effect of PTB on tau splicing in ECII neurons in vivo, we modulated PTB level in the mouse EC by stereotaxically injecting recombinant adeno-associated viruses (AAV). Here we overexpressed PTB, as significant silencing of PTB in vivo could not be achieved in the mouse EC (Figure 5D, F; Figure S3E–H), likely because of the multiple mechanisms tightly regulating endogenous PTB expression (Wollerton et al., 2004; Yeom et al., 2018). We found that PTB overexpression affected exon usage of a number of genes involved in microtubule dynamics or microtubule-based transport (e.g. Cdc42, Clip1, Dnm1, Eml4, Klc1, Kif1b), supporting our earlier findings of the saliency of microtubule-related processes in vulnerable neurons (Figure S4).
Crucially, we discovered that PTB had a significant effect on human exon 10 usage in transgenic mice expressing the full human wild-type tau gene (htau PAC mice) (Andorfer et al., 2003). Specifically, the overexpression of PTB increased the 3R/4R ratio for human tau, as measured by fluorescent RT-PCR experiments using primers surrounding exon 10 (Figure 5E, G, Figure S3I, one-tailed paired Student’s t-test, p-value = 0.012), consistent with the decreased 3R/4R ratio observed when we silenced PTB in vitro. We further validated these results using quantitative PCR on total EC RNA. (Figure S3J, one-tailed paired Student’s t-test, p-value = 0.012). Importantly, NFTs in ECII neurons have been shown to be devoid of 4R-tau, in contrast to NFTs in other hippocampal neurons that have both tau isoforms (Hara et al., 2013; Iseki et al., 2006). This regional imbalance, previously unexplained, could contribute to the preferential NFT deposition in ECII neurons. An ECII-specific dysregulation of PTB could thus be one of the driving factors for the early initiation of tau pathology in these neurons, potentially contributing to their vulnerability.
Discussion
Little is known about the molecular basis of selective neuronal vulnerability in AD and the molecular pathways that lead to NFT formation and neurodegeneration. Furthermore, no animal model comprehensively recapitulates every aspect of human AD pathogenesis. While the templated spread of tau along axonal routes (Kaufman et al., 2018) could explain the chronology of NFT appearance later in the disease, pathological tau can only form ex nihilo in the most vulnerable neurons due to intrinsic properties of these neurons. Kaufman et al. recently showed that EC isolates from AD patients were the first to gain tau seeding potential, before isolates from any other region of the brain, further demonstrating that pathology is not passed on to EC from another region (Kaufman et al., 2018). The molecular dissection of the initial step whereby tau pathology forms in the EC has long remained elusive.
Here, we provide an integrative and unbiased framework for the study of AD that combines advantages of both mouse models and human data. Our approach 1) models AD vulnerable and resistant human neurons in silico with high-quality cell-type specific molecular profiles generated in the non-diseased mouse and a compendium of publicly available human data, 2) leverages human quantitative genetics to identify genes and pathways relevant for AD pathology within these in silico models, and 3) experimentally tests in the mouse the effect of age and Aβ, a major AD endophenotype, on the genes we predict to be associated with AD pathology, elucidating the pathological cascade of AD. Our approach is general and applicable to any complex disease with selective cell vulnerability where relevant human GWAS data are available. For neurodegenerative diseases with a complex multicellular pathogenesis, the approach also allows for the identification of cell-type specific pathological pathways.
Previous network-based approaches (Miller et al., 2008, 2013; Mostafavi et al., 2018; Zhang et al., 2013) provided successful attempts at leveraging functional genomics for the molecular dissection of AD. Their use of bulk genomics data allowed them to study gene expression patterns from all cell types, but also limited their ability to precisely identify neuronal processes. Our framework focused on leveraging neuron-type specific network models, enabled us to further pinpoint an axon plasticity module responsible for the vulnerability of ECII neurons. Furthermore, the use of a rich array of functional genomics data for the construction of the networks allows us to identify interactions between genes that are not purely based on coexpression, including modulation of tau splicing by PTB. Our study is also the first one which shows the centrality of the tau and alpha synuclein genes in the disease process within vulnerable neurons, though these genes are not enriched in vulnerable neurons.
Using this approach, we identify molecular mechanisms underlying neuronal vulnerability in AD. In addition to significantly predicting many of the genes previously associated with AD, we also outline novel pathways linking Aβ and tau pathology. Of the numerous genes revealed by our framework, one of the most striking ones is PTB, a splice factor dysregulated in AD, which we show modulates the 3R- to 4R-tau balance. An excess of 3R-tau is thought to be the cause of a number of familial frontotemporal dementia cases (Liu and Gong, 2008). But in other tauopathies like corticobasal syndrome, NFTs are constituted of 4R-tau exclusively (Liu and Gong, 2008). Deviation from the physiological 3R/4R equimolar balance is thus considered to contribute to tau aggregation. In AD, vulnerable neuron-specific dysregulation of PTB could precipitate a 3R/4R imbalance in these neurons and explain their premature accumulation of NFTs.
More generally, our unbiased data-driven analyses place microtubule dynamics at the center of AD pathogenesis. We find that this process is both one of the most salient characteristics of the most vulnerable neuronal subtype and closely associated with NFT formation. As key regulators of neuronal architecture and intraneuronal trafficking, microtubules are the endpoint of many neuronal processes. Thus, it is important to determine which specific pathways lead to dysregulation of microtubule dynamics in the context of AD. While a conclusive answer to this question requires further study, our analyses of ECII vulnerability highlight two potential candidate processes: axonogenesis and synaptic vesicle release. Both have been previously linked to microtubule remodeling (Bodaleo and Gonzalez-Billault, 2016; T. L. Lewis et al., 2013) and are connected to microtubule genes within the vulnerability-specific module. Interactions between structural and electrophysiological properties of the axon could be more prominent in ECII neurons (known to display considerable axon arborization (Tamamaki and Nojyo, 1993)) than in other cell types – which could confer exceptional axonal plasticity to ECII neurons, but could also be responsible for ECII vulnerability.
Successful therapeutic strategies for treating AD will likely involve diverse approaches. This might include molecular interventions that prevent neuronal pathology and eventual degeneration, acting on the glial response that accompanies the neuropathology and contributes to the clinical symptoms (microglia and astrocytes), or on the psychological and lifestyle interventions that can mitigate the symptoms. In any case, it is indispensable to gain an understanding of how the neuropathology begins, and in particular, how NFTs, present in every single AD patient, form in the most vulnerable neurons of the brain before spreading to further regions. Addressing this question is crucial to the design of novel therapeutics for the earliest stages of AD.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information should be directed to and will be fulfilled by the Lead Contact, Olga troyanskaya (ogt@cs.princeton.edu).
Specific questions regarding experimental details and reagents should be directed to Jean-Pierre Roussarie (jroussarie@rockefeller.edu).
Materials Availability
All modified BACs and embryos for some of the bacTRAP mice generated in this study (Sh3bgrl2-, Rasgrp2-, Sstr4#7-, Sstr4#19-, and Calca- are frozen at Rockefeller University and available on request). For the other bacTRAP mice generated in this study, transgene expression was lost across generations, as is often the case with BAC transgenic lines, and the lines were discontinued. The materials are available from corresponding author Jean-Pierre Roussarie (jroussarie@rockefeller.edu) with a completed Materials Transfer Agreement.
Data and Code Availability
The datasets generated during this study are available at GEO: GSE151460 for the bacTRAP profiles at 5, 12 and 24 months of age, and the APP/PS1 ECII-bacTRAP profiles, and GSE151356 for the ECII-bacTRAP data upon silencing and overexpression of Ptbp1.
