

Abstract
Background:
Cachexia, a syndrome of muscle atrophy, adipose loss, and anorexia, is associated with reduced survival in cancer patients. The colon adenocarcinoma C26c20 cell line secretes the cytokine leukemia inhibitory factor (LIF) which induces cachexia. We characterized how LIF promotes cachexia-associated weight loss and anorexia in mice through JAK-dependent changes in adipose and hypothalamic tissues.
Methods:
Cachexia was induced in vivo with the heterotopic allotransplanted administration of C26c20 colon adenocarcinoma cells or the intraperitoneal administration of recombinant LIF in the absence or presence of JAK inhibitors. Blood, adipose, and hypothalamic tissues were collected and processed for cyto/adipokine ELISAs, immunoblot analysis, and quantitative RT-PCR. Cachexia-associated lipolysis was induced in vitro by stimulating differentiated adipocytes with recombinant LIF or IL-6 in the absence or presence of lipase or JAK inhibitors. These adipocytes were processed for glycerol release into the media, immunoblot analysis, and RT-PCR.
Results:
Tumor-secreted LIF induced changes in adipose tissue expression and serum levels of IL-6 and leptin in a JAK-dependent manner influencing cachexia-associated adipose wasting and anorexia. We identified two JAK inhibitors that block IL-6 family-mediated adipocyte lipolysis and IL-6 induction using an in vitro cachexia lipolysis assay. JAK inhibitors administered to the in vivo C26c20 cancer cachexia mouse models led to 1) a decrease in STAT3 phosphorylation in hypothalamic and adipose tissues, 2) a reverse in the cachexia serum cyto/adipokine signature, 3) a delay in cancer cachexia-associated anorexia and adipose loss, and 4) an improvement in overall survival.
Conclusions:
JAK inhibitors suppress LIF-associated adipose loss and anorexia in both in vitro and in vivo models of cancer cachexia.
Keywords: cachexia, cancer, leukemia inhibitory factor, IL-6, leptin, janus kinase
INTRODUCTION
Cachexia represents a wasting syndrome consisting of muscle atrophy, adipose loss, and anorexia. It is observed in up to 50% of patients with solid tumors.1 Cancer patients with cachexia have greater than a 50% reduction in overall survival when compared to stage matched patients without cachexia.1 Even when cachexia is identified in early stage cancer patients, survival is not improved because of a potential lack of effective therapeutic interventions.2 Although cachexia has been recognized for more than half a century, most preclinical studies and clinical trials targeting the immune system (TNF-α, IL-6), appetite stimulation, and muscle regeneration have failed to durably improve the syndrome.3 The majority of studies on cachexia have focused on the sarcopenia of this wasting syndrome, with less emphasis on adipose loss or anorexia. Recent evidence suggests that blocking adipocyte lipolysis using global lipase null mice limited not only the adipose wasting but also the sarcopenia observed in murine models of cancer cachexia.4 Additionally, Zimmers and colleagues demonstrated that pancreatic cancer cachexia patients can have adipose wasting without sarcopenia and an associated decrease in survival.5 Therefore, identification and complete characterization of cachexia factors and the common mechanisms that they utilize to induce adipose wasting and anorexia may lead to an effective treatment for patients with cancer cachexia.
When transplanted into syngeneic mice, the murine colon adenocarcinoma cell line C26c20 promotes cachexia-associated adipose wasting and anorexia.6 Previously, we created an in vitro cachexia screen to identify tumor-secreted molecules that can contribute to cachexia-associated adipose wasting. This screen identified leukemia inhibitory factor (LIF) as a cachexia-inducing molecule secreted from the C26c20 colon adenocarcinoma line.6 In an analysis of more than 30 cancers in the TCGA database, LIF is most highly expressed in gastrointestinal, thoracic, and genitourinary cancers6 which are all associated with cachexia. Two recent papers suggested that LIF is critical to pancreatic cancer development,7, 8 further illustrating the importance of this molecule to cachexia-associated cancers. LIF is a 21 kDa protein in the IL-6 family of cytokines that binds to its receptor, LIFR-α, and the co-receptor gp130 inducing JAK-STAT signaling.9 Recombinant LIF (rLIF) administered to wild-type mice causes adipose and body weight loss reproducing the cachexia phenotype.6 LIF induction of cachexia through adipose wasting and anorexia is associated with JAK/STAT signaling peripherally in the adipose tissue and centrally in the hypothalamus.6 As rLIF induces the cachexia-associated adipose loss, there is a corresponding decrease in serum levels of leptin. Leptin is an adipokine, a cytokine-like molecule produced by adipose tissue, which regulates appetite through JAK/STAT signaling of the hypothalamus in the setting of changes in adipose levels.10, 11 As rLIF induces cachexia-associated adipose loss, the reduction of adipose leptin compensates for rLIF’s anorexic effect.
In the present study, we show that the colon adenocarcinoma C26c20 and rLIF-driven cachexia mouse models have increased serum LIF and IL-6 with decreased leptin, defining a cachexia signature in mice. Consistent with these serum findings, adipose mRNA levels of LIF, IL-6, and leptin from both cachexia models were similarly altered. By showing that LIF was still able to induce cachexia in IL-6−/− mice, we demonstrated that both LIF and IL-6 could independently promote anorexia and adipose loss. We therefore hypothesized that inhibition of the JAK signaling pathway would suppress the cachexia phenotype, since the molecules that are altered in the cachexia cyto/adipokine signature (LIF, IL-6, and leptin) all signal through this pathway.12 A screen of candidate JAK inhibitors in our in vitro cachexia adipocyte lipolysis assay led us to test tofacitinib and ruxolitinib in vivo. These JAK inhibitors are effective in the treatment of ulcerative colitis, rheumatoid arthritis, and myelofibrosis.13–15 The independent administration of either JAK inhibitor to rLIF- or tumor-driven models of cancer cachexia led to decreased STAT3 phosphorylation in adipose and hypothalamic tissues with a concomitant suppression of cachexia-associated changes to adipose mRNA expression and serum levels of IL-6 and leptin. The net effect of these changes resulted in a mitigation of rLIF-induced anorexia and adipose/body weight loss and additionally led to an improvement of survival in the heterotopic allotransplanted C26c20 cancer cachexia model. These studies suggest that: 1) Tumor-secreted LIF induces cachexia through JAK-STAT signaling in multiple tissues altering levels of IL-6 and leptin, and 2) JAK inhibition of these signaling events suppresses cachexia-associated adipose loss and anorexia long enough to lead to an improvement in cancer cachexia survival.
METHODS
See Supplementary Materials for a more detailed Materials and Methods.
Mouse Studies.
Male wild-type Balb/c mice and C57BL/6J were obtained from Charles River Laboratories or Jackson Laboratories at approximately 8 weeks of age. IL-6−/− mice (B6.129S2-IL6tm1Kopf/J, 002650) were obtained from Jackson Laboratories at approximately 7 weeks of age. All mice were allowed to acclimate in UT Southwestern animal facilities before experimentation for at least 1 week. Animals were kept in a temperature-controlled facility with a 12 h light/dark cycle and were fed normal chow diet and provided water ad libitum. Approximately 100 g of standard chow diet was placed in each cage. When food reached approximately 50 g per cage, it was replenished to approximately 100 g. Food was weighed at the same time daily and compared to the previous day’s weight to calculate the 24 h food intake per cage. Body weight was measured using a standard balance (digital Soenhle scale). Adipose tissue mass and lean tissue mass were measured longitudinally using ECHO MRI (ECHO Medical Systems) at 9AM at the indicated time points. Whole blood was drawn from tail vein bleed longitudinally or by cardiac puncture at terminal time points. Serum was obtained by subjecting the whole blood to centrifugation at 960 × g at 4 ˚C for 10 min. Supernatant was removed followed by protein concentration quantification using a bicinchonianic acid kit (Pierce). For analysis of serum cytokine changes, 25 μl of mouse serum was diluted with 25 μl of PBS and sent to Eve Technologies (Calgary, Canada) for ELISA analysis of multiple cytokines. For analysis of serum leptin or IL-6, serum dilution and ELISA analysis was performed as performed per kit directions from Crystal Chem and Sigma, respectively. The rest of the serum was stored at −80 °C for future blood analysis. For tumor studies, 0.75–1 × 107 C26c20 cells in 100 μl PBS were injected into the right hind flank of mice at day 0, and tumor volume was calculated by taking half of the product of the caliper (VWR) measurements of length, width and breath at the indicated time points. At the end of the experiments, mice were euthanized at the indicated time point in non-tumor experiments or within 12 hours of expected death in tumor studies as recommended by IACUC using a CO2 chamber and organs were collected and snap frozen.
Adipocyte Lipolysis Assay.
Media glycerol concentration from differentiated adipocytes was measured for each condition in triplicate as previously described.6 Pre-adipocytes from the stromal vascular fraction were differentiated into adipocytes using the established and characterized protocol used to characterize adipocyte physiology.16, 17
Statistical Analysis.
Data is presented as mean ± SEM, dot plots ± SEM, or dot plots with bars representing mean ± SEM. A Student’s t test was used to determine differences between groups at distinct time points. A Generalized Estimating Equation approach was used to determine differences between groups over time. Kaplan Meier analysis was conducted for survival with statistical evaluation using the Gehan-Breslow-Wilcoxon approach. For some animal studies, the ROUT method was used to remove outliers followed by ANOVA with a multiple comparison post-test (Dunnett’s or Tukey’s). Significance was considered if p < 0.05.
