

ABSTRACT
Evidence has been accumulating to indicate that extracellular vesicles (EVs), including exosomes, released by cancer cells can foster tumour progression. The molecular chaperones – CDC37, HSP90α and HSP90β play key roles in cancer progression including epithelial-mesenchymal transition (EMT), although their contribution to EVs-mediated cell–cell communication in tumour microenvironment has not been thoroughly examined. Here we show that triple depletion of the chaperone trio attenuates numerous cancer malignancy events exerted through EV release. Metastatic oral cancer-derived EVs (MEV) were enriched with HSP90α HSP90β and cancer-initiating cell marker CD326/EpCAM. Depletion of these chaperones individually induced compensatory increases in the other chaperones, whereas triple siRNA targeting of these molecules markedly diminished the levels of the chaperone trio and attenuated EMT. MEV were potent agents in initiating EMT in normal epithelial cells, a process that was attenuated by the triple chaperone depletion. The migration, invasion, and in vitro tumour initiation of oral cancer cells were significantly promoted by MEV, while triple depletion of CDC37/HSP90α/β reversed these MEV-driven malignancy events. In metastatic oral cancer patient-derived tumours, HSP90β was significantly accumulated in infiltrating tumour-associated macrophages (TAM) as compared to lower grade oral cancer cases. HSP90-enriched MEV-induced TAM polarization to an M2 phenotype, a transition known to support cancer progression, whereas the triple chaperone depletion attenuated this effect. Mechanistically, the triple chaperone depletion in metastatic oral cancer cells effectively reduced MEV transmission into macrophages. Hence, siRNA-mediated knockdown of the chaperone trio (CDC37/HSP90α/HSP90β) could potentially be a novel therapeutic strategy to attenuate several EV-driven malignancy events in the tumour microenvironment.
Abbreviations
CDC37: cell division control 37; EMT: epithelial-mesenchymal transmission; EV: extracellular vesicles; HNSCC: head and neck squamous cell carcinoma; HSP90: heat shock protein 90; TAM: tumour-associated macrophage
KEYWORDS: Extracellular vesicles, epithelial-mesenchymal transition, tumour-associated macrophage, CDC37, HSP90, tetraspanin, oral cancer
Introduction
Extracellular vesicles (EVs) are extracellular nano-particles surrounded by lipid membranes containing a variety of molecular cargos such as proteins, small and long RNA, DNA molecules, glycan and metabolites that are secreted by cells [1,2]. Major EV subtypes are endosome-originated exosomes and plasma membrane-derived ectosomes (aka microvesicles or microparticles) while additional types of EVs consisting of large oncosomes (1–10 μm), matrix vesicles, migrasomes, exopheres (~4 μm), exomeres (~35 nm) and bacterial outer membrane vesicles have been reported [3,4]. EVs can play roles in discarding redundant intracellular materials while also mediating cell–cell communication when their cargos reached recipient cells/organs [5,6]. These vesicles also play pivotal roles in influencing the local tumour microenvironment, enter body fluids, and initiate distant organ modifications such as pre-/pro-metastatic niche formation [5,6]. Recent studies, including ours, have shown that cancer cell-derived EVs could also alter the properties of non-cancerous host cells such as epithelial cells and macrophages, through molecular transfer [7,8]. Tumour-derived EVs are often enriched with heat shock proteins (HSPs) [9–12], tetraspanins such as CD9 and CD63 (aka LAMP3), and CD326 (aka epithelial cell-adhesion molecule [EpCAM], a marker of cancer-initiating cells) [1,12,13]. We showed that high-metastatic oral squamous cell carcinoma cells secrete EVs enriched with HSP90α, HSP90β, CD326 and CD9 compared to low-metastatic oral cancer cell-derived EVs [12]. Moreover, the elevated expression of HSP90 family members was found to correlate with a poor prognosis for head and neck cancer cases [12]. Double knockdown of HSP90α and HSP90β, using small interfering RNA (siRNA) significantly reduced the survival of highly metastatic oral cancer cells, although single knockdown of each HSP90 was ineffective [12].
HSP90 is an essential protein in intracellular protein folding, wound healing and cancer progression [14–16]. However, more recent studies, including ours, have revealed the secretion of HSP90, leading to novel functions in cell–cell communication. More recently, HSP90 has been found also as a major cargo contained in EVs [10–12,17]. The HSP90 family consists of four members including the proteotoxic stress-inducible HSP90α encoded by HSP90AA1, the constitutively expressed HSP90β encoded by HSP90AB1, the mitochondrially localized TRAP1, and an ER-resident paralog Gp96 (aka Grp94 or endoplasmin) [12,17]. Notably, HSP90α is often overexpressed in cancer cells and can be secreted to the extracellular space as a soluble protein or as a cargo protein of EVs [10,12,18]. Recently, the EV-proteome analysis of oral cancer cells has revealed abundant secretion of EVs enriched with HSP90 from high-metastatic oral cancer cells [12].
Epithelial-mesenchymal transition (EMT) is a key biological event potentially regulated by extracellular HSP90 [19,20]. EMT involves phenotypic changes in cells from epithelial to mesenchymal properties and promotes motility [21], migration, invasion, stemness [22], and drug resistance [23–25] in various types of cancer cells [26]. Numerous EMT-promoting factors have been identified, although recent studies, including ours, have revealed that cancer cell-derived EVs often initiate EMT in epithelial cells [8,10] and promote EMT in tumour cells [27]. Notably, cell division control 37 (CDC37) plays a pivotal role in stabilizing HSP90 client proteins, thereby facilitating numerous intracellular oncogenic signalling pathways [14,28–31]. CDC37 functions primarily in a complex with both HSP90 paralogs- HSP90α and HSP90β to mediate the three-dimensional (3D) folding and structural integrity of client proteins kinases essential for EMT [32–35]. Moreover, knockdown of CDC37 reduced cellular proteostasis and release of EV proteins such as CD9 [10].
Macrophages resident in the tumour microenvironment may play substantial roles in deciding the delicate balance between cancer progression and suppression. Macrophages are highly heterogeneous cells that can rapidly change their properties in response to local microenvironmental signals [36]. The tumour microenvironment often contains infiltrating tumour-associated macrophages (TAM) that affect many processes, including tumour initiation, migration, invasion [37], angiogenesis, metastasis [38], chemoresistance [39] and immune checkpoint [40,41]. An elevated number of TAM bearing an M2-type polarity is associated with a poor prognosis for cancers [40]. M2-type macrophages play a key role in adhesion, migration, invasion and cytokine production of oral cancer cells favouring tumour growth [42]. We conjecture that the roles of TAM could be modified by EV bioactive molecules secreted by tumour cells.
In the present study, we aimed to examine: (i) if high-metastatic oral cancer-derived EVs could promote cancer malignancy properties such as EMT, enhanced migration, increased invasion, tumour initiation and macrophage M2 polarization, (ii) if triple depletion of the chaperone trio – CDC37, HSP90α and HSP90β could lessen the effects of EVs on tumour progression. We also aimed to investigate (iii) HSP90 expression and distribution in tumours, their stroma and in TAM in different malignancy grades of oral cancer, to elucidate potential roles of HSP90-EVs in the tumour microenvironment.
Materials and methods
Cell culture
HSC-3, HSC-3-M3, Ca9-22, HO-1-u-1, SAS, HSC-2, HSC-4 and THP-1 were obtained from JCRB Cell Bank and maintained in DMEM containing 10% FBS. An immortalized oral mucosa epithelial cell line RT7 was provided by Dr Nobuyuki Kamata at Hiroshima University and maintained in KGM-gold (Lonza). For EV preparation, cells were cultured in serum-free DMEM. THP-1 was maintained in RPMI-1640 with 10% FBS and for macrophage-like differentiation, stimulated with 200 nM of phorbol 12-myristate 13-acetate (PMA) for 24 h.
siRNA transfection
We designed siRNA sequences targeting mRNA of CDC37, HSP90AA1 or HSP90AB1 (Supplementary Table 1) [10,12]. For targeting each mRNA, a mixture of two types of siRNA duplex was used. The control non-targeting siRNA was purchased from Nippon Gene. siRNA was transfected as described previously [10]. Briefly, cells were pre-cultured in DMEM containing 10% FBS for 1 day or until 60–80% confluency and then transfected with siRNA at a final concentration of 5 nM using Lipofectamine RNAi MAX (ThermoFisher). The medium was replaced with serum-free one at 24 h post-transfection. Cells were then cultured for 48 h before EV preparation, ATP-based cell viability assay, and lactate dehydrogenase release (LDH) assay. For Gluc assay, cell culture supernatants were collected at 6, 24 and 48 h post-medium replacement. At the same time points, cells were detached using Trypsin/EDTA and stained with Trypan Blue for cell counting. The number of Trypan Blue-negative and -positive cells were counted.
EV and EV-free fractions
EV fraction and soluble fraction were prepared from serum-free culture supernatants at 48 h post-medium replacement using a modified polymer-based precipitation method as described [10,12]. Simultaneously, whole cell lysates were prepared as described below. Briefly, cell culture supernatant was centrifuged at 2,000 × g for 30 min at 4°C. The supernatant was centrifuged at 10,000 × g for 30 min at 4°C. The supernatants were filtered with a 0.2-μm pore filter in a few experiments, as shown in supplemental items. The pass-through was concentrated using an ultrafiltration device for MW.100 k to separate an EV fraction and a soluble EV-free fraction. The concentrate was applied to polymers of Total Exosome Isolation (ThermoFisher). The pass-through was concentrated using an ultrafiltration device for MW.10 k and used as EV-free conditioned media. The EV fractions were suspended in 100–200 μL PBS (−). For protein assay, 10 μL of 10× RIPA buffer and 100× a protease inhibitor cocktail (Sigma) were added to the 100 μL of the EV fraction and incubated on ice for 15 min. Protein concentration was analysed using micro BCA protein assay (ThermoFisher).
Whole cell lysate
The whole cell lysate (WCL) was prepared as described previously [5,10]. Briefly, cells cultured in a 6-cm dish were lysed in 150–200 μL/dish of a RIPA buffer (1% NP-40, 0.1% SDS, and 0.5% deoxycholate, and EDTA-free protease inhibitor cocktail in PBS) and collected by using a cell scraper. Cells were further lysed by a 25-gauge syringes for 10 strokes and then incubated for 30 min on ice. The lysate was centrifuged at 12,000 × g for 20 min at 4°C and the supernatant was used as a WCL. Protein concentration of the WCL was analysed using micro BCA protein assay (ThermoFisher).
Western blotting
Western blotting was performed as described [10,13,43]. The same protein amounts of WCL were subjected to SDS-PAGE, followed by transfer to a PVDF membrane using wet- and semi-dry methods where appropriate. The membranes were blocked in 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 for 60 min, incubated with primary antibodies, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. For CD63, blocking was performed in 10% overnight and the primary antibody was reacted for 2 days. Blots were visualized with ECL substrate. We used antibodies against CD9 (MEX001-3, Medical and Biological Laboratories), CD63 (EXOAB-CD63A-1, System Biosciences), HSP90α (GTX109753, GeneTex), HSP90β (GTX101448, GeneTex), MMP9 (ab38898, Abcam) and HRP-conjugated anti-β-actin antibody (clone 2F3, Fujifilm/Wako) as optimized previously [5,8,10,12,23,44]. Antibodies against CD326 (VU1D9), CDC37 (D11A3), E-cadherin (24E10), β-catenin (D10A8), Vimentin (D21H3), Snail (C15D3), Claudin-1 (D5H1D) and COX-4 (3E11) were purchased from Cell Signaling Technology.
