

Supplemental Digital Content is available in the text.
Keywords: apolipoproteins, atherosclerosis, gastrointestinal microbiome, hepatocytes, lipoproteins, pharmacology, Toll-like receptors
Abstract
Rationale:
Dysbiosis of gut microbiota plays an important role in cardiovascular diseases but the molecular mechanisms are complex. An association between gut microbiome and the variance in HDL-C (high-density lipoprotein-cholesterol) level was suggested in a human study. Besides, dietary fat was shown to increase both HDL-C and LDL-C (low-density lipoprotein-cholesterol) levels. We speculate that certain types of gut bacteria responding to dietary fat may help to regulate HDL-C level and potentially affect atherosclerotic development.
Objective:
We aimed to investigate whether and how high-fat diet (HFD)-associated gut microbiota regulated HDL-C level.
Methods and Results:
We found that HFD increased gut flagellated bacteria population in mice. The increase in HDL-C level was adopted by mice receiving fecal microbiome transplantation from HFD-fed mouse donors. HFD led to increased hepatic but not circulating flagellin, and deletion of TLR5 (Toll-like receptor 5), a receptor sensing flagellin, suppressed HFD-stimulated HDL-C and ApoA1 (apolipoprotein A1) levels. Overexpression of TLR5 in the liver of TLR5-knockout mice was able to partially restore the production of ApoA1 and HDL-C levels. Mechanistically, TLR5 activation by flagellin in primary hepatocytes stimulated ApoA1 production through the transcriptional activation responding to the binding of NF-κB (nuclear factor-κB) on Apoa1 promoter region. Furthermore, oral supplementation of flagellin was able to stimulate hepatic ApoA1 production and HDL-C level and decrease atherosclerotic lesion size in apolipoprotein E-deficient (Apoe−/−) mice without triggering hepatic and systemic inflammation. The stimulation of ApoA1 production was also seen in human ApoA1-transgenic mice treated with oral flagellin.
Conclusions:
Our finding suggests that commensal flagellated bacteria in gut can facilitate ApoA1 and HDL-C productions in liver through activation of TLR5 in hepatocytes. Hepatic TLR5 may be a potential drug target to increase ApoA1.
In This Issue, see p 1217
Meet the First Author, see p 1218
HDL (High-density lipoprotein) is strongly associated with reduced cardiovascular risk.1–3 The apolipoprotein on HDL, ApoA1, facilitates reverse cholesterol transport by acquiring phospholipids and cholesterol through its interaction with ATP-binding cassette transporters on macrophages, thus preventing foam cell formation, a key etiological feature of atherogenesis.4 Low-fat diet (LFD) aiming to decrease LDL-C (low-density lipoprotein-cholesterol) has been advocated for decades to improve cardiovascular health, but several clinical studies fail to show any reduced risk.5–7 One of the reasons is due to the parallel decrease in HDL-C (high-density lipoprotein-cholesterol) level by low fat intake.8,9 People with normolipidemia are not benefited upon LFD compared with those with dyslipidemia owing to the unchanged LDL-C/HDL-C ratio.10 In mice, high-fat diet (HFD) treatment increases both HDL-C and LDL-C levels.11 How dietary fat leads to increased HDL-C remains unclear.
Gut microbiota refers to the trillions of bacteria residing in our gastrointestinal tract which exerts commensal effects to us as host. Dysbiosis of gut microbiota is associated with various diseases, including metabolic and cardiovascular diseases. HFD can weaken gut integrity leading to metabolic endotoxemia, and such effect is partially due to the decrease in a particular strain of gut bacteria, Akkermansia muciniphila.12,13 Oral supplementation of A muciniphila by preserving gut integrity ameliorates systemic inflammation and subsequently protects against obesity and atherosclerosis.12,13 Bacteroides vulgatus and Bacteroides dorei are able to inhibit atherosclerosis through reducing gut microbial lipopolysaccharide production.14 Conversely, a positive correlation between circulating trimethylamine N-oxide, a bacteria-derived metabolite from dietary choline, and cardiovascular risk is observed in human.15 These studies suggest that different types of gut microbiota can yield different medical outcomes.
Human gut microbiome is found to partially explain the variance in triglyceride and HDL-C, but not LDL-C and total cholesterol levels, independent of age, gender, and genetic risk factors.16 As gut microbiota is readily changed upon diet, the LFD-lowered HDL-C level may be associated with the alteration of gut bacteria. Here, we boldly hypothesized that the HFD-induced change of gut microbiota facilitates the increase in HDL-C level, possibly to counterbalance the metabolic adversity. In this study, we used various in vivo and in vitro models to unveil a unique mechanism of how HFD induces HDL-C level. HFD favors the growth of flagellated bacteria in mice, resulting in increased hepatic content of flagellin and HDL-C level, and the activation of TLR5 (Toll-like receptor 5) by flagellin in hepatocytes leads to ApoA1 production. Hepatic TLR5 may serve as a therapeutic target to increase HDL-C level.
Methods
Data Availability
A detailed description of materials and methods is provided in the Materials and the Major Resource Table in the Data Supplement.17–28 The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Statistical Analysis
Statistical analyses were performed using Statistical Package for Social Sciences Version 23.00 (IL, United States) and R software Version 3.6.2. A power of 95% was achieved for this study, on the 2 independent group design, 5% significance, and the difference of 10% for HFD-induced HDL-C level with SD of 5 mg/dL. Data were presented as mean±SE of mean (SEM) or SD as appropriate. An independent Student t test was applied for 2 groups comparison with normal distribution, or Mann Whitney U test for nonparametric test. The difference in gut microbiota pattern was evaluated by the permutational ANOVA using the Adonis function from the vegan package in R programme. One-way ANOVA was applied for comparisons among different antibiotics treatment groups, followed by post hoc analysis using Tukey HSD for data with equal variance or Games-Howell for data with unequal variance. Two-way ANOVA followed by post hoc analysis using Tukey HSD was conducted for comparison in Figure 4. P values were adjusted by the Benjamini-Hochberg method when multiple comparison was applied for Figures 1C, 2G, and 5A. Normality was checked using the Shapiro-Wilk test. Partial Spearman correlation analysis adjusted with age and body mass index was applied for human data. All significance tests were 2-tailed and the statistical significance level was defined as 0.05 presented by P value.
Figure 4.
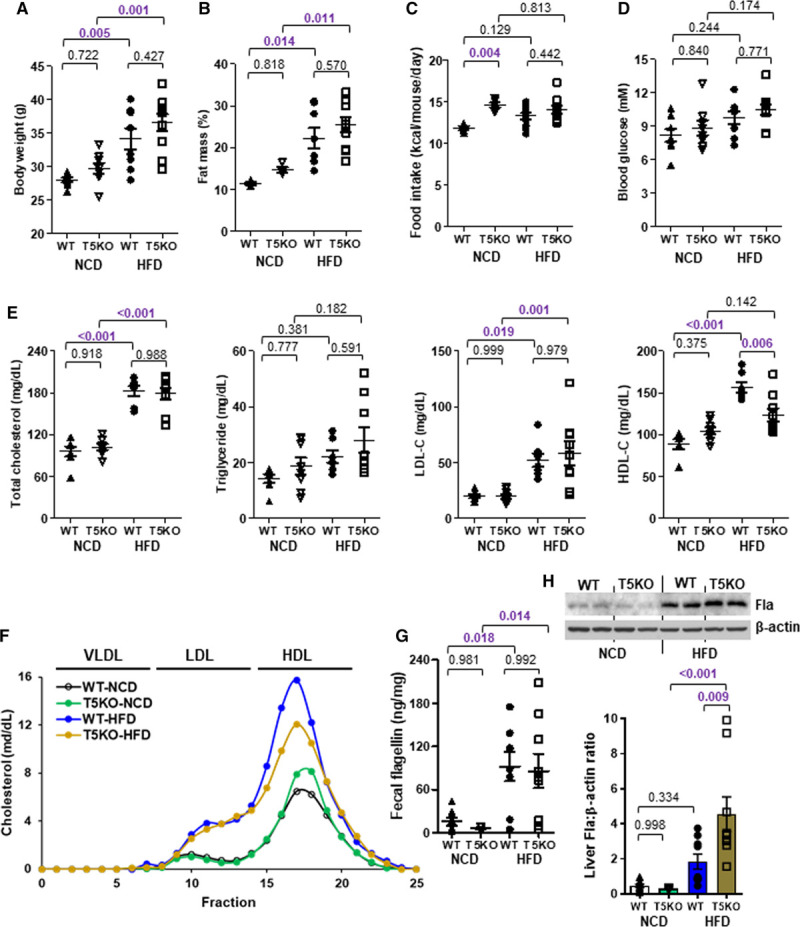
TLR5 (Toll-like receptor 5) deficiency decreases high-fat diet (HFD)-induced HDL-C (high-density lipoprotein-cholesterol) level. Six-week-old wild-type (WT) and T5KO (TLR 5-knockout) mice were cohoused and fed a normal chow diet (NCD) or HFD for 10 wk. A, Body weight, (B) fat mass percentage, (C) food intake, (D) 5-h fasting blood glucose, and (E) serum total cholesterol, triglyceride, LDL-C (low-density lipoprotein-cholesterol) and HDL-C levels were measured. F, Serum samples were pooled and separated by fast protein liquid chromatography, followed by cholesterol measurement for each fraction. G, Fecal and (H) hepatic flagellin (Fla) contents were determined. Representative images are shown. Data are represented as mean±SEM (n=9 for each group).