Study Approval.
All animal studies were conducted under an Institutional Animal Care and Use Committee approved protocol at UT Southwestern Medical Center (Dallas, Texas).
RESULTS
Serum and Adipose mRNA Cyto/Adipokine Levels in the C26c20 Cachexia Mouse Model.
To verify that the increased LIF and decreased leptin serum levels observed previously in the rLIF-induced cachexia model6 were also altered similarly in the C26c20 cancer cachexia model, we evaluated the cytokine and leptin levels in serum from the C26c20 mouse model. The C26c20 cell line is a subclone of the parental C26 colon adenocarcinoma line and induces a cachexia phenotype when administered in vivo.18 Serum was collected from cancer-bearing mice for cyto/adipokine ELISA analysis after each animal lost 30–40% of its adipose mass. These mice had a significant serum increase in LIF (Figure 1A) and a decrease in leptin (Figure 1B), similar to findings from the rLIF-induced cachexia model.6 In addition to the expected changes in LIF and leptin levels in the C26c20 cancer cachexia model, we also observed a 10-fold increase in serum IL-6 levels. Other groups have demonstrated an association between altered serum IL-6 levels and cachexia progression.18–20 Furthermore, we generalized these findings of increased serum IL-6 and LIF across multiple syngeneic murine cancer cachexia models, including those created with LLC and 4T1 tumor cells (data not shown).
Figure 1. Serum and Adipose mRNA Levels of Cyto/Adipokines in a Colon Cancer Cachexia Mouse Model.
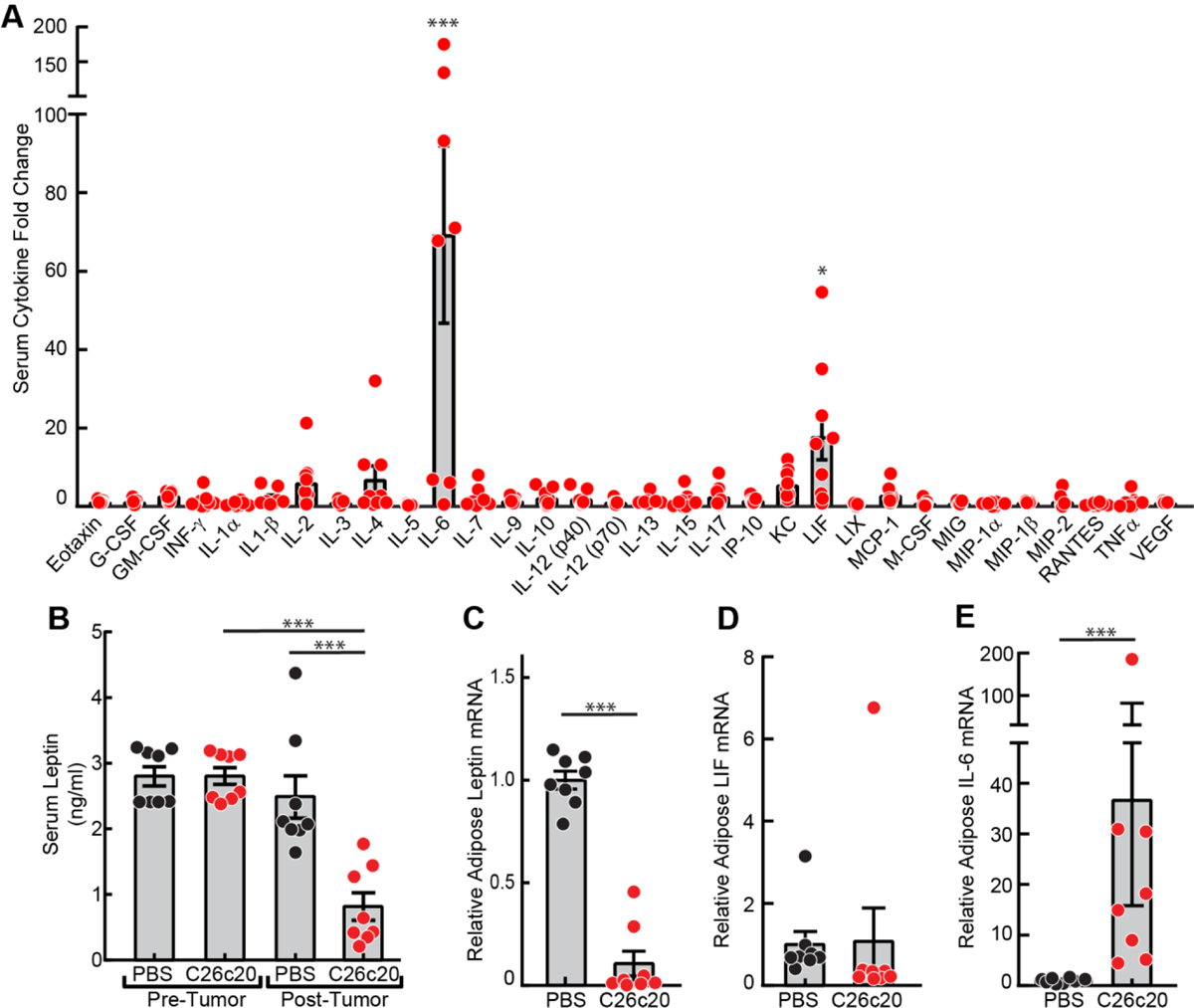
Chow-fed Balb/c mice (10-week-old males) were injected s.c. in the right flank with PBS in the absence or presence of C26c20 cells. A-B) Serum Cyto/Adipokine levels. Blood was collected by tail vein bleed on day 0 and by cardiac puncture after each mouse lost 30–50% of its adipose mass as measured by ECHO MRI. Serum was isolated and the cytokine and adipokine levels were measured as described in Methods. C-E) Adipose leptin, LIF and IL-6 mRNA levels. Epididymal white adipose tissue was harvested at sacrifice for measurement of the indicated mRNA by quantitative RT-PCR. For each gene, the amount of mRNA from PBS-treated mice is set to 1, and mRNA amounts from C26c20-bearing mouse adipose tissue are expressed relative to this reference value. The average Ct values for β-actin (invariant control) for PBS- and C26c20-administered mice were 18.0 and 18.7, respectively. The average PBS-administered mice Ct values for leptin, LIF, and IL-6 were 22.1, 28.2, and 33.1, respectively. Data is shown as dot plots with bars representing mean ± SEM (A-E) of five (A) or eight (B-E) mice. *p<0.05 and ***p<0.001 based on use of a ROUT method (Q=0.001) to remove outliers followed by an ANOVA and Dunnett’s multiple comparison post-test comparing the relative change in the indicated serum cytokine relative to VEGF (A) or to PBS control (B-E). These results were confirmed in at least two independent experiments.
Considering that adipose is the primary source of leptin,21 we next tested whether the cancer-induced changes in serum cyto/adipokines paralleled the changes in adipose mRNA expression levels. As expected, mice bearing C26c20 tumors demonstrated a decrease in mRNA expression of leptin in their white adipose tissue (WAT) compared to mice receiving PBS alone (Figure 1C). Although WAT from tumor-bearing mice did not exhibit an increase in their LIF mRNA expression (Figure 1D), there was a ~ 10-fold increase in the mRNA expression of IL-6 in WAT from tumor-bearing mice compared to vehicle-treated mice (Figure 1E). This data suggests that cancer secreted factors, such as LIF, not only cause adipose wasting/lipolysis, but also change the expression profile of other cyto/adipokines, IL-6 and leptin, that can also contribute to the adipose wasting and anorexia observed in cachexia.
Serum and Adipose mRNA LIF, IL-6, and Leptin Levels in the rLIF Cachexia Mouse Model.
Knowing that rLIF is able to alter serum leptin levels, we hypothesized that it also increases IL-6 serum levels matching changes observed in in vivo cancer cachexia models. To test this hypothesis, we evaluated serum levels and adipose mRNA expression levels of LIF, IL-6, and leptin in our rLIF-driven cachexia model (Figure 2). As rLIF-injected mice lose fat mass (Figure 2A), there was a parallel increase in serum LIF (Figure 2B) and IL-6 (Figure 2C) with a corresponding decrease in serum leptin (Figure 2D).
Figure 2. Serum and Adipose mRNA Levels of LIF, IL-6 and Leptin in the rLIF Cachexia Mouse Model.