We loaded the same protein amount per lane in each Western blotting experiment. To obtain relative expression levels, relative band intensities were measured in the same membrane using ImageJ. The raw values were converted into relative values as control = 1.0. For normalization, relative levels of the protein of interest per loading control were calculated.
Transmission electron microscopy (TEM)
TEM and particle diameter distribution analysis were carried out as described previously [10,12]. Briefly, a 400-mesh copper grid coated with formvar/carbon films was hydrophilically treated. The EV suspension (5–10 μL) was placed on Parafilm, and the grid was floated on the EV liquid and left for 15 min. The sample was negatively stained with 2% uranyl acetate solution for 2 min. EVs on the grid were visualized with 20,000 times magnification with an H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
Particle size distribution
A part of EV fraction was diluted within PBS (−) to volume up to 40 μL and then analysed using Zetasizer nano ZSP (Malvern Panalytical, Malvern, UK) in a range of 0.3–10,000 nm-diameters, as described [10,12]. Alternatively, a part of EV fraction was mixed with PBS (−) to volume up to 250 µL and then particle diameters of EV fractions were analysed using ELS-8000 (Otsuka Electronics, Hirakata, Japan) between 30 and 1000 nano-diameters, as described [8].
Fluorescent ceramide-labelled EV transmission
As described [8,45,46], 20 μg of EV fraction was incubated with 10 μM BODIPY TR Ceramide (ThermoFisher) for 20 min at 37°C. Excessive dye was removed using Exosome Spin Columns (MW. 3000) (ThermoFisher). The labelled EVs were added to the culture medium of HSC-3 cells in 96-well plates at 25 μg/mL. Cells were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.5% Tween-20 in PBS for 5 min. Cells were incubated with ActinGreen488 (ThermoFisher) for 30 min and with DAPI for 5 min. Fluorescence images of random four fields were taken using a Floid® Imaging Station (ThermoFisher) and fluorescence-positive cells in each field were counted.
EV reporters
The lentiviral reporter constructs of CSCW-palmitoylation signal-tandem dimer Tomato (palmT), CSCW-palmitoylation signal-EGFP (palmG) [47] and CSCW-Gaussia luciferase-biotin acceptor-PDGFR transmembrane-IRES-GFP (mbGluc) [48] were kindly gifted from Dr Charles P. Lai. For virus production, HEK293T cells at 70–80% confluence were transfected with PalmT or PalmG constructs, psPAX2 packaging plasmid and pMD2.G envelope plasmid using PEI max (Polysciences). HSC-3, HSC-3-M3 or THP-1 cells were infected by using spinfection method with the viral solution. Infected/transduced stable cells were selected using puromycin. Isolation of single clones was carried out by limiting dilution method. We established palmtdTomato-expressed HSC-3-M3 cells (designated HSC3M3/palmT), palmGFP-expressed HSC-3-M3 cells (designated HSC3M3/palmG), palmGFP-expressed HSC-3 cells (designated HSC3/palmG) and palmGFP-expressed THP-1 cells (designated THP1/palmG) [46].
Luciferase assay
Gaussia luciferase activities were quantified using a Gaussia luciferase glow kit (Pierce). Culture supernatants of cells transfected with siRNA were collected at 6, 24 and 48 h after the replacement of media. Cell culture supernatant (10 or 20 μL) were mixed with 50 μL of coelenterazine working solution in a white 96-well plate. Luminescence was measured in a plate reader Gemini XP SOK (Molecular Devices).
Confocal laser-scanning microscopy
CLSM was carried out as described previously [5,8,49]. Briefly, recipient cells were seeded on a type I collagen-coated coverslip in a 24-well plate at a density of 1 × 104 cells per well and cultured for 24 h in a serum-containing culture media. EVs (HSC3 M3/palmT-EV; 5 μg/mL) or PBS were added to the culture media. After the incubation for 24 h, cells were fixed with 4% paraformaldehyde in PBS for 10 min. Fluorescence images were taken using a CLSM system (LSM 780 META; Carl Zeiss).
Reporter vesicle transmission
As recipients, HSC3/palmG cells or THP1/palmG cells treated with PMA for 24 h were seeded in a 96-well plate at a concentration of 1 × 104 cells/well and cultured for 24 h in a serum-containing media. EV fractions (2.5 and 25 μg/mL) or PBS were added to the culture media. The transmission of the palmT-EVs was monitored every 1 h for 8 h in ArrayScan High Content Screening (HCS) System (ThermoFisher). The fluorescence intensity of each cell was determined using a filter set (485/594) for (GFP/Tomato). The fluorescence intensity values less than 85 and 60 were defined as the background values in recipient HSC3/palmG and macrophage-like THP1/palmG cells, respectively, and subtracted from the raw values. The fluorescence-positive cells were counted in one field (2,632 μm2). The transmission rate was calculated as a percent of exogenous palmT-positive cells per palmG-positive recipient cells with a fluorescence intensity 35 or more.
Cell viability
Cell viability was measured as described previously [50–52]. ATP content was quantified using a CellTiterGlo (CTG) Luminescent Cell Viability Assay (Promega Co., Madison, WI). Cells were detached using Trypsin/EDTA and suspended with 150 μL of CTG solution and incubated for 10 min at 37°C. Luminescent measurements were done on the plate reader Gemini XP SOK (Molecular Devices).
Lactate dehydrogenase release assay
Cytotoxicity was measured using the index of lactate dehydrogenase (LDH) release from cells, as described previously [10]. The LDH activity was measured using the LDH cytotoxicity assay kit (Nacalai Tesque, Kyoto, Japan) according to the manufacturer’s instructions. The culture supernatants of cells transfected with siRNA were collected at 48 h after the replacement of media (72 h after siRNA transfection). The collected supernatants were transferred to another 96-well plate and incubated with substrate solution at RT for 20 min. The cells were lysed with cell lysis buffer, then incubated with substrate solution at RT for 20 min. The stop solution was added and absorbance (490 nm) was measured. Each absorbance was converted into a relative value to the control. For normalization, the relative level of LDH release per cell number was calculated.
Morphological analysis of EMT
Cellular EMT morphology was analysed as described previously [8,10]. Briefly, HSC-3-M3 cells were transfected with siRNAs. Then, culture media were replaced with serum-free ones. Cells were further cultured for 48 h. Then photomicrographs of random four fields were taken using a Floid® Imaging Station (ThermoFisher). Spindle-shaped cells were defined as the cells with more than two-fold length/width ratio.
RT7 cells were cultured in KGM-2 in 96-well plates and EV (50 μg/mL) or PBS were added to the culture media. Spindle-shaped cells longer than 80 μm in random three fields were counted using ImageJ at 48 h post-EV addition.
Immunocytochemistry
THP-1 cells were treated with PMA for 24 h for macrophage-like differentiation. EVs were added to culture media of the differentiated THP-1 in 96-well plates at 25 μg/mL. Cells were further cultured for 48 h and fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.5% Tween-20 in PBS for 5 min. Immunocytochemistry was carried out as described previously [5,49]. The fixed cells were blocked in normal goat serum solution for 30 min, incubated with rabbit anti-CD206 (ab64693, Abcam) and mouse anti-CD68 IgG (M0814, Dako/Agilent) in PBS, and incubated with anti-rabbit IgG AlexaFluor488 and anti-mouse IgG AlexaFluor568. Cells were incubated with DAPI. Fluorescence images of random nine fields were taken using Floid® Imaging Station. To define CD206 or CD68 positivity, the fluorescence intensity of the cells without the primary antibody was subtracted as background.
Wound healing assay
Confluent HSC-3 cells in 48-well plates were wounded by scratching with pipette tips. EVs (50 µg/mL) or PBS were added to culture media. Images were captured immediately and subsequently at 8 h using Floid® Imaging Station. The percentage of the area closed in 8 h was measured using Image J.
Migration assay and invasion assay
Migration/invasion assays were carried out as described previously [5,51]. Briefly, uncoated and Matrigel‐coated culture systems were used for in vitro migration and invasion assays, respectively. HSC-3 cells (25 k cells) in serum-free DMEM were mixed with EVs (10 μg/mL) or PBS and immediately seeded to an upper chamber of Transwell® 24‐well (Corning). Cells migrated/invaded to the lower surface of filters were fixed, stained using Diff‐Quick, and counted at 33 h post-seeding period.
Tumoroid formation
Tumoroid (or tumorsphere) formation assay was performed as described [13,45,50,51]. Briefly, HSC-3 cells were cultured with EVs (50 μg/mL) or PBS in serum-free DMEM in 96-well NanoCulture Plates (Medical and Biological Laboratories) for 2 days. Lox-1 probe (Medical and Biological Laboratories) was added to culture media at 2 μM and then cells/tumorspheres were cultured for 24 h. Tumorspheres with red fluorescence were measured using the ArrayScan HCS System (ThermoFisher). The fluorescence intensity was determined using a filter set for TRITC. For counting spheroids, the size of each tumorsphere (spheroid) in the entire cell population was measured. The size of the top 300 tumorspheres from three wells in each experimental group was counted and dot-plotted. Fluorescent areas greater than 300 μm2 were counted in each well and calculated the average number of spheroids in three wells. A single well of a 96-well plate was sectionized to 9 fields, and the area of 1 field is 2,632 μm2.
Patient-derived samples
Sixteen oral squamous cell carcinoma tissues (stage I: n = 8, stage IV: n = 8) resected at Okayama University Hospital Oral Surgery were examined. The study was approved by the Okayama University Human Ethics Committee (Approval number: 1608–018) and informed consent was obtained from all patients.
Immunohistochemistry
IHC was carried out as described previously [53,54]. Paraffin blocks of specimens were cut at 3-µm thickness. The sections were deparaffinized and then autoclaved in a 0.2% citrate buffer for 15 min. Sections were incubated with 3% hydrogen peroxide for 30 min. Sections were treated with 10% normal goat serum for 30 min, incubated with anti-HSP90α (GTX109753, GeneTex) and anti-HSP90β (GTX101448, GeneTex) antibodies at 4°C overnight, and visualized with the avidin-biotin complex method (PK4001, Vector Laboratories). To rate HSP90-positive cancer cells, five areas were randomly chosen in each tumour specimen at high magnification (×200) and HSP90α or HSP90β-positive cancer cells were counted. To rate HSP90-positive stromal cells, five areas were randomly chosen in each specimen and HSP90α or HSP90β-positive stromal cells per 100 mm2 stromal area were counted. The criteria of positive cells were cells that exhibited DAB colouration determined by two pathologists.
Co-expression correlation
The data set of head and neck squamous cell carcinoma (HNSCC) with 523 patient-derived 523 samples (TCGA, Pan-Cancer Atlas) was analysed using cBioPortal.