The Sleipnir library for functional genomics used for network integration is available at https://libsleipnir.bitbucket.io/
Uncropped original images for Western blot and fluorescent PCR for Figures 5 and S3 in the paper are available on Mendeley: http://dx.doi.org/10.17632/g67bhh7zsj.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
All experiments were approved by the Rockefeller University Institutional Animal Care and Use Committee (RU-IACUC protocols #07057, 10053, 13645-H, 16902), and were performed in accordance with the guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were housed in groups of up to 5 animals on a 12 h dark/light cycle at 22 °C and provided with rodent diet (Picolab) and water ad libitum. All animals were drug and test naïve (other than genotyping) when they were used for the described experiment. Littermates of the same sex were randomly assigned to the different groups. All bacTRAP mice and APP/PS1 mice (B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax purchased from the Jackson laboratories) were maintained in a heterozygous state by crossing them with non-transgenic C57Bl/6J mice (Jackson Lab). Human tau phage artificial chromosome (PAC) mice (B6.Cg-Mapttm1(EGFP)Klt Tg(MAPT)8cPdav/J purchased from the Jackson lab) express the full wild-type human MAPT gene transgenically, as part of a phage artificial chromosome (Andorfer et al., 2003). In these mice, both 3R and 4R human tau isoforms are present in adult neurons, while in wild-type mice, adult neurons express only 4R mouse tau. Htau PAC mice were maintained by crossing (htau PAC +/−, mtau KO/KO) with wild-type C57Bl/6J mice. For cell-type specific profiling in wild-type mice, only male mice were used, and the tissue from two males was pooled. Each type of neuron was profiled at 4–5 months, 12 months, and 24 months. For comparing ECII neurons in wild-type and APP/PS1 mice, both male and female mice were used, and each sample corresponded to the tissue of one mouse. (see Key Resource Table for genotyping primers). Baseline PTB expression was tested in 3-month old C57Bl/6J mice. Stereotaxic injections were performed in 8 to 11 month-old female and male (htau PAC +/, mtau KO/+) mice.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat anti PTBP1 polyclonal antibody | abcam | Abcam Cat# ab5642, RRID:AB_305011 |
| monoclonal anti GFP antibody Htz-19C8 (for bacTRAP), bioreactor supernatant | Memorial Sloan-Kettering Cancer Center - Monoclonal Antibody Facility | Cat# HtzGFP-19C8, RRID : NA |
| monoclonal anti GFP antibody Htz-19F7 (for bacTRAP), bioreactor supernatant | Memorial Sloan-Kettering Cancer Center - Monoclonal Antibody Facility | Cat# HtzGFP-19F7, RRID : NA |
| chicken anti GFP polyclonal antibody (for immunofluorescence) | abcam | Abcam Cat# ab13970, RRID:AB_300798 |
| Alexa 488-donkey anti-chicken secondary antibody | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A-11039, RRID:AB_2534096 |
| IRDye 680RD Donkey anti-Goat IgG, secondary antibody | Li-COR Biosciences | LI-COR Biosciences Cat# 926–68074, RRID:AB_10956736 |
| IRDye 800CW Donkey anti-Mouse IgG, secondary antibody | Li-COR Biosciences | LI-COR Biosciences Cat# 926–32212, RRID:AB_621847 |
| IRDye 800CW Donkey anti-Goat IgG, secondary antibody | Li-COR Biosciences | LI-COR Biosciences Cat# 926–32214, RRID:AB_621846 |
| IRDye 680RD Donkey anti-Mouse IgG, secondary antibody | Li-COR Biosciences | LI-COR Biosciences Cat# 926–68072, RRID:AB_10953628 |
| mouse anti-β actin monoclonal antibody | Cell Signaling Technology | Cell Signaling Technology Cat# 3700, RRID:AB_2242334 |
| Bacterial and Virus Strains | ||
| AAV1-hSyn1-mPTBP1-IRES-mCherry carrying the NM_008956 cDNA for PTBP1 | Vector biolabs | RRID : NA |
| AAV1-mCherry-U6-scrmb-shRNA 5′-CCGG-CAACAAGATGAAGAGCACCAACTCGAGTT GGTGCTCTTCATCTTGTTG-TTTTT-3′ | Vector biolabs | RRID : NA |
| AAV1-mCherry-U6-mPTBP1-shRNA - shRNA sequence 5′-CCGG-CTCAATGTCAAGTACAACAATCTCGAGATT GTTGTACTTGACATTGAG-TTTTT −3′ | Vector biolabs | RRID : NA |
| AAV1-hSyn1-mCherry-WPRE | Vector biolabs | RRID : NA |
| Critical Commercial Assays | ||
| human MAPT, FAM/MGB Taqman probe spanning exons 11 and 12 (total tau) | Applied Biosystems | Assay ID Hs00902193_m1, RRID : NA |
| human SNCA, FAM/MGB Taqman probe | Applied Biosystems | Assay ID Hs00240906_m1, RRID : NA |
| mouse Ptbp1, FAM/MGB Taqman probe | Applied Biosystems | Assay ID Mm01731480_gH, RRID : NA |
| human PTBP1, FAM/MGB Taqman probe | Applied Biosystems | Assay ID Hs00738538_g1, RRID : NA |
| mouse gapdh, endogenous control, FAM/MGB Taqman probe | Applied Biosystems | Cat# 4352932E, RRID : NA |
| human gapdh, endogenous control, FAM/MGB Taqman probe | Applied Biosystems | Cat# 4333764F, RRID : NA |
| Accell non-targeting control pool | Horizon discovery (Dharmacon) | Cat# D-001910–10, RRID : NA |
| Accell human PTBP1 SMARTpool siRNA | Horizon discovery (Dharmacon) | Cat# E-003528–00, RRID : NA |
| human MAPT, FAM/MGB Taqman probe spanning exons 9 and 11 (3R-tau) | Applied Biosystems | Assay ID Hs00902192_m1, RRID : NA |
| human MAPT, FAM/MGB Taqman probe spanning exons 9 and 10 (4R-tau) | Applied Biosystems | Assay ID Hs00902312_m1, RRID : NA |
| Deposited Data | ||
| Adult Changes in Thought, dataset of hippocampal samples of elderly individuals at risk for dementia with paired gene expression data and pathology measurements. Used for testing correlation between NetWAS 2.0 results and amyloid plaques and tau. | Miller et al., 2017 | http://aging.brain-map.org/download/index |
| Liang et al. dataset of lasercapture microdissected ECII neurons in AD vs. control: used for testing differential expression of NetWAS genes in ECII neurons of AD patients. | GEO, Liang et al., 2008 | GSE5281, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE5281 |
| Agora AMP-AD : used for testing differential expression of PTB in the brain of AD patients. | https://agora.ampadportal.org/genes/(genes-router:gene-details/ENSG00000011304)) | |
| JASPAR, part of the human data compendium for functional network construction. | Mathelier et al., 2014 | http://jaspar.genereg.net/ |
| MSigDB, part of the human data compendium for functional network construction. | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb/ |
| BioGRID, part of the human data compendium for functional network construction. | Chatr-Aryamontri et al., 2013 | https://thebiogrid.org/ |
| IntAct, part of the human data compendium for functional network construction. | Orchard et al., 2014 | https://www.ebi.ac.uk/intact/ |
| MINT, part of the human data compendium for functional network construction. | Licata et al., 2012 | https://mint.bio.uniroma2.it/ |
| MIPS, part of the human data compendium for functional network construction. | Mewes et al., 2011 | https://mips.helmholtz-muenchen.de/proj/ppi/ |
| Uncropped gel images for Figure 5 and Figure S3 | Mendeley | http://dx.doi.org/10.17632/g67bhh7zsj.1 |
| RNAseq data for the bacTRAP profiling of 7 neuron types at 5, 12 and 24 months of age, and the APP/PS1 ECII-bacTRAP profiles | GEO | GSE151460, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151460 |
| RNAseq data for bacTRAP profiling of ECII neurons upon silencing and overexpression of Ptbp1 in the mouse EC. | GEO | GSE151356, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151356 |
| Experimental Models: Cell Lines | ||
| SH-SY5Y neuroblastoma cell line | ATCC | ATCC Cat# CRL-2266, RRID:CVCL_0019 |
| Experimental Models: Organisms/Strains | ||
| Mouse: wild-type mice: C57Bl/6J | Jackson lab | IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: APP/PS1: B6.Cg- Tg(APPswe,PSEN1dE9)85Dbo/Mmjax | Jackson lab | MMRRC Stock No: 34832-JAX, RRID : NA |
| Mouse: Cck-bacTRAP: B6;FVB-Tg(Cck-EGFP/Rpl10a)GM391Htz/J |
Jackson lab | IMSR Cat# JAX:030249, RRID:IMSR_JAX:030249 |
| Mouse: Gprin3-bacTRAP | described in Gray et al., Mol. Psy., 2018 | RRID: NA |
| Mouse: htau PAC: B6.Cg-Mapttm1(EGFP)Klt Tg(MAPT)8cPdav/J | Jackson lab | IMSR Cat# JAX:005491, RRID:IMSR_JAX:005491 |
| Oligonucleotides | ||
| all mouse genotyping primers | See Table S7 for sequences | IDT-DNA, RRID : NA |
| fluorescent PCR for human tau splicing - forward primer | IDT-DNA | HPLC purified 5’-IRDye 800 – CTCCAAAATCAGGGGATCGC – 3’, RRID: NA |
| fluorescent PCR for human tau splicing - reverse primer | IDT-DNA | unlabeled 5’ –CCTTGCTCAGGTCAACTGGT – 3’, RRID: NA |
| fluorescent PCR for mouse tau splicing - forward primer | IDT-DNA | HPLC purified 5’-IRDye 800 – CACCAAAATCCGGAGAACGA – 3’, RRID: NA |
| fluorescent PCR for mouse tau splicing - reverse primer | IDT-DNA | unlabeled 5’ –CTTTGCTCAGGTCCACCGG – 3’, RRID: NA |
| Recombinant DNA | ||
| RP23–329L1 unmodified BAC | CHORI | RRID : NA |
| RP23–181A2 unmodified BAC | CHORI | RRID : NA |
| RP24–68J22 unmodified BAC | CHORI | RRID : NA |
| RP23–307B16 unmodified BAC | CHORI | RRID : NA |
| RP23–199D5 unmodified BAC | CHORI | RRID : NA |
| RP24–344N1 unmodified BAC | CHORI | RRID : NA |
| RP23–126C5 unmodified BAC | CHORI | RRID : NA |
| RP23–329L1 - eGFP-L10a | This paper | Cacng5-bacTRAP - modified BAC , RRID : NA |
| RP23–181A2 - eGFP-L10a | This paper | Calca-bacTRAP - modified BAC , RRID : NA |
| RP24–68J22- eGFP-L10a | This paper | Cartpt - modified BAC , RRID : NA |
| RP23–307B16 - eGFP-L10a | This paper | Sh3bgrl2-bacTRAP - modified BAC , RRID : NA |
| RP23–199D5 - eGFP-L10a | This paper | Rasgrp2–199D5-bacTRAP - modified BAC, RRID : NA |
| RP24–344N1 - eGFP-L10a | This paper | Rasgrp2–344N1-bacTRAP – modified BAC, RRID : NA |
| RP23–126C5 - eGFP-L10a | This paper | Sstr4-bacTRAP - modified BAC , RRID : NA |
| mouse Ptbp1 untagged cDNA clone NM_008956 | Origene | Cat# MG223224, RRID : NA |
| Software and Algorithms | ||
| htseq | Anders et al., 2015 | https://github.com/htseq/htseq |
| FIMO | Grant et al., 2011 | http://meme-suite.org/doc/fimo.html |
| edgeR | Robinson et al., 2010 | http://bioconductor.org/packages/devel/bioc/html/edgeR.html |
| The Sleipnir Library for Computational Functional Genomics | Huttenhower et al., 2008 | https://libsleipnir.bitbucket.io/ |
| VEGAS2 | Mishra and Macgregor, 2015 | https://vegas2.qimrberghofer.edu.au/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
Mouse genotyping
All mice were genotyped by PCR on tail clips using the following primers: GFP-forward 5’-GACGTAAACGGCCACAAGTTCAG-3’ and GFP-reverse 5’-ATGGTGCGCTCCTGGACGTAG-3’ for all bacTRAP mice and for htau-PAC mice (testing for the presence of the mouse tau knockout allele), APP-forward 5’- AGGACTGACCACTCGACCAG-3’, APP-reverse 5’-CGGGGGTCTAGTTCTGCAT-3’, PSEN1-forward 5’-AATAGAGAACGGCAGGAGCA-3’ and PSEN1-reverse 5’- GCCATGAGGGCACTAATCAT-3’ for APP/PS1 mice, and htau-forward 5’- ACTTTGAACCAGGATGGCTGAGCCC-3’, htau-reverse 5’-CTGTGCATGGCTGTCCACTAACCTT-3’ (testing for the presence of the human tau PAC allele), mtau-forward 5’- CTCAGCATCCCACCTGTAAC −3’, mtau-reverse 5’ CCAGTTGTGTATGTCCACCC 3’ (testing for the presence of the endogenous mouse tau allele) (Andorfer et al., 2003).