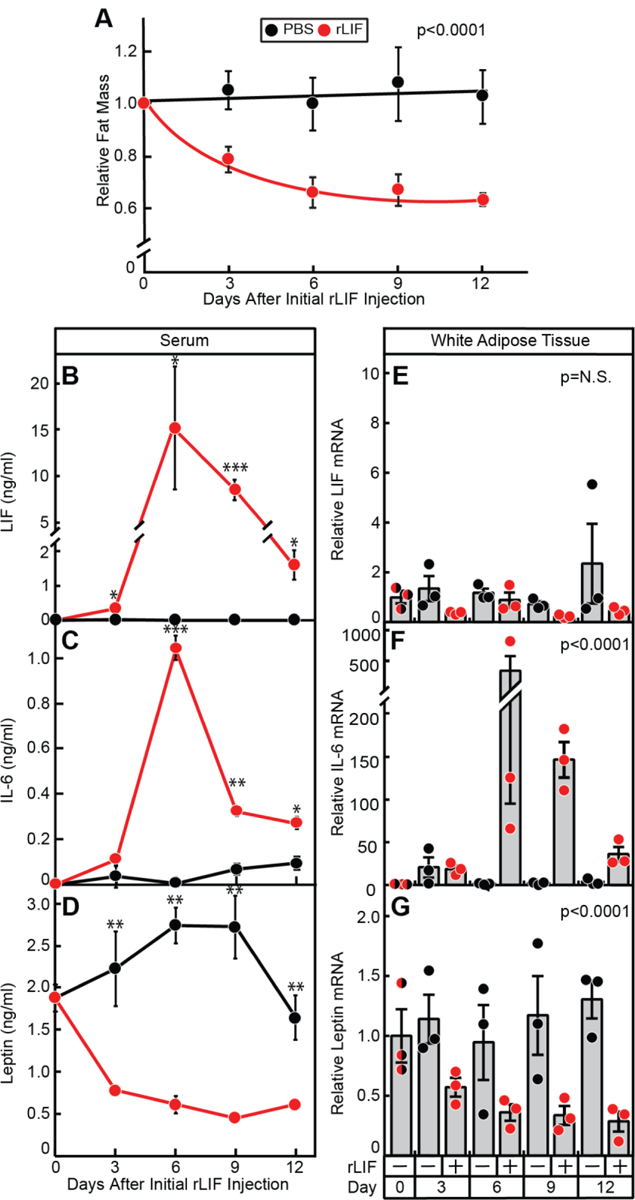
Chow-fed C57BL/6J mice (10-week-old males) were injected i.p. with PBS in the absence or presence of recombinant LIF at 80 μg/kg body weight twice daily and fat mass (A) was measured by ECHO MRI. Fat mass is shown relative to the average day 0 reference value for each respective cohort which was 3.07 and 3.16 g for the PBS- and rLIF-treated mice, respectively. B-G) Serum levels and adipose tissue mRNA expression of LIF, IL-6, and leptin. Every 3 days, 3 mice from each cohort were sacrificed for harvesting of serum for the respective cyto/adipokine ELISA (B-D) and epididymal white adipose tissue for measurement of the indicated mRNA by quantitative RT-PCR (E-G) as described in Methods. For each gene, the amount of mRNA from day 0 is set to 1, and mRNA amounts from adipose tissue from the indicated days are expressed relative to this reference value (E-G). The average Ct values for β-actin (invariant control) for day 0, 3, 6, 9, and 12 for PBS-treated mice were 18.8, 18.8, 18.9, 18.5, and 18.7, respectively, and for rLIF-treated mice were 18.8, 18.0, 18.5, 17.9, and 17.8, respectively. The average day 0 Ct values for LIF, IL-6, and leptin were 26.4, 33.2, and 21.8, respectively. Data is shown as mean ± SEM (A-D) or dot plots with bars representing mean ± SEM (E-G) of three mice. *p<0.05, **p<0.01, and ***p<0.001 based on use of Student’s t-test (B-D) or Generalized Estimating Equation approach comparing the two groups over time with rLIF-treated mice as the reference value (A, E-G). These results were confirmed in at least two independent experiments.
To determine whether adipose mRNA expression correlated with rLIF-induced changes in serum LIF, IL-6 and leptin, we collected RNA from WAT of rLIF- and vehicle-treated mice for quantitative RT-PCR analysis. Adipose tissue from mice treated with rLIF had a greater than 50-fold increase in IL-6 mRNA expression (Figure 2F) and an ~5-fold decrease in leptin mRNA expression (Figure 2G) with no significant change in LIF expression (Figure 2E). The changes of IL-6 and leptin adipose mRNA expression correlated with the changes observed in their respective serum levels (compare Figures 2C and 2F, 2D and 2G). Overall, the simple model of rLIF-induced cachexia had a similar serum and adipose signature to that of the complex C26c20 colon cancer cachexia model (see Figure 1) with a net result of increased serum LIF and IL-6, with a corresponding decrease in leptin, making it a powerful system to understand the biology of cachexia.
Evaluation of rLIF-induced Cachexia in an IL-6−/− Mouse Model.
As described previously, rLIF injected into C57BL/6J mice causes a ~5–10% muscle loss and an ~30–40% adipose loss resulting in an ~10–15% body weight loss mimicking a cachexia phenotype.6 IL-6 has also been reported to induce cachexia in vivo.18–20 We have shown that LIF treatment leads to an increase in serum IL-6 (Figure 2C) and adipose IL-6 expression (Figure 2F) in vivo. Therefore, we next determined whether rLIF-associated cachexia is dependent on its induction of IL-6. PBS in the absence or presence of rLIF was injected into IL-6+/+ or IL-6−/− mice and changes in food intake, body weight, and fat and lean mass by ECHO MRI were measured over time (Figure 3). Figure 3A shows that circulating concentrations of LIF were elevated in both IL-6+/+ and IL-6−/− mice administered rLIF at day 21 compared to day 0. As expected, circulating levels of IL-6 were also increased with rLIF administration in IL-6+/+ mice, but not in IL-6−/− or PBS-treated IL-6+/+ mice at day 21 (Figure 3B). Mice receiving rLIF had reduced food intake in both IL-6+/+ and IL-6−/− mice compared to mice receiving PBS during the first 9 days of the experiment (Figure 3C). Both IL-6+/+ and IL-6−/− mice also demonstrated decreased fat mass (Figure 3D) and body weight (Figure 3E) when treated with recombinant rLIF compared to control conditions treated with PBS. Although an increase in IL-6 can promote a cachexia phenotype, LIF’s induction of cachexia was not dependent on its ability to increase serum IL-6. Therefore, both molecules are likely driving the cachexia phenotype in cancer models.
Figure 3. LIF Induces Cachexia-Associated Anorexia and Adipose/Body Weight Loss in IL-6−/− mice.
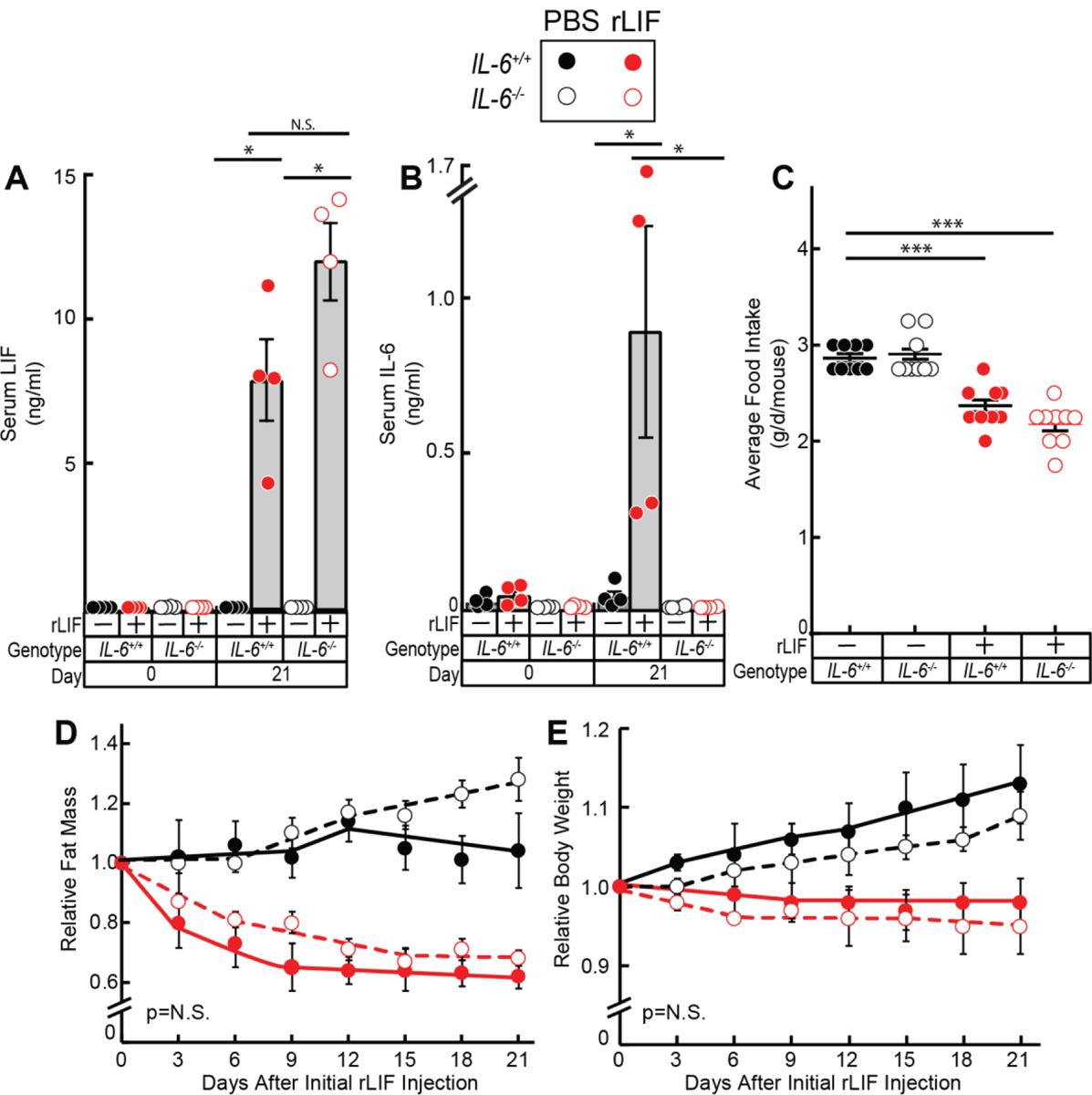
Chow-fed IL-6+/+ and IL-6−/− C57BL/6J mice (8-week-old males) were injected i.p. with PBS in the absence or presence of rLIF at 80 μg/kg body weight twice daily. Serum was collected on day 0 and day 21 for evaluation of LIF (A) and IL-6 (B) levels by ELISA as described in Methods. Food intake (C), ECHO MRI measurements of fat mass (D), and body weight (E) were measured at the indicated time points. Fat mass and body weight are shown relative to the average day 0 reference value for each respective cohort. The average values for fat mass at day 0 of the PBS- and rLIF- treated IL-6+/+ mice were 2.8 and 2.4 g, respectively, and for the IL-6−/− mice were 3.6 and 3.5 g, respectively. The average values for body weight at day 0 of the PBS- and rLIF- treated IL-6+/+ mice were 23 and 24 g, respectively, and for the IL-6−/− mice were 24 and 24 g, respectively. Data is shown as dot plots with mean ± SEM (A-C) or each value represents mean ± SEM (D-E) of four mice. *p<0.05 and ***p<0.001 based on Student’s t-test (A-C) or based on use of Generalized Estimating Equation approach comparing IL-6+/+ to IL-6−/− mice treated with either PBS or rLIF (D-E).