Statistical analysis
Statistical significance was calculated using GraphPad Prism and Microsoft Excel. The difference between the two sets of data was examined by unpaired student’s t-test. P < 0.05 was considered to indicate statistical significance. Data were expressed as means±SD unless otherwise specified. The size of tumorspheres was statistically analysed using ANOVA Tukey’s multiple comparisons test and expressed as mean±SD.
Results
Metastatic oral cancer-derived EVs enriched with HSP90 promote cancer cell migration
We asked firstly whether HSP90α and HSP90β were secreted in EVs released by seven oral squamous cell carcinoma cell lines and one oral epithelial cell line, known as RT7. HSP90α was found in EVs secreted by the seven cell lines (Ca9-22, HO-1-u-1, SAS, HSC-2, HSC-3, HSC-4 and HSC-3-M3) and by RT7 (Fig. S1). By contrast, HSP90β was markedly found in EVs secreted by the highly metastatic HSC-3-M3, but not detected in EVs secreted by the other seven cell lines. High-metastatic HSC-3-M3-derived EVs (M3-EV) contained significantly higher levels of HSP90α and HSP90β compared to HSC-3-derived EVs (HSC3-EV) (Figure 1a,b; Fig. S2). Additionally, M3-EV contained a higher level of CD326 compared to HSC3-EV. Moreover, M3-EV tended to contain a higher level of CD63 compared to HSC3-EV. Both HSC3-EV and M3-EV possessed cup-like shapes (Figure 1c, Fig. S3), although M3-EV (average 141.8 nm) tended to be larger than HSC3-EV (average 119.2 nm) (Figure 1d–g; Fig. S4). The main populations of HSC3-EV and M3-EV were between 50 and 200 nm diameters.
Figure 1.
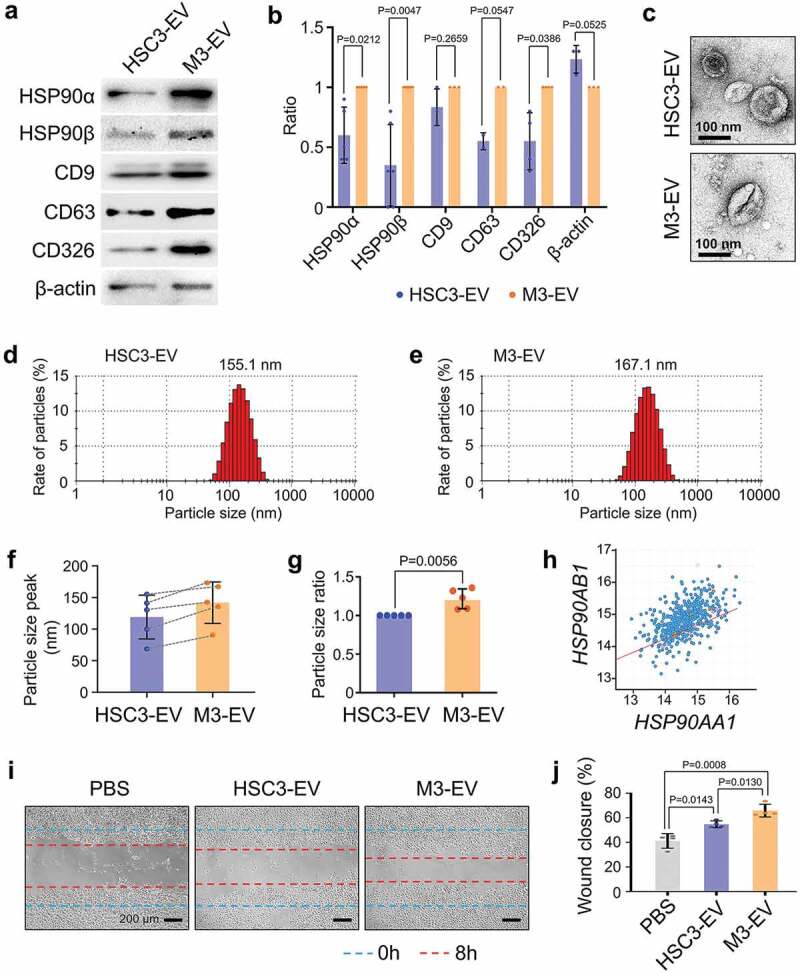
Different characters of high- and low-metastatic oral cancer-derived EVs. (a) Western blotting showing HSP90α, HSP90β, tetraspanins (CD9, CD63), and CD326 in HSC-3-derived EVs (HSC3-EV) and HSC-3-M3-derived EVs (M3-EV). Similar data were obtained from three independent experiments. (b) Column scatters plotting to compare protein levels in HSC3-EV vs. M3-EV. Relative band intensities of Western blotting were plotted. (c) Representative TEM images of HSC3-EV and M3-EV. Scale bars, 100 nm. (d,e) Representative particle diameter distribution of (d) HSC3-EV and (e) M3-EV. (f) EV size peaks from five independent experiments. Dotted lines indicate the same sets of experiments. (g) The ratio of EV size peaks. (h) Scatter plot showing co-expression correlation between HSP90AA1 and HSP90AB1 in HNSCC. A regression line was shown in red. Data were represented as log2. The data set of head and neck squamous cell carcinoma (HNSCC) with 523 patient-derived 523 samples (TCGA, Pan-Cancer Atlas) was analysed using cBioPortal. (i,j) Wound healing assay of HSC-3 cells treated with PBS, HSC3-EV or M3-EV. (i) Representative images at 8 h after the wounding. --- (blue) initial wounds. --- (red) closed wounds at 8 h. Scale bars, 200 μm. (j) Rate of the wound closure. n = 4.
We next examined the co-expression correlation between the HSP90AA1 gene, which encodes HSP90α and the HSP90AB1 gene encoding HSP90β, among 523 tumour samples derived from 523 HNSCC patients registered in the cancer genome atlas. A high-level co-expression correlation was found between HSP90AA1 and HSP90AB1 with Spearman’s correlation score 0.52 (p = 2.59e−37), Pearson’s correlation score 0.50 (p = 2.71e−34) and a regression line (y = 0.44x±8.39) (Figure 1h). The positive correlation of co-expression between HSP90AA1 and HSP90AB1 may be consistent with the co-expression of HSP90α and HSP90β observed in M3-EV derived from high-metastatic oral cancer in Figure 1a,b. To query the pro-migratory potential of oral cancer-derived HSC3-EV and M3-EV, we next performed a scratch wound-healing assay. Treatment with either M3-EV or HSC3-EV promoted significant levels of migration of HSC-3 cells, effectively closing the scratched wound, while M3-EV was a more potent pro-migratory agent than HSC3-EV (Figure 1i,j).
These data indicate that metastatic oral cancer-derived EVs are enriched with HSP90α and HSP90β and promote cancer cell migration.
siRNA-mediated knockdown of CDC37/HSP90α/HSP90β diminishes the EMT phenotype in metastatic oral cancer cells
We have shown that M3-EV tended to be enriched with HSP90α and HSP90β [12]. We also reported that the double-knockdown of HSP90α and HSP90β significantly reduced the survival of metastatic oral cancer cells, although single knockdown of each HSP90 was ineffective in this regard [12]. Next, we examined whether the knockdown of CDC37/HSP90α/β could alter the survival of metastatic oral cancer cells. Single knockdown of each chaperone did not alter the survival of HSC-3-M3 cells, while the triple knockdown of CDC37/HSP90α/β reduced the survival of the metastatic oral cancer cells (Fig. S5). It was thus suggested that triple targeting of the molecular chaperone trio could be antitumorigenic and potentially damage targeted cells.
Increases in cellular migration and motility have been associated with a phenotypic change of the cells from epithelial to mesenchymal properties – EMT [26]. CDC37, a key cochaperone for both paralogs of HSP90, is essential in chaperoning the EMT-promoting kinase signals in cancer cells [28,44]. Moreover, knockdown of CDC37 diminished EV protein release and the EMT properties of prostate cancer cells [10,55]. We therefore next asked whether CDC37 and HSP90α/β could act cooperatively in the EMT process. This experiment involved targeting of CDC37, HSP90α and/or HSP90β and determining the levels of EMT markers in HSC-3-M3. We first confirmed that individual siRNA aimed at either HSP90α, HSP90β or CDC37 could successfully silence the translated protein levels of their mRNA targets in HSC-3-M3 (Figure 2a, top to third raw blot; Fig. S6). However, siRNA-mediated depletion of HSP90α triggered simultaneous increases in HSP90β, while depletion of HSP90β triggered simultaneous increases in HSP90α as well, suggesting a compensatory response within the CDC37/HSP90 system. This observed compensatory response therefore rationalized the use of double and triple depletion of CDC37, HSP90α and HSP90β in this study to effectively neutralize the HSP90/CDC37 chaperone system. Additionally, the siRNA-mediated silencing of HSP90β reduced the level of CDC37, and vice versa, the silencing of CDC37 led to reductions in both CDC37 and HSP90β. Nevertheless, double and triple targeting of CDC37, HSP90α and/or HSP90β successfully diminished the levels of each of the chaperone trio in the metastatic oral cancer cells (Figure 2a, top to third raw blot; Fig. S6).
Figure 2.
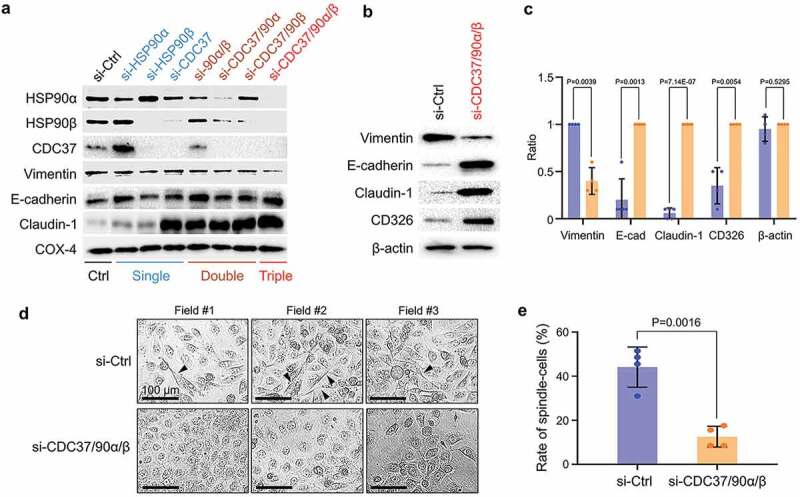
The siRNA-mediated knockdown of CDC37/HSP90α/HSP90β diminishes the EMT phenotype in metastatic oral cancer cells. (a) Representative Western blotting showing HSP90α, HSP90β, CDC37, and EMT markers. HSC-3-M3 cells were transfected with siRNAs targeting CDC37, HSP90α and/or HSP90β or non-targeting siRNA (si-Ctrl) for 48 h. COX4, loading control. Similar data were obtained from two independent experiments. (b–e) Anti-EMT effect of triple chaperone knockdown. HSC-3-M3 cells were transfected with siRNAs targeting CDC37/HSP90α/HSP90β or si-Ctrl. Medium was replaced with serum-free one at 24 h post-transfection. Cells were further cultured 48 h prior to protein sampling and photo taking. (b) Western blotting showing EMT markers. β-actin, loading control. Similar data were obtained from four independent experiments. (c) Alteration of EMT markers. The band intensities of Western blotting were quantified. (d,e) Morphological analysis of EMT. (d) Representative photomicrographs of HSC-3-M3 cells transfected with siRNA. Arrowheads indicate spindle-shaped cells. Scale bars, 100 μm. Similar data were obtained from three independent experiments. (e) Rates of spindle-shaped cells. Spindle shaped cells were defined as the cells with more than two-fold length/width ratio. n = 4 random fields.