Cell culture
The SH-SY5Y neuroblastoma cell line were purchased from ATCC (sex: female), and used directly after purchase. It was grown in DMEM/F-12 medium supplemented with 10% fetal bovine serum, at 37°C in 5% CO2. For mouse entorhinal cortex neuron primary cultures, we used timed pregnant C57Bl/6J female mice (Jackson laboratory) at E17 in utero. Briefly, entorhinal cortices from embryos were dissected and incubated at 37°C in 0.05% Tripsine/EDTA (Life Technologies, USA) for 10 min. After centrifugation the tissue pellet was dissociated in HBSS containing 0.5mg/ml DNAse I (Roche) with a Pasteur pipette. Cells were seeded at 25,000 cells/cm2 in Neurobasal media (Thermo Fisher) with 2% B-27 supplement (Thermo Fisher) and 2 mM GlutaMAX (Thermo Fisher), and incubated at 37 °C in a humidified 5% CO2-containing atmosphere. Neuron treatments were performed after a minimum of 10 DIV.
METHOD DETAILS
bacTRAP transgene construction
To construct cell type-specific bacTRAP mice, we searched for drivers specific to each type of neuron. For that purpose, we mined the ABA and GENSAT databases for genes expressed selectively in the different cell types of interest. We selected the following genes: Rasgrp2 and Sh3bgrl2 (ECII principal neurons), Sstr4 (CA1 pyramidal neurons), Cacng5 (CA2 pyramidal neurons), Gprin3 (CA3 pyramidal neurons), Calca (V1 pyramidal neurons), Cartpt (S1 pyramidal neurons) for enriched expression in the cell type of interest compared to neighboring cell types. Regulatory regions of these genes should drive expression in the corresponding neuron types. We thus used these genes to construct corresponding bacTRAP mice according to previously described procedures (Gong et al., 2003). Specifically, we obtained the bacterial artificial chromosomes (BACs) where the open reading frame (ORF) for each of these genes is most centrally located, ensuring that both upstream and downstream regulatory sequences are driving the expression of the bacTRAP construct: RP23–307B16 (Sh3bgrl2), RP23–199D5 and RP24–344N1 (Rasgrp2), RP23–126C5 (SSTR4), RP23–329L1 (Cacng5), RP23–181A2 (Calca), RP24–68J22 (Cartpt) (Children’s Hospital and Research Center at Oakland, CHORI). We then set out to modify each BAC to place the eGFP-L10a cDNA under the control of each gene’s regulatory sequences (S. Gong et al., 2003). For each gene, we first amplified by PCR on the non-modified BAC a small homology arm corresponding to approximately 500 bp of sequence upstream of the gene ORF, stopping 5 bp before the ORF (sequences of all small homology arms in the Table S7), and cloned it in the S296 shuttle vector (a pLD53.SC2 plasmid containing the cDNA for eGFP-L10a). Integration of eGFP-L10a in the BACs takes place when the non-modified BAC, the corresponding shuttle vector carrying the small homology arm upstream of eGFP-L10a, and recA are co-expressed in the same bacteria. For that purpose, we first made the BAC host cells competent for transformation: BAC host cells obtained from CHORI were grown overnight at 37°C with chloramphenicol (20 μg/ml). A single colony was then grown for each BAC to an optical density of 0.8 in chloramphenicol. Cells were harvested by centrifugation, resuspended in an ice-cold solution of 50 mM CaCl2 and kept on ice for 15 minutes. Cells were then pelleted again and resuspended in an ice-cold solution of 50 mM CaCl2 and 20% glycerol before being snap-frozen. Competent BAC host cells were then chemically transformed with PSV1.recA plasmid coding for the recA recombinase and grown overnight at 30°C with chloramphenicol and tetracycline (10 μg/ml) to an optical density of 0.8. Cells were then made electro-competent by pelleting them, resuspending them in 10% glycerol, washing them twice in 10% glycerol and snap freezing them. Lastly each type of BAC-recA competent cells was electroporated with the S296 vector containing the corresponding small homology arm. Cells were then grown overnight at 30°C on chloramphenicol, tetracycline and ampicillin (50 μg/ml) to allow for BAC recombination. Cells were then grown overnight on chloramphenicol and ampicillin plates at 43°C, to select for cells with co-integrate BAC clones. Co-integrates were first screened by PCR on individual colonies. The proper integration of eGFP-L10a at the beginning of each ORF was monitored using southern blot on the colonies that came positive from PCR screening. Purified BAC stocks were prepared using CsCl2 gradient and BACs were linearized the BAC with PI-SceI before injection. Linearized BACs were dialyzed on 0.025 μm filter membranes (Millipore). Their quality and concentration was assessed on a pulse-field gel. The Rockefeller University Transgenic Services then performed pronuclear injection of the linearized BACs in fertilized oocytes of C57Bl/6J mice (Jackson Lab). BacTRAP mice were crossed at all subsequent generations with C57Bl/6J mice. F1 and F2 of the different founder lines were then tested for proper expression pattern. One of the founder lines with the Sstr4 BAC (Sstr4#19 line) presented ectopic expression in granule cells from the dentate gyrus and no expression in CA1 neurons. We thus used Sstr4#19 for granule cell profiling. We used another founder line (Sstr4#7) for CA1 neurons. We also separately obtained Cck- and Gprin3-bacTRAP mice, which were previously described (Doyle et al., 2008; Gray et al., 2018).
Detail of the bacTRAP mice analyzed
Table S7 lists the bacTRAP lines used for each of the different cell types.
Cell type-specific molecular profiling
To isolate cell type-specific mRNA, bacTRAP mice from the different transgenic lines were decapitated after slight CO2 intoxication, and brains were promptly taken out. For each transgenic line, we dissected the minimal area where transgene expression is restricted to the cell type of interest (for ECII bacTRAP lines, we made a coronal cut around −3.3 mm anteroposterior (AP); for Sh3bgrl2-bacTRAP, we then scooped the hippocampus off the tissue caudal to the cut, discarded it, and kept the tissue located ventral to the rhinal fissure; for Rasgrp2-bacTRAP, we took all the tissue caudal to the −3.3 mm AP cut, and ventral to a horizontal cut around −3 mm dorsoventral (DV); for SSTR4#7- and SSTR4#19-bacTRAP lines, we used all the hippocampus; for the CCK-, CACNG5- and Gprin3-bacTRAP lines, we used all the hippocampus rostral from a coronal cut around −3.3mm AP; for CALCA-bacTRAP, we made a sagittal cut around +3.6 mm mediolateral (ML) on each side, a coronal cut around −3 mm AP and we extracted the cortex respectively dorsal and caudal to these cuts, while cutting out the mEC; for CARTPT-bacTRAP we made coronal cuts around 1.75 mm AP, −0.25 mm AP, and −2.25 mm AP and for each slice, we dissected out the part of the cortex that contains the somatosensory cortex).
We then performed bacTRAP purification following the previously described procedure (Heiman et al., 2008) except for two differences. First the volume of lysis buffer used for tissue homogenization depends on the size of each particular brain region. The buffer volumes for each bacTRAP line are shown in Table S7. Second, we used RNeasy Plus Micro Kit (Qiagen) to purify RNA after bacTRAP, and RNA was thus detached from beads using the RLT Plus buffer supplemented with 1% β-mercaptoethanol (MP biomedicals). RNA integrity was evaluated with a bioanalyzer RNA 6000 pico chip (Agilent) and RNA quantified by fluorescence detection with Quant-It Ribogreen RNA reagent (ThermoFisher). All samples included in the study had RNA Integrity Numbers above 7. Five ng of RNA were then used for reverse-transcription with Ovation RNAseq v2 kit (NuGEN). cDNAs were cleaned up using a QIAquick PCR purification kit (Qiagen). Double-stranded cDNAs were quantified by fluorescence detection using Quant-IT Picogreen dsDNA reagent (ThermoFisher). cDNAs (200 ng) were sonicated in 120 μl volume using a Covaris S2 ultrasonicator (duty cycle, 10%; intensity, 5; cycles/burst, 100; time, 5 minutes) to generate 200 bp fragments on average. The fragmented cDNAs were then used to construct sequencing libraries using TruSeq RNA sample prep kit v2 (Illumina). Library concentration was evaluated using bioanalyzer, and libraries were multiplexed. Multiplexes were then sequenced at the Rockefeller University genomics resource center with a HiSeq 2500 sequencer (Illumina).
Histology
To study the expression pattern of the bacTRAP transgene, bacTRAP mice were transcardially perfused with 4% paraformaldehyde. Dissected brains were fixed by immersion for one hour in 4% paraformaldehyde, frozen embedded in OCT compound (TissueTek), and cut in 40 μm-thick sections on a CM3050 S cryostat (Leica). Sections were permeabilized in PBS with 0.1% Fish Gelatin (Sigma), 2% normal goat serum (Jackson ImmunoResearch), and 0.1% triton X-100 and then stained overnight at 4°C in PBS with 0.1% fish gelatin and 2% normal goat serum with a chicken anti-GFP antibody (1/300). The primary antibody was detected with an Alexa 488-donkey anti-chicken secondary antibody (1/300). After the last wash, sections were mounted with Prolong Gold Medium containing DAPI. Sections were imaged using a Zeiss LSM 510 META laser scanning confocal microscope. Images were minimally processed using Photoshop (Adobe Systems) to enhance brightness and contrast for optimal representation of the data.