IL-6 mRNA Expression in IL-6 Family-stimulated Differentiated Adipocytes
To better understand how LIF and the subsequent increase in IL-6 can stimulate adipose tissue to promote cachexia-associated wasting, we studied the effect of these cytokines on in vitro differentiated adipocytes in relation to lipolysis and changes in mRNA expression of IL-6, LIF, and TNFα. As shown in Figure 4A, both wild-type rLIF and recombinant IL-6 (rIL-6) increased adipocyte lipolysis, whereas the mutant rLIF K159A had no effect. The point mutation (K159A) in LIF disrupts its interaction with it receptor, LIFR-α.22 The mutant rLIF K159A is unable to stimulate lipolysis of differentiated adipocytes in vitro or promote cachexia-associated adipose wasting when administered in mice.6 Compared to rIL-6, rLIF stimulated lipolysis at much lower concentrations. However, rIL-6 caused ~ 3-fold higher level of maximum lipolysis than rLIF. Wild type rLIF and rIL-6, but not rLIF K159A, stimulated the phosphorylation of STAT3 in adipocytes (Figure 4B, top immunoblot) at similar concentrations to those necessary to stimulate lipolysis (see Figure 4A).
Figure 4. Lipolysis, JAK/STAT Activation, and mRNA Expression of IL-6 Family-stimulated Adipocytes.
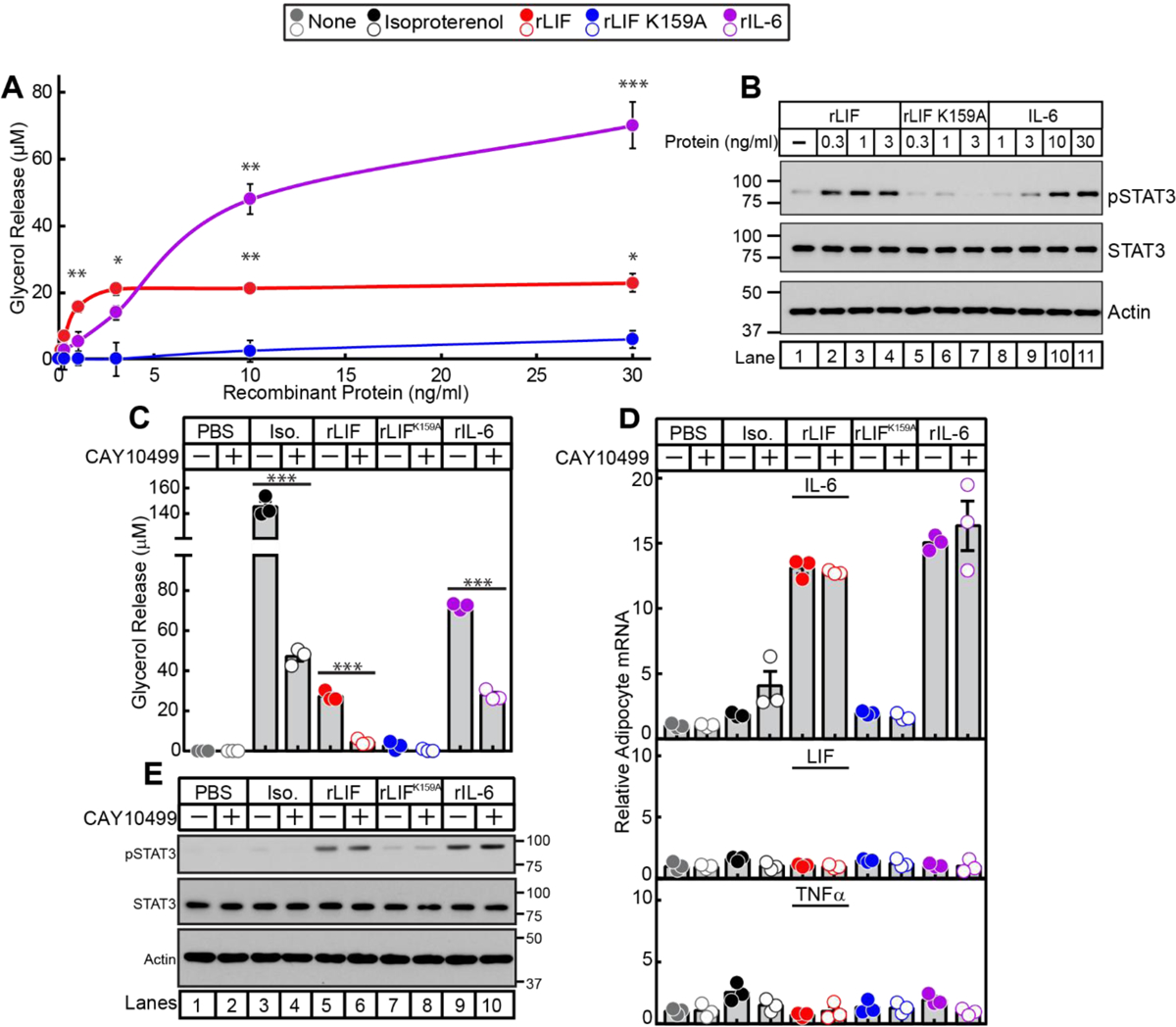
Differentiated adipocytes in a 12-well format were treated in a final volume of 1.5 ml of medium E supplemented with the indicated concentration of recombinant LIF, LIF K159A, or IL-6 (A-B) or 10 nM isoproterenol, 3 ng/ml rLIF, 3 ng/ml rLIF K159A, or 50 ng/ml rIL-6 (C-E) in the absence or presence of 10 μM CAY10499. After incubation for 20 h at 37 ˚C, medium was collected and glycerol concentration was measured (A,C) and cells were harvested for measurement of the indicated mRNA by quantitative RT-PCR (D) or subjected to IB analysis (B,E) with the indicated antibody as described in Methods. For each gene (D), the amount of mRNA from PBS-treated adipocytes in the absence or presence of CAY10499 is set to 1. The average Ct values for cyclophilin (invariant control) for the PBS-, isoproterenol-, rLIF-, rLIF K159A-, and IL-6-treated adipocytes in the absence of CAY10499 were 20.3, 20.9, 20.4, 20.3, and 20.7, respectively, and in the presence of CAY10499 were 20.4, 21.4, 20.6, 20.5, and 20.8, respectively. The average reference Ct values for IL-6 (upper panel), LIF (middle panel), and TNFα (lower panel) in PBS-treated adipocytes in the absence of CAY10499 were 28.3, 29.2, and 33.1, respectively, and in the presence of CAY10499 were 28.6, 29.2, and 33.8, respectively. Data is shown as mean ± SEM (A) or as dot plots with mean ± SEM (C,D) of three wells. *p<0.05, **p<0.01, and ***p<0.001 based on Student’s t-test comparing conditions to rLIF K159A-treated adipocytes (A) or the PBS-treated adipocytes (C-D). These results were confirmed in at least three independent experiments.
