

Abstract
We have recently investigated the reactivity of aryl-fluorosulfates as warheads to form covalent adducts with Lys, Tyr, and His residues. However, the rate of reaction of aryl-fluorosulfates seemed relatively slow, putting into question their effectiveness to form covalent adducts in cell. Unlike the previously reported agents that targeted a relatively remote Lys residue with respect to the target’s binding site, the current agents were designed to more directly juxtapose an aryl-fluorosulfate with a Lys residue that is located within the binding pocket of the BIR3 domain of XIAP. We found that such new agents can effectively and rapidly form a covalent adduct with XIAP-BIR3 in vitro and in cell, approaching the rate of reaction, cellular permeability, and stability that are similar to what attained by acrylamides when targeting Cys residues. Our studies further validate aryl-fluorosulfates as valuable Lys-targeting electrophiles, for design of inhibitors of both enzymes and protein-protein interactions.
Keywords: Drug discovery, lysine covalent, PPIs, protein-protein interactions, XIAP, IAP, covalent inhibitors, aryl-fluorosulfates
Graphical Abstract
Introduction
Covalent inhibitors targeting Cys residues have recently provided drug designers the ability to attain potent and selective irreversible inhibitors with proper pharmacological properties. Hence, the design of targeted irreversible inhibitors is currently heavily pursued1–14 and it has resulted in the recent FDA approval of several covalent drugs in oncology. Acrylamide-based Cys-covalent kinase inhibitors, osimertinib, ibrutinib, neratinib and afatinib, for example, have been all approved by the FDA in very recent years, to cite a few. The success of these agents likely resides in their uniquely high potency (irreversible) and the sustained inhibition of their targets; these are highly desirable properties in oncology drugs that are difficult to obtain with reversible compounds, making the approach of deriving irreversible drugs very appealing. While it has been well established that acrylamides offer the proper balance of Cys-reactivity and selectivity, similar studies on Lys targeting electrophiles are ongoing by us15, 16 and others in the field.17–24 Recent studies aimed at probing the ability of small molecule-containing aryl-fluorosulfates to capture potential drug targets after long exposure (24 h) to cell lysates,17 but specific studies on their drug-likeness, including cell penetration and studies demonstrating direct covalent bond formation in cell, for example, have not yet been directly and/or fully investigated. Moreover, most studies on aryl-fluorosultates focused on enzyme inhibitors,17, 25 while our recent efforts in regard focused on investigating the possibility of generating effective covalent antagonists of protein-protein interactions (PPIs) for therapeutic use,15, 16 given that targeting PPIs remains a difficult task for new therapeutics development. While PPIs targeting short peptides can be relatively easily identified, their evolution into potent and selective, cell permeable agents remains a challenging task. For a limited number of targets that present a Cys residue within their binding surfaces, increased potency and selectivity of binding peptides can be accomplished by introducing properly juxtaposed acrylamides or chloroacetamides.26–28 However, Cys residues are relatively infrequent in binding interfaces, hence, in the search of similarly suitable warheads to target other residues including Lys, Tyr, and His, we have recently compared the reactivity of aryl-sulfonyl fluorides10, 18, 29 and aryl-fluorosulfates17, 25, 30, 31 warheads when inserted in binding peptides targeting the Inhibitors of Apoptosis Proteins (IAPs)32–34. Several agents based on the tetrapeptide of sequence Ala-Val-Pro-Phe (AVPF, or also the peptide AVPI) that interacts with various members of the IAP family, including XIAP, cIAP1, and cIAP2,35–38 have been developed as potential therapeutic agents39–58, including clinical candidates AT406,39, 59–62 and LCL161,63–67 (Table 1). We recently reported on compound 1 (Table 1, Figure 1A) as the first XIAP covalent agent targeting Lys 311 in its BIR3 domain.16 As Lys 311 is not present in cIAP1 nor cIAP2, we expect that this agent can form a covalent adduct only for XIAP-BIR3 (Figure 1).16 However, likely due to the flexibility of both the side chain carrying the electrophile and the targeted residue Lys 311 (Figure 1A), we found that the rate of the reaction in vitro was relatively slow (> 6 h for the reaction to go to completion at physiological pH, in vitro)16, which shed some doubts on whether such reaction could take place also in cell. Here we report on the design of new agents that juxtapose more directly an aryl-fluorosulfate with a Lys residue within the binding pocket of the BIR3 domain of XIAP. We demonstrate that such proper juxtaposition of the aryl-fluorosulfate with Lys 297 in XIAP-BIR3 results in agents (compounds 2 and 3; Table 1) that more readily react in vitro and in cells. Hence, we found for the first time that the reaction rate, cellular permeability, aqueous solubility and stability, and plasma stability, of the designed Lys-targeting aryl-fluorosulfates targeting a PPI appear on par with those generally observed for Cys targeting acrylamides. The stability data are also in agreement with previous studies in targeting Ser residues with ary-lfluorosulfates.68 In that article, the authors had noted a slow reactivity of the agents and anticipated that enhanced binding interactions could lead to accelerated reaction,68 as indeed we demonstrated here experimentally. Therefore, our studies provide critical experimental proof of concept that aryl-fluorosulfates, when properly placed on a binding agent targeting a PPI, can provide Lys-covalent agents with favorable pharmacological properties. When applied to the IAP family, the approach resulted in novel and effective pan-IAP inhibitors with cellular affinity that is comparable to that observed with current clinical candidates for these targets.