Comparison of the bacTRAP profiles with Allen Brain Atlas
In order to cross-validate the bacTRAP profiles, we compared them to Allen Brain Atlas in situ hybridization (ISH) pictures. Based on cell type-specific bacTRAP data, we first calculated an ontology z-score for each gene in each neuron type. For each neuron type, we obtained the 50 genes with the highest ontology z-scores. We then analyzed coronal - whenever possible - or sagittal ISH sections from the Allen Brain Atlas, for each of these genes (50 genes per 7 types of neuron). We scored expression in the seven types of neuron using the Allen Brain Atlas “expression” tool, blind to the identity of the gene, and to the region where it is enriched. Similarly to what Cembrowski et al. had done to cross-validate their data, we verified that each gene predicted to be enriched in a given neuron type with our bacTRAP data indeed presented expression in the corresponding neuron type in the Allen Brain Atlas (ABA). We found excellent correspondence between our data and the ABA data, as 98%, 100%, 96%, 100%, 98%, 94% and 87% of the genes predicted to be enriched in ECII, CA1, CA2, CA3, DG, S1 and V1 neurons respectively indeed showed expression in the correct regions in the ABA (we disregarded genes that do not show expression in any ISH section that are probably expressed below the detection level). In addition, we show detectable signal in our bacTRAP data for some genes with no ABA ISH signal, like Pkib for example. ISH pictures for the five genes with the highest ontology z-scores for each neuron type, that were available on the ABA, and that showed expression in some parts of the brain, are shown in Figure S1. The reference for the ABA Images shown on Figure 2 and on Figure S1 are the following: for Figure 2: Reln (Image series: 890, image: 135), Lamp5 (70927827; 313), Whrn (77371813; 168), Wfs1 (74881161l; 260), Ptpn5 (74743293; 253), Bok, (71064032; 252), Prox1 (73520980; 237); for Figure S1: Nr2f2 (308055507; 9), Apaf1 (68745275; 8), Camk2d (68668030; 5), Reln (79394359; 13), Rab3c (69816745; 13), Rasd1 (2521; 219), Neurod6 (698; 224), Arhgap12 (71836846; 258), Kcnab2 (1754; 221), Hpca (72129291; 248), Fam19a5 (69059974; 97), Prkca (77869816; 268), Scrg1 (71924331; 268), Syce2 (70609150; 74), Nrip3 (73520999; 270), Ociad2 (75041527; 269), Rnf182 (70719034; 92), Mpped2 (73497744; 74), Prox1 (73520980; 237), Synpr (1862; 229), Lhfpl2 (72007934; 258), Slc39a6 (73930852; 246), Smad3 (70593360; 72), Kcnh5 (77620826; 61), Rorb (79556597; 161), Pak7 (75988567; 165), Sytl2 (73520979; 316), Rspo1 (73636101; 152), Lamp5 (70927827; 313), Cbln2 (70231306; 300), Tbc1d30 (72283432; 1), Kalrn (73930821; 302).
Cell culture
For siRNA experiments, cells were seeded at a density of 4 × 105 cells per well in 12- well plates and treated the following day with 1 μM of Accel siRNA (Dharmacon) – either non-targeting control pool or PTBP1 directed SMARTpool. We harvested cells three days after treatment with PureLink Mini Lysis buffer supplemented with β- mercaptoethanol. The first column flow-through from the PureLink Mini kit was precipitated with acetone for protein analysis. For RNA-sequencing, RNA was purified from the column-bound material following the manufacturer’s instructions, with an in-column DNase digestion (Qiagen RNAse-free DNAse set). The resulting RNA was quantified using a Nanodrop One. Sequencing libraries were synthesized using 400 ng of RNA and an Illumina TruSeq RNA sample prep kit v2. Library concentration was evaluated using high-sensitivity DNA tapestation (Agilent), and libraries were multiplexed. Multiplexes were then sequenced at the Rockefeller University genomics resource center with a NextSeq sequencer and 75 bp paired-end reads (Illumina). Libraries from five independent samples were sequenced for each condition. For verifying the successful downregulation of PTBP1, we probed proteins from the same cells by western blot. We cleaned up the acetone precipitates obtained above with ice-cold ethanol, resuspended the precipitates in RIPA supplemented with Complete mini protease inhibitors (Roche), and sonicated them to resuspend them properly.
Protein and RNA extraction from brain lysates
For extracting bulk protein and RNA from the mouse brain, brains were dissected out, and EC, neocortex, hippocampus and spleen were snap frozen. For investigating Ptb protein expression, tissue was sonicated for 1 minute in ice-cold RIPA buffer supplemented with complete protease inhibitors. For investigating both protein and RNA contents in mice stereotaxically injected with control or Ptb viruses, the EC was homogenized mechanically in a small volume of PBS 1X supplemented with complete mini protease inhibitors (Roche), PhosSTOP phosphatase inhibitors (Roche), RNasin (40 U/ml) and Superasin (20 U/ml), using a motorized homogenizer. Part of the homogenate was then used for RNA extraction, and part for protein. For the RNA fraction, Purelink RNA mini lysis buffer was added to the homogenate and RNA was purified following the manufacturer’s instructions. For the protein fraction, ice-cold RIPA buffer supplemented with protein and phosphatase inhibitors was added. The protein extracts were then centrifuged for 10 minutes at 10,000 g to pellet insoluble debris.
Tau splicing analysis
For studying tau splicing on bulk EC lysates, or on SH-SY5Y lysates, RNA was quantified using a nanodrop. RNA (500 ng) was reverse-transcribed for these samples, using Superscript III first strand synthesis system (ThermoFisher) with a 1:1 mix of oligo-dT primers and random hexamers from the kit. RNA was degraded using RNAse H after reverse-transcription. qPCR was then run on these cDNA using TaqMan Universal PCR mastermix and the following FAM-labeled TaqMan assays in a Life Technologies QuantStudio 12K-flex machine: Human GAPDH endogenous control, Hs00240906_m1 (human α-synuclein), Hs00902312_m1 (exon 10-containing human tau – primers and probe spanning exon 9 and exon 10), Hs00902192 (non-exon-10-containing human tau – primers and probe spanning exon 9 and exon 11). Hs00738538 (human PTBP1), mouse GAPDH endogenous control, Mm01731480 (mouse Ptbp1). Ct values were analyzed with default settings in the Quantstudio software. For testing tau splicing with fluorescent PCR, we ran a regular PCR on carefully titrated cDNA amounts and PCR cycle numbers to ensure linear amplification range, following previously published methods (Andorfer et al., 2003; Furlanis et al., 2019; Wamsley et al., 2018), using the following HPLC-purified primers (designed by (Duff et al., 2000)): for human tau, forward primer : 5′-IRDye 800 – CTCCAAAATCAGGGGATCGC – 3′, reverse primer: unlabeled 5′ – CCTTGCTCAGGTCAACTGGT – 3′ (IDT-DNA), for mouse tau, forward primer: 5′-IRDye 800 – CACCAAAATCCGGAGAACGA – 3′, reverse primer: unlabeled 5′ – CTTTGCTCAGGTCCACCGG – 3′. For both human and mouse tau, the PCR amplifies a 297-nucleotide band from 3R-tau, and a 390-nucleotide band from 4R-tau. Labeled PCR products were then loaded on a 1 mm Novex TBE 10% polyacrylamide gel. The 1 kb plus DNA ladder was stained with SYTO 60 for evaluating fragment size. The gels were imaged with a Li-COR Odyssey with an offset of 0.5 mm and quantified in the Odyssey software.
Western blot
For verifying Ptbp1 downregulation protein concentrations were determined using a BCA assay. Ten μg of protein were denatured at 85°C for 3 minutes and loaded in a 4– 12% Bis-Tris gel in MOPS buffer. The gel was transferred to nitrocellulose membranes. Membranes were blocked for 1 h in Li-COR blocking buffer, incubated overnight with goat anti-PTBP1 (ab5642, Abcam, 1/1,500) and mouse anti-actin (8H10D10, Cell Signaling, 1/1,000) primary antibodies diluted in Li-COR blocking buffer supplemented with 0.1% of Tween-20, and incubated for 1 h in secondary antibody (donkey anti-goat IRDye 680 antibody, donkey anti-mouse IRDye 800) in TBS-Tween. Blots were imaged using a Li-COR Odyssey.
Stereotaxic injections
Adeno-associated virus (AAV1 subtype) were injected in the EC of adult mice using an Angle Two mouse stereotaxic frame with a motorized nanoinjector (Leica). Animals were anesthetized with xylazine (4.5 mg/kg body weight) and ketamine (90 mg/kg body weight) injected peritoneally. An ophthalmic ointment was applied to the anesthetized animals to prevent corneal drying during the procedure. AAVs were loaded in a 10 μl syringe (Hamilton, 7653–01) with a 33-gauge needle (Hamilton, 7803–05). Virus stocks were produced by Vector biolabs. 2 × 1013 genome copies of AAV1 particles were injected in 2 μl, in the mouse EC (AP: −3.70; ML: −4.65; DV: −4.50) with the nanoinjector tilted −4.05°. The contralateral EC (AP: −3.70; ML: +4.65; DV: −4.50) was injected with a nanoinjector tilt of +4.05°. Injection rate was 0.4 ml/min. For testing the effects of Ptb silencing or overexpression in ECII neurons specifically, ECII-bacTRAP mice (Sh3bgrl2- bacTRAP) were injected (the same virus was injected on both sides). All viruses were prepared by Vector Biolabs. The viruses used for Ptb silencing were the following: AAV1-mCherry-U6-mPTBP1-shRNA (carrying a small hairpin RNA (shRNA directed against Ptb under the control of a U6 promoter, shRNA sequence: 5'-CCGG-CTCAATGTCAAGTACAACAATCTCGAGATTGTTGTACTTGACATTGAG-TTTTT −3' ) and AAV1-mCherry-U6-scrmb-shRNA (control for the Ptb silencing virus, shRNA sequence : 5′-CCGG-CAACAAGATGAAGAGCACCAA-CTCGAG-TTGGTGCTCTTCATCTTGTTG-TTTTT-3′).