Next, we asked whether EMT properties could be altered by the chaperone depletion. The concentration of the most prominent mesenchymal marker vimentin was reduced by either a combination of double depletion or most profoundly by the triple knockdown of the chaperone trio (Figure 2a–c, Fig. S7). The epithelial marker E-cadherin was increased by double depletion of HSP90α/β and by triple chaperone depletion (Figure 2a–c). The most prominent alteration was observed in the levels of claudin-1, an epithelial tight junction component. Claudin-1 was profoundly elevated by either single depletion of CDC37 alone or double/triple depletion of CDC37/HSP90α/β (Figure 2a–c), suggesting that silencing CDC37 could lead to the claudin-1-positive epithelial phenotype. CD326 aka EpCAM has been shown as a type of epithelial marker as well as tumour-initiating cell marker [13,56,57]. In our study, CD326 tended to be increased by the triple knockdown of the chaperones, consistently with other epithelial markers (Figure 2b,c). Thus, triple depletion of CDC37/HSP90α/β reduced the EMT properties of metastatic oral cancer cells more effectively than both single and double chaperone depletion.
We next examined morphological changes in the metastatic oral cancer cells upon chaperone silencing. To evaluate morphological changes coupled with EV release, the culture medium was replaced with serum-free one at 24 h post-transfection and cells were further cultured for 48 h before photo taking and EV sampling. Cell viabilities were not much altered in this protocol between the control and triple knockdown (Fig. S8). The most noticeable difference was observed between control siRNA-transfected and triple-silenced cells. A spindle-like shaped morphology, one of the principal characteristics of EMT, was observed in the control cells, whereas cells significantly reverted to a more typically epithelial morphology upon triple chaperone depletion (Figure 2d,e; Fig. S8).
These findings indicate that the siRNA-mediated triple chaperone knockdown of CDC37, HSP90α and HSP90β could diminish the EMT phenotype in metastatic oral cancer cells.
Triple chaperone knockdown reduced HSP90α, HSP90β and tetraspanins in metastatic EVs
It has been shown that tetraspanins such as CD9 and CD63 play crucial roles in intercellular communication [58–60]. Besides, we recently reported that a single knockdown of CDC37 reduced EV-associated CD9 release from prostate cancer cells [10]. We therefore hypothesized that molecular chaperones could regulate the stability and release of tetraspanins with EVs. We queried whether siRNA-mediated triple depletion of CDC37/HSP90α/β could alter the incorporation of HSP90 and tetraspanins (CD9 and CD63) from cancer cells to EVs. We here used the same protocol of cell culture used in Figure 2d,e and Fig. S8. We observed depletion of HSP90α levels in cell lysates, EV fractions and the EV-free soluble culture supernatant by the triple chaperone depletion (si-Ctrl vs si-CDC37/HSP90α/β) (Figure 3a–c, Fig. S9).
Figure 3.
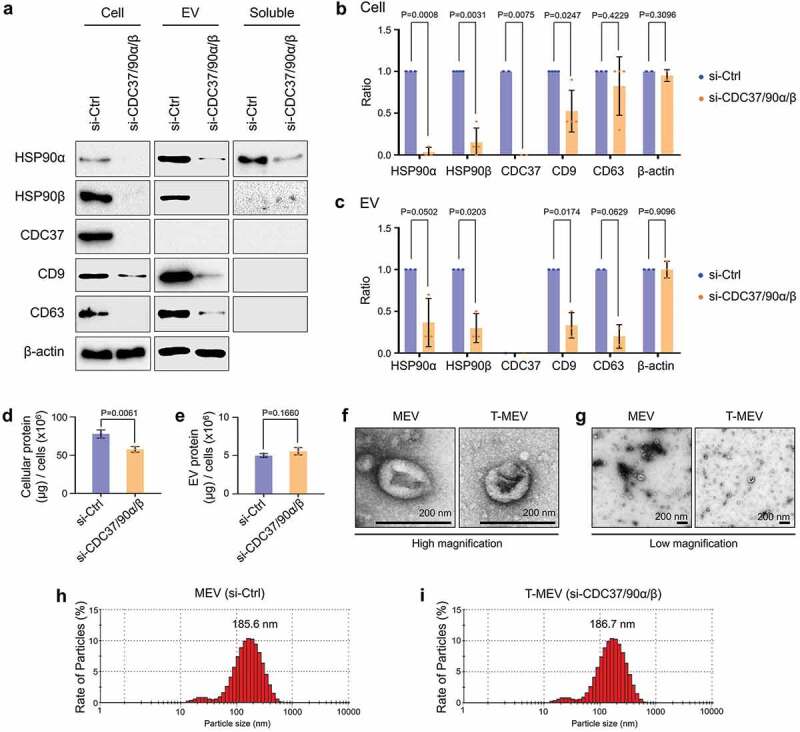
Triple knockdown of CDC37/HSP90α/HSP90β inhibits proteostasis and alters properties of EVs. HSC-3-M3 cells were transfected with non-targeting siRNA (si-Ctrl) or siRNA mixture targeting CDC37, HSP90α, and HSP90β (si-CDC37/90α/β). Medium was replaced with serum-free one at 24 h post-transfection. Cell lysates, EV and non-EV soluble fractions of culture supernatants were collected at 48 h post-medium change. (a) Representative Western blotting showing HSP90, CDC37, CD9, CD63 and β-actin. (b,c) Column scatters plotting to compare protein levels between si-Ctrl vs. si-CDC37/90α/β groups in cells (b) and EVs (c). Relative band intensities of Western blotting were plotted. (d,e) Protein quantification of the whole cell lysates and EV fractions. (d) Total cellular protein levels per 106 cells. n = 3. (e) Total EV protein levels per 106 cells. n = 3. (f,g) Representative TEM images of EV fractions at (f) high and (g) low magnifications. Scale bars, 200 nm. (h,i) Particle diameter distribution of EV fractions from HSC-3-M3 transfected with si-Ctrl (h) or si-CDC37/90α/β (i), called MEV and T-MEV, respectively.
We recently reported that cell stress triggered stressome release including extracellular HSP90α and damaged membrane vesicles [10]. It is conceivable that siRNA transfection could also lead to membrane damage and the release of cytosolic proteins and intra-EV cargos. The release of EV-free HSP90α was inhibited by the triple knockdown, while extracellular β-actin was increased in the soluble fraction of the triple chaperone knockdown compared to the control (Fig. S9).
The depletion of HSP90β was also confirmed in the cellular and EV fractions by the triple chaperone targeting, although HSP90β was not found in the EV-free culture supernatant (Figure 3a–c, Fig. S9). These data were consistent with previous reports showing that tumour-derived EVs could often carry HSP90α and HSP90β as cargo [10,12] while secretion of EV-free extracellular HSP90α was also observed [10,13,61].
CDC37 was reduced in metastatic oral cancer cells by triple chaperone silencing, although this chaperone was not found in the extracellular fractions, consistent with our recent report [10]. Notably, tetraspanins CD9 and CD63 were reduced by the triple chaperone silencing in metastatic oral cancer-derived EVs (Figure 3a–c).
These data indicated that CDC37/HSP90 might play a crucial role in the stability and vesicular incorporation of tetraspanins CD9 and CD63.
As CDC37 and HSP90 have been shown to regulate proteostasis [10,30], we next examined whether protein levels in HSC-3-M3 cells and their EVs were altered by triple chaperone depletion compared to the control. Indeed, triple chaperone depletion significantly reduced overall protein levels in the cells, although not altering the EV protein level (Figure 3d,e). These data suggest that reduction in CDC37/HSP90 may increase cellular proteolysis of unfolded proteins in the cytoplasm, although effects on protein synthesis and maturation may also be involved.
Hereafter, we define EVs derived from control siRNA-transfected HSC-3-M3 cells as MEV, and EVs derived from triple siRNA (CDC37/HSP90α/β) transfected HSC-3-M3 cells as T-MEV. Next, we compare the different properties of MEV and T-MEV. The morphologies and sizes of EVs were similar between MEVs and T-MEVs. MEVs and T-MEVs had cup-like morphologies (Figure 3f,g; Fig. S10). The main populations of MEV and T-MEV sized between 50 and 400 nm with a peak at approx. 185 nm, while minor smaller particles (20–30 nm) were detected (Figure 3h,i; Fig. S10).
These data suggest that the chaperone trio CDC37/HSP90α/β could be crucial in intracellular proteostasis and extracellular release of EV proteins, including tetraspanins and thereby triple chaperone knockdown altered proteome signature including CD9 and CD63 in the cells and EVs.
Triple chaperone knockdown reduced Gluc-EV release
We recently showed that cell stress triggers the release of stressome including EVs, HSP90 and LDH, a marker of cell membrane damage, from cancer cells [10]. CDC37 was also induced intracellularly by cell stress [10]. We therefore hypothesize that the chaperone trio is involved in membrane damage-related release of EVs.
Moreover, a previous study reported that Gaussia princeps-derived luciferase (Gluc) fused with PDGFR transmembrane domain was anchored to the lipid-bilayered membrane and then released from producer cells [48]. To quantify the release of EVs, we established a stable cell line designated as HSC3 M3/mbGluc that produce membrane-bound Gluc (mbGluc) (Figure 4a,b). The morphology of vesicles released by the HSC3 M3/mbGluc cells was cup-shaped and sized between 50 and 200 nm (Figure 4c, Fig. S11). Gluc activity was detectable and quantitative in the culture supernatant of HSCM3/mbGluc cells (Figure 4d,e). mbGluc was released from HSC-3-M3/mbGluc cells into culture media in a time-dependent manner at 6, 24 and 48 h culture period (Figure 4f). Triple targeting of CDC37/HSP90α/β lowered the release of mbGluc at 6–24 h post-medium replacement (Figure 4f). These data indicate that the chaperone trio is involved in EV release.
Figure 4.