Table 1.
Structures and Inhibitory Data for Select Compounds That Interact with Various Members of the IAP Familya,b,c
| ID | STRUCTURE | XIAP-BIR3 | cIAP1-BIR3 IC50 (nM) | cIAP2-BIR3 IC50 (nM) | |
|---|---|---|---|---|---|
| IC50 [nM]a | ΔTm [°C]b | ||||
| LCL161 | 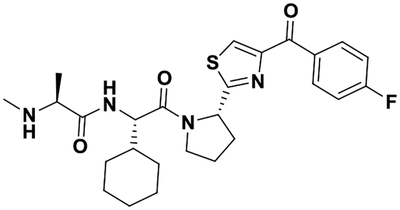 |
50 ± 5 40 ± 14 |
14.0 18.5 |
18.6 ± 0.1 21 ± 3 |
11 ± 4 19 ± 2 |
| AT406 | 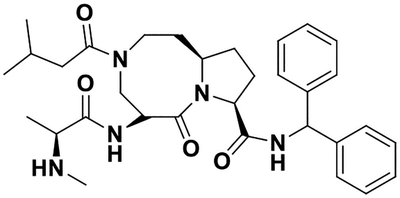 |
42 ± 7 47.8 ± 0.2 |
17.0 19.0 |
34± 1 35± 2 |
44 ± 5 133.0 ± 0.2c |
| 1 | 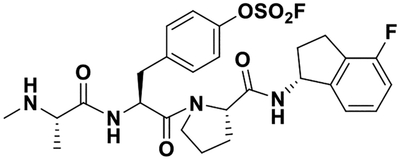 |
63 ± 6 28 ± 6 |
9.5 33.5 |
24 ± 1 20 ± 5 |
40 ± 3 47 ± 23 |
| 2 | 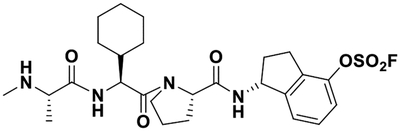 |
38 ± 1 12 ± 2 |
37.5 38.0 |
25 ± 1 14 ± 2 |
28 ± 1 9 ± 3 |
| 3 | 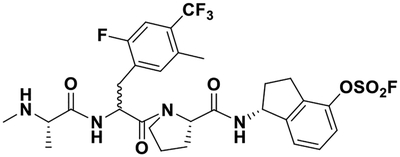 |
74 ± 2 11.0 ± 0.2 |
37.5 35.5 |
50 ± 2 15 ± 2 |
120 ± 24 12.5 ± 0.3 |
IC50 values calculated from dose-response curves obtained without pre-incubation and after 6 h pre-incubation of protein and test ligands, at room temperature.
Thermal shift (ΔTm) data were obtained from pre-incubating the proteins and test ligands for 2 h at room temperature or 6 h at 37 °C.
The increased IC50 value is likely due to compound precipitation after 6-h pre-incubation.
Figure 1. Molecular docking of compounds 1 and 2 with the BIR3 domains of XIAP, cIAP1, and cIAP2.