Figure 1.
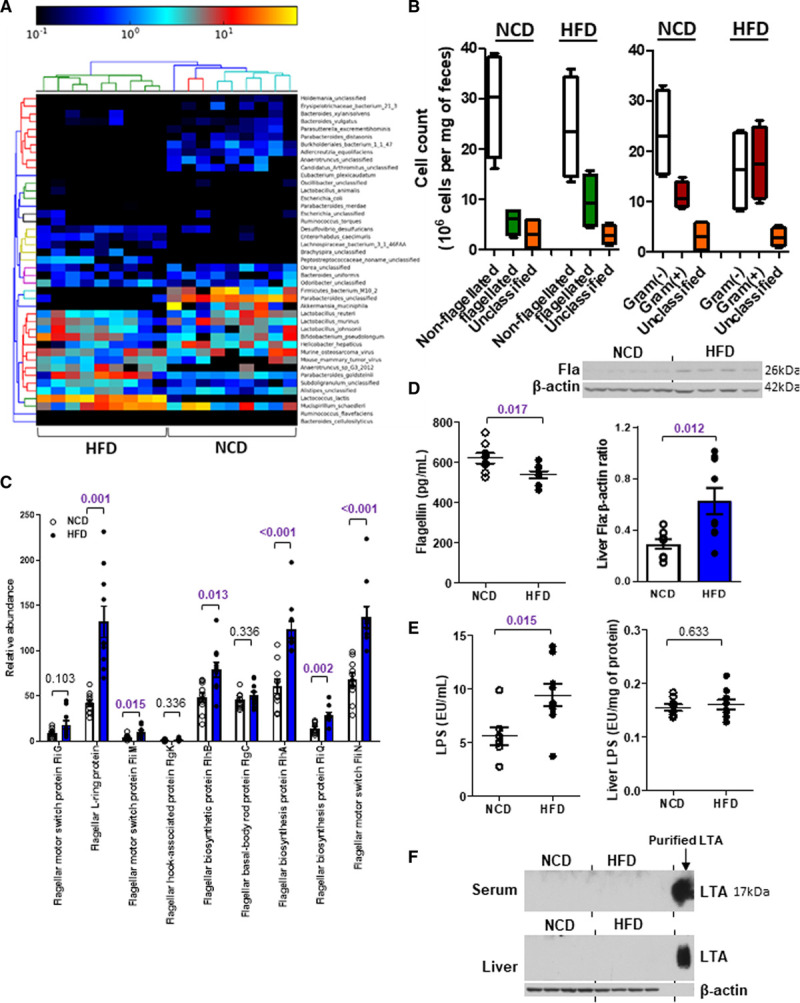
High-fat diet (HFD) increases flagellated bacteria in gut. Six-week-old wild-type mice were fed a normal chow diet (NCD) or HFD for 10 wk. A, The heat map shows the relative abundance and strains of bacteria in feces commonly present in both dietary groups (permutational ANOVA; PERMANOVA: P=0.001, df=17). B, The abundances of flagellated and nonflagellated (PERMANOVA: R2=0.106, P=0.186, df=17) or Gram(−) and Gram(+) bacteria (PERMANOVA: R2=0.331, P=0.003, Df=17) were calculated. C, The relative abundances of various genes representing flagellated bacteria were determined by MEGAN6. P values were adjusted for multiple testing. D–F, The concentrations of (D) flagellin (Fla), (E) lipopolysaccharide (LPS) and (F) lipoteichoic acid (LTA) were measured in serum and liver. Purified LTA served as the positive control in F. Representative images are shown. Data are represented as mean±SEM (n=9 for each group).
Figure 2.
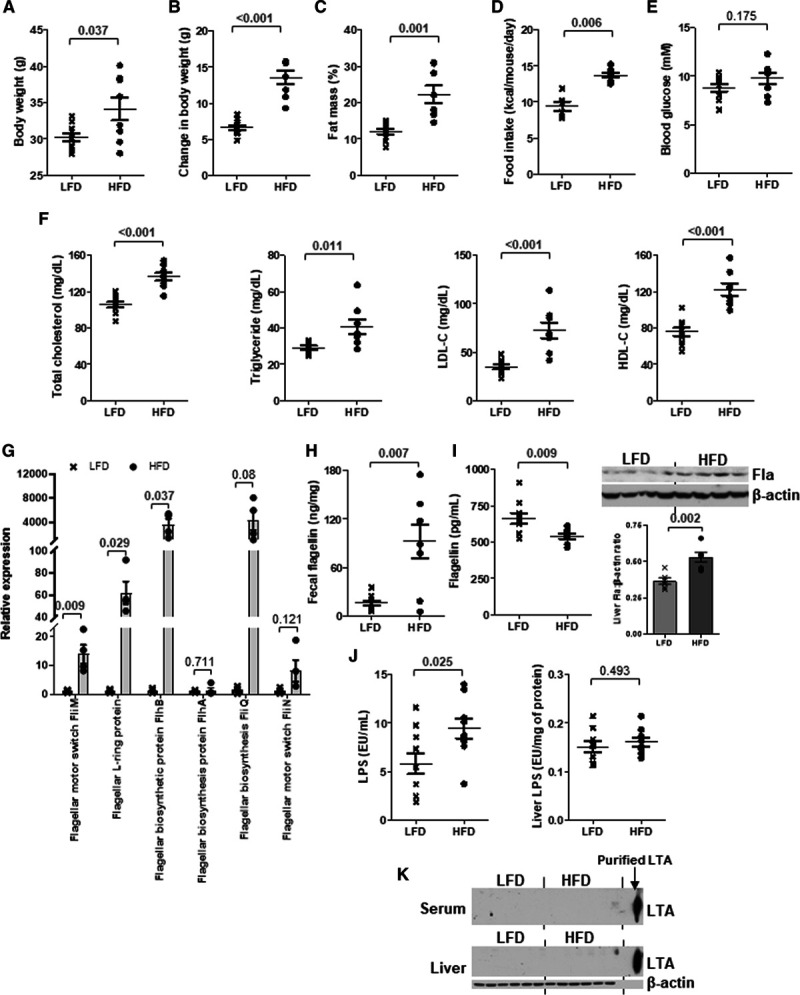
Dietary fat induces flagellar genes and hepatic flagellin (Fla). Six-week-old wild-type mice were fed a low-fat diet (LFD) or high-fat diet (HFD) for 10 wk. A, Body weight, (B) change in body weight, (C) fat mass percentage, (D) food intake, (E) 5-h fasting blood glucose, (F) serum total cholesterol, triglyceride, LDL-C (low-density lipoprotein-cholesterol), and HDL-C (high-density lipoprotein-cholesterol) levels were measured. G, The relative expressions of various genes representing flagellated bacteria in feces were examined by real-time polymerase chain reaction with 16S rRNA gene as housekeeping gene. H, Fecal, (I) serum and Fla levels, (J) lipopolysaccharide (LPS), and (K) lipoteichoic acid (LTA) levels in serum and liver were determined. Purified LTA served as positive control in K. Representative images are shown. Data are represented as the mean±SEM (n=10 for each group).
Figure 5.

The high-fat diet (HFD)-induced HDL-C (high-density lipoprotein-cholesterol) level is associated with ApoA1 (apolipoprotein A1) expression in liver. A–C, Six-week-old wild-type (WT) and T5KO (TLR 5-knockout) mice were cohoused and fed HFD for 10 wk. A, The mRNA expressions of key genes regulating lipoprotein trafficking and reverse cholesterol transport were determined. Actb was used as housekeeping gene. P values were adjusted for multiple testing. B, ApoA1 and ApoE proteins in liver, and (C) ApoA1 in serum were examined. D and E, Eight-week-old WT mice received fecal microbiome transplantation from normal chow diet (NCD)- or HFD-fed donor mice. The expressions of ApoA1 in (D) liver and (E) serum were examined. F–H, Six-week-old T5KO mice were fed HFD for 5 wk, and received one injection of Ad-GFP (Ad-green fluorescent protein) or Ad-TLR5 (5×109 viral particles) at the fourth week. F, Reconstitution was confirmed by the GFP signal in liver. Nuclei were stained with DAPI (blue). ApoA1 protein levels in (G) liver and (H) serum were determined. Representative images are shown. Data are represented as mean±SEM (A–C, n=10; D–E, n=7; F–H, n=5 for each group).