The viruses used for overexpression were the following: AAV1-hSyn1-mCherry-WPRE (empty control expressing mCherry under the control of the human synapsin-1) and AAV1-hSyn1-mPTBP1-IRES-mCherry (expressing the cDNA encoding mouse Ptb Δexon 9 isoform, under the control of the human synapsin-1 promoter, NM_008956 from Origene, and mCherry). For testing tau splicing in vivo, AAV1-hSyn1-mPTBP1-IRES-mCherry and AAV1-hSyn1-mPTBP1-IRES-mCherry were injected contralaterally. To avoid side-specific bias, test and control viruses were injected in alternating sides. Bacitracin antibiotic gel was applied to the surgery wound, that was sutured with a non-absorbable monofilament. Warm, sterile saline solution was injected intraperitoneally (3% of the body weigh) to compensate fluid loss, and animals were monitored until complete recover from anesthesia. Mice were sacrificed for RNA and protein isolation 1 to 2 weeks after virus injections. For all injections, we tested the presence of mCherry by qPCR. We excluded 1 bacTRAP mouse in the Ptbp1 silencing experiment, and 1 bacTRAP mouse in the Ptb overexpression experiment, which did not have mCherry expression in the EC.
RNA-seq analysis
RNA-sequencing reads were mapped to the mouse genome (Ensembl 75) using STAR (version 2.3.0e, default parameters) (Dobin et al., 2013), and gene-level counts were quantified using htseq-count (version 0.9.1) (Anders et al., 2015). Genes were subjected to an expression detection threshold of 1 count per million reads per gene in more than 3 samples and oligodendrocyte, endothelial, and ependymal cell gene clusters were excluded to focus on the neuronal signal. These clusters were identified by constructing coexpression matrices, and the gene clusters with canonical oligodendrocyte, endothelial, and ependymal cell genes (olig: Olig1 (OPC), Mag (mature oligos); epend: Foxj1, Sntn; endot: Kdr, Vwf, Tek (pericytes)) were excluded. Differential expression and multidimensional scaling analysis were performed using edgeR (version 3.8.6) (Robinson et al., 2010).
Spatial homology analysis
Human brain microarray data were downloaded from the ABA (http://human.brain-map.org/static/download) (Hawrylycz et al., 2012). Brain regions that were measured in fewer than 3 out of the 6 subjects profiled were excluded from downstream analysis to ensure robustness.
We calculated an ontology-aware spatial homology score between each of our 7 mouse neuron types and each of the 205 human brain regions robustly measured by the ABA, as follows: , , , for mouse neuron type i and human brain region j (thus Tm = 7 and Th = 205), human gene g.
where μig, σig are respectively the mean and standard deviation of expression for the closest mouse functional ortholog(Park et al., 2013) of gene g in mouse neuron type i (in log2(rpkm)). Ni is the number of samples for neuron type Ti, while , are the mean and standard deviation of expression values for the mouse ortholog for unrelated neuron types (e.g., for neuron type hippocampus CA2, would be the mean expression of all non-hippocampus neuron types). The quantile used was q = 0.9. Normalized microarray expression values as processed by the Allen Institute of Brain Science were used to calculate the corresponding scores (μjg, σjg, etc.) for gene g in human. Intuitively, is a normalized enrichment score for the mouse ortholog of gene g in neuron type Ti of the mouse. Si is the set of genes that are both highly expressed (high μig) and highly specific (high ) to tissue Ti, thus providing a strong molecular signature for that tissue. This signature is combined with the enrichment scores from human to produce a final spatial homology score.
To provide a quantitative summary for the spatial homology mapping, we also calculated a mouse-human tissue match score , the average of individual match scores per mouse neuron type, , where and .
Briefly, for mouse neuron type i, Mi is the set of ‘perfect’ human brain region matches based on anatomy (Table S2), and rip is the rank (in the event of ties, i.e., identical spatial homology scores, the mean of the tied ranks is used) of human brain region p for mouse neuron i among all possible human brain regions (1 corresponds to the best ranked, and Th= 205 is the worst ranked). To facilitate comparisons, we transform the simple rank score to be between 0 and 1, where 1 corresponds to the best rank. When there are multiple human brain regions that match to the same mouse neuron type (e.g., ECII neurons are located in the human parahippocampal gyrus, and the Allen Brain Atlas had data for both parahippocampal gyrus, bank of the collateral sulcus, as well as parahippocampal gyrus, lateral bank of gyrus), the maximum transformed rank score was taken as the individual match score (vi).
To evaluate the spatial homology mappings, we permuted the spatial homology scores to create a null distribution of mouse-human tissue match scores (n = 10,000) and found that the spatial homology mapping was highly significant (p-value < 0.0001). To examine how sensitive the homology mapping was to the choice of quantile threshold, we varied the quantile threshold from 0.1 to 0.97 (q = 0.97 was the highest for which all mouse neuron types still had expression signatures that passed the threshold). Permutation tests at each threshold (n = 1,000) showed that the spatial homology mapping was significantly better than random for thresholds at higher than q=0.4 and that the mappings stabilized after q=0.7 (Figure S2A).
Construction of functional networks
We used the cell type-specific molecular signatures to construct a cell-type specific gold standard (see Gold standard section below), which we then used to integrate a human genome-scale data compendium (see Human data compendium section below) to construct cell-type specific functional networks based on our tissue-specific regularized Bayesian integration method (Greene et al., 2015) (see Data integration section below).
Gold standard
The cell type-specific gold standard was constructed by combining a functional interaction standard and cell type-specific signatures. The functional interaction gold standard was constructed based on either the presence or absence of gene co-annotations to expert-selected biological process terms from the Gene Ontology (GO) based on whether the term could be experimentally verifiable through targeted molecular experiments. For each of these 337 selected GO terms, we obtained all experimentally derived gene annotations (i.e., annotations with GO evidence codes: EXP, IDA, IPI, IMP, IGI, IEP). After gene propagation in the GO hierarchy, gene pairs co-annotated to any of the selected terms were considered positive examples, whereas gene pairs lacking co-annotation to any term were considered negative examples, except in cases where the two genes were (i) separately annotated to highly overlapping GO terms (hypergeometric p-value < 0.05) or (ii) co-annotated to higher-level GO terms that may still indicate the possible presence of a functional relationship.
We then combined our expanded cell type-specific molecular gene signature sets (q = 0.75) with this functional interaction standard by defining the four classes of edges (C1, C2, C3, and C4) as described in (Greene et al., 2015), with the adjustment of allowing genes annotated to nervous system tissues to be considered for the C2 negative example class (to emphasize cell-type specificity in relation to other general nervous system genes, rather than excluding them based on the hierarchical tissue ontology as in Greene et al.). More specifically, for a neuron type i, C1 is the class of positive functional edges between genes that are also specifically coexpressed in i; C2 is the class of positive functional edges between a gene that is expressed in neuron type i and in an unrelated tissue or cell type (in our case, we consider general nervous system genes to also be in this class); C3 is the class of negative functional edges between genes that are coexpressed in neuron type i; C4 is the class of negative functional edges between one gene expressed in neuron type i and another gene specifically expressed in an unrelated tissue. C1 edges are considered positive examples (genes are both functionally related and expressed in the neuron type of interest), and C2–4 edges are negative examples (at least one of the conditions of functional relationship or tissue-specificity is violated).
Human data compendium
We downloaded and processed 31,157 human interaction measurements and brain expression-based profiles from over 24,000 publications, as well as experimentally defined transcription factor binding motifs, chemical and genetic perturbation data, and microRNA target profiles.
Physical interaction data were downloaded from BioGRID (version 3.2.118)(Chatr-Aryamontri et al., 2013; Stark et al., 2006), IntAct (Nov 2014) (Orchard et al., 2014), MINT (2013–03-26) (Licata et al., 2012), and MIPS (Nov 2014) (Mewes et al., 2011). Interaction edges from BioGRID were discretized into five bins (0–4), depending on the number of experiments supporting the interaction. For all other interaction databases, edges were discretized based on the presence or absence of an interaction.
A total of 6,907 expression profiles from 268 human brain expression datasets were downloaded from the Gene Expression Omnibus (GEO) (Barrett et al., 2013). Duplicate samples were collapsed, and genes with values missing in over 30% of the samples were removed. All other missing values were imputed (Troyanskaya et al., 2001) . Normalized Fisher’s z-transformed expression scores were calculated per pair of genes and discretized into the corresponding bin: (−∞, −1.5), [−1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, 4.5), [4.5, ∞).
Experimentally defined transcription factor binding motifs were downloaded from JASPAR (Mathelier et al., 2014), and the 1-kb upstream region of each gene was scanned for presence of binding motifs using FIMO (Grant et al., 2011) from the MEME software suite (Bailey et al., 2015). For each pair of genes, the Fisher z-transformed Pearson correlation of binding profiles was calculated and discretized into one of the corresponding bins: (−∞, −1.5), [−1.5, −0.5), [−0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, 4.5), [4.5, ∞).
Chemical and genetic perturbation and microRNA target profiles were downloaded from the Molecular Signatures Database (MSigDB, c2:CGP and c3:MIR gene sets, respectively) (Subramanian et al., 2005). For each pair of genes, similarity based on the weighted mean of number of shared profiles (weighted by the specificity of the profile (1/len(genes)) was calculated and discretized into the corresponding bin: (−∞, −1.5), [−1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, 4.5), [4.5, ∞).
Data integration
We applied our tissue-specific regularized Bayesian integration method (Greene et al., 2015) for each of the 7 neuron types to train a naïve Bayesian classifier by comparing against the positive and negative examples from the cell-type specific gold standard. For each cell type, we constructed a binary class node representing the indicator function for whether a pair of genes has a cell type-specific functional relationship, conditioned on additional nodes representing each of the datasets in the data compendium. Each model was then applied to all pairs of genes in the data compendium to estimate the probability of tissue-specific functional interactions. All code for data integration is available in our open-source Sleipnir library for functional genomics (Huttenhower et al., 2008).
Network connectivity analysis
We calculated a z-score for cohesiveness of various biological process GO terms in each of the neuron-specific networks: , where XGO is the mean posterior probability of all gene pairs within a particular GO term, and Xnull, SEnull are respectively the mean and standard error of the null distribution (based on gene sets randomly sampled within all genes with a GO annotation, with equivalent size to the GO term in question).
NetWAS 2.0 on AD GWAS
Here, using an AD GWAS for Braak stages (NFT pathology-based staging) (Beecham et al., 2014) as gold standard and the ECII-specific functional network neighborhoods as features, we applied NetWAS 2.0 with n = 10,000 to rank each of the 23,950 genes for potential association to AD.