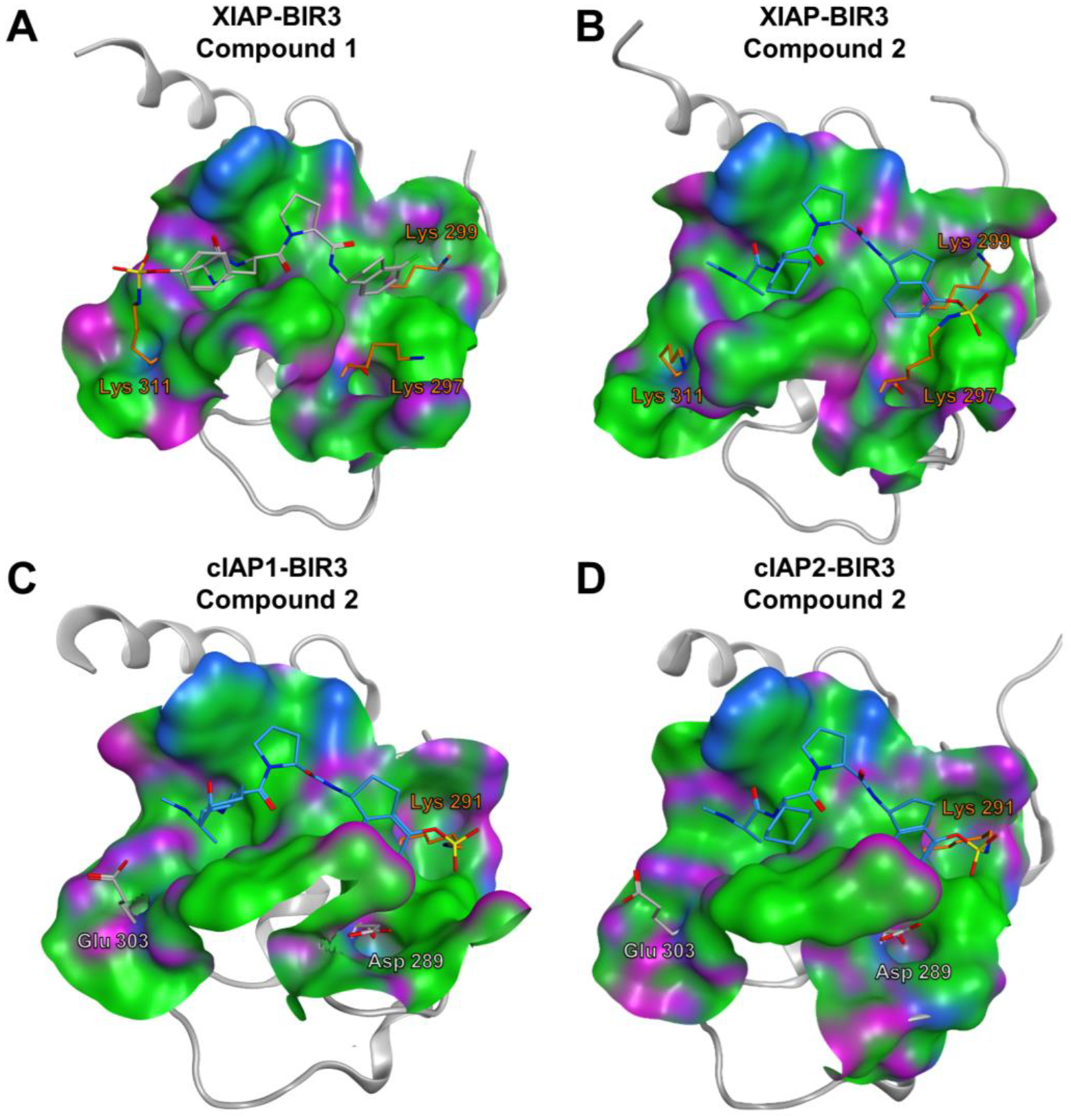
Covalent docking pose of compound 1 into the binding pocket of the BIR3 domain of XIAP (PDB ID 3HL5). Lys 311, forming the covalent bond with compound 1, and lysine residues 297, and 299 are highlighted in orange. B) Covalent docking pose of compound 2 into the binding pocket of the BIR3 domain of XIAP (PDB ID 3HL5). Lys 297, forming the covalent bond with compound 2, and lysine residues 311 and 299 are displayed as stick models and labeled. C) Covalent docking pose of compound 2 into the binding pocket of the BIR3 domain of cIAP1 (PDB ID 3UW4). Lys 291, forming the covalent bond with compound 2, is highlighted as stick model, while the residues glutamic acid 303 and aspartic acid 289, corresponding to the Lys 311 and Lys 297 of XIAP, respectively, are also displayed and labeled. D) Covalent docking pose of compound 2 into the binding pocket of the BIR3 domain of cIAP2 (PDB ID 2UVL). Lys 291, forming the covalent bond with compound 2, is displayed, while the residues glutamic acid 303, and Aspartic acid 289, corresponding to the Lys 311 and Lys 297 of XIAP, respectively, are also displayed and labeled.