Results
HFD-Induced HDL-C and Gut Flagellated Bacteria Population in Mice
Six-week-old wild-type C57BL/6J mice were fed a normal chow diet (NCD) or an HFD (45% kcal fat diet) for 10 weeks. We observed an elevation of HDL-C level in the HFD group, along with higher body weight, fat mass, serum cholesterol, and LDL-C levels compared with the NCD group (Figure IA through ID in the Data Supplement). Fecal DNA was isolated from both groups of mice and subjected to whole-genome shotgun metagenomic analysis. The nonmetric multidimensional scaling plot using the Bray-Curtis dissimilarity index indicated a distinct pattern of gut microbiota for each dietary group (Figure IE in the Data Supplement). Numerous strains were predominantly present in one diet but not the other (Figure 1A). Among these bacteria, the nonflagellated bacteria dominated regardless of the diet, but HFD increased the population of flagellated bacteria (Figure 1B). The linear discriminant analysis effect size revealed that the abundances of 22 strains were significantly different between NCD and HFD groups. Among those with higher abundance in NCD group, ≈33% of the species were flagellated, while for those higher in HFD group, it was ≈70% (Figure IF in the Data Supplement). The functional analysis of these bacterial DNAs using MEGAN6 and HUMAnN2 congruently demonstrated an increased abundance of genes representing flagellated bacteria upon HFD (Figure 1C; Figure IIA in the Data Supplement). Furthermore, a higher level of flagellin, a structural monomer of bacterial flagellum, in liver and a lower level in serum were detected in the HFD-treated mice than in the NCD group (Figure 1D). The levels of flagellin in spleen and intestine were undetectable (Figure IIB through IIC in the Data Supplement). By contrast, HFD increased circulating lipopolysaccharide levels but the hepatic level remained the same as that of NCD-fed mice, whereas lipoteichoic acid level was undetectable in both liver and serum (Figure 1E through 1F). Owing to the concern that the alteration of gut microbiota might be contributed by the different fiber contents between grain-based NCD and purified HFD,29 we included another group of mice fed a purified LFD (10% kcal fat diet). The differences in body weight, fat mass, food intake, blood glucose, serum lipids, LDL-C, and HDL-C levels between LFD and HFD groups were similar to those between NCD and HFD groups (Figure 2A through 2F). The HFD-increased fecal flagellin level, flagellar genes in gut bacteria, hepatic flagellin (Fla) content, and circulating lipopolysaccharide level were still observed when comparing with LFD-fed mice (Figure 2G through 2J), while lipoteichoic acid level remained undetected (Figure 2K). These results implicate that dietary fat favors the growth of flagellated bacteria in gut and gut bacteria-derived flagellin tends to distribute in liver instead of circulation.30
HFD-Induced HDL-C Mediated by TLR5 and Gut Flagellated Bacteria
To validate whether there was a cause-effect relationship between gut microbiota and HDL-C level through HFD, we adopted a previous protocol of fecal microbiome transplantation with modifications to repopulate the gut microbiome in recipients with feces from donor mice fed either HFD or NCD.31 The mice receiving fecal microbiome of HFD donor showed a higher flagellin content in feces and liver than those with microbiome of NCD donor, and the level in circulation remained unchanged (Figure 3A through 3C). The body weight, fat mass, and blood glucose of recipients were similar in both groups (Figure 3D through 3F). The HFD-induced HDL-C level but not triglyceride, total cholesterol, and LDL-C levels was adopted by the recipient mice (Figure 3G). Moreover, when HFD-fed wild-type mice were partially depleted of flagellated bacteria using a selective nonabsorbable antibiotic, aztreonam (Table I in the Data Supplement), the HFD-induced HDL-C level was suppressed without affecting body weight, fat mass, blood glucose, total serum lipids, and LDL-C levels (Figure IIIA through IIID in the Data Supplement). Aztreonam was able to decrease Fla but not lipopolysaccharide content (Figure IIIE and IIIF in the Data Supplement). Conversely, mice were treated with a 100% orally absorbable antibiotic, linezolid (Table I in the Data Supplement), and there was no inhibition of HDL-C level (Figure IIIA through IIIF in the Data Supplement). Given the fact that TLR5 is the plasma membrane-associated pattern recognition receptor that recognizes flagellin in innate immunity,30,32 we then examined whether TLR5 participated in the HFD-induced HDL-C level. Wild-type and Tlr5−/− mice were fed a NCD and HFD for 10 weeks. The Tlr5−/− mice showed a higher body weight, fat mass, food intake, and blood glucose compared with the wild-type littermates for both types of diet, which is congruent with a previous study,33 but only food intake reached statistical difference (Figure 4A through 4D). Without altering the HFD-elevated triglyceride, total cholesterol, and LDL-C levels, deletion of TLR5 partially abolished the HFD-induced HDL-C level despite an increased level of Fla (Figure 4E through 4H). The aztreonam-suppressed HDL-C level was also diminished in Tlr5−/− mice and LDL-C level remained unaltered. The increases in triglyceride and total cholesterol levels were possibly owing to some off-target effects (Figure IIIG in the Data Supplement). These data suggest that both flagellated bacteria and host TLR5 are required to increase HDL-C level.
Figure 3.
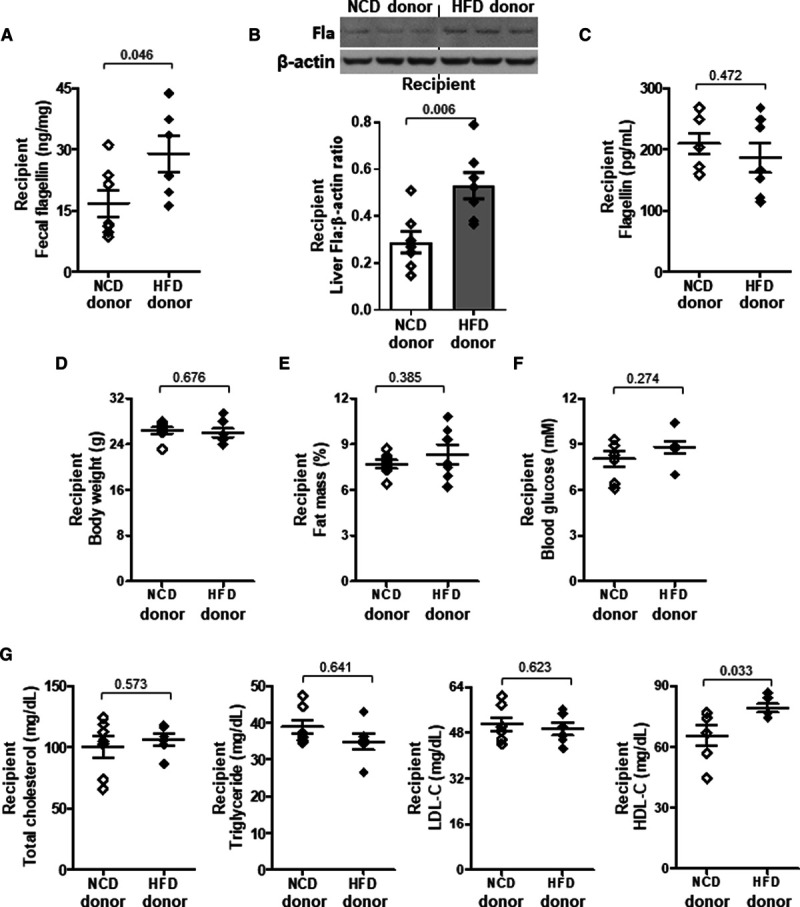
HDL-C (High-density lipoprotein-cholesterol) level is elevated by the transplantation of fecal microbiome from high-fat diet (HFD)-fed mice. Eight-week-old wild-type mice received fecal microbiome transplantation from normal chow diet (NCD)- or HFD-fed donor mice. Mice were euthanized 24 h after the last dose of fecal suspension. A, Fecal, (B) hepatic, and (C) serum flagellin (Fla) levels were examined. D, Body weight, (E) fat mass percentage, (F) 5-h fasting blood glucose, and (G) serum total cholesterol, triglyceride, LDL-C (low-density lipoprotein-cholesterol), and HDL-C levels were determined. Representative images are shown. Data are represented as mean±SEM (n=7 for each group).