In adipocytes, triglycerides are sequentially hydrolyzed by adipose triglyceride lipase, hormone sensitive lipase (HSL), and monoacylglycerol lipase, each of which sequentially removes one fatty acid molecule to produce glycerol and fatty acids. CAY10499 is a commercially available pan inhibitor of these lipases.23, 24 To evaluate if IL-6- or LIF-stimulated adipocyte lipolysis is required for the induction of IL-6 mRNA expression, we performed quantitative RT-PCR to assess the fold change of mRNA expression in differentiated adipocytes that were treated with either vehicle, isoproterenol, wild-type rLIF, mutant rLIF K159A, or rIL-6 in the absence or presence of lipase inhibitor CAY10499. Isoproterenol is a β-adrenergic agonist that enhances lipolysis by increasing cAMP stimulating the phosphorylation and activation of the lipase HSL.25 CAY10499 was able to suppress adipocyte lipolysis induced by isoproterenol, rLIF, and rIL-6 (Figure 4C). However, these cytokines were still able to activate adipocytes in the presence of CAY10499 as demonstrated by cytokine-induced phosphorylation of STAT3 (Figure 4E). Although isoproterenol-treated adipocytes had a greater than 2-fold increase in lipolysis relative to rIL-6- or rLIF-treated adipocytes (Figure 4C), there was no significant increase in IL-6 mRNA expression (Figure 4D, upper panel). Contrarily, adipocytes treated with wild-type rLIF or rIL-6 had a greater than 10-fold increase in relative IL-6 mRNA expression, which remained elevated even in the presence of lipase inhibitor CAY10499. The mRNA expression of LIF (middle panel) and TNFα (lower panel) were unchanged in β−adrenergic-induced or IL-6 family-induced adipocytes in the absence or presence of CAY10499. IL-6 family of cytokines did not affect the mRNA expression of leptin in the well-established in vitro differentiated adipocyte model we utilized due to its inherent low baseline leptin mRNA expression (data not shown). Combined, the results of these experiments demonstrated that IL-6 family of cytokines are able to stimulate IL-6 expression in adipocytes independent of their induction of lipolysis.
JAK Inhibition Blocks Lipolysis, JAK/STAT Activation, and IL-6 mRNA Expression in IL-6 Family-stimulated Differentiated Adipocytes.
Having demonstrated that cachexia factors LIF and IL-6 are both increased in cancer cachexia, an ideal inhibitor would block a common pathway these cytokines activate to induce adipose loss, anorexia, and muscle wasting. Since LIF and IL-6 both induce JAK/STAT activation in adipocytes (see Figure 4B) and in the hypothalamus6, 26, we next screened JAK inhibitors for their ability to block in vitro IL-6 family cytokine-stimulated adipocyte lipolysis, JAK/STAT activation, and IL-6 mRNA expression. We incubated adipocytes with isoproterenol or rIL-6 in the presence of increasing concentrations of multiple JAK inhibitors including tofacitinib, ruxolitinib, decernotinib, and filgotinib (Figure 5A). Beta-adrenergic agonist isoproterenol-induced adipocyte lipolysis was not affected by any JAK inhibitor. Adipocyte lipolysis induced by rIL-6 was significantly inhibited when adipocytes were incubated with tofacitinib or ruxolitinib.
Figure 5. IL-6 Family Cytokine Stimulation of Adipocytes in the Presence of Janus Kinase Inhibitors.
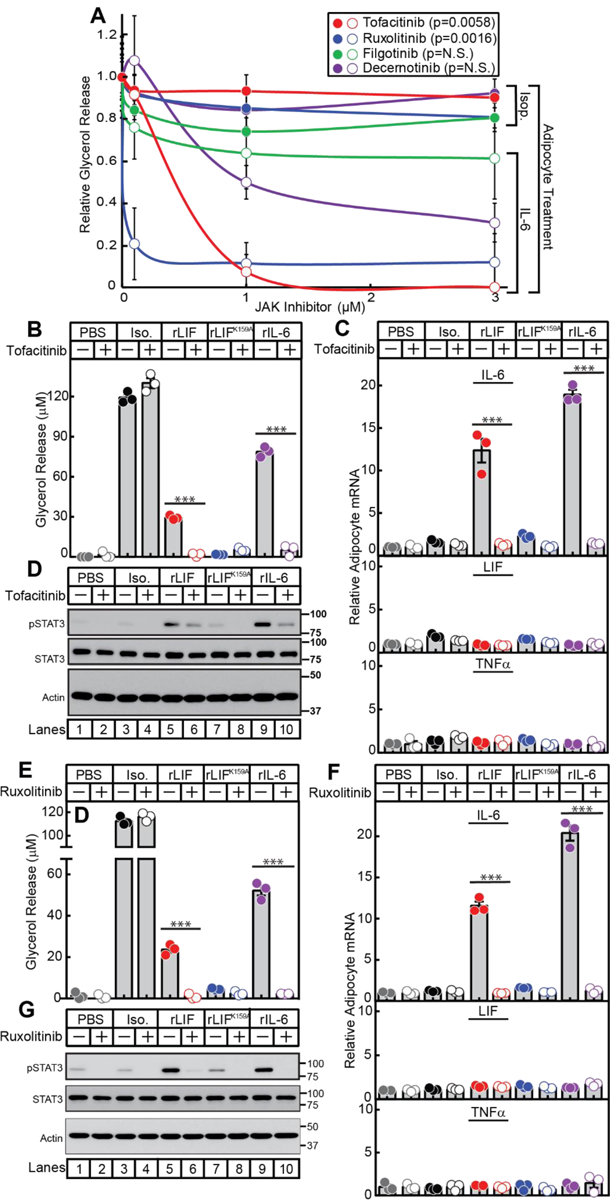
Differentiated adipocytes in a 12-well format were treated in a final volume of 1.5 ml of medium E with 10 nM isoproterenol, 50 ng/ml rIL-6, or 3 ng/ml rLIF in the absence or presence of the indicated concentration of JAK inhibitor (A), 10 μM tofacitinib (B-C), or 3 μM ruxolitinib (E-F). After incubation for 20 h at 37 ˚C, medium was collected and glycerol concentration was measured (A,B,E) and cells were harvested for measurement of the indicated mRNA by quantitative RT-PCR (C,F) or subjected to IB analysis (D,G) with the indicated antibody as described in Methods. For each gene (C,F), the amount of mRNA from PBS-treated adipocytes in the absence or presence of the JAK inhibitor are set to 1. The average Ct values for cyclophilin (invariant control) for the PBS-, isoproterenol-, rLIF-, rLIF K159A-, and IL-6-treated adipocytes in the absence of tofacitinib were 20.3, 20.7, 20.4, 20.3, and 20.4, respectively, and in the presence of tofacitinib were 20.5, 20.9, 20.6, 20.6, and 20.6, respectively. The average reference Ct values for IL-6 (upper panel), LIF (middle panel), and TNFα (lower panel) in PBS-treated adipocytes in the absence of tofacitinib were 28.5, 29.1, and 33.5, respectively, and in the presence of tofacitinib were 30.2, 29.2, and 33.7, respectively. F) The average Ct values for cyclophilin (invariant control) for the PBS-, isoproterenol-, rLIF-, rLIF K159A-, and IL-6-treated adipocytes in the absence of ruxolitinib were 20.5, 20.9, 20.5, 20.6, and 20.8, respectively, and in the presence of ruxolitinib were 20.8, 21.1, 20.9, 20.9, and 21.2, respectively. The average reference Ct values for IL-6 (upper panel), LIF (middle panel), and TNFα (lower panel) in PBS-treated adipocytes in the absence of ruxolitinib were 28.4, 28.5, and 32.7, respectively, and in the presence of ruxolitinib were 30.2, 29.1, and 33.5, respectively. Data is shown as mean ± SEM (A) or as dot plots with mean ± SEM (B-C,E-F) of three wells. ***p<0.001 based on Student’s t-test comparing cytokine-treated adipocytes to isoproterenol-treated adipocytes at 1 μM (A) or comparing conditions to the PBS-treated adipocytes (B-C,E-F). These results were confirmed in at least three independent experiments.
To evaluate whether cytokine-induced adipocyte IL-6 expression was dependent on JAK-STAT activation, we incubated adipocytes with PBS, isoproterenol, rLIF, mutant rLIF K159A, or rIL-6 in the absence or presence of tofacitinib or ruxolitinib. As shown in Figure 5B, tofacitinib significantly suppressed rIL-6 and rLIF-mediated, but not isoproterenol-mediated adipocyte lipolysis. To verify that tofacitinib inhibited JAK-mediated STAT activation, immunoblot analysis of adipocyte lysates demonstrated reduced IL-6 family cytokine-mediated STAT3 phosphorylation in the presence of tofacitinib (Figure 5D, top immunoblot). Tofacitinib also completely suppressed cytokine-mediated induction of adipocyte IL-6 expression (Figure 5C, top panel). Adipocyte mRNA expression of other cytokines, LIF (middle panel) and TNFα (bottom panel), were unchanged in adipocytes treated with PBS, isoproterenol, or cytokines in the absence or presence of tofacitinib. Another JAK inhibitor, ruxolitinib, also suppressed IL-6 family cytokine-mediated lipolysis (Figure 5E), phosphorylation of STAT3 (Figure 5G), and IL-6 mRNA expression (Figure 5F, top panel).
JAK Inhibition Suppresses Anorexia and Adipose Loss in the rLIF Cachexia Mouse Model.
To demonstrate that JAK inhibition blocks anorexia and adipose wasting in the rLIF-induced cachexia model, we injected mice with PBS or rLIF in the absence or presence of tofacitinib (Figures 6A–D) or ruxolitinib (Figures 6E–J). The addition of tofacitinib or ruxolitinib to rLIF-treated mice restored food intake (Figure 6A and 6E), body weight (Figure 6B and 6F), and fat mass (Figure 6C and 6G) towards levels observed in vehicle-treated mice. There was no change in lean mass among all cohorts (Figure 6D and 6H) within the limited time frame of the experiment. To confirm that JAK inhibition suppressed STAT activation in target tissues relevant to appetite and wasting, STAT3 phosphorylation was measured in hypothalamic and adipose tissues, respectively, by immunoblot analysis. Mice treated with rLIF and ruxolitinib (lanes 10–12) had decreased STAT3 phosphorylation compared to rLIF-treated mice in the absence of ruxolitinib (lanes 7–9) in hypothalamic (Figure 6I) and adipose (Figure 6J) tissues.