Results
Design and synthesis of novel BIR3 targeting aryl-fluorosulfates.
In order to design Lys-covalent BIR3 antagonists targeting XIAP, cIAP1, and cIAP2, we examined the respective X-ray structures of these domains in complex with various antagonists (XIAP and cIAP1) or in the apo-form (cIAP2) (Figure 1). These domains bind tetrapeptides of general sequence Aϕ03C5Pϕ where ϕ represents hydrophobic residues in positions P2 and P4 of the tetrapeptide, while Ala and Pro occupy positions P1 and P3, respectively. Starting with a positional scanning library of tetrapeptides of sequence AXXX (where X is one of 42 between natural and non-natural aminoacids) we previously identified optimal residues for positions P2-P4.69–71 In particular, using an enthalpy-based screening approach, we identified a 4-fluoro-1-amino indane as an effective P4 substituent, while P2 substituents included a DL-2F,4CF3,5CH3-Phe71 instead of the L-cyclohexyl-Gly, commonly found in IAP inhibitors (e.g LCL161, Table 1) or, after further optimizations, also a L-Phe(OSO2F) (compound 1), that we reported targeting Lys 311 of XIAP-BIR3 (Figure 1A).16, 71 Here, using simple molecular docking strategies we identified XIAP-BIR3 residues Lys 297 and Lys 299 as possible targets for possible covalent interactions (Figure 1B). Correspondingly, Lys 291 cIAP1 (Figure 1C), and Lys 291 in cIAP2 (Figure 1D), are also located in correspondence of the P4 position of binding tetrapeptides in these two other members of the IAP family. Hence, we designed novel agents that contained a 4-fluorosulfate on the P4 1-amino indane, and that in P2 contained either cyclohexyl-Gly (compound 2) or the DL-2F,4CF3,5CH3-Phe (compound 3). Unfortunately, our attempts to separate the two diasteroisomers of 3 were not successful, and the agent was tested a racemic mixture. However, modeling studies suggested that both compound 2 and the L-isomer of compound 3 could form a covalent adduct with XIAP Lys 297 (Figure 1B), and possibly also with Lys 291 in cIAP1 (Figure 1C), and Lys 291 in cIAP2 (Figure 1D).
The synthesis of compounds 2 and 3 was accomplished using the scheme of Figure 2. Briefly, a BAL resin (0.05 mmol scale) was loaded using a solution of (R)-1-amino-2,3-dihydro-1H-inden-4-ol (3 eq.) in DMF added to the reactor and shaken for 30 min, followed by reduction using sodium triacetoxyborohydride (3 eq., overnight reaction at room temperature). The resin was subsequently filtered, washed three times with DMF, three times with DCM (3x) and again three times with DMF. For the coupling of Fmoc-proline on the secondary amine reaction time was increased to 2 h. Fmoc deprotection and peptide elongation then followed standard procedures described in the general chemistry section. [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF)30 reagent (1.2 eq., 2.2 eq. of DBU in THF, overnight reaction at room temperature) was used to introduce the fluorosulfate reactive group on the compounds while still on the resin. Peptides were cleaved from the BAL resin with a cleavage cocktail containing TFA/TIS/water/phenol (94:2:2:2) for 3 h. The cleaving solution was filtered from the resin, evaporated under reduced pressure, and the peptides precipitated in Et2O, centrifuged and dried in high vacuum. The crude peptides were purified by preparative RP-HPLC using a Luna C18 column (Phenomenex) and water/acetonitrile gradient (5% to 100%) containing 0.1% TFA. The final compounds were characterized by HRMS, and subsequently tested in a variety of in vitro and cellular assays as reported below.
Figure 2. General scheme for the synthesis of compounds 2 and 3.