Stimulation of ApoA1 Production and HDL-C Level by Hepatic TLR5
To explore how flagellin/TLR5 affected HDL-C level, the key genes that regulate lipoprotein trafficking and reverse cholesterol transport in liver were examined in HFD-fed mice. The mRNA expressions of Apoa1 and Apoe were inhibited in the liver of Tlr5−/− mice (Figure 5A). These 2 TLR5-associated genes were stimulated by dietary fat (Figure IV in the Data Supplement). Nonetheless, at the protein level, only ApoA1 but not ApoE was decreased in the liver of Tlr5−/− mice (Figure 5B), and the hepatic ApoA1 level projected to the decrease in circulation (Figure 5C). Increases in ApoA1 levels were also observed in the liver and circulation of mice receiving fecal microbiome from HFD-fed donors (Figure 5D and 5E). Moreover, aztreonam was able to suppress ApoA1 mRNA and protein expressions in liver of wild-type but not Tlr5−/− mice (Figure V in the Data Supplement). Next, to validate that the induction of ApoA1 by flagellin/TLR5 pathway took place in liver, we reconstituted TLR5 in the liver of Tlr5−/− mice using adenoviral gene delivery. Tlr5−/− mice received a single dose of either GFP (Ad-green fluorescent protein) or Ad-TLR5 at the fourth week of a 5-week HFD treatment. The positive GFP signal in the liver demonstrated the successful infection (Figure 5F). The hepatic and circulating ApoA1 expressions, and HDL-C level but not LDL-C or other serum lipids levels in Tlr5−/− mice were significantly increased by the reconstitution of TLR5 (Figure 5G and 5H; Figure VIA in the Data Supplement). When the TLR5 overexpression was applied to wild-type mice, similar results including higher ApoA1 and HDL-C levels were observed (Figure VIB through VIE in the Data Supplement). Altogether, our data support that gut bacteria-derived flagellin interacts with hepatic TLR5 and stimulates ApoA1 production.
Induction of ApoA1 Production in Hepatocytes by Flagellin-Activated TLR5/MyD88/NF-κB Pathway
Hepatocytes are the major cells that produce ApoA1 in liver. The treatment of flagellin (derived from Salmonella typhimurium) increased the protein and mRNA expressions of ApoA1 in wild-type but not in Tlr5−/− hepatocytes, without stimulating the expressions of Il1b and Il18, 2 key flagellin-sensitive cytokines (Figure 6A; Figure VIIA in the Data Supplement).34 Suppression of MyD88, the cytosolic adaptor transducing TLR5 signal, by siRNA inhibited the flagellin-induced ApoA1 expression in wild-type hepatocytes, and such inhibition was absent in Tlr5−/− cells (Figure 6B). MyD88/NF-κB (nuclear factor-κB) pathway can also be induced by the interaction between lipopolysaccharide and TLR4 or lipoteichoic acid and TLR2, but neither treatment with lipopolysaccharide nor lipoteichoic acid affected ApoA1 expression in both wild type and Tlr5−/− hepatocytes, indicating that the effect on ApoA1 was selective upon TLR5 activation (Figure VIIB and VIIC in the Data Supplement). Next, we constructed 3 reporter plasmids encoding different lengths of the promoter region of Apoa1 gene and transfected into Hela cells followed by flagellin treatment. The reporter signal was induced by flagellin when cells were transfected with pGL3/-1600+327mApoA1 but not the other 2 plasmids (Figure VIII in the Data Supplement). Chromatin immunoprecipitation analysis further validated the binding of the active subunit of NF-κB, p65, to the promoter region of Apoa1 gene in wild-type hepatocytes (Figure 6C). As the expressions of TLRs vary among different species, we repeated the in vitro experiment using human hepatocytes. Human HepG2 cells did not express TLR5 (Figure 6D), which was likely owing to the structural abnormalities in chromosome 1, where the TLR5 locus is located.35 Flagellin failed to stimulate ApoA1 expressions in such TLR5-knockout-like HepG2 cells (Figure 6E and 6F). Overexpression of TLR5 using pCMV-TLR5 was able to restore the flagellin-induced APOA1 mRNA and protein expressions (Figure 6E and 6F). By contrast, in human primary hepatocytes in which TLR5 was highly expressed (Figure 6G), flagellin was able to stimulate APOA1 mRNA expression and secretion in these cells (Figure 6H and 6I). Collectively, we showed that the flagellin-induced ApoA1 production was facilitated through the classical TLR5/MyD88/NF-κB pathway in hepatocytes.
Figure 6.
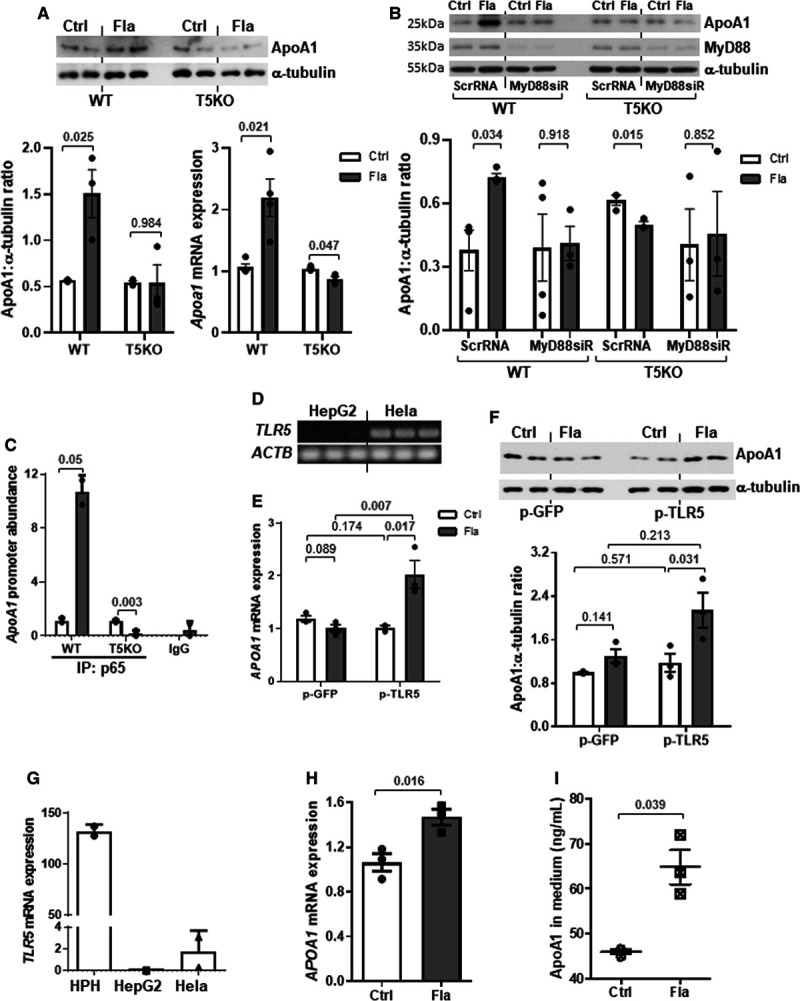
Flagellin stimulates ApoA1 (apolipoprotein A1) production in primary hepatocytes through activation of TLR5 (Toll-like receptor 5). A, Wild-type (WT) and T5KO (TLR 5-knockout) mouse hepatocytes were treated with or without flagellin (Fla, 100 ng/mL; Ctrl, control) for 8 h, and ApoA1 protein and mRNA expressions were examined. B, WT and T5KO hepatocytes were transfected with scrambled RNA (ScrRNA) or MyD88 siRNA (siR) for 24 h followed by flagellin treatment (100 ng/mL) for 8 h. ApoA1 and MyD88 proteins were determined. C, Mouse hepatocytes were treated with flagellin for 3 h, and the abundance of promoter region of Apoa1 was determined after chromatin immunoprecipitation with antibody against p65 or IgG. D, The TLR5 expressions in Hela and HepG2 cells were shown. HepG2 cells were transfected with GFP (green fluorescent protein) or TLR5 plasmid followed by treatment with or without flagellin, and (E) mRNA and (F) protein expressions of ApoA1 were determined. G, The TLR5 expression in human primary hepatocytes (HPH) was shown. HPH was treated with flagellin for 8 h and the (H) mRNA expression and (I) medium level of ApoA1 were determined. Actb was used as housekeeping gene. Representative images are shown. Data are represented as mean±SEM (A and B, n=4; C–F, n=3 independent experiments; G–I, n=3 individual donors).
Intestinal TLR5 Unable to Stimulate ApoA1 Protein Expression
Despite that flagellin-induced ApoA1 expression in hepatocytes (Figure 6A), we could not rule out that HFD might downregulate hepatic TLR5 and cause flagellin to act on other organs. Thus, we examined the TLR5 expression in liver, and no difference between NCD- and HFD-fed mice was found (Figure IX in the Data Supplement). ApoA1 is produced by both liver and intestine. We found that HFD conversely decreased ApoA1 level in intestine, but there was no difference between wild-type and Tlr5−/− mice (Figure XA and XB in the Data Supplement). The intestinal and circulating expressions of an intestine-specific and HDL-associated apolipoprotein A, ApoA4, was also unaffected by the deletion of TLR5 (Figure XC in the Data Supplement). In addition, we examined the effect of flagellin on ApoA1 expression ex vivo in mouse ileum and in vitro in human Caco-2 cell, which is an intestinal epithelial cell line. We did not observe a consistent TLR5-dependent alteration of ApoA1 in flagellin-treated mouse ileum and Caco-2 cells. Flagellin treatment stimulated only Apoa1 mRNA but not protein in both situations (Figure XD through XI in the Data Supplement). Although the HFD-suppressed intestinal ApoA1 expression requires further investigation, it indicates that the HFD-induced HDL-C was unlikely owing to the intestinal ApoA1 production or TLR5 expression.