Figure 6. JAK Inhibition Suppresses rLIF-mediated Cachexia.
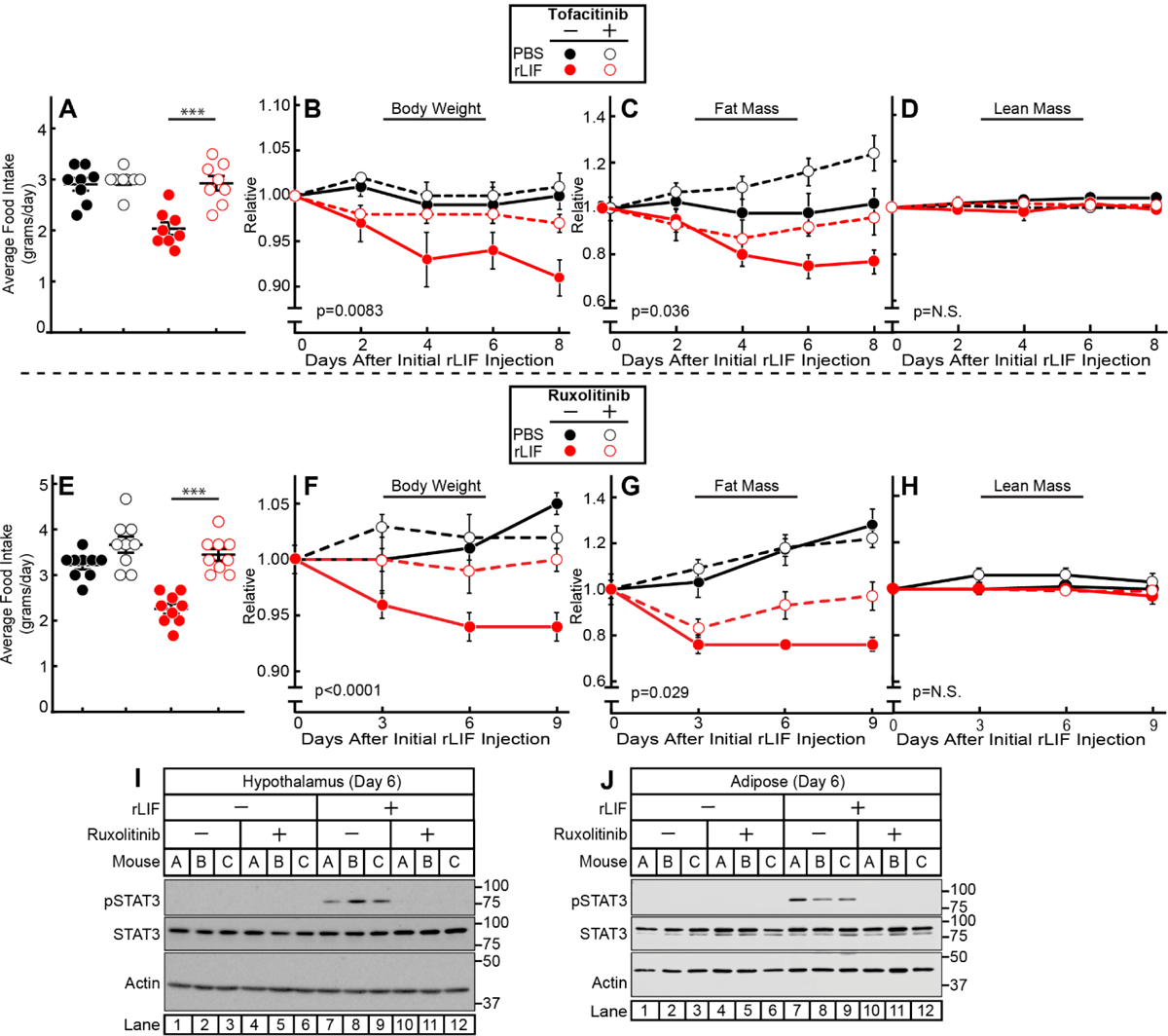
Chow-fed C57BL/6J mice (10-week-old males) were were injected i.p. with PBS in the absence or presence of rLIF at 80 μg/kg body weight twice daily. Ninety minutes after each rLIF injection, mice also received either oral gavage of 200 μl PBS containing 0.5% (v/v) methylcellulose and 0.1% (v/v) Tween 20 in the absence or presence of 25 mg/kg tofacitinib (A-D) or i.p. of 150 μl PBS containing 2% DMSO (v/v) and 30% PEG300 (v/v) in the absence or presence of 25 mg/kg ruxolitinib (E-J) twice daily. Food intake (A,E), body weight (B,F), ECHO MRI measurement of fat mass (C,G) and lean mass (D,H), or IB analysis of harvested hypothalamic (I) or adipose (K) tissue were measured at the indicated time points. Body weight, fat mass, and lean mass are shown relative to the average day 0 reference value for each respective cohort. The average values for day 0 for the PBS with vehicle, PBS with tofacitinib, rLIF with vehicle, and rLIF with tofacitinib were as follows: body weight (27, 25.4, 26.7, and 26.8 g), fat mass (3.6, 3.4, 3.7, and 3.6 g) and lean mass (19, 18, 18.7, and 19 g), respectively. The average values for day 0 for the PBS with vehicle, PBS with ruxolitinib, rLIF with vehicle, and rLIF with ruxolitinib were as follows: body weight (26.5, 28, 26.4, and 27 g), fat mass (2.7, 3.0, 3.0, and 2.8 g) and lean mass (20, 21, 20, and 21 g), respectively. Data is shown as dot plots with mean ± SEM (A and E) or each value represents the mean ± SEM (B-D,F-H) of four (A-D) or three (E-H) mice. These results were confirmed in at least three independent experiments.
JAK Inhibition Suppresses Anorexia and Adipose Loss Improving Survival in the C26c20 Cachexia Mouse Model
To test if JAK/STAT signaling pathways are an integral component of cachexia, we tested JAK inhibitors in the in vivo C26c20 cancer cachexia model. Mice were injected with PBS or C26c20 cells on day 0 in the absence or presence of ruxolitinib (Figure 7). Mice implanted with colon adenocarcinoma C26c20 cells and treated with ruxolitinib had an ~20–30% increase in median survival compared to mice receiving C26c20 in the absence of ruxolitinib (Figure 7A), despite the absence of any significant ruxolitinib effect on tumor growth (Figure 7B). Mice receiving both C26c20 cells and ruxolitinib had a reduction in the cachexia-associated-adipose loss at intermediate time points (Figure 7D, right panel), coinciding with decreased adipose tissue STAT3 phosphorylation (Figure 7F, top blot, compare lanes 4 and 5). At the time that animals were sacrificed due to cachexia morbidity, there was no longer a significant difference in adipose mass either in the absence or presence of ruxolitinib (Figure 7D, right panel). This end point coincided with increasing adipose tissue STAT3 phosphorylation even in the ruxolitinib-treated cancer-bearing mice (Figure 7F, top blot, compare lanes 9,10). Mice receiving both C26c20 cells and ruxolitinib also had a reduction in the cachexia-associated anorexia between days 5 and 8 (Figure 7C, middle panel), coinciding with decreased hypothalamic tissue STAT3 phosphorylation (Figure 7F, 4th blot, compare lanes 4 and 5). However, these mice reached the same levels of anorexia before sacrifice (days 9–12) as the cancer-bearing mouse cohort receiving vehicle (Figure 7C, right panel), coinciding with increasing adipose tissue STAT3 phosphorylation even in the ruxolitinib-treated cancer-bearing mice (Figure 7F, 4th blot, compare lanes 9,10). During intermediate time points when JAK inhibition was effective at blocking STAT3 phosphorylation in adipose tissue, there was also a significant suppression of cachexia-associated changes in circulating leptin (Figure 7G) and IL-6 (Figure 7H). As STAT3 phosphorylation returned in adipose tissue even in the presence of JAK inhibition, the serum levels of IL-6 and leptin approached those observed in a cachexia mouse without JAK inhibition.
Figure 7. JAK Inhibition Suppresses Colon Cancer Cachexia.