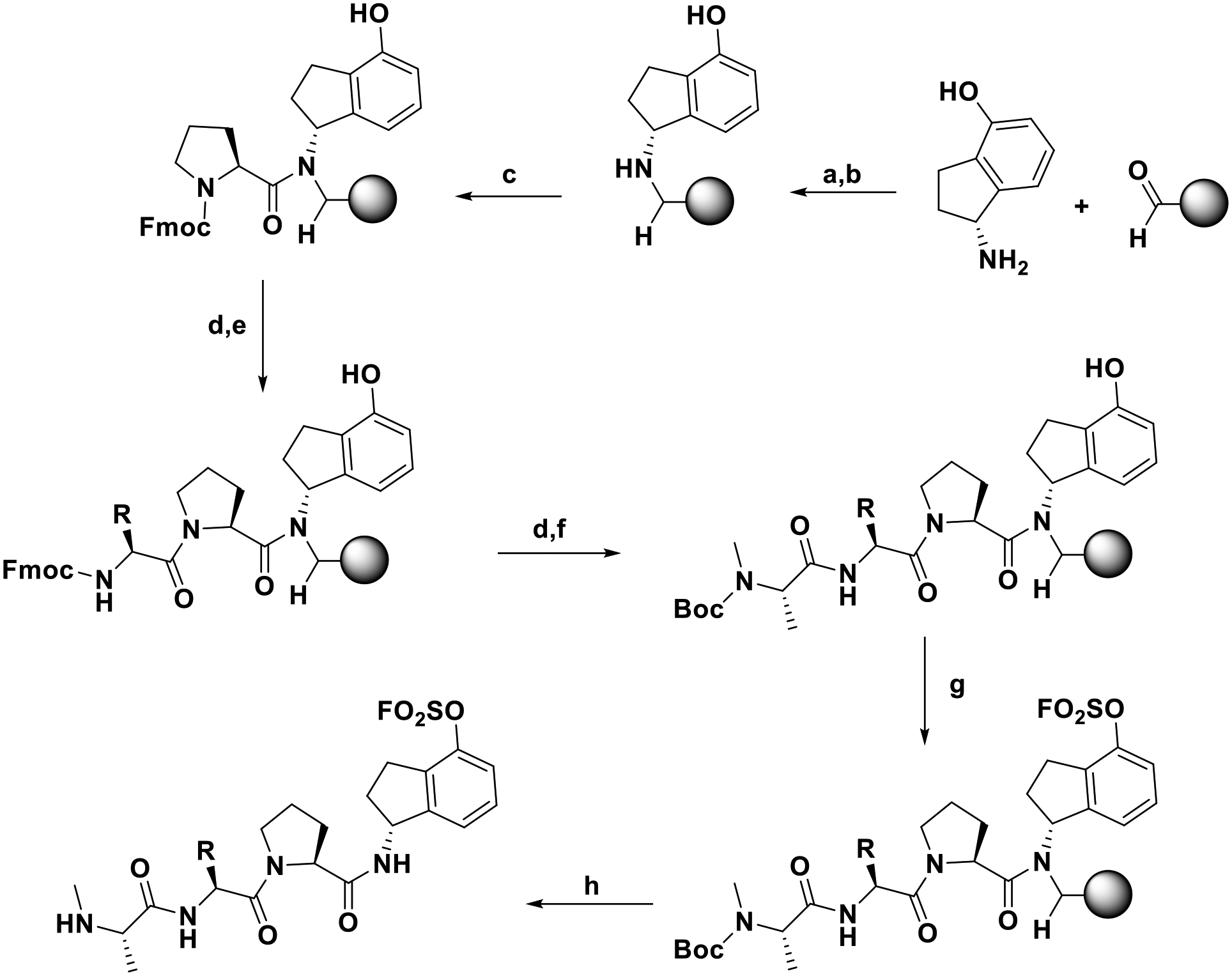
Conditions: (a) DMF, 30 min, rt; (b) Sodium triacetoxyborohydride (3 eq), o/n, rt; (c) Fmoc-Pro-COOH (3 eq), HATU (3 eq), DIPEA (5 eq), 2 h, rt; (d) 20% 4-methylpiperidine in DMF, rt; (e) Fmoc-Chg-COOH or Fmoc-2-amino-3-(2-fluoro-5-methyl-4-(trifluoromethyl)phenyl)propanoic acid (3 eq), HATU (3 eq), DIPEA (5 eq), 50 min, rt; (f) Boc-NMe-Ala-COOH, HATU (3 eq), DIPEA (5 eq), 50 min, rt; (g) AISF (1.2 eq), DBU (2.2 eq), THF, o/n, rt; (h) TFA/TIS/water/phenol (94:2:2:2), 3 h, rt. The stereochemistry for the group R is L for compound 2 and racemic DL for compound 3.