Decreased Atherosclerotic Lesion by Oral Supplementation of Flagellin in Apoe−/− Mice
We next conducted a proof-of-concept study to examine whether flagellin could provide protection against atherosclerosis through stimulating ApoA1 using the classical atherosclerotic mouse model, Apoe−/− mice. In-house flagellin was prepared by shearing Salmonella typhimurium, and its induction on ApoA1 expression was blocked by pretreatment with neutralizing antibody (Figure XIA in the Data Supplement). To ensure a fixed amount of flagellin to be received and minimize its variation caused by diet, these mice were fed the NCD instead of HFD. To examine the tissue distribution of flagellin, mice were orally given fluorophore-labeled flagellin (0.2 mg/kg) or fluorophore alone and euthanized after 30 minutes, 90 minutes, and 6 hours. We found that fluorescent signal was transiently present in intestine from 30 to 90 minutes and the location of signal was similar for both mice treated with flagellin and dye alone. Meanwhile, fluorescent signals appeared in liver starting from 30 minutes and persisted until 6 hours in both groups, but increasing intensity over time was detected in the flagellin-treated group only (Figure 7A). No fluorescent signal was found in spleen. These suggest that flagellin is absorbed through intestine without retention and accumulates in liver. Moreover, the dose-response curve of the in-house flagellin demonstrated that 0.1 mg/kg (≈2.5 μg per mouse) and 0.2 mg/kg of flagellin were able to increase HDL-C level (Figure XIB in the Data Supplement). Higher dose might potentially trigger inflammation, so the dose of 0.1 mg/kg was used in the following study. Since residue of lipopolysaccharide was present in the in-house flagellin, 179 ng/kg of lipopolysaccharide (≈4.5 ng per mouse) was added into the vehicle group to match the amount in flagellin group. Supplementation of flagellin (≈2.5 µg per mouse) by oral gavage for 4 weeks to Apoe−/− mice was sufficient to increase HDL-C level (Figure 7B). No significant change in body weight, fat mass, serum triglyceride, total cholesterol levels was induced by flagellin in these Apoe−/− mice, except an increase in VLDL (very-low-density lipoprotein)/LDL-C level (Figure 7B; Figure XIC and XID in the Data Supplement). Such dose of in-house flagellin only increased hepatic, but not circulating and fecal flagellin levels in these mice, and the intestinal flagellin level was undetectable (Figure XIE through XIH in the Data Supplement), which is in line with the biodistribution of fluorophore-conjugated flagellin. The expressions of ApoA1 in liver and circulation of Apoe−/− mice were augmented by orally supplemented flagellin (Figure 7C and 7D). Because of the increased VLDL/LDL-C level, we also measured both ApoB and ApoA1 levels in the VLDL fraction. A higher level of ApoA1 but not ApoB level in this fraction was observed in the treatment group (Figure XIIA in the Data Supplement). There was no difference in serum ApoB levels between control and experimental groups in the HFD, fecal microbiome transplantation and Apoe−/− models (Figure XIIB through XIID in the Data Supplement). Thus, unlike ApoA1, there is no evidence to show a direct relationship between flagellin or TLR5 and ApoB level. With a higher level of ApoA1, the serum samples from mice treated with flagellin induced a higher efficiency in cholesterol efflux in macrophages, which was blocked by pretreatment of the serum with anti-ApoA1 antibody (Figure 7E). The lesion size and area were smaller in Apoe−/− mice treated with flagellin (Figure 7F), and fewer macrophages indicated by F4/80 staining were found in the lesions (Figure 7G). At last, we examined the flagellin effect in human ApoA1-transgenic mice which were generated by infecting Apoe−/− mice with adenovirus encoding human ApoA1 along with its promoter, and oral flagellin treatment was also able to increase human ApoA1 production (Figure 7H). Taken together, flagellin-induced ApoA1 production in liver is sufficient to amelioriate lesion formation.
Figure 7.
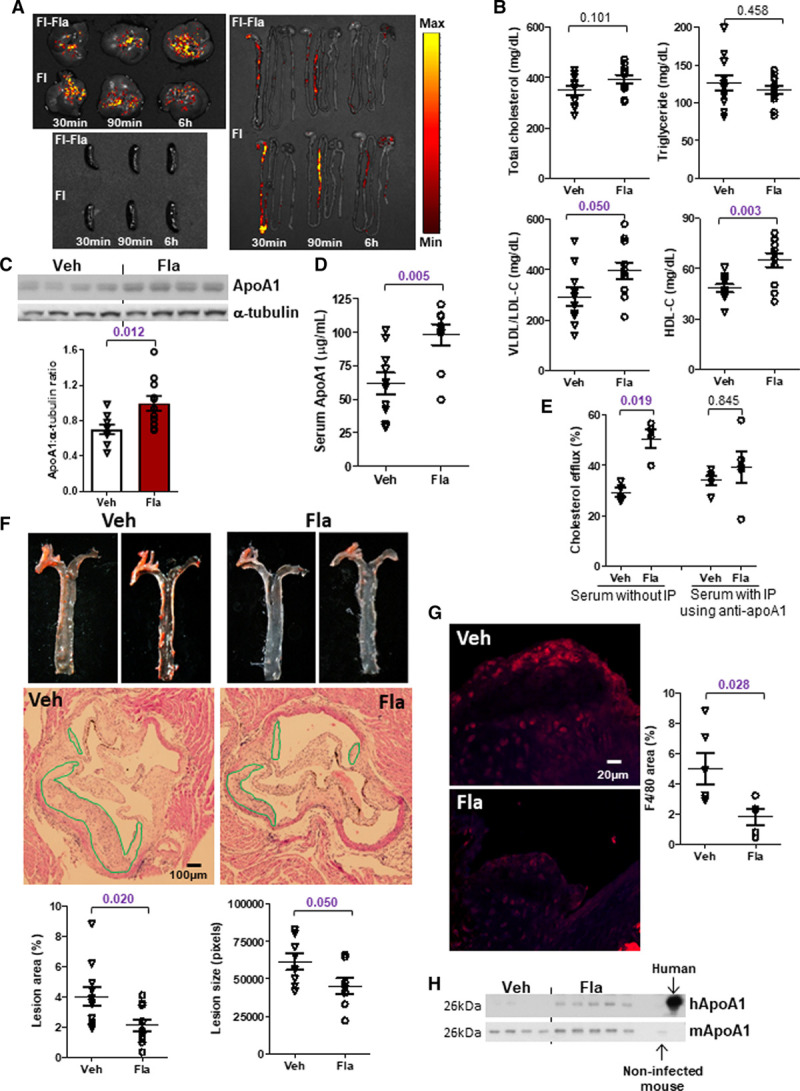
Oral supplementation of flagellin leads to smaller atherosclerotic lesions in ApoE-deficient mice. A, Fluorophore-conjugated flagellin (Fl-Fla) and the fluorophore alone (Fl) were orally administrated in 20-week old Apoe−/− mice, and distributions of fluorescence in liver, spleen, and gastrointestinal tract following various time points were shown. B, Serum total cholesterol, triglycerides, LDL-C (low-density lipoprotein-cholesterol), and HDL-C (high-density lipoprotein-cholesterol) levels, and ApoA1 (apolipoprotein A1) expressions in (C) liver and (D) serum were determined in 18- to 20–week-old normal chow diet (NCD)-fed Apoe−/− mice treated with flagellin (0.1 mg/kg) or vehicle (Veh) every other day for 4 wk. Mice were euthanized 24 h after the last dose of flagellin. E, Mouse primary macrophages were loaded with radiolabeled cholesterol, followed by the treatment with serum samples of mice treated as in B. Serum was pretreated with or without anti-ApoA1 immunoprecipitation (IP) and the percentage of intracellular cholesterol effluxed to medium was determined. F, The atherosclerotic lesions along the thoracic aorta and at the root of aortic arch were visualized by Oil Red O and H&E staining. G, Macrophage content in the atherosclerotic lesions was evaluated with immunofluorescent staining of F4/80 (red color). The quantification of lesion area and size, and red fluorescent signal were shown. H, Human and mouse ApoA1 expressions in serum were determined in 22-week-old NCD-fed human ApoA1-transgenic Apoe−/− mice treated with or without flagellin for 2 wk. Serum samples from noninfected mouse and human served as the positive controls. Representative images are shown. Data are represented as mean±SEM (A, n=2; B–D and F, n=10; E, G–H, n=5 for each group).