We trained support vector machine classifiers (Joachims, 2005) using (i) nominally significant (p-value < 0.01) GWAS genes as positive examples, (ii) randomly sampled non-significant genes with probability proportional to their GWAS p-value as negatives, and (iii) the network neighborhoods of genes as features. Thus, genes with lower p-values (i.e., more significant) would have a lower chance of being chosen as a negative example than genes with higher p-values. Gene-level p-values were obtained using the versatile gene-based association study 2 (VEGAS2, version:16:09:002) software (Mishra and Macgregor, 2015).
To ensure robustness, we independently sampled n such sets of negatives and trained n support vector machines. After applying each of the support vector machines to re-rank genes, we aggregated the n rankings into a final NetWAS 2.0 gene ranking. Intuitively, the key advance of the NetWAS 2.0 method is that it leverages the GWAS p-values as opposed to treating all non-significant genes as having equal probability of being negative examples as in the original NetWAS method (Greene et al., 2015).
Establishment of the expert-curated gene set
To establish amyloid and NFT gene sets, we recruited independent curators who were unaware of any of the NetWAS 2.0 results: a laboratory member who is an AD expert (doctoral degree in biology, 20 years of experience in the AD field), for the amyloid and NFT-1 gene sets, and separately, two professional curators (doctoral degrees in biology, combined 28 years of experience in curation of biomedical literature) for the NFT-2 gene set.
We asked the AD-expert curator to search for genes involved in tau phosphorylation, aggregation, cleavage, folding, localization, clearance (for the NFT-1 set), and in Aβ production, clearance, aggregation (for the amyloid set). The searches were done with PubMed, and included publications released between January 2000 and April 2017. In order to build the NFT-2 gene list, the professional curators reviewed biological resources and the published literature for evidence of tau modification, which included tau phosphorylation, aggregation, cleavage, folding, localization, and clearance. tau association was not sufficient, rather a causal link for the modification was required. For example, proteins that affected tau modification when activated/inhibited/knocked-out/over produced or expressed in a mutant form were included on the gene list. Due to the requirement of a clear causal link, only low-throughput experiments carried out in vitro or in vivo were assessed whereas high-throughput studies were not considered. The main focus was on the modification of human tau, but relevant results shown in various cell lines and animal models were also evaluated. If the results were shown in any species other than human, then the corresponding human ortholog was added to the list. To avoid duplication of effort, multiple curated resources were initially explored for potential tau modifiers present in different database download files from November, 2019. Custom download files for human tau were obtained from iPTMnet (Huang et al., 2017) to identify proteins responsible for tau phosphorylation/dephosphorylation. Only genes with experimental evidence were extracted and their associated PubMed references were verified. tau modifying gene products were also extracted from relevant Gene Ontology (GO) annotations created by the Alzheimer’s Research United Kingdom — University College London (ARUK—UCL) team (Kramarz et al., 2018). Tau-related data already captured in BioGRID (Oughtred et al., 2018; Stark et al., 2006) was also examined by screening the biochemical activity class of interactions in which tau was the hit/prey protein. These interactions represent in vitro experiments in which a protein has been demonstrated to directly modify tau with post-translational modifications (PTMs) or via proteolytic processing. Additional Tau modifiers were added to the gene list based on a recently published review of the biological roles of the tau protein by Guo et al. (Guo et al., 2017) and the original research articles cited in this review were further prioritized for curation. Curators then branched out to review the primary literature in more detail and include proteins with a more indirect Tau modifying role. To this end, online text-mining resources were used to triage the scientific literature, i.e. RLIMS-P (Torii et al., 2015) and PIE the search (Kim et al., 2011). RLIMS-P is an information retrieval and extraction tool that highlights text relevant to phosphorylation interactions and was used to triage papers using tau as the query gene. Curators also used PIE the search, which scores publications based on their likelihood of containing protein interaction data, by running queries containing the following tau modification terms: tau cleavage, tau aggregation, tau folding, Tau localization, or tau clearance. These searches resulted in lists of publications that were reviewed for evidence that the interaction had an effect on tau modification. All together, these curation efforts generated a list of 174 unique genes which were associated with their relevant tau modifications, UniProt protein accession IDs, and original references. Approximately 52% of the 174 unique genes on the tau modifier gene list were found using various literature searches or extracted from relevant reviews indicating this gene list could not be autogenerated from existing annotations found in various biological resources.
Analysis of NetWAS 2.0 predictions
Comparison against the ACT study
We downloaded paired RNA-seq trascriptomes and neuropathological quantifications from the ACT study (http://aging.brain-map.org/download/index) (Miller et al., 2017). We then calculated, for every gene, the Fisher’s z-transformed absolute Spearman’s correlation between its expression in the hippocampus, and IHC amyloid plaque load across all samples.
To aggregate the scores without selecting an arbitrary cutoff, we calculated an amyloid plaque association score for each percentile cutoff averaging the transformed correlation scores for the top x% of NetWAS 2.0 genes (with x = 1%, 5%, 10%, 15%, …,100%). We compared these scores against the counterparts calculated based on ranking by the p-values in the Braak GWAS study. For the background distribution, we sampled an equivalent number of genes 1000 times per percentile cutoff.
To calculate bootstrapped 95% confidence intervals for the NetWAS 2.0 amyloid plaque association scores, we subsampled genes with replacement within each percentile cutoff.
Analysis of Liang et al. ECII dataset
We downloaded microarray expression profiles measuring LCM ECII neurons in control and AD patients (Liang et al., 2008). Data normalization and differential expression analysis were performed using limma (version 3.22.7) (Smyth, 2004). Genes with Benjamini-Hochberg multiple hypothesis test-corrected FDR ≤ 0.05 were considered significantly differentially expressed.
Identification of functional modules
To identify functional modules represented in our top NetWAS 2.0 genes, we created an ECII subnetwork using the top 10% (i.e., top 2,395) of NetWAS 2.0 ranked genes. Then, we used an approach based on shared k-nearest-neighbors (SKNN) and the Louvain community-finding algorithm (Blondel et al., 2008) to cluster the network into distinct modules. This approach alleviates the effect of high-degree genes and accentuates local network structure by connecting genes that are likely to be functionally clustered together in the ECII network. We calculated the ECII SKNN network by using the number of shared top k-nearest neighbors between genes as edge weights and taking the subnetwork defined by the top 5% of edge weights as the subnetwork for downstream analysis. The clustering presented here was calculated with k = 50, but we confirmed that the clustering was robust for k between 10 and 100. Enrichment of Gene Ontology biological process terms and of other experiment-derived gene sets of interest in each module were calculated using one-sided Fisher’s exact tests, with Benjamini-Hochberg multiple hypothesis test correction to calculate FDR.
Gene connectivity analysis
For each gene g in each cell-type specific functional network, we calculated a z-score for gene connectivity, a measure of how central a gene is in the network: , where is the average posterior probability of edges incident on gene g. μ, σ, and n are respectively the mean, standard deviation, and number of all edges in the network.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for all experiments can be found in figure legends. The only samples excluded were: a) samples from two htau PAC misgenotyped mice in the Ptbp1 OE experiment (figure 5D–G), which were homozygous mouse tau knockout rather than heterozygous mouse tau knockout like all of the other mice of the experiment, b) samples from bacTRAP mice in the Ptbp1 silencing and Ptbp1 OE experiment which did not show any read mapping to mCherry in the RNAseq data, indicating that these samples did not have any detectable presence of the transduction virus – one mouse in the Ptbp1 silencing experiment, one mouse in the Ptbp1 OE experiment (Figure S3E–G).
ADDITIONAL RESOURCES
We have made the following data available at http://alz.princeton.edu:
Gene expression levels in the mouse across the 7 different types of neurons, at 5, 12 and 24 months of age (“Expression values”).
The seven in silico human genome-wide network models, each representing one AD-vulnerable or resistant neuron type in the non-disease state, both for download and dynamic, query-based exploration (“Tissues relating to AD”).
Supplementary Material
Table S1: Comparisons between types of neurons. Related to Figure 2. Sheet 1: Top enriched genes in each of the seven mouse neuron types compared to the six other neuron types. Genes with the top 50 normalized enrichment score (z_ont) are shown. Sheets 2 – 4: Comparisons between AD resistant and vulnerable neurons. Sheet 2: Genes enriched in resistant and vulnerable neurons. Negative logFC corresponds to expression enriched in vulnerable neurons, whereas positive logFC corresponds to expression enriched in resistant neurons. Sheet 3: Processes enriched in resistant and vulnerable neurons: scores are z-scores corresponding to FDR for PAGE (Parametric Analysis of Gene Set Enrichment) enrichment; sign corresponds to direction of enrichment (e.g., −1.65 corresponds roughly to FDR = 0.05 enrichment in lower numbers, such as smaller p-values or lower log-fold changes). Sheet 4: pathway cohesiveness measures in resistant and vulnerable neurons: mean cohesiveness is shown. ‘Vulnerable – resistant’ shows the difference between mean vulnerable cohesiveness and mean resistant cohesiveness. Scores are z-scores (compared with network background) for cohesiveness of pathway genes in each network.
Table S2: Mouse-human spatial homology. Related to Figure 3. Sheet 1: Spatial homology scores for the 1,435 pairs of mouse neuron type (as profiled in our bacTRAP experiments) and human brain region (as profiled in the ABA human microarray data). Sheet 2: Human brain regions from the Allen Brain Atlas that are anatomically homologous to the mouse neuron types studied here.
Table S3: NetWAS 2.0 predictions. Related to Figure 4. Sheet 1: Ranks for reprioritized genes as attributed by the NetWAS 2.0 algorithm using ECII functional network in concert with GWAS for Braak stage. Sheet 2: Processes most enriched in NetWAS 2.0 prioritized genes. Scores shown are z-scores of enrichment FDRs, with positive numbers representing enrichment at the top of the list.