In vitro evaluations of the designed Lys-targeting agents
In order to assess the potential of the proposed agents in targeting covalently the BIR3 domains, we tested the agents in biochemical and biophysical assays in vitro. First, we used a Dissociation-Enhanced Lanthanide Fluorescence Immunoassay (DELFIA) displacement assay as we described previously71 that measures the ability of test agents to compete for the binding of a reference biotinylated AVPI peptide. IC50 values were obtained from dose-response curves measured after no pre-incubation or with 6-h pre-incubation of the test ligand and protein. Depending on the reaction rate of the agent, a large decrease in IC50 value with pre-incubation can be interpreted as possible slow covalent interaction (Table 1; Figure 3A). As controls, the pan-IAP clinical candidates AT406 and LCL161 were also tested, as well as our previously reported XIAP-BIR3 covalent agent compound 1 (Table 1). Compound 1, targeting XIAP Lys311 as we recently reported, is covalent only for XIAP-BIR3, but not for cIAP1 or cIAP2 that present a glutamic acid in correspondence of Lys 311 in their BIR3 domains (Figure 1).15, 16 For each agent we also measured their induced denaturation thermal shifts (ΔTm) on the BIR3 domain of XIAP. Non-covalent agents, such as AT406 or LCL161, displayed a ΔTm < 20 °C (regardless of the incubation times of either 2 h or 6 h), while putative covalent compounds showed significantly larger shifts (ΔTm of > 30 °C; Table 1, and supplementary Figure S1).16 Finally, covalent adduct formation was verified by SDS gel electrophoresis and mass spectrometry data (Figures 3B,C). These data clearly suggested that compounds 2 and 3 formed a stable covalent adduct with the BIR3 domain of XIAP. In addition, both 1D 1H NMR and 2D [15N,1H] NMR spectra with a 15N-Lys-selectively labeled BIR3 sample and compound 2 revealed time-dependent chemical shift changes for the backbone amide of Lys 297, presumably due to the covalent bond formation with its side chain over time (Figure 3D,E).72 These data collectively demonstrated that compounds 2 and 3 are effective Lys-covalent agents for the BIR3 domain of XIAP likely targeting Lys 297.
Figure 3. In vitro characterization of covalent binding for compounds 2 and 3 to the BIR3 domain of XIAP.
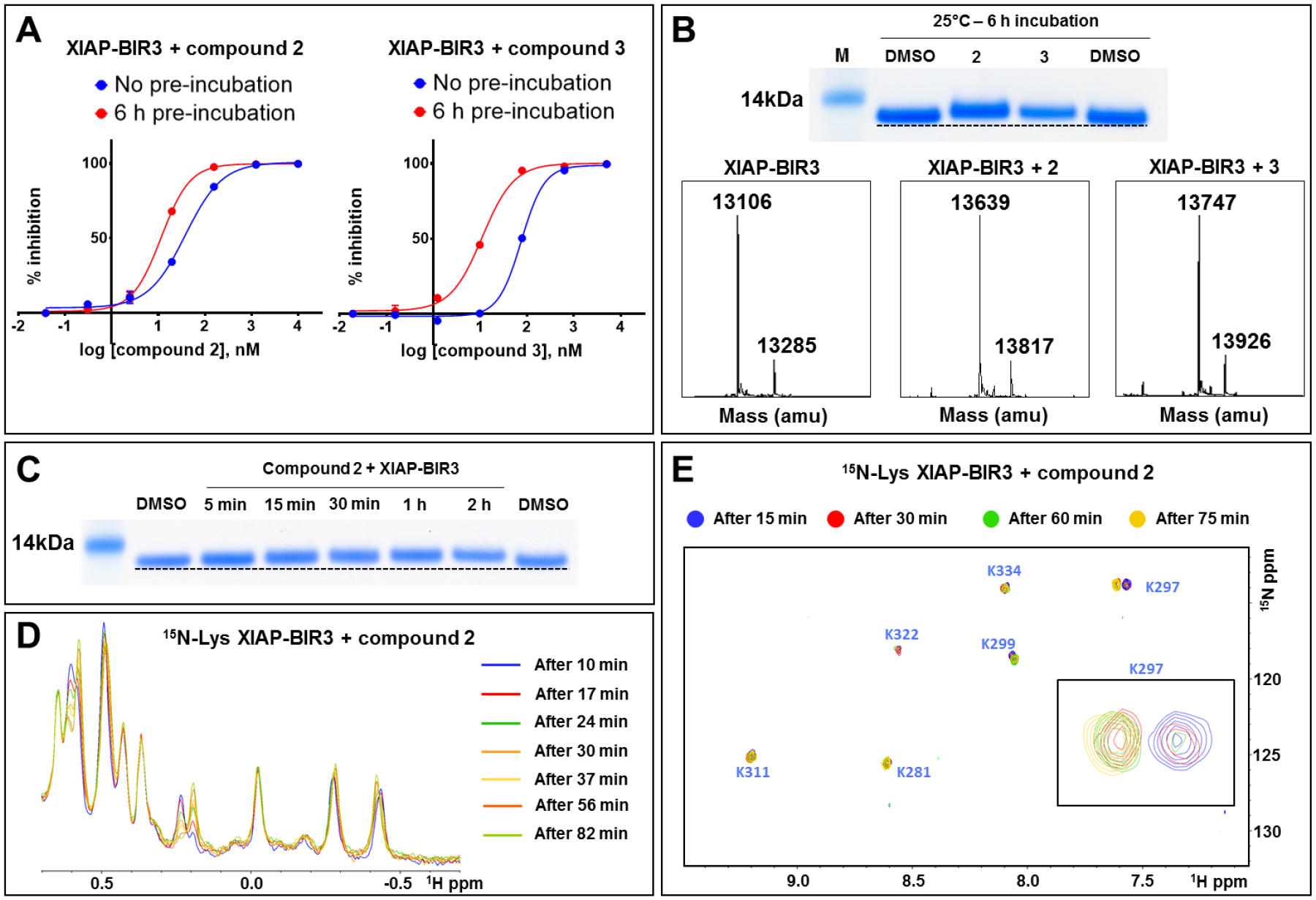