No Hepatic or Systemic Inflammation Elicited by Oral Supplementation of Flagellin in Apoe−/− Mice
The observation of NF-κB activation by flagellin in vitro raised a concern in the trigger of inflammation in the atherogenic mice. In line with the increase in flagellin level in liver (Figure XIG in the Data Supplement), we observed a higher degree of NF-κB activation indicated by nuclear p65 subunit in liver but not intestine in the Apoe−/− mice treated with the oral flagellin (Figure 8A). Other than TLR5, flagellin may also interact with NLRC4 (NLR family CARD domain-containing protein 4) to stimulate inflammation.34 However, there was no expression of NLRC4 in the liver of flagellin-treated mice, and NLRC4 expression in hepatocytes was negligible compared with macrophages (Figure 8B). Consistently, flagellin treatment did not induce caspase 1 activity in the liver of Apoe−/− mice (Figure 8C). Furthermore, the hepatic Il1b, Il18, and Ccl2 mRNA expressions and circulating IL (interleukin)-1β, sTNFRII (soluble tumor necrosis factor receptor type II), and CCL2 (C-C motif chemokine ligand 2) and SAA (serum amyloid A protein) levels were not affected by flagellin treatment (Figure 8D and 8E). As HDL-C is considered as anti-inflammatory molecule, we examined whether there was a direct binding of ApoA1 to flagellin resulting in inflammatory suppression. After the pull-down of ApoA1 with antibody, no detection of flagellin in the serum samples in these Apoe−/− mice was observed, suggesting the ApoA1 dose not bind to flagellin (Figure 8F). Altogether, these results suggest that activation of TLR5 in hepatocytes does not lead to hepatic and systemic inflammation.
Figure 8.
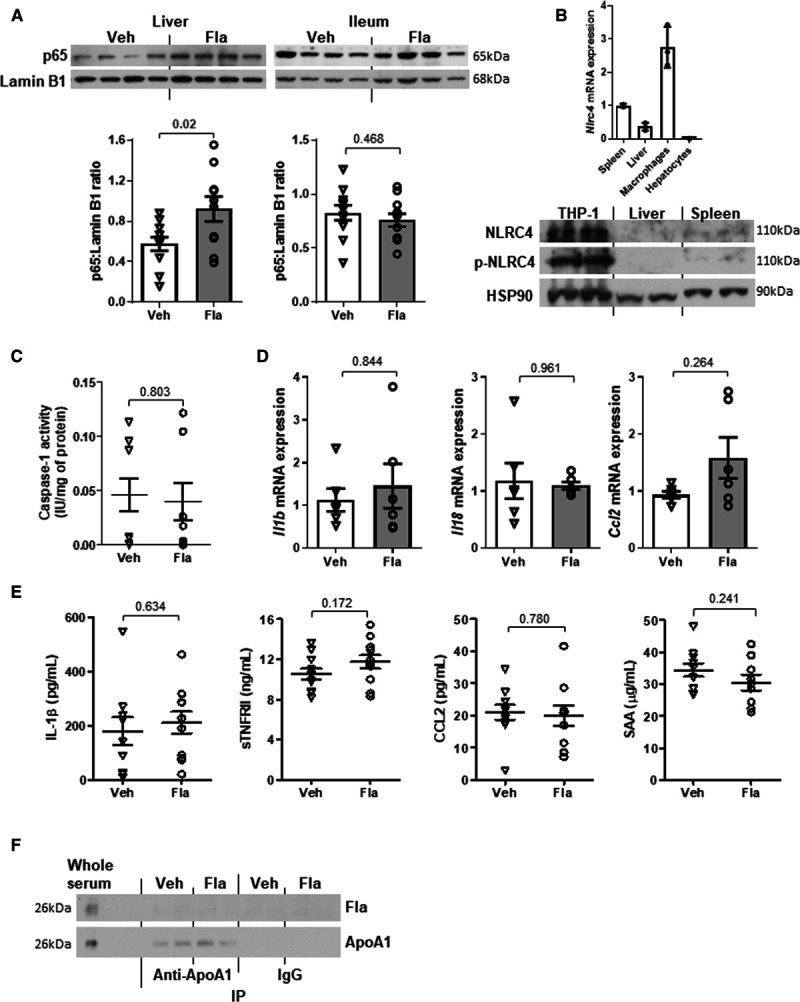
Oral supplementation of flagellin leads to hepatic activation of NF-κB (nuclear factor-κB) but not hepatic or systemic inflammation in ApoE-deficient mice. Eighteen- to twenty-week-old Apoe−/− mice fed a normal chow diet (NCD) were orally gavaged with vehicle (Veh) or flagellin (Fla, 0.1 mg/kg) every other day for 4 wk. A, The p65 expressions in nuclear fraction of liver and ileum were examined. B, The expression of NLRC4 (NLR family CARD domain-containing protein 4) in liver of flagellin-treated mice was evaluated in relation to untreated spleen, macrophages, and hepatocytes. C, Caspase 1 activity was measured in liver. D, Hepatic mRNA expressions of Il1b, Il18, and Ccl2 were determined and Actb was used as housekeeping gene. E, The circulating levels of cytokines, including IL (interleukin)-1β, sTNFRII (soluble tumor necrosis factor receptor type II), CCL2 (C-C motif chemokine ligand 2), and SAA (serum amyloid A protein), were measured. F, Co-immunoprecipitation (IP) was performed using serum samples, and flagellin content and pull-down of ApoA1 (apolipoprotein A1) were examined. Representative images are shown. Data are represented as the mean±SEM (n=10 for each group).
Positive Correlations of HDL-C Level and Flagellin-Based Flagellum Genes in Human Gut Microbiota
To explore whether an association of HDL-C level with gut flagellated bacteria was present in human, we examined data derived from a previous study.18 Owing to the potential interference of estrogen, we chose a study with male participants.36 Among all 50 samples with complete lipid profile and shotgun metagenomics sequencing data, 2 outliers with the relative abundance of flagellin-based flagellum genes exceeding 2 SD were excluded in further analysis. A trend of correlation was observed between flagellum genes and HDL-C and total cholesterol levels (Figure XIII in the Data Supplement). Gut microbiota was expected to be one of the contributors to lipoprotein levels, so we separated out the cases with potential strong genetic interference by applying the clinical cutoff of HDL-C level at 40 mg/dL (=1.0 mmol/L) based on the 2018 Guideline from American Heart Association.37 The correlation between the abundance of genes representing the flagellin-based flagellum in feces and HDL-C level was stronger and statistically significant in individuals with HDL-C≥1 mmol/L but not for triglyceride, total cholesterol and LDL-C levels, and no correlation was observed between HDL-C and serum flagellin levels (Table II in the Data Supplement). Next, we also compared the correlations in the subgroup with HDL-C<1 mmol/L, and the abovementioned positive correlation disappeared (Table II in the Data Supplement). Taken together, although the direct relationship between HFD and flagellated bacteria in human gut remains to be clarified, our data suggest a positive correlation between gut flagellated bacteria and HDL-C level in human.
Discussion
High fat intake decreases the diversity in gut microbiome and decrease in the variety of food source for omnivores possibly limits the types of gut bacteria. Nonetheless, it lacks evidence to support that decreased diversity leads to pathogenic effect. By contrast, a parallel decrease in HDL-C level by low fat intake, and a lower HDL-C level in germ-free mice than conventionally raised mice have been reported.9,38 These suggest that an unique change in gut microbiota induced by HFD prompts the increase in HDL-C level. Our findings reveal that TLR5 in host hepatocytes is the missing link between gut microbiota and HFD-induced HDL-C level.
In a previous study, HDL-C level was augmented after 5 weeks of HFD.11 Here, we included a 10-week treatment and such elevation was reproduced, suggesting the increase in HDL-C level was not a transient metabolic adaptation. In our microbiome transplantation and antibiotics models, the dosing frequency as every other day was estimated based on a study showing that gut microbiota reverted to original pattern 2 days after the termination of dietary change.39 Nonetheless, how long the elevation of flagellated bacteria lasts after quitting HFD and whether the host will develop resistance against such increase remains to be investigated. The HFD-induced HDL-C level not only depends on the response of commensal bacteria but also the intrinsic factor of the host. The whole body genetic deletion of TLR5 resulted in elevation of flagellated bacteria in gut24 and Fla. However, owing to the loss of TLR5 in hepatocytes, HFD was unable to induce ApoA1 production and HDL-C elevation. A short-term reconstitution of hepatic TLR5 in the knockout mice restored the HFD-induced elevation of ApoA1 expression, manifesting the important counterpart from the host.