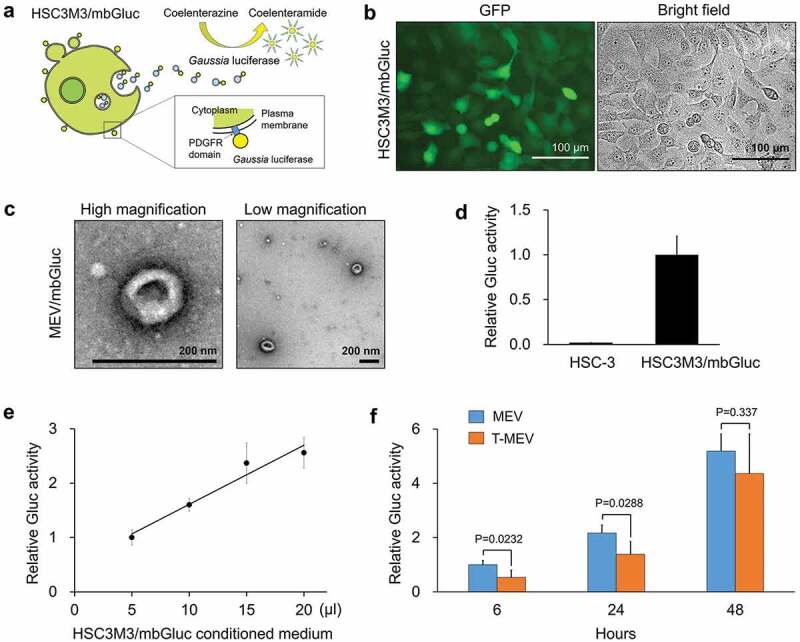
Triple knockdown of CDC37/HSP90α/β inhibited the release of membrane-bound Gaussia luciferase (mbGluc) reporter. The expression construct of mbGluc fused with the PDGFR transmembrane domain was stably transfected to HSC-3-M3 cells. GFP was co-expressed by an internal ribosomal entry sequence (IRES) for labelling the donor cells designated as HSC3M3/mbGluc. (a) Schemes showing a concept of Gluc-labelled EVs. (b) Representative images of HSC3M3/mbGluc cells. Left, an image of GFP-expressed HSC3M3/mbGluc cells. Right, a bright-field image. Scale bars, 100 μm. (c) A representative TEM images of mbGluc-MEV. Scale bars, 200 nm. (d, e) Establishment of mbGluc quantification. (d) Relative Gluc activities of 20 μL culture media of HSC-3 (as negative control) and HSC3M3/mbGluc cells. (e) A standard curve of Gluc activities. Culture media were prepared at 48 h after the medium change. (f) Relative Gluc activities of MEV and T-MEV. HSC3M3/mbGluc cells were transfected with control siRNA or si-CDC37/HSP90α/β for 24 h and then culture media were replaced with fresh ones. Gluc activities in the culture supernatants were measured at 6, 24 and 48 h after the medium changes. Values were normalized with the number of cells. n = 3.
The total protein concentrations of the EV fractions were not different between the triple knockdown versus the control, as shown in Figure 3e. However, mbGluc release was delayed by the triple knockdown. Therefore, it is conceivable that EVs containing mbGluc, HSP90, CD9 and CD63 were reduced by the triple knockdown, while other components must be increased instead.
We also examined whether cellular growth, viability, and damage were altered by the triple knockdown of CDC37/HSP90α/β in HSC3 M3/mbGluc cells. Cellular growth and viability were not altered between the triple siRNA-transfected cells and control cells (Fig. S12a–d), while LDH release was inhibited by the triple knockdown of CDC37/HSP90α/β (Fig. S12e–g).
These data indicated that triple knockdown of CDC37/HSP90α/β could lower the release of EVs from cancer cells.
Triple depletion of CDC37/HSP90α/HSP90β attenuated cancer EV-driven malignancy events
We next examined the effects of MEV and T-MEV on properties associated with processes involved in malignant progression, including the initiation of EMT in normal epithelial cells, advanced migration, invasion and tumorigenesis of oral cancer cells. MEV treatment of epithelial RT7 cells significantly increased the number of spindle-shaped cells compared to the PBS-treated control, whereas the EMT-initiating properties of MEV were significantly reduced by triple chaperone depletion (Figure 5a,b; Fig. S13). To confirm the alteration of EMT at the level of protein markers, we performed western blot analysis. Claudin-1 and E-cadherin were markedly reduced by MEV treatment of the epithelial cells compared to the PBS-treatment control, whereas triple chaperone depletion prevented the reduction in claudin-1 and E-cadherin (Figure 5c; top and second raw blot; Fig. S14). Consistent with the reduced epithelial properties, a representative mesenchymal marker protein, vimentin was increased by the MEV treatment (Figure 5c; third raw blot). Meanwhile, Snail, a protein whose stability is important for the establishment of the EMT phenotype was increased by the MEV treatment, although also increased by T-MEV treatment (Figure 5c; fourth raw blot). β-catenin, whose activity is also important in the induction of EMT, was also increased by the MEV treatment of the epithelial cells (Figure 5c; bottom second blot). Triple chaperone depletion inhibited MEV-triggered increases in vimentin and β-catenin. Therefore, these data indicate that: (i) metastatic oral cancer-derived MEV was a potent inducer of EMT in epithelial cells and (ii) triple chaperone depletion effectively reduced EMT-initiating activity of metastatic EVs.
Figure 5.
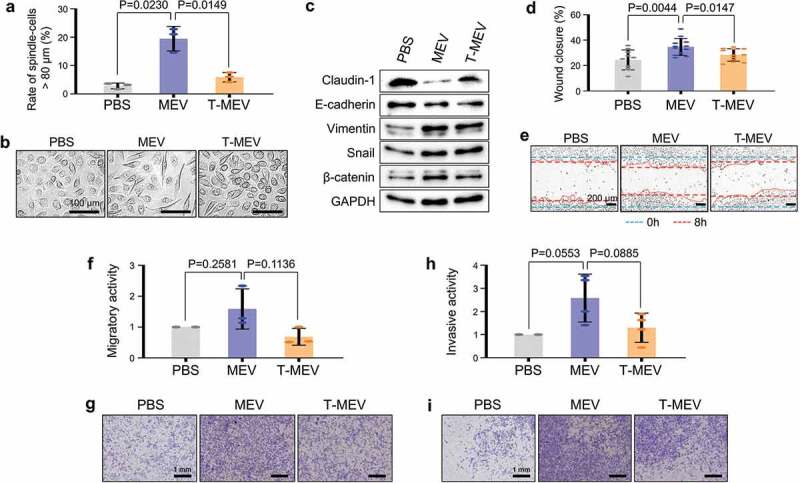
Triple depletion of CDC37/HSP90α/HSP90β inhibited EV-driven malignancy events. (a-c) EMT levels altered by MEV and T-MEV. Epithelial cell line RT7 was treated with MEV, T-MEV or PBS. (a) Rate of spindle-shaped cells longer than 80 μm. n = 3. (b) Representative images of RT7 cells. Scale bars, 100 μm. (c) Western blotting showing EMT markers. (d,e) Wound healing assay. HSC-3 cells were treated with MEV, T-MEV or PBS. (d) Rate of wound closure. n = 12. (e) Representative photomicrographs of closing wounds. --- (blue) initial wounds. --- (red) closed wounds at 8 h. Scale bars, 200 μm. (f,g) Migration assay using a transwell system. (f) Migration activities of HSC-3 cells treated with MEV, T-MEV or PBS. n = 3. (g) Representative images of migrated cells. Cells were stained with Diff-Quik. Scale bars, 1 mm. (h,i) Invasion assay using Transwell system. (h) Invasion activities of HSC-3 cells treated with MEV, T-MEV or PBS. n = 3. (i) Representative images of invaded cells. Cells were stained with Diff-Quik. Scale bars, 1 mm.
We next hypothesized that the EMT-inducing activity of MEV could potentially lead to augmented migration, invasion, and tumorigenesis of oral cancer cells. MEV promoted migration of HSC-3 cells as demonstrated by the scratch wound healing assay and by the chamber-well migration assay (Figure 5d–i). The depletion of the chaperone trio inhibited the MEV-driven migration of oral cancer cells (Figure 5d–g, MEV vs. T-MEV). MEV also promoted oral cancer cells to invade through matrices, whereas triple chaperone depletion attenuated the MEV-driven invasion (Figure 5h,i).
These data indicate that MEV initiates EMT in oral epithelial cells, an event attenuated by triple chaperone depletion. Additionally, MEV enhanced migration and invasion of oral cancer cells, events inhibited by triple chaperone depletion.
Triple depletion of CDC37/HSP90α/HSP90β attenuated cancer EV-driven tumorsphere formation
We have originally developed a 3D culture system for the evaluation of in vitro tumorigenesis with enhanced expression of cell adhesion molecules such as E-cadherin and CD326/EpCAM and cancer stem cell markers [13]. Next, we determined whether MEV could promote in vitro tumorigenesis and asked if depletion of the chaperone trio could cancel the MEV-driven tumorsphere formation. As TGFβ pro-migratory activity can promote intercellular attachment leading to sphere formation [62], we used TGFβ-promoting tumorsphere formation as a positive control. Tumorsphere formation by HSC-3 cells was significantly enhanced by MEV treatment (50 μg/mL) and by TGFβ treatment (5 ng/mL) compared to the PBS-treated control (Figure 6a,b; Fig. S15). Triple chaperone depletion significantly lowered the tumorsphere forming potential of MEV (Figure 6a,b; MEV vs. T-MEV). Moreover, T-MEV significantly inhibited tumorsphere formation compared to the control. The number of tumorspheres was significantly less in the T-MEV group than that in the MEV group (Figure 6c).
Figure 6.
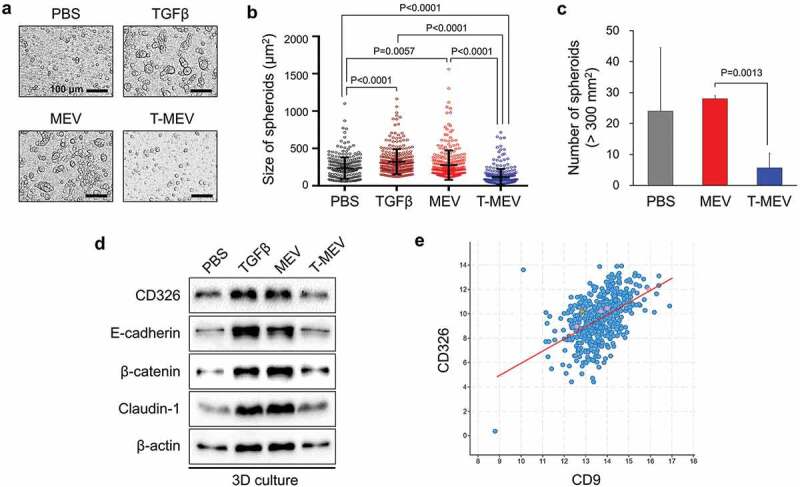
Metastatic EVs promote tumorsphere formation, a property diminished by triple chaperone knockdown. (a–d) In vitro tumorigenesis assay in the three-dimensional (3D) culture system. HSC-3 cells were treated with MEX (50 μg/mL), T-MEX (50 μg/mL), TGFβ (5 ng/mL) or PBS in nano-culture plates and then (a–c) tumorsphere formation and (d) protein expression levels were examined. (a) Representative photomicrographs of tumorspheres. Scale bars, 100 μm. (b) Column scatter plotting showing tumorsphere size. The size of the top 300 tumorspheres was measured in each group using ArrayScan HCS System. The horizontal lines indicate mean ± SD analysed by ANOVA Turkey’s multiple comparisons test. (c) The number of tumorspheres altered by MEX or T-MEX. Tumorspheres larger than 200 mm2 were counted. n = 3, P = 0.0201. (d) Western blotting showing CD326/EpCAM, E-cadherin, β-catenin, claudin-1 and β-actin. The cell lysates were prepared from the tumorspheres. (e) Scatter plot showing co-expression correlation between CD326/EpCAM and CD9 in head and neck squamous cell carcinoma (HNSCC). A regression line was shown in red. Data were represented as log2. The data set of HNSCC with 523 patient-derived 523 samples (TCGA, Pan-Cancer Atlas) was analysed.