A) DELFIA displacement curves relative to the BIR3 domain of XIAP without pre-incubation (blue), and after 6 h pre-incubation (red) with compounds 2 (left), and 3 (right). The decrease in IC50 values with the pre-incubation time is typical for compounds that form a covalent bond. B) (top panel) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP incubated for 6 h at 25 °C with compounds 2 and 3 with a protein-ligand ratio of 1:10. (bottom panel) LC-MS spectra of the BIR3 domain of XIAP in the absence (left), in the presence of compound 2 (center), and in the presence of compound 3 (right). The mass of the BIR3 domain of XIAP is 13,106 Da, and the mass increases by 533 Da in the presence of compound 2, and by 641 Da in the presence of compound 3, corresponding to those of the respective covalent adducts. C) SDS-PAGE gel electrophoresis followed by Coomassie staining of the BIR3 domain of XIAP incubated at different times at 25 °C with compound 2 with a protein-ligand ratio of 1:10. D) 1H-1D NMR spectra of the aliphatic region of 20 μM BIR3 domain of XIAP with 100 μM of compound 2 recorded at different times as indicated. The spectra are changing with time, suggesting the presence of an ongoing reaction between the protein and the covalent agent. E) [1H, 15N]-sofastHMQC spectra of 20 μM BIR3 domain of XIAP selectively labeled with 15N-Lysine in the presence of 100 μM of compound 2 recorded after 15 min (blue), 30 min (red), 60 min (green), and 75 min (yellow). The intensity of the peak corresponding to the Lys 297 is time-dependent, typical of an ongoing reaction, suggesting the covalent binding to this Lysine residue.
To extend these observations to cIAP1 and cIAP2, a similar set of data was collected with the agents and their respective BIR3 domains (Table 1, Figure 4). First, dose-response DELFIA curves with and without pre-incubation (Figure 4A,B) revealed decreased IC50 values only for compounds 2 (in particular) and 3, that presumably can form covalent adducts with Lys residues present in correspondence to the 4-fluorosulfate (Figure 1). Subsequently, mass spectrometry data with these complexes releveled covalent adduct formation between compound 2 and 3 and both cIAP1 and cIAP2 (Figure 4D,E), but not compound 1 that was designed to interact only with the BIR3 domain of XIAP.16 These data collectively suggest that compounds 2 and 3 are putative P4 covalent agents for XIAP-BIR3 in particular, but can also target cIAP1 and cIAP2.
Figure 4. In vitro validation of covalent binding of compounds 1, 2, and 3, to the BIR3 domain of cIAP1 and cIAP2.