Flagellin can be derived from both commensal and pathogenic bacteria. The flagellin we used as treatment was derived from Salmonella typhimurium, which is a type of flagellated bacteria in the phylum of γ-Proteobacteria. An investigational drug, CBLB502, which is a polypeptide drug derived from Salmonella flagellin has been invented as TLR5 agonist for cancer treatment (URL: https://www.clinicaltrials.gov. Unique identifiers: NCT02715882 and NCT01527136). Flagellin is composed of 4 domains, where D0 and D1 are the conserved domains which interact with TLR5, and D2 and D3 are highly variable among bacteria. Glycosylation takes place at D2 and D3 domains and can affect inflammatory potency of flagellin. For instance, the glycosylation of flagellin in Pseudomonas is required for full inflammatory activity, whereas in Lactobacillus agilis contributes to the motility but attenuates the immunologic potency toward TLR5.40,41 However, some strains do not glycosylate their flagellin, such as Escherichia coli and Salmonella typhimurium.41,42 In our study, flagellin derived from other sources including E coli and Bacillus subtilis showed a similar degree of stimulation of ApoA1 expression in hepatocytes as from Salmonella typhimurium in a TLR5-dependent manner (Figure XIV in the Data Supplement), suggesting a universal effect which is not strain-specific. However, whether and how host can distinguish the flagellin produced by different bacteria are unclear.
The flagellin-induced ApoA1 production is mediated by the classical TLR5/MyD88/NF-κB pathway. A microarray analysis shows that Apoa1 is among the top 50 downregulated genes in gastric tissue of MyD88-deficient mice upon infection from Helicobacter pylori, a flagellated bacterium.43 MyD88/NF-κB signaling is a common pathway shared among TLRs except TLR3. Activation of TLR4 by lipopolysaccharide in hepatocytes is also possible. A basal level of lipopolysaccharide was detected in the liver but such level was not altered by HFD, so the HFD-induced hepatic ApoA1 production and HDL-C level were unlikely because of the stimulation of TLR4. We compared various NF-κB-sensitive cytokine expressions in hepatocytes and macrophages treated with flagellin or lipopolysaccharide in vitro and found that the expressions of these cytokines responded more robustly in macrophages upon stimulation by lipopolysaccharide (Figure XVA through XVC in the Data Supplement). Comparing with macrophages, the mRNA expressions of these cytokines in hepatocytes were much lower, while the Apoa1 mRNA expression showed the opposite (Figure XVD in the Data Supplement). Although these cytokines and Apoa1 are NF-κB-responsive genes, it is possible that additional cell-specific transcription factors may be needed for activation of various genes, or TLR sensitivity is cell-type dependent.
In the atherogenic model of this study, when fluorophore-conjugated flagellin was given orally, flagellin transiently appeared in intestine and accumulated in liver (Figure 7A). Moreover, an increase in flagellin in liver but not serum upon high fat feeding led us to presume that gut bacteria-derived flagellin travels to the liver followed by elimination. Hepatocyte-specific, but not dendritic cell-specific, TLR5-deficient mice showed an impaired clearance of intravenously loaded E coli (MG1655, a commensal strain), leading to its accumulation in spleen and liver.30 Together with our data, activation of TLR5 and the breakdown of flagellin are suggested to take place in liver. However, we observed a significantly decrease of serum flagellin in HFD groups in the 10-week dietary models (Figures 1D and 2I). In the fecal microbiome transplantation and Apoe−/− models which involved shorter duration of treatment (4 weeks), such decrease was subdued (Figure 3C; Figure XIF in the Data Supplement). The decrease in serum flagellin may be the result of time- or dose-dependent auto-induction of its elimination. Nonetheless, the elimination of flagellin may or may not be TLR5-dependent. The detailed pharmacokinetics of flagellin requires further study.
The oral dose of flagellin used in this study was determined based on the fecal content in the HFD-treated mice. Such dose of flagellin concurrently increased VLDL/LDL-C level despite an elevation of HDL-C level and a decrease in atherosclerotic lesion size. The reason why flagellin caused the increase in VLDL/LDL-C level is unclear. Such increased VLDL/LDL-C level may be due to the limitation of Apoe−/− model itself. In Apoe−/− mice overexpressing human ApoA1, ApoA1 shifted its distribution from HDL to ApoB-containing lipoproteins.44 In fact, we also detected a much higher level of ApoA1 in the VLDL fraction of flagellin-treated Apoe−/− mice compared with the vehicle group, without affecting ApoB level (Figure XIIA in the Data Supplement). As ApoA1 and ApoE are exchangeable apolipoproteins, the observed increase in ApoB-containing lipoproteins upon flagellin treatment might be a secondary effect upon stimulating ApoA1 production in the absence of ApoE. Nevertheless, the ApoA1-transgenic Apoe−/− mice with ≈30% higher HDL-C level have smaller atherosclerotic lesions,45 and here the oral treatment of flagellin also increased HDL-C level in a similar degree. It is possible that an increase in HDL-C level yields a more preferential outcome than altering LDL-C level. Whether fine-tuning the dose of flagellin can avoid the undesired effect on VLDL/LDL-C level needs further examination.
In conclusion, the commensal flagellated bacteria in gut readily responds to dietary fat and triggers an increase of HDL-C level, which may serve as an adaptive mechanism to prevent high-fat induced dyslipidemia. TLR5 may serve as a potential therapeutic target to increase ApoA1/HDL level, and the search for synthetic TLR5 agonists with selective action on hepatocytes to stimulate ApoA1 production should be warranted.
Acknowledgements
We thank Dr Pak-Leung Ho (Department of Microbiology, LKS Faculty of Medicine, HKU) for providing Salmonella typhimurium and Staphylococcus aureus.
Sources of Funding
This work was supported by the Health and Medical Research Fund (05162606), Hong Kong Research Grants Council/Area of Excellence (AoE/M-707/18), Collaborative Research Fund (C7037-17W) and matching grant for the State Key Laboratory of Pharmaceutical Biotechnology from the University of Hong Kong.
Disclosures
None.
Supplemental Materials
Expanded Material and Methods
Online Figures I–XV
Online Table I–III
Supplementary Material
Footnotes
Nonstandard Abbreviations and Acronyms
- Apo
- apolipoprotein
- CCL2
- C-C motif chemokine ligand 2
- HDL-C
- high-density lipoprotein-cholesterol
- HFD
- high-fat diet
- IL
- interleukin
- LDL-C
- low-density lipoprotein-cholesterol
- LFD
- low-fat diet
- NCD
- normal chow diet
- NF-κB
- nuclear factor-κB
- NLRC4
- NLR family CARD domain-containing protein 4
- SAA
- serum amyloid A protein
- sTNFRII
- soluble tumor necrosis factor receptor type II
- T5KO
- TLR 5-knockout
- TLR
- Toll-like receptor
- VLDL
- very-low-density lipoprote
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.317362.
For Sources of Funding and Disclosures, see page 1251.
Novelty and Significance
What Is Known?
Diet high in saturated fat and cholesterol increases HDL-C (high-density lipoprotein-cholesterol) level in mice and humans.
Gut microbiome explains ≤25.9% of HDL-C variance independent of age, sex, and host genetics in human.
What New Information Does This Article Contribute?
Elevation of HDL-C level by high-fat diet is mediated by the change of gut microbiota, and dietary fat increases flagellated bacteria in feces.
Activation of TLR5 (Toll-like receptor 5) by flagellin in hepatocytes leads to increased ApoA1 (apolipoprotein A1) and HDL-C productions.
Treatment with flagellin can ameliorate lesion formation in atherogenic mice by stimulating ApoA1 production in liver without triggering hepatic and systemic inflammation.
Previous studies have reported dietary fat and cholesterol increase HDL-C level in human and mouse models. We found that flagellated bacteria in the gut was increased upon high-fat diet. These bacteria produced flagellin which is a TLR5 agonist. Genetic deletion of TLR5 suppressed ApoA1 production in hepatocytes and decreased HDL level in circulation, that was restored by specific reconstitution of TLR5 in the liver. Activation of TLR5 stimulated the production of ApoA1 in hepatocytes, leading to increased circulating HDL levels. TLR5 agonist acted on the receptor and turned on the MyD88 pathway in hepatocytes, resulting in the activation of a transcription factor, NF-κB (nuclear factor-κB), and subsequently stimulation of the transcription of ApoA1, the key component of HDL. Oral administration of flagellin, a natural agonist of TLR5, increased ApoA1 levels in both liver and serum, as well as HDL, and ameliorated atherosclerosis. These data indicate the potential to use TLR5 agonists with specific action on hepatocytes to increase ApoA1 and HDL levels.