Table S4: Gene sets used for the validation of the NetWAS data. Related to Figure 4. Sheets 1–3: Expert-curated amyloid, NFT-1 and NFT-2 sets. All sets were curated by curators independent from the study and unaware of the analysis results. The amyloid and NFT-1 sets were curated by an AD expert. The NFT-2 set was curated by curation experts. Sheets 4 and 5: genes significantly changed by age in ECII neurons, and by Aβ accumulation in ECII neurons of APP/PS1 mice. For differential expression by age (tab 4), comparison was between 24-month old mice and 5-month old mice, with logFC < 0 corresponding to lower expression in old mice (each independent sample is a pool of 2 mice, n = 17 samples for 5 months and 12 samples for 24 months). For Aβ accumulation (tab 5), logFC<0 corresponds to lower expression in APP/PS1 mice (n = 8 APP/PS1 mice).
Table S5: Top NetWAS 2.0 gene modules. Related to Figure 4. Sheet 1: Genes in the different modules. Sheet 2: processes enriched in each of the modules. Sheet 3: enrichment of pathology-associated genes in the different modules (values shown are Benjamini-Hochberg-adjusted FDRs).
Table S6: Genes with the largest differential network centralities between ECII neurons and resistant neurons. Related to Figure 4. The network centrality for each gene is the sum of connectivity to all other genes. The differential centrality was calculated by subtracting to the centrality in ECII the average centrality in all resistant neurons. Genes were then ranked by differential centrality.
Highlights :
Ribosomal profiling of AD vulnerableƒresistant neurons in 5, 12, 24-month old mice
Using human neuron-type functional networks and GWAS to model vulnerability
Identification of axon plasticity genes linking Aß, aging, tau in vulnerable neurons
PTB, regulator of tau exon 10 splicing, might contribute to selective vulnerability
Acknowledgments
We thank A. Milosevic, R. Chottekalapanda, H. Rebholz, M. Riessland, B. Kolisnyk, R. Sealfon, C. Theesfeld, R. Zhang, for critical reading of the manuscript, E. Griggs for assistance with graphic design, R. Norinsky and Rockefeller University (RU) Transgenic Services for all pronuclear injections, C. Zhao and RU Genomics Resource Center for all sequencing, RU Bioimaging Resource Center.
Funding: J.-P.R., M.F. and P.G. were supported by the Fisher Center for Alzheimer’s Disease research. V.Y. was supported in part by US NIH grant T32 HG003284. O.G.T. is a senior fellow of the Genetic Networks program of the Canadian Institute for Advanced Research (CIFAR). P.R-R. was supported in part by the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 799638. This study was supported by the JPB Foundation and Cure Alzheimer’s Fund to P.G. and M.H., United States Army Medical Research and Material Command (USAMRMC), Award No. W81XWH-14–1-0046 to J.-P.R., the National Institute on Aging of the NIH, Awards RF1AG047779 to P.G. and J.-P.R., RF1AG054564 to P.G., J.-P.R. and A.B.-C., and P50 AG005138 to P.R.H., NIH R01 GM071966 to O.G.T, the Office of Research Infrastructure Programs of the NIH Award R01OD010929 to K.D, the National Institute of Neurological Disorders and Stroke of the NIH, Award R01NS091722 to E.F.S, the Howard Hughes Medical Institute to N.H. The results published here are in part based on data obtained from Agora, a platform initially developed by the NIA-funded AMP-AD consortium. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the sponsors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, Silva R. de, Tucker KL, Barde Y-A, Duff K, and Davies P (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of Neurochemistry 86, 582–590. [DOI] [PubMed] [Google Scholar]
- Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Abdullatif AA, Ferragud A, Botelho E, Ballance HI, Dhawan U, et al. (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nature Neuroscience 21, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, and Hyman BT (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. [DOI] [PubMed] [Google Scholar]
- Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, et al. (2018). Epigenetic regulation of brain region-specific microglia clearance activity. Nature Neuroscience 21, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Research 43, W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, and Gallagher M (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. (2013). NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Research 41, D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, Corneveaux JJ, Hardy J, Vonsattel J-P, Younkin SG, et al. (2014). Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genetics 10, e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008, P10008. [Google Scholar]
- Borchelt DR, Ratovitski T, Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, and Sisodia SS (1997). Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19, 939–945. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin C-H, Chawla G, Ostrow K, Shiue L, Ares M, and Black DL (2007). A post-transcriptional regulatory switch in polypyrimidine tract- binding proteins reprograms alternative splicing in developing neurons. Genes & Development 21, 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap E-L, Walker Z, Zhang J, JP J-P, Alvarez MJ, Califano A, et al. (2015). Identification of neurodegenerative factors using translatome-regulatory network analysis. Nature Neuroscience 18, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, et al. (2016). Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Science Translational Medicine 8, 338ra66–338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussière T, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, and Hof PR (2003). Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: stereologic analysis of prefrontal cortex area 9. The Journal of Comparative Neurology 463, 281–302. [DOI] [PubMed] [Google Scholar]
- Caraci F, Battaglia G, Busceti C, Biagioni F, Mastroiacovo F, Bosco P, Drago F, Nicoletti F, Sortino MA, and Copani A (2008). TGF-beta 1 protects against Abeta-neurotoxicity via the phosphatidylinositol-3-kinase pathway. Neurobiology of Disease 30, 234–242. [DOI] [PubMed] [Google Scholar]
- Carlyle BC, Kitchen RR, Kanyo JE, Voss EZ, Pletikos M, Sousa AMM, Lam TT, Gerstein MB, Sestan N, and Nairn AC (2017). A multiregional proteomic survey of the postnatal human brain. Nature Neuroscience 20, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, and Spruston N (2016). Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. ELife 5, e14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Breitkreutz B-J, Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas N, O’Donnell L, et al. (2013). The BioGRID interaction database: 2013 update. Nucleic Acids Research 41, D816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibnik LB, White CC, Mukherjee S, Raj T, Yu L, Larson EB, Montine TJ, Keene CD, Sonnen J, Schneider JA, et al. (2018). Susceptibility to neurofibrillary tangles: role of the PTPRD locus and limited pleiotropy with other neuropathologies. Molecular Psychiatry 23, 1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, et al. (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Human Molecular Genetics 21, 3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J, et al. (2017). Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathologica 133, 839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK, Holland D, Brewer JB, Chen C-H, Thompson WK, et al. (2015). Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol Psychiatr 20, 1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. (2008). Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, and Yan SS (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proceedings of the National Academy of Sciences of the United States of America 107, 18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, et al. (2000). Characterization of Pathology in Transgenic Mice Over-Expressing Human Genomic and cDNA Tau Transgenes. Neurobiol Dis 7, 87–98. [DOI] [PubMed] [Google Scholar]
- Dugas-Ford J, Rowell JJ, and Ragsdale CW (2012). Cell-type homologies and the origins of the neocortex. Proceedings of the National Academy of Sciences of the United States of America 109, 16974–16979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, LaFleur BJ, Coon KD, Brown KM, Caselli R, et al. (2006). Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiology of Aging 27, 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou AG, and Goate AM (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmer KL, Waxman EA, Covy JP, and Giasson BI (2011). E46K human alpha- synuclein transgenic mice develop Lewy-like and tau pathology associated with age- dependent, detrimental motor impairment. The Journal of Biological Chemistry 286, 35104–35118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlanis E, Traunmüller L, Fucile G, and Scheiffele P (2019). Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nature Neuroscience 22, 1709–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, and Hof PR (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, and Lee VM-Y (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics (Oxford, England) 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JD, Rubin TG, Kogan JF, Marrocco J, Weidmann J, Lindkvist S, Lee FS, Schmidt EF, and McEwen BS (2018). Translational profiling of stress-induced neuroplasticity in the CA3 pyramidal neurons of BDNF Val66Met mice. Molecular Psychiatry 23, 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, et al. (2015). Understanding multicellular function and disease with human tissue-specific networks. Nature Genetics 47, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Noble W, and Hanger DP (2017). Roles of tau protein in health and disease. Acta Neuropathol 133, 665–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RL (2000). Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathology (Zurich, Switzerland) 10, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Hirokawa K, Kamei S, and Uchihara T (2013). Isoform transition from four- repeat to three-repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathologica 125, 565–579. [DOI] [PubMed] [Google Scholar]
- Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, Jegga AG, Aronow BJ, Lee C-K, Bernard A, et al. (2015). Canonical genetic signatures of the adult human brain. Nature Neuroscience 18, 1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, Lagemaat L.N. van de, Smith KA, Ebbert A, Riley ZL, et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez- Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Arighi CN, Ross KE, Ren J, Li G, Chen S-C, Wang Q, Cowart J, Vijay-Shanker K, and Wu CH (2017). iPTMnet: an integrated resource for protein post-translational modification network discovery. Nucleic Acids Res 46, D542–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Schroeder M, Chikina MD, and Troyanskaya OG (2008). The Sleipnir library for computational functional genomics. Bioinformatics (Oxford, England) 24, 1559–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki E, Yamamoto R, Murayama N, Minegishi M, Togo T, Katsuse O, Kosaka K, Akiyama H, Tsuchiya K, Silva R. de, et al. (2006). Immunohistochemical investigation of neurofibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neuroscience Letters 405, 29–33. [DOI] [PubMed] [Google Scholar]