Some aspects of tumorigenesis, as well as tumorsphere formation, may require intercellular adhesion by CD326/EpCAM and E-cadherin and tight junctions formed by claudins [13,63,64]. Moreover, MEV tended to be enriched with CD326, as indicated in Figure 1. We therefore next asked whether the addition of MEV, T-MEV or TGFβ could alter CD326, E-cadherin, β-catenin and claudin-1 in tumorspheres. The addition of MEV or TGFβ stimulation increased levels of CD326, E-cadherin, β-catenin and claudin-1 in tumorspheres, while T-MEV tended to show lesser effects (Figure 6d, Fig. S16). Consistently, co-expression correlation of CD326/EpCAM and tetraspanin CD9 was found in HNSCC clinical cases with Spearman’s correlation score 0.51 (p = 1.43e−33), Pearson’s correlation score 0.50 (p = 1.49e−32), and a regression line (y = 0.99x±3.91) (Figure 6e), suggesting that CD326/EpCAM-related tumour-initiating activity could be involved in tetraspanins-bearing EV transmission.
These findings indicate that metastatic oral cancer-derived EVs increase cancer malignancy by promoting EMT, migration, invasion and tumorigenesis of oral cancer cells, whereas the siRNA-mediated triple depletion of CDC37/HSP90α/β could inhibit the MEV-driven oral cancer malignancy progression.
HSP90α is dominantly expressed in cancer cells, while HSP90β is accumulated in tumour-associated macrophages in metastatic oral cancer
To examine the expression and tissue localization of HSP90α and HSP90β in the tumour microenvironment, we next performed immunohistochemistry (IHC) on tumour specimens derived from patients suffering from stage IV (with metastasis) vs. stage I (without metastasis) oral squamous cell carcinoma. The HSP90α-positive cancer cell rate in the stage IV group was significantly higher than that in stage I (Figure 7a,b; Fig. S17). HSP90α-positive cells were also found in the stroma, although the levels were not significantly different between stage I and stage IV (Figure 7c). In contrast, the HSP90β-positive cancer cell rate in the stage I group was significantly higher than that in stage IV (Figure 7a,d). However, the HSP90β-positive stromal cell rate in stage IV oral cancer group was significantly higher than that in stage I (Figure 7a,e; Fig. S17). Additionally, we found that HSP90β-positive round-shaped cells and dendritic cells were infiltrating into tumours in stage IV oral cancer and were morphologically reminiscent of macrophages (Figure 7f, arrowheads).
Figure 7.
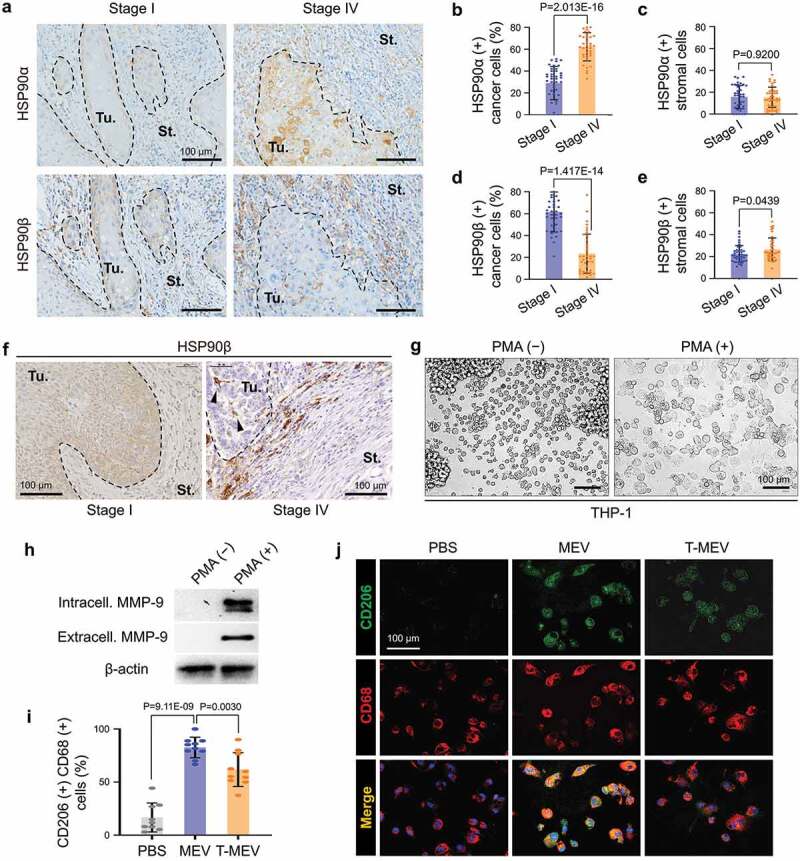
Metastatic oral cancer-derived MEVs induced macrophages M2 polarization. (a–f) Immunohistochemistry (IHC) of oral cancer patient-derived specimens. (a) Representative IHC showing HSP90α and HSP90β in stage I vs. stage IV oral cancers. Tu, tumour. St, stroma. --- borders between tumour and stroma. (b–e) Rate of (b,c) HSP90α+ or (d,e) HSP90β+ cancer cells or stromal cells. The rates were calculated by counting immuno-positive cells per total cell number in each random field in (b,d) tumour areas or (c,e) 100 mm2 stromal areas. Five random fields each in 8 oral cancer cases. N = 40. (f) IHC for HSP90β in stage I vs. stage IV oral cancer. Arrowheads, HSP90β-positive infiltrating tumour-associated macrophages (TAM). (g–j) Macrophage M2 polarity was altered by MEV and T-MEV. THP-1 cells were stimulated with PMA for the differentiation to macrophages and then treated with MEV, T-MEV (25 μg/mL) or PBS. (g) Representative images of THP-1 cells treated with or without PMA for 24 h. (h) Western blotting of extracellular and intracellular MMP-9 in THP-1 cells with or without PMA treatment. (i,j) Immunocytochemistry of macrophage markers with or without MEV or T-MEV. (i) Rate of CD206/CD68 double-positive M2-like macrophages. N = 9. (j) Representative staining images of CD206 (green) and CD68 (red). Blue, DAPI. Scale bars, 100 μm.
These data suggested that HSP90 abundantly released by cancer cells could be taken TAM.
Metastatic EVs induce macrophages M2 polarization
Next, we asked whether macrophage M2 polarization could be altered by metastatic oral cancer-derived MEVs. As it has been shown that pro-tumorigenic metalloproteinase MMP9 was produced by M2 macrophages [65], we queried whether PMA and MEV could alter the MMP9 levels and release by macrophages. The monocytic cell line THP-1 was first differentiated to macrophage-like cells by stimulation with the phorbol ester PMA before EV treatment. THP-1 cells were morphologically monocytic and non-adherent to tissue culture plates without PMA stimulation while the differentiated macrophage-like THP-1 cells adhered to tissue culture plates after the PMA treatment (Figure 7g, Fig. S18). Moreover, MMP9 was detectable in the PMA-treated THP-1, although not detectable without PMA treatment (Figure 7h). MMP9 secretion by THP-1-derived macrophages was enhanced by the MEV treatment, compared to PBS-treated control cells. Triple chaperone targeting of HSC-3-M3 reduced the MEV-driven MMP9 production by the macrophage-like cells (Fig. S19). The treatment with MEV tended to increase HSP90β in the macrophage-like cells, while T-MEX treatment showed a lesser effect.
To query whether the exposure to MEV could alter macrophage polarity, we next examined the expression of CD206, an M2 polarity marker, and CD68, a general macrophage marker. CD206/CD68 double-positive macrophages were significantly increased by the MEV treatment compared to PBS-treated control cells (Figure 7i,j; Fig. S20). Triple depletion of CDC37/HSP90α/β in HSC-3-M3 reduced the MEV-driven macrophage M2 polarization (Figure 7i,j; MEV vs. T-MEV).
These data show that metastatic oral cancer-derived EVs induce macrophage M2 polarization, a phenotype inhibited by triple depletion of CDC37/HSP90α/HSP90β. In addition, MEV-derived HSP90β could accumulate in macrophages and was involved in the M2 polarization.
Triple depletion of CDC37/HSP90α/HSP90β lowered EV transmission potentials
Next, we compared the transmission potentials of MEV and T-MEV to recipient cells. We have established and tested two methods of the EV labelling: (i) labelling EVs with red-fluorescent sphingolipid [8,45] and (ii) establishment of stable cells that produced membrane-bound palmitoylation signal-linked GFP or tdTomato red-fluorescent proteins [46]. Using the first method, we found that the transmission potential of T-MEV was significantly lower than that of MEV at 1.5 and 3 h after the EV addition to HSC-3 cells, although it was not significant at 6 and 12 h, indicating that there was a time-dependent reduction of the rate of transmission of EVs (Figure 8a,b).
Figure 8.
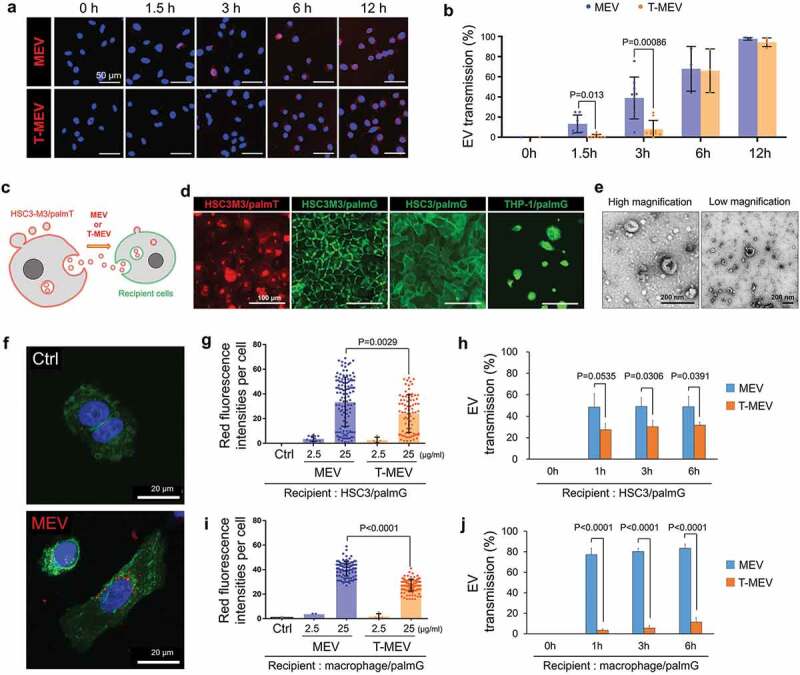
Triple knockdown of CDC37/HSP90α/HSP90β attenuates EV transmission potential into recipient macrophages and cancer cells. (a,b) Transmission of MEV and T-MEV into recipient HSC-3 cells. MEV was prepared from the culture supernatant of HSC-3-M3 transfected with si-Ctrl. T-MEV was prepared from the culture supernatant of HSC-3-M3 transfected with si-CDC37/90α/β. MEV and T-MEV were labelled with red-fluorescent ceramide and added to the culture media of HSC-3 cells. Cells were fixed at the indicated incubation period and analysed under fluorescent microscopy. (a) Representative images of vesicular transmission. Scale bars, 50 μm. Blue, DAPI staining. (b) The rate of EV transmission per total cells. N = 3. (c-j) Monitoring EVs labelled with membrane-bound palmitoylation (palm) signal-fused fluorescent proteins. (c) A scheme of the concept of fluorescent EV transmission. (d) Representative fluorescent images of HSC3M3/palmT, HSC3M3/palmG, HSC3/palmG and THP1/palmG cells. THP1/palmG was stimulated with PMA. Scale bars, 100 μm. (e) Representative TEM images of MEV released by HSC3M3/palmT cells. Scale bars, 200 nm. (f) Confocal laser scanning microscopy of HSC3/palmG cells treated with or without palmT-MEV. MEV (red) were prepared from culture media of HSC3 M3/palmT cells and added to the culture media of HSC3/palmG cells (green). Blue, DAPI staining. Scale bars, 20 μm. (g–j) MEV transmission rate into (g,h) HSC3/palmG cells or (i,j) THP1/palmG macrophages. For macrophage differentiation, THP1/palmG cells were stimulated with PMA. (g,i) Column scatters plotting of MEV transmission. The values indicate palmT fluorescence intensity per cell at 1 h after the addition of MEV or T-MEV. The upper end of each column indicates mean. (h,j) Transmission efficiencies of MEV vs. T-MEV into (h) HSC3/palmG cells and (j) THP1/palmG-derived macrophages. n = 3.