References
- 1.Boekholdt SM, Arsenault BJ, Hovingh GK, Mora S, Pedersen TR, Larosa JC, Welch KM, Amarenco P, Demicco DA, Tonkin AM, et al. Levels and changes of HDL cholesterol and apolipoprotein A-I in relation to risk of cardiovascular events among statin-treated patients: a meta-analysis. Circulation. 2013;128:1504–1512. doi: 10.1161/CIRCULATIONAHA.113.002670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR, Jr, Bangdiwala S, Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8 [DOI] [PubMed] [Google Scholar]
- 3.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB, Sr, Davidson MH, Davidson WS, Heinecke JW, et al. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 4.Duong PT, Weibel GL, Lund-Katz S, Rothblat GH, Phillips MC. Characterization and properties of pre beta-HDL particles formed by ABCA1-mediated cellular lipid efflux to apoA-I. J Lipid Res. 2008;49:1006–1014. doi: 10.1194/jlr.M700506-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siri-Tarino PW, Sun Q, Hu FB, Krauss RM. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am J Clin Nutr. 2010;91:535–546. doi: 10.3945/ajcn.2009.27725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. J Clin Endocrinol Metab. 2003;88:1617–1623. doi: 10.1210/jc.2002-021480 [DOI] [PubMed] [Google Scholar]
- 7.de Souza RJ, Mente A, Maroleanu A, Cozma AI, Ha V, Kishibe T, Uleryk E, Budylowski P, Schünemann H, Beyene J, et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ. 2015;351:h3978 doi: 10.1136/bmj.h3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meckling KA, O’Sullivan C, Saari D. Comparison of a low-fat diet to a low-carbohydrate diet on weight loss, body composition, and risk factors for diabetes and cardiovascular disease in free-living, overweight men and women. J Clin Endocrinol Metab. 2004;89:2717–2723. doi: 10.1210/jc.2003-031606 [DOI] [PubMed] [Google Scholar]
- 9.Brinton EA, Eisenberg S, Breslow JL. A low-fat diet decreases high density lipoprotein (HDL) cholesterol levels by decreasing HDL apolipoprotein transport rates. J Clin Invest. 1990;85:144–151. doi: 10.1172/JCI114405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asztalos B, Lefevre M, Wong L, Foster TA, Tulley R, Windhauser M, Zhang W, Roheim PS. Differential response to low-fat diet between low and normal HDL-cholesterol subjects. J Lipid Res. 2000;41:321–328 [PubMed] [Google Scholar]
- 11.Hayek T, Ito Y, Azrolan N, Verdery RB, Aalto-Setälä K, Walsh A, Breslow JL. Dietary fat increases high density lipoprotein (HDL) levels both by increasing the transport rates and decreasing the fractional catabolic rates of HDL cholesterol ester and apolipoprotein (Apo) A-I. Presentation of a new animal model and mechanistic studies in human Apo A-I transgenic and control mice. J Clin Invest. 1993;91:1665–1671. doi: 10.1172/JCI116375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Lin S, Vanhoutte PM, Woo CW, Xu A. Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in apoe-/- mice. Circulation. 2016;133:2434–2446. doi: 10.1161/CIRCULATIONAHA.115.019645 [DOI] [PubMed] [Google Scholar]
- 14.Yoshida N, Emoto T, Yamashita T, Watanabe H, Hayashi T, Tabata T, Hoshi N, Hatano N, Ozawa G, Sasaki N, et al. Bacteroides vulgatus and bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation. 2018;138:2486–2498. doi: 10.1161/CIRCULATIONAHA.118.033714 [DOI] [PubMed] [Google Scholar]
- 15.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JA, Brandsma E, Marczynska J, Imhann F, Weersma RK, et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res. 2015;117:817–824. doi: 10.1161/CIRCRESAHA.115.306807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434–438. doi: 10.1073/pnas.88.2.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Wang Y, Ni Y, Cheung CKY, Lam KSL, Wang Y, Xia Z, Ye D, Guo J, Tse MA, et al. Gut microbiome fermentation determines the efficacy of exercise for diabetes prevention. Cell Metab. 2020;31:77–91 e5. doi: 10.1016/j.cmet.2019.11.001 [DOI] [PubMed] [Google Scholar]
- 19.Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–511. doi: 10.1038/nature24460 [DOI] [PubMed] [Google Scholar]
- 20.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–903. doi: 10.1038/nmeth.3589 [DOI] [PubMed] [Google Scholar]
- 21.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- 23.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962–968. doi: 10.1038/s41592-018-0176-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, Aderem A. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol. 2003;4:1247–1253. doi: 10.1038/ni1011 [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–2224. doi: 10.1172/JCI32057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang MD, Kiss RS, Franklin V, McBride HM, Whitman SC, Marcel YL. Different cellular traffic of LDL-cholesterol and acetylated LDL-cholesterol leads to distinct reverse cholesterol transport pathways. J Lipid Res. 2007;48:633–645. doi: 10.1194/jlr.M600470-JLR200 [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Li J, Yiu JHC, Lam JKW, Wong CM, Dorweiler B, Xu A, Woo CW. TRIF-dependent Toll-like receptor signaling suppresses Scd1 transcription in hepatocytes and prevents diet-induced hepatic steatosis. Sci Signal. 2017;10:eaal3336 doi: 10.1126/scisignal.aal3336. [DOI] [PubMed] [Google Scholar]
- 29.Pellizzon MA, Ricci MR. The common use of improper control diets in diet-induced metabolic disease research confounds data interpretation: the fiber factor. Nutr Metab (Lond). 2018;15:3 doi: 10.1186/s12986-018-0243-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease. Cell Mol Gastroenterol Hepatol. 2016;2:584–604. doi: 10.1016/j.jcmgh.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youngster I, Russell GH, Pindar C, Ziv-Baran T, Sauk J, Hohmann EL. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA. 2014;312:1772–1778. doi: 10.1001/jama.2014.13875 [DOI] [PubMed] [Google Scholar]
- 32.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882 [DOI] [PubMed] [Google Scholar]
- 33.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vijay-Kumar M, Carvalho FA, Aitken JD, Fifadara NH, Gewirtz AT. TLR5 or NLRC4 is necessary and sufficient for promotion of humoral immunity by flagellin. Eur J Immunol. 2010;40:3528–3534. doi: 10.1002/eji.201040421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong N, Lai P, Pang E, Leung TW, Lau JW, Johnson PJ. A comprehensive karyotypic study on human hepatocellular carcinoma by spectral karyotyping. Hepatology. 2000;32:1060–1068. doi: 10.1053/jhep.2000.19349 [DOI] [PubMed] [Google Scholar]
- 36.Herrington DM, Reboussin DM, Brosnihan KB, Sharp PC, Shumaker SA, Snyder TE, Furberg CD, Kowalchuk GJ, Stuckey TD, Rogers WJ, et al. Effects of estrogen replacement on the progression of coronary-artery atherosclerosis. N Engl J Med. 2000;343:522–529. doi: 10.1056/NEJM200008243430801 [DOI] [PubMed] [Google Scholar]
- 37.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2019;139:e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabot S, Membrez M, Bruneau A, Gérard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24:4948–4959. doi: 10.1096/fj.10-164921 [DOI] [PubMed] [Google Scholar]
- 39.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kajikawa A, Midorikawa E, Masuda K, Kondo K, Irisawa T, Igimi S, Okada S. Characterization of flagellins isolated from a highly motile strain of lactobacillus agilis. BMC Microbiol. 2016;16:49 doi: 10.1186/s12866-016-0667-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma A, Arora SK, Kuravi SK, Ramphal R. Roles of specific amino acids in the N terminus of Pseudomonas aeruginosa flagellin and of flagellin glycosylation in the innate immune response. Infect Immun. 2005;73:8237–8246. doi: 10.1128/IAI.73.12.8237-8246.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horstmann JA, Lunelli M, Cazzola H, Heidemann J, Kühne C, Steffen P, Szefs S, Rossi C, Lokareddy RK, Wang C, et al. Methylation of salmonella typhimurium flagella promotes bacterial adhesion and host cell invasion. Nat Commun. 2020;11:2013 doi: 10.1038/s41467-020-15738-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozano-Pope I, Sharma A, Matthias M, Doran KS, Obonyo M. Effect of myeloid differentiation primary response gene 88 on expression profiles of genes during the development and progression of helicobacter-induced gastric cancer. BMC Cancer. 2017;17:133 doi: 10.1186/s12885-017-3114-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsukamoto K, Hiester KG, Smith P, Usher DC, Glick JM, Rader DJ. Comparison of human apoA-I expression in mouse models of atherosclerosis after gene transfer using a second generation adenovirus. J Lipid Res. 1997;38:1869–1876 [PubMed] [Google Scholar]
- 45.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1994;91:9607–9611. doi: 10.1073/pnas.91.20.9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of materials and methods is provided in the Materials and the Major Resource Table in the Data Supplement.17–28 The data that support the findings of this study are available from the corresponding authors upon reasonable request.