- Kaufman SK, Tredici KD, Thomas TL, Braak H, and Diamond MI (2018). Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathologica 136, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Feng LR, Maguire-Zeiss K, Rebeck G, Lashuel HA, and Moussa CE (2010). Parkinson-related parkin reduces a-Synuclein phosphorylation in a gene transfer model. Molecular Neurodegeneration 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, and Moussa CE-H (2012). Wild type and P301L mutant Tau promote neuro-inflammation and a-Synuclein accumulation in lentiviral gene delivery models. Molecular and Cellular Neurosciences 49, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kwon D, Shin S-Y, and Wilbur WJ (2011). PIE the search: searching PubMed literature for protein interaction information. Bioinformatics 28, 597–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung H-Y, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, et al. (2014). Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nature Neuroscience 17, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramarz B, Roncaglia P, Meldal BHM, Huntley RP, Martin MJ, Orchard S, Parkinson H, Brough D, Bandopadhyay R, Hooper NM, et al. (2018). Improving the Gene Ontology Resource to Facilitate More Informative Analysis and Interpretation of Alzheimer’s Disease Data. Genes-Basel 9, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, Volfovsky N, Packer A, Lash A, and Troyanskaya OG (2016). Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nature Neuroscience 19, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. (2013). Meta- analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Zhao X, and Gage FH (2004). Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 24, 3879–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Campbell MJ, Terry RD, and Morrison JH (1987). Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: a quantitative study of visual and auditory cortices. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 7, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC, et al. (2007). Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiological Genomics 28, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, et al. (2008). Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiological Genomics 33, 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, et al. (2012). MINT, the molecular interaction database: 2012 update. Nucleic Acids Research 40, D857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, and Gong C-X (2008). Tau exon 10 alternative splicing and tauopathies. Molecular Neurodegeneration 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-C, Tsai C-W, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, et al. (2014). Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron 84, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Pleasure SJ, Collins AE, Noebels JL, Naya FJ, Tsai MJ, and Lowenstein DH (2000). Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proceedings of the National Academy of Sciences of the United States of America 97, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorian M, Schwartz S, Clark TA, Hollander D, Tan L-Y, Spellman R, Gordon A, Schweitzer AC, Grange P de la, Ast, G., et al. (2010). Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nature Structural & Molecular Biology 17, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Ruepp A, Theis F, Rattei T, Walter M, Frishman D, Suhre K, Spannagl M, Mayer KFX, Stümpflen V, et al. (2011). MIPS: curated databases and comprehensive secondary data resources in 2010. Nucleic Acids Research 39, D220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, and Geschwind DH (2008). A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 28, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Woltjer RL, Goodenbour JM, Horvath S, and Geschwind DH (2013). Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Medicine 5, 48–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Guillozet-Bongaarts A, Gibbons LE, Postupna N, Renz A, Beller AE, Sunkin SM, Ng L, Rose SE, Smith KA, et al. (2017). Neuropathological and transcriptomic characteristics of the aged brain. ELife 6, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, and Macgregor S (2015). VEGAS2: Software for More Flexible Gene- Based Testing. Twin Research and Human Genetics : The Official Journal of the International Society for Twin Studies 18, 86–91. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nature Neuroscience 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaud S, Jones DR, Moussaud-Lamodière EL, Delenclos M, Ross OA, and McLean PJ (2014). Alpha-synuclein and tau: teammates in neurodegeneration? Molecular Neurodegeneration 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, et al. (2009). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nature Medicine 15, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. (2014). The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Research 42, D358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R, Stark C, Breitkreutz B-J, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, et al. (2018). The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, and Mucke L (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nature Neuroscience 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A, and Caruncho HJ (1998). Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proceedings of the National Academy of Sciences of the United States of America 95, 3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, and Parker R (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell- type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, and Caroni P (2011). Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71, 35–48. [DOI] [PubMed] [Google Scholar]
- Scheckel C, Drapeau E, Frias MA, Park CY, Fak J, Zucker-Scharff I, Kou Y, Haroutunian V, Ma’ayan A, Buxbaum JD, et al. (2016). Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. ELife 5, 4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulcre J, Grothe MJ, Sabuncu M, Chhatwal J, Schultz AP, Hanseeuw B, Fakhri GE, Sperling R, and Johnson KA (2017). Hierarchical Organization of Tau and Amyloid Deposits in the Cerebral Cortex. JAMA Neurology 74, 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, et al. (2016). Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Reports 7, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel J-P, and Kim T-W (2005). Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Annals of Neurology 58, 909–919. [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3, Article3–25. [DOI] [PubMed] [Google Scholar]
- Song A, Yan J, Kim S, Risacher SL, Wong AK, Saykin AJ, Shen L, Greene CS, and Initiative ADN (2016). Network-based analysis of genetic variants associated with hippocampal volume in Alzheimer’s disease: a study of ADNI cohorts. BioData Mining 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, Baquet ZC, Hodges A, Cunningham P, Holmans P, Jones KR, Jones L, Kooperberg C, and Olson JM (2007). Conservation of regional gene expression in mouse and human brain. PLoS Genetics 3, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sun Y, Ling S-C, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S, et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1- mediated ALS. Proceedings of the National Academy of Sciences of the United States of America 112, E6993–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, and Nojyo Y (1993). Projection of the entorhinal layer II neurons in the rat as revealed by intracellular pressure-injection of neurobiotin. Hippocampus 3, 471–480. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, et al. (2006). Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. The Journal of Clinical Investigation 116, 3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, and Braak H (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. [DOI] [PubMed] [Google Scholar]
- Torii M, Arighi CN, Li G, Wang Q, Wu CH, and Vijay-Shanker K (2015). RLIMS- P 2.0: A Generalizable Rule-Based Information Extraction System for Literature Mining of Protein Phosphorylation Information. Ieee Acm Transactions Comput Biology Bioinform Ieee Acm 12, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, and Altman RB (2001). Missing value estimation methods for DNA microarrays. Bioinformatics (Oxford, England) 17, 520–525. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurology 70, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, and Jucker M (2017). The Exceptional Vulnerability of Humans to Alzheimer’s Disease. Trends in Molecular Medicine 23, 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B, Jaglin XH, Favuzzi E, Quattrocolo G, Nigro MJ, Yusuf N, Khodadadi-Jamayran A, Rudy B, and Fishell G (2018). Rbfox1 Mediates Cell-type-Specific Splicing in Cortical Interneurons. Neuron 100, 846–859.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gao Q-S, Wang Y, Lafyatis R, Stamm S, and Andreadis A (2004). Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. Journal of Neurochemistry 88, 1078–1090. [DOI] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, and Smith CWJ (2004). Autoregulation of Polypyrimidine Tract Binding Protein by Alternative Splicing Leading to Nonsense-Mediated Decay. Molecular Cell 13, 91–100. [DOI] [PubMed] [Google Scholar]
- Yeom K-H, Mitchell S, Linares AJ, Zheng S, Lin C-H, Wang X-J, Hoffmann A, and Black DL (2018). Polypyrimidine tract-binding protein blocks miRNA-124 biogenesis to enforce its neuronal-specific expression in the mouse. Proceedings of the National Academy of Sciences of the United States of America 115, E11061–E11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Shen EH, Hohmann JG, Oh SW, Bernard A, Royall JJ, Glattfelder KJ, Sunkin SM, Morris JA, Guillozet-Bongaarts AL, et al. (2012). Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell 149, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, and Black DL (2012). PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nature Neuroscience 15, 381–8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Comparisons between types of neurons. Related to Figure 2. Sheet 1: Top enriched genes in each of the seven mouse neuron types compared to the six other neuron types. Genes with the top 50 normalized enrichment score (z_ont) are shown. Sheets 2 – 4: Comparisons between AD resistant and vulnerable neurons. Sheet 2: Genes enriched in resistant and vulnerable neurons. Negative logFC corresponds to expression enriched in vulnerable neurons, whereas positive logFC corresponds to expression enriched in resistant neurons. Sheet 3: Processes enriched in resistant and vulnerable neurons: scores are z-scores corresponding to FDR for PAGE (Parametric Analysis of Gene Set Enrichment) enrichment; sign corresponds to direction of enrichment (e.g., −1.65 corresponds roughly to FDR = 0.05 enrichment in lower numbers, such as smaller p-values or lower log-fold changes). Sheet 4: pathway cohesiveness measures in resistant and vulnerable neurons: mean cohesiveness is shown. ‘Vulnerable – resistant’ shows the difference between mean vulnerable cohesiveness and mean resistant cohesiveness. Scores are z-scores (compared with network background) for cohesiveness of pathway genes in each network.
Table S2: Mouse-human spatial homology. Related to Figure 3. Sheet 1: Spatial homology scores for the 1,435 pairs of mouse neuron type (as profiled in our bacTRAP experiments) and human brain region (as profiled in the ABA human microarray data). Sheet 2: Human brain regions from the Allen Brain Atlas that are anatomically homologous to the mouse neuron types studied here.
Table S3: NetWAS 2.0 predictions. Related to Figure 4. Sheet 1: Ranks for reprioritized genes as attributed by the NetWAS 2.0 algorithm using ECII functional network in concert with GWAS for Braak stage. Sheet 2: Processes most enriched in NetWAS 2.0 prioritized genes. Scores shown are z-scores of enrichment FDRs, with positive numbers representing enrichment at the top of the list.
Table S4: Gene sets used for the validation of the NetWAS data. Related to Figure 4. Sheets 1–3: Expert-curated amyloid, NFT-1 and NFT-2 sets. All sets were curated by curators independent from the study and unaware of the analysis results. The amyloid and NFT-1 sets were curated by an AD expert. The NFT-2 set was curated by curation experts. Sheets 4 and 5: genes significantly changed by age in ECII neurons, and by Aβ accumulation in ECII neurons of APP/PS1 mice. For differential expression by age (tab 4), comparison was between 24-month old mice and 5-month old mice, with logFC < 0 corresponding to lower expression in old mice (each independent sample is a pool of 2 mice, n = 17 samples for 5 months and 12 samples for 24 months). For Aβ accumulation (tab 5), logFC<0 corresponds to lower expression in APP/PS1 mice (n = 8 APP/PS1 mice).
Table S5: Top NetWAS 2.0 gene modules. Related to Figure 4. Sheet 1: Genes in the different modules. Sheet 2: processes enriched in each of the modules. Sheet 3: enrichment of pathology-associated genes in the different modules (values shown are Benjamini-Hochberg-adjusted FDRs).
Table S6: Genes with the largest differential network centralities between ECII neurons and resistant neurons. Related to Figure 4. The network centrality for each gene is the sum of connectivity to all other genes. The differential centrality was calculated by subtracting to the centrality in ECII the average centrality in all resistant neurons. Genes were then ranked by differential centrality.
Data Availability Statement
The datasets generated during this study are available at GEO: GSE151460 for the bacTRAP profiles at 5, 12 and 24 months of age, and the APP/PS1 ECII-bacTRAP profiles, and GSE151356 for the ECII-bacTRAP data upon silencing and overexpression of Ptbp1.
The Sleipnir library for functional genomics used for network integration is available at https://libsleipnir.bitbucket.io/
Uncropped original images for Western blot and fluorescent PCR for Figures 5 and S3 in the paper are available on Mendeley: http://dx.doi.org/10.17632/g67bhh7zsj.1