A previous study had reported that GFP, as well as tdTomato RFP, fused with protein palmitoylation signal (designated palmtdTomato and palmGFP, respectively), were associated with the EV membrane, thus being released from producer cells into the culture supernatant [46,47]. To monitor the transmission of EVs, we established palmtdTomato-expressed HSC-3-M3 cells (designated HSC3M3/palmT), palmGFP-expressed HSC-3-M3 cells (designated HSC3M3/palmG), palmGFP-expressed HSC-3 cells (designated HSC3/palmG) and palmGFP-expressed THP-1 cells (designated THP1/palmG) (Figure 8c,d; Fig. S21). We collected EVs (palmT-MEV) from the culture supernatant of HSC3M3/palmT cells (Figure 8e). We then confirmed that the palmT-MEV were produced from HSC3M3/palmT cells and then being transmissive into HSC3M3/palmG cells, under CLSM (Figure 8f; Fig. S22a,b). Dose-dependent transmission of EVs was found in palmT-labelled MEV and T-MEV (Figure 8g). Triple targeting of CDC37/HSP90α/β significantly lowered the transmission of the palmT-labelled MEV into the recipient HSC3/palmG cells at 1, 3, and 6 h after the addition of the EVs (MEV vs. T-MEV) (Figure 8g,h; Fig. S22 c). To monitor the transmission of palmT-MEV into macrophages, THP1/palmG cells were differentiated into macrophage-like cells with PMA. Triple targeting of CDC37/HSP90α/β significantly lowered the uptake of the palmT-MEV by macrophages (Figure 8i,j; Fig. S22d). T-MEV uptake by macrophages was significantly lower than MEV and this lasted at least 24 h after the addition of EVs (Fig. S22e).
Thus, triple depletion of CDC37/HSP90α/HSP90β in metastatic oral cancer cells inhibited cancer vesicular transmission to recipient cells such as macrophages.
Discussion
Our data indicate the potency of metastatic tumour-derived EVs in changing the tumour microenvironment into a more pro-malignant state. We demonstrate that oral cancer-derived EVs possess the potential to initiate epithelial cell transformation – EMT, to promote the tumorigenic, migratory and invasive abilities of oral cancer cells and to polarize macrophages into an M2 phenotype. To potentially target these pro-malignant events, our data, for the first time, indicate the validity of triple depletion of the interacting CDC37/HSP90α/HSP90β complex (Figure 9, graphical abstract). siRNA-mediated triple depletion of CDC37/HSP90α/β significantly diminished the pro-malignant activities of oral cancer cells and MEVs. Although earlier studies had shown that intracellular HSP90 promotes cancer progression, indicating its targeting by small molecule inhibitors in clinical trials, we have uncovered novel roles for the co-chaperone CDC37 in cancer [10,14,28–30,44,55]. Notably, our data indicate that individual depletion of either HSP90α, HSP90β or CDC37 triggers simultaneous increases in the other chaperones. This observed compensatory response within the CDC37/HSP90 system was the rationale behind our original approach of depleting the three chaperones. Indeed, our data indicated that cancer-initiation and EMT markers were attenuated by the triple chaperone depletion more profoundly than double depletion. Consistently, our recent study showed that triple depletion of the chaperone trio inhibited tumorigenesis of prostate cancer cells in vivo [10]. As a potential mechanism, a recent study showed that directional cell movement through tissues is controlled by EV secretion [11,66]. Consistently, our data indicate that HSP90-high oral cancer-EVs promote migration-dependent tumorigenesis of cancer cells; triple chaperone depletion significantly diminished the pro-migratory activities of EVs. Thus, our studies could offer a potential solution to the pro-migratory and pro-tumorigenic activities of cancer EVs.
Figure 9.

Graphical abstract. (a) Metastatic oral cancer cells secreted HSP90-high EVs transmitting to recipient cells. Metastatic oral cancer-EVs initiated EMT in normal epithelial cells and promoted migration, invasion, and tumorigenesis in oral cancer cells and M2-like polarity in tumour-associated macrophages. (b) The siRNAs-mediated triple silencing of CDC37/HSP90α/HSP90β diminished EMT markers in oral cancer cells. The HSP90-depleted EVs-reduced activities for transmission and pro-malignancy in recipient cells.
Our studies also, for the first time, indicate a potent role of HSP90-rich EVs in trans-cellular communication in the tumour microenvironment. It has been shown that cancer cell-derived EVs induce macrophage M2 polarity in breast cancer [67], colon cancer [37], pancreatic cancer [38], ovarian cancer [68], brain cancer [69], and head and neck cancer [42]. It was also reported that TAM showed an M2-type polarity in oral cancer and the number of such infiltrating TAM was correlated with poor prognosis [70]. Our studies were consistent with these reports, although, in oral cancer patient-derived tumours, we found that HSP90β-positive TAMs were profoundly associated with and infiltrating into the tumours. Distinctively, HSP90α might be processed during phagocytosis and antigen presentation, as HSPs play key roles in capturing tumour antigens and permitting their uptake and processing by antigen-presenting cells prior to activation of cytotoxic lymphocytes [17,71–74]. Relevantly, it has been shown that complexes of antigenic peptides with HSP90, calreticulin, and HSP70 were taken up by macrophages and dendritic cells and re-presented by MHC class I molecules [73,75]. Importantly, HSP90 mediates efficient antigen cross-presentation through scavenger receptors such as SCARF1 and LOX-1 [76,77]. Moreover, circulating EVs containing HSP90 were demonstrated to mediate the activation of macrophages and Kupper cells in alcoholic liver disease [78]. Our current study indicated that loss of HSP90α/β significantly inhibited EV uptake by macrophages. Thus, it is conceivable that HSP90 is crucial for EV uptake, activation and M2-type polarization of macrophages. Our findings support one of our conclusions, namely that metastatic oral cancer releases HSP90-rich EVs leading to polarization of macrophages to an M2 phenotype, while triple chaperone knockdown is effective in inhibiting EV uptake by TAM and their M2 polarization.
Our studies also suggest that tetraspanins (CD9 and CD63) might be crucial mediators for cell-cell communication in tumour progression and programming of the microenvironment. It has been shown that tetraspanins are crucial for intercellular communications required for cancer progression [64,79,80], while CD9 and CD63 are exosome markers and potentially contained in other EV types [1]. In our study, the total protein concentrations of the EV fractions were not different between the triple knockdown versus the control, while EVs that contained HSP90, CD9, CD63 and mbGluc appeared to be reduced by the triple chaperone knockdown, also suggesting that the number of EVs (or exosomes) that contained HSP90, CD9 and CD63 might be reduced by this targeting, while the other components must be increased instead. Such a loss of tumour-promoting EVs can be a mechanism by which triple chaperone knockdown effectively inhibits protumorigenic events and EV uptake by macrophages.
In conclusion, we have demonstrated that the siRNA-mediated triple silencing of CDC37/HSP90α/HSP90β could potentially lead to a novel therapeutic strategy for attenuating cancer EV-driven macrophage M2 polarization, EMT, migration, invasion and tumorigenesis. We also unveiled the novel finding that the triple chaperone depletion weakens the EV transmission activity required for pro-tumorigenic cell-cell communication in the tumour microenvironment.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of one of our mentors, Professor Ken-ichi Kozaki, who passed away on 29 May 2016. The authors thank Haruo Urata for the operation of TEM, Tomonari Kasai and Masaharu Seno for particle size analysis using ELS-8000, Kazuko Kobayashi for the operation of Zetasizer, Eriko Aoyama for maintenance of HCS analyzer. The authors thank Manabu Itoh and Kazuya Arai at JSR for technical advice in NCP. The authors thank Keisuke Nakano for illuminating discussion and encouraging support.
Author contributions
TE, KOn and SKC conceptualized and administrated the study. TE acquired funding. HN, AS, KK, MT, KOk, HO, SI and TE prepared resources. TE, CS, MTT, HK, KOn, KK, HO and KK devised methodology. KOn, HK, MTT, EAT, YL, YO, CS and MWO performed the experimentation. KOn, HK, TE, MTT and CS curated, validated and visualized data. TE, CS, KOk, HO and KK supervised. TE, KOn and HK wrote the original draft. TE and SKC reviewed and edited, and revised the manuscript. All authors reviewed the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Supplementry material
Supplemental data for this article can be accessed here.