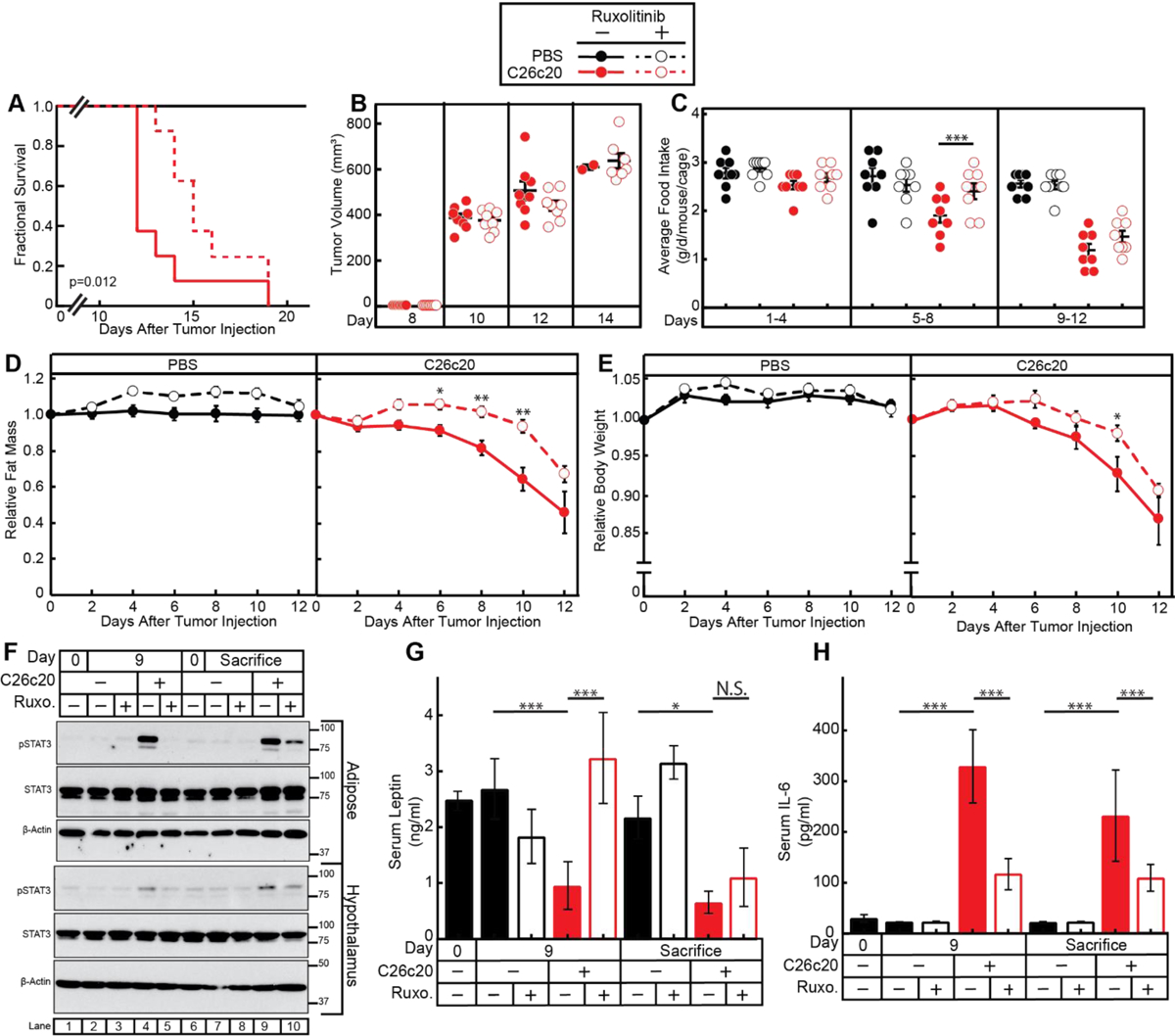
Chow-fed Balb/c mice (10-week-old males) were injected s.c. in right flank with PBS in the absence or presence of C26c20 cells on day 0 followed by i.p. administration of 150 μl PBS containing 2% DMSO (v/v) and 30% PEG300 (v/v) in the absence or presence of 25 mg/kg ruxolitinib twice daily thereafter. Survival (A), tumor volume (B), food intake (C), ECHO MRI of fat mass (D), body weight (E), IB analysis of harvested hypothalamic and adipose tissue with the indicated antibodies (F), and ELISA of the serum of the indicated cyto/adipokine (G-H) were measured at the indicated time points as described in Methods. Fat mass and body weight are shown relative to the average day 0 reference value for each respective cohort. The average values for day 0 for the PBS with vehicle, PBS with ruxolitinib, C26c20 with vehicle, and C26c20 with ruxolitinib were 3.3, 2.8, 3.2, and 3.3 g for fat mass and 24, 23, 23, and 23 g for body weight, respectively. Data is shown as Kaplan-Meier survival curve (A), dot plots with mean ± SEM (B-C, G-H), or each value represents the mean ± SEM (D-E) of eight mice (A-G) or four mice (G-H). The Gehan-Breslow-Wilcoxon approach was used to compare survival curves (A) of C26c20-bearing mice in the absence or presence of ruxolitinib. *p<0.05, **p<0.01, and ***p<0.001 based on Student’s t-test comparing the absence to the presence of ruxolitinib in PBS- and C26c20-bearing mice (B-E) or ANOVA and Tukey’s multiple comparison post-test (G-H). These results were confirmed in at least three independent experiments.
DISCUSSION
LIF is a tumor-secreted factor that induces cachexia-associated anorexia and adipose loss through actions on the hypothalamus and increased lipolysis in adipose tissue.6 In this study, we demonstrated that the murine colon adenocarcinoma C26c20 model has a serum signature of increased LIF and IL-6 and decreased leptin, generalizable across multiple in vivo cachexia tumor models. Consistent with the serum findings, there was also an increase in IL-6 and a decrease in leptin mRNA expression in adipose tissue of cancer bearing mice. These IL-6 and leptin changes in serum and adipose mRNA expression levels were consistently altered in both the C26c20 cancer cachexia model and the rLIF-administered cachexia model. We next considered whether LIF’s induction of cachexia was a consequence of its direct effect on target tissues or due to the upregulation of the other cachexia factor, IL-6. LIF’s ability to induce cachexia in vivo was independent of IL-6 since it still promoted cachexia-associated anorexia and wasting in IL-6−/− mice. However, this LIF-induced systemic increase of IL-6 likely enhanced LIF’s overall contribution to cachexia development, with IL-6 able to also stimulate adipocyte lipolysis (Figure 4A), alter appetite through hypothalamic signaling,26 and promote the further amplification of systemic IL-6 expression (Figure 4C). The cytokine-mediated induction of IL-6 mRNA expression was also observed in vitro when differentiated adipocytes were stimulated by LIF or IL-6. We used this model to show that cytokine-mediated IL-6 mRNA induction was not dependent on lipolysis, but rather on JAK-STAT pathway activation. Use of inhibitors of JAK, a common signaling pathway of LIF and IL-6, suppressed cachexia development in rLIF-treated mice and C26c20 tumor-bearing mice, resulting in decreased cachexia-associated anorexia (Figure 6A, 6E, and 7C) and adipose loss (Figures 6C, 6G, and 7D) that correlated with a parallel decrease in STAT3 phosphorylation in the hypothalamus (Figures 6I and 7F) and adipose tissue (Figures 6J and 7F). Furthermore, JAK inhibition in these cachexia models normalized cytokine-driven alterations in IL-6 and LIF serum levels (Figures 7G and 7H) during time points of effective inhibition of STAT3 phosphorylation of the adipose tissue (Figure 7F). Inhibiting the JAK-STAT pathway in target tissues resulted in an approximate 20–30% increase in median survival (Figure 7A), importantly, without significant change in primary tumor size (Figure 7B).
Targeting single molecules such as TNFα, IL-6, or ghrelin have not met the threshold for becoming standard of care treatments for cachexia.27–30 The lack of a durable therapy could be due to the following: 1) the previously targeted molecules are not relevant to all types of cachexia; 2) each patient’s cachexia may be driven by a unique set of factors; and/or 3) there are multiple factors upregulated in cachexia that contribute to the cachexia phenotype. Our results favor the third hypothesis. They suggest how a single cytokine, such as LIF, amplifies its own signal through targeting of multiple tissues including the hypothalamus and adipose, changing circulating levels of multiple cytokines including IL-6 and the adipokine leptin that work synergistically to promote the cachexia-associated appetite and body composition changes. We hypothesize that cancer cachexia-associated tumors and/or chronic inflammation exploit an intrinsic signaling axis between the immune system, adipose and the brain to enhance anorexia and wasting, reducing survival in cancer models. Tumor- or immune-secreted cytokines, such as LIF, act on the adipose tissue to promote lipolysis and alter the release of appetite regulating molecules IL-6 and leptin. LIF can also induce expression of IL-6 in other cell types, such as fibroblasts31 and myotubules.32 These altered levels of cyto/adipokines can also act directly on the hypothalamus and other parts of the brain to regulate appetite.26, 33 Additionally, others have shown that IL-6 can also cause cachexia-associated muscle atrophy in a STAT3 dependent manner.34 We are encouraged that a simplified rLIF-injected mouse model is appropriate to study cachexia since a recent publication showed that genetic silencing of LIF from the C26 parental tumor line led to an anticipated decrease in systemic levels of IL-6 with suppression of the cachexia phenotype.35 Therefore, our studies support the premise that cachexia patients have multiple circulating cachexia-inducing factors at any given time explaining why therapeutic interventions targeting a single molecule have been ineffective.
Because of the heterogeneity in potential factors driving the cachexia phenotype, there is a need to identify the common downstream signaling pathways to elucidate targets to permit sustained responses. To block LIF and its activation of target tissues, an inhibitor would have to block not only LIF’s direct effects centrally on anorexia and peripherally on adipose tissue but also its indirect effects on these tissues from changing other cyto/adipokine serum levels. Therefore, we inhibited JAK since both LIF and IL-6 use this pathway when signaling target tissues. With evidence that both tofacitinib and ruxolitinib could block lipolysis and induction of IL-6 in our in vitro cachexia adipocyte assay, we tested these compounds in our in vivo cachexia models. Both JAK inhibitors independently blocked rLIF- and cancer- induced cachexia, with suppression of anorexia and adipose/body weight loss. In the C26c20 cancer cachexia model, the JAK inhibitors improved median overall survival. The compounds were most effective in blocking cachexia-induced anorexia and adipose loss when they were able to suppress adipose and hypothalamic STAT3 phosphorylation. This data supports the importance of both anorexia and adipose loss to the cachexia phenotype. As shown in the aggressive C26c20 cancer cachexia model, these JAK inhibitors were not as effective sustaining the cachexia suppression durably. There are several reasons why these JAK inhibitors are less effective long term: 1) the pharmacokinetics of these JAK inhibitors are not optimized, 2) these JAK inhibitors are not optimally blocking the activated JAK subclass up regulated by the cachexia factors, 3) these JAK inhibitors could be adversely affecting the negative regulators of JAK-STAT signaling, and 4) JAK independent pathways are being up regulated in the cancer cachexia model. Further studies will need to be done to optimize the pharmacokinetics of the JAK inhibitors used in these studies and to identify other selective inhibitors that block the subclass of JAK molecules regulating the cytokine-mediated signaling. It will also be important to evaluate the effect of these JAK inhibitors on the STAT3 negative feedback regulators including protein tyrosine phosphatases, suppressors of cytokine signaling, and protein inhibitor of activated STAT. The effect of each JAK inhibitor on tumor growth kinetics and immune modulation will also need to be evaluated.
Despite the completion of hundreds of prospective clinical trials, there are no universally accepted therapies for cancer cachexia. One hypothesis for this absence of clinical options is that patients accrued on these studies had cachexia that was end-stage, i.e. treatment refractory. These failed outcomes were part of our rationale in treating our cancer cachexia models with JAK inhibition at tumor induction and also why it could be a consideration when managing patients with cancers highly associated with cachexia. Obviously, being able to use biomarkers, such as LIF, IL-6, and leptin, to predict cancer patients at very high risk for subsequent cachexia development could potentially allow us to optimize JAK inhibition use clinically. With the work from this paper and further studies, we anticipate identifying such a cachexia signature that could be validated in future clinical trials to risk stratify therapeutic use.
In patients with myelofibrosis, ruxolitinib improved clinical symptoms and survival.14 Those who received ruxolitinib also had an ~3% weight gain compared to those receiving placebo who had ~2% weight loss. The patients receiving ruxolitinib who gained weight also had a decrease in serum IL-6 and an increase in serum leptin. The changes in serum cyto/adipokine levels of ruxolitinib-treated patients are consistent with a reversed cachexia cyto/adipokine serum signature. These clinical findings support the importance of cytokine signaling to human physiology and pathology.
In summary, our studies demonstrate the inherent complexity of treating patients with cancer cachexia, since the presence of even one cachexia factor can amplify its signal by altering the levels of other independently-acting molecules that induce cachexia/anorexia. Due to these changes of serum cyto/adipokines in cancer cachexia, our data offers an explanation for why therapies targeting single molecules have been ineffective in curtailing cachexia progression. Our current findings indicate that targeting common pathways, such as JAK, of cytokine-mediated signaling in the adipose and centrally will suppress the cancer cachexia phenotype of hypophagia, muscle atrophy, adipose loss, and body weight loss, resulting in improved quality of life and survival.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michael Brown and Joseph Goldstein for their continued mentorship and valuable suggestions. We also thank Jay Horton and Philipp Scherer for their valuable suggestions. We appreciate the laboratory of Joel Elmquist for help with hypothalamic processing. We thank Ijeoma Dukes and Lisa Beatty for cell culture assistance. This work was supported by the Burroughs Wellcome Fund Career Awards for Medical Scientists (1019692); American Cancer Society grants 133889-RSG-19-195-01-TBE and IRG-17-174-13; American Gastroenterological Association grant 2019AGARSA3; Cancer Prevention and Research Initiative of Texas (RP200170); V Foundation V Scholar Program (V2019-014); and National Institute of Health grants 5P01-HL20948, P30CA142543, and 5T32GM007062-44.
Abbreviations:
- IL-6
interleukin 6
- JAK
janus kinase
- LIF
leukemia inhibitory factor
- rLIF
recombinant leukemia inhibitory factor
- STAT3
signal transducer and activator of transcription 3
Footnotes
ETHICAL STANDARDS
All authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle: update 2017.36
Conflict of Interest: The authors disclose no conflicts
REFERENCES
- 1.Gannavarapu BS, Lau SKM, Carter K, et al. Prevalence and Survival Impact of Pretreatment Cancer-Associated Weight Loss: A Tool for Guiding Early Palliative Care. J Oncol Pract 2018;14:e238–e250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lau SKM, Gannavarapu BS, Carter K, et al. Impact of Socioeconomic Status on Pretreatment Weight Loss and Survival in Non-Small-Cell Lung Cancer. J Oncol Pract 2018;14:e211–e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–66. [DOI] [PubMed] [Google Scholar]
- 4.Das SK, Eder S, Schauer S, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011;333:233–8. [DOI] [PubMed] [Google Scholar]
- 5.Kays JK, Shahda S, Stanley M, et al. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J Cachexia Sarcopenia Muscle 2018;9:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arora GK, Gupta A, Narayanan S, et al. Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang MT, Fer N, Galeas J, et al. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat Commun 2019;10:3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Gao W, Lytle NK, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019;569:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol 2016;54:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bates SH, Stearns WH, Dundon TA, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003;421:856–9. [DOI] [PubMed] [Google Scholar]
- 11.Vaisse C, Halaas JL, Horvath CM, et al. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 1996;14:95–7. [DOI] [PubMed] [Google Scholar]
- 12.Heinrich PC, Behrmann I, Muller-Newen G, et al. Interleukin-6-type cytokine signalling through the gp130/ Jak/STAT pathway. Biochem J 1998;334 (Pt 2):297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan K, Chen J, Xu A. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;371:1163–4. [DOI] [PubMed] [Google Scholar]
- 14.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med 2017;376:1723–1736. [DOI] [PubMed] [Google Scholar]
- 16.Reed BC, Lane MD. Insulin receptor synthesis and turnover in differentiating 3T3-L1 preadipocytes. Proc Natl Acad Sci U S A 1980;77:285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao M, Vishvanath L, Busbuso NC, et al. De novo adipocyte differentiation from Pdgfrbeta(+) preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat Commun 2018;9:890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soda K, Kawakami M, Kashii A, et al. Characterization of mice bearing subclones of colon 26 adenocarcinoma disqualifies interleukin-6 as the sole inducer of cachexia. Jpn J Cancer Res 1994;85:1124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller A, McLeod L, Alhayyani S, et al. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 2016. [DOI] [PubMed] [Google Scholar]
- 20.Petruzzelli M, Schweiger M, Schreiber R, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab 2014;20:433–47. [DOI] [PubMed] [Google Scholar]
- 21.Green ED, Maffei M, Braden VV, et al. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res 1995;5:5–12. [DOI] [PubMed] [Google Scholar]
- 22.Hudson KR, Vernallis AB, Heath JK. Characterization of the receptor binding sites of human leukemia inhibitory factor and creation of antagonists. J Biol Chem 1996;271:11971–8. [DOI] [PubMed] [Google Scholar]
- 23.Muccioli GG, Labar G, Lambert DM. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem 2008;9:2704–10. [DOI] [PubMed] [Google Scholar]
- 24.Iglesias J, Lamontagne J, Erb H, et al. Simplified assays of lipolysis enzymes for drug discovery and specificity assessment of known inhibitors. J Lipid Res 2016;57:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lass A, Zimmermann R, Oberer M, et al. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 2011;50:14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timper K, Denson JL, Steculorum SM, et al. IL-6 improves energy and glucose homeostasis in obesity via enhanced central IL-6 trans-signaling. Cell Rep 2017;19:267–280. [DOI] [PubMed] [Google Scholar]
- 27.Marcora SM, Chester KR, Mittal G, et al. Randomized phase 2 trial of anti-tumor necrosis factor therapy for cachexia in patients with early rheumatoid arthritis. Am J Clin Nutr 2006;84:1463–72. [DOI] [PubMed] [Google Scholar]
- 28.Wu C, Fernandez SA, Criswell T, et al. Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013;42:813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Temel JS, Abernethy AP, Currow DC, et al. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol 2016;17:519–531. [DOI] [PubMed] [Google Scholar]
- 30.Bayliss TJ, Smith JT, Schuster M, et al. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther 2011;11:1663–8. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen HN, Noss EH, Mizoguchi F, et al. Autocrine Loop Involving IL-6 Family Member LIF, LIF Receptor, and STAT4 Drives Sustained Fibroblast Production of Inflammatory Mediators. Immunity 2017;46:220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seto DN, Kandarian SC, Jackman RW. A key role for leukemia inhibitory factor in C26 cancer cachexia. J Biol Chem 2015;290:19976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao S, Zhu Y, Schultz RD, et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab 2019;30:706–719.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonetto A, Aydogdu T, Jin X, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandarian SC, Nosacka RL, Delitto AE, et al. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J Cachexia Sarcopenia Muscle 2018;9:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Haehling S, Morley JE, Coats AJS, et al. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.