References
- [1].Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Colombo M, Raposo G, Thery C.. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–21. [DOI] [PubMed] [Google Scholar]
- [3].van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018. April;19(4):213–228. [DOI] [PubMed] [Google Scholar]
- [4].Seyama M, Yoshida K, Yoshida K, et al. Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochim Biophys Acta Mol Basis Dis. 2020. February 20;1866(6):1–12. [DOI] [PubMed] [Google Scholar]
- [5].Okusha Y, Eguchi T, Tran MT, et al. Extracellular vesicles enriched with moonlighting metalloproteinase are highly transmissive, pro-tumorigenic, and trans-activates cellular communication network factor (CCN2/CTGF): CRISPR against cancer. Cancers (Basel). 2020;12(4):1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015. November 19;527(7578):329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Franzen CA, Blackwell RH, Todorovic V, et al. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis. 2015. August;17(4):e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fujiwara T, Eguchi T, Sogawa C, et al. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018;86:251–257. [DOI] [PubMed] [Google Scholar]
- [9].Clayton A, Turkes A, Navabi H, et al. Induction of heat shock proteins in B-cell exosomes. J Cell Sci. 2005. August 15;118(Pt 16):3631–3638. [DOI] [PubMed] [Google Scholar]
- [10].Eguchi T, Sogawa C, Ono K, et al. Cell stress induced stressome release including damaged membrane vesicles and extracellular HSP90 by prostate cancer cells. Cells. 2020;9(3):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Eguchi T, Ono K, Kawata K. Regulatory roles of HSP90-Rich extracellular vesicles In: Asea AAA and Kaur P, editors. heat shock protein 90 in human diseases and disorders. Heat shock proteins. Vol. 19 Cham, Swizerland: Springer Nature; 2019. p. 3–17. [Google Scholar]
- [12].Ono K, Eguchi T, Sogawa C, et al. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J Cell Biochem. 2018 Sep;119(9):7350–7373. [DOI] [PubMed] [Google Scholar]
- [13].Eguchi T, Sogawa C, Okusha Y, et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. Plos One. 2018. February 7;13(2):e0191109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Calderwood SK. Molecular cochaperones: tumor growth and cancer treatment. Scientifica (Cairo). 2013;2013:217513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8(4 Suppl):S55–61. [DOI] [PubMed] [Google Scholar]
- [16].Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005. October;5(10):761–772. [DOI] [PubMed] [Google Scholar]
- [17].Taha EA, Ono K, Eguchi T. Roles of extracellular HSPs as biomarkers in immune surveillance and immune evasion [Review]. Int J Mol Sci. 2019. September 17;20(18):1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eustace BK, Sakurai T, Stewart JK, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004. 6;Jun(6):507–514. [DOI] [PubMed] [Google Scholar]
- [19].Nolan KD, Franco OE, Hance MW, et al. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem. 2015. March 27;290(13):8271–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hance MW, Dole K, Gopal U, et al. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J Biol Chem. 2012. November 02;287(45):37732–37744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009. June;28(1–2):15–33. [DOI] [PubMed] [Google Scholar]
- [22].Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008. May 16;133(4):704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fujiwara T, Eguchi T, Sogawa C, et al. Anti-EGFR antibody cetuximab is secreted by oral squamous cell carcinoma and alters EGF-driven mesenchymal transition. Biochem Biophys Res Commun. 2018;503(3):1267–1272. 2018 Available online 13 July. [DOI] [PubMed] [Google Scholar]
- [24].Eguchi T, Taha EA, Calderwood SK, et al. A novel model of cancer drug resistance: oncosomal release of cytotoxic and antibody-based drugs [Review]. Biology (Basel). 2020. March 5;9(3):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015. November 26;527(7579):525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nieto MA, Huang RY, Jackson RA, et al. Emt: 2016. Cell. 2016. June 30;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- [27].Blackwell RH, Foreman KE, Gupta GN. The role of cancer-derived exosomes in tumorigenicity & epithelial-to-mesenchymal transition. Cancers (Basel). 2017. 10 9(8):Aug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gray PJ, Prince T, Cheng J, et al. Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer. 2008;8:491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gray PJ Jr., Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007. December 15;67(24):11942–11950. [DOI] [PubMed] [Google Scholar]
- [30].Calderwood SK. Cdc37 as a co-chaperone to Hsp90. Subcell Biochem. 2015;78:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kimura Y, Rutherford SL, Miyata Y, et al. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997;11:1775–1785. [DOI] [PubMed] [Google Scholar]
- [32].Roe SM, Ali MMU, Meyer P, et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50 cdc37. Cell. 2004;116:87–98. [DOI] [PubMed] [Google Scholar]
- [33].Chua HL, Bhat-Nakshatri P, Clare SE, et al. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007. February 01;26(5):711–724. [DOI] [PubMed] [Google Scholar]
- [34].Wu Y, Deng J, Rychahou PG, et al. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009. May 05;15(5):416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li CW, Xia W, Huo L, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012. March 01;72(5):1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011. October 14;11(11):723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lan J, Sun L, Xu F, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019. January 1;79(1):146–158. [DOI] [PubMed] [Google Scholar]
- [38].Wang X, Luo G, Zhang K, et al. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kgamma to promote pancreatic cancer metastasis. Cancer Res. 2018. August 15;78(16):4586–4598. [DOI] [PubMed] [Google Scholar]
- [39].Zheng P, Chen L, Yuan X, et al. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017. April 13;36(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Taha EA, Lu Y, Eguchi T. Exosomes“The little thing that matter the most”in tumor immunology and microenvironment In: kagiya T, editor. Exosomes and microvesicles: role in disease and clinical applications. Hauppauge, New York: Nova Science Publishers; 2020. [Google Scholar]
- [41].Wang F, Li B, Wei Y, et al. Tumor-derived exosomes induce PD1(+) macrophage population in human gastric cancer that promotes disease progression. Oncogenesis. 2018. May 25;7(5):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pirila E, Vayrynen O, Sundquist E, et al. Macrophages modulate migration and invasion of human tongue squamous cell carcinoma. PLoS One. 2015;10(3):e0120895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ono K, Eguchi T, Sogawa C, et al. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J Cell Biochem. 2018. May 16;119(9):7350–7362. [DOI] [PubMed] [Google Scholar]
- [44].Eguchi T, Prince TL, Tran MT, et al. MZF1 and SCAND1 Reciprocally Regulate CDC37 Gene Expression in Prostate Cancer. Cancers (Basel). 2019. June 8;11(6):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Namba Y, Sogawa C, Okusha Y, et al. Depletion of lipid efflux pump ABCG1 triggers the intracellular accumulation of extracellular vesicles and reduces aggregation and tumorigenesis of metastatic cancer cells. Front Oncol. 2018;8(376):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Taha EA, Sogawa C, Okusha Y, et al Knockout of MMP3 weakens solid tumor organoids and cancer extracellular vesicles. Cancers, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lai CP, Kim EY, Badr CE, et al. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun. 2015. May;13(6):7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lai CP, Mardini O, Ericsson M, et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano. 2014. January 28;8(1):483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Eguchi T, Kubota S, Kawata K, et al. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol Cell Biol. 2008. April;28(7):2391–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Arai K, Eguchi T, Rahman MM, et al. A novel high-throughput 3D screening system for EMT inhibitors: a pilot screening discovered the EMT inhibitory activity of CDK2 inhibitor SU9516. PLoS One. 2016;11(9):e0162394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sogawa C, Eguchi T, Tran MT, et al. Antiparkinson drug benztropine suppresses tumor growth, circulating tumor cells, and metastasis by acting on SLC6A3/DAT and reducing STAT3. Cancers (Basel). 2020;12(2):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sogawa C, Eguchi T, Okusha Y, et al. A reporter system evaluates tumorigenesis, metastasis, beta-catenin/MMP regulation, and druggability. Tissue Eng Part A. 2019. February 8;25(19–20):1413–1425. [DOI] [PubMed] [Google Scholar]
- [53].Yoshida S, Kawai H, Eguchi T, et al. Tumor angiogenic inhibition triggered necrosis (TAITN) in oral cancer. Cells. 2019. July 22;8(7):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Okusha Y, Eguchi T, Sogawa C, et al. The intranuclear PEX domain of MMP involves proliferation, migration, and metastasis of aggressive adenocarcinoma cells. J Cell Biochem. 2018. September;119(9):7363–7376. [DOI] [PubMed] [Google Scholar]
- [55].Eguchi T, Sogawa C, Ono K, et al. CDC37 and HSP90 are essential for stressome release and tumor progression in resistant prostate cancer. 11 February 2020. by MDPI (Preprints). doi: 10.20944/preprints202002.0148.v1. [DOI] [Google Scholar]
- [56].Litvinov SV, Velders MP, Bakker HA, et al. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J Cell Biol. 1994. April;125(2):437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009. July 15;69(14):5627–5629. [DOI] [PubMed] [Google Scholar]
- [58].Zona L, Tawar RG, Zeisel MB, et al. CD81-receptor associations–impact for hepatitis C virus entry and antiviral therapies. Viruses. 2014. February 18;6(2):875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].van Niel G, Charrin S, Simoes S, et al. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell. 2011. October 18;21(4):708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res. 2009. May 15;315(9):1584–1592. [DOI] [PubMed] [Google Scholar]
- [61].Li W, Li Y, Guan S, et al. Extracellular heat shock protein-90alpha: linking hypoxia to skin cell motility and wound healing. Embo J. 2007. March 7;26(5):1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].de la Mare JA, Jurgens T, Edkins AL. Extracellular Hsp90 and TGFbeta regulate adhesion, migration and anchorage independent growth in a paired colon cancer cell line model. BMC Cancer. 2017. March 16;17(1):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Capaldo CT, Nusrat A. Claudin switching: physiological plasticity of the Tight Junction. Semin Cell Dev Biol. 2015. June;42:22–29. [DOI] [PubMed] [Google Scholar]
- [64].Le Naour F, Zoller M. The tumor antigen EpCAM: tetraspanins and the tight junction protein claudin-7, new partners, new functions. Front Biosci. 2008. May 1;13:5847–5865. [DOI] [PubMed] [Google Scholar]
- [65].Luo Y, Zhou H, Krueger J, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006. August;116(8):2132–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sung BH, Ketova T, Hoshino D, et al. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015. May;13(6):7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ham S, Lima LG, Chai EPZ, et al. Breast cancer-derived exosomes alter macrophage polarization via gp130/STAT3 Signaling. Front Immunol. 2018;9:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kanlikilicer P, Bayraktar R, Denizli M, et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine. 2018;38:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].de Vrij J, Maas SL, Kwappenberg KM, et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer. 2015. October 1;137(7):1630–1642. [DOI] [PubMed] [Google Scholar]
- [70].Fujii N, Shomori K, Shiomi T, et al. Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance. J Oral Pathol Med. 2012. July;41(6):444–451. [DOI] [PubMed] [Google Scholar]
- [71].Calderwood SK, Gong J, Murshid A. Extracellular HSPs: the Complicated Roles of Extracellular HSPs in Immunity. Front Immunol. 2016;7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Murshid A, Gong J, Calderwood SK. Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology. 2014. December;219(12):924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Furuta K, Eguchi T. Roles of heat shock proteins on antigen presentation In: Asea AAA, Kaur P, editors. Heat shock proteins in cancer therapeutics. heat shock proteins. 2020, in press. Cham, Switzerland: Springer Nature. [Google Scholar]
- [74].Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol. 1994. June 1;152(11):5398–5403. [PubMed] [Google Scholar]
- [75].Basu S, Binder RJ, Ramalingam T, et al. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001. March;14(3):303–313. [DOI] [PubMed] [Google Scholar]
- [76].Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol. 2010. September 1;185(5):2903–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Murshid A, Borges TJ, Bonorino C, et al. Immunological outcomes mediated upon binding of heat shock proteins to scavenger receptors SCARF1 and LOX-1, and endocytosis by mononuclear phagocytes. Front Immunol. 2019;10:3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Saha B, Momen-Heravi F, Furi I, et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology. 2018. May;67(5):1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer. 2014. January;14(1):49–60. [DOI] [PubMed] [Google Scholar]
- [80].Charrin S, Jouannet S, Boucheix C, et al. Tetraspanins at a glance. J Cell Sci. 2014. September 1;127(Pt 17):3641–3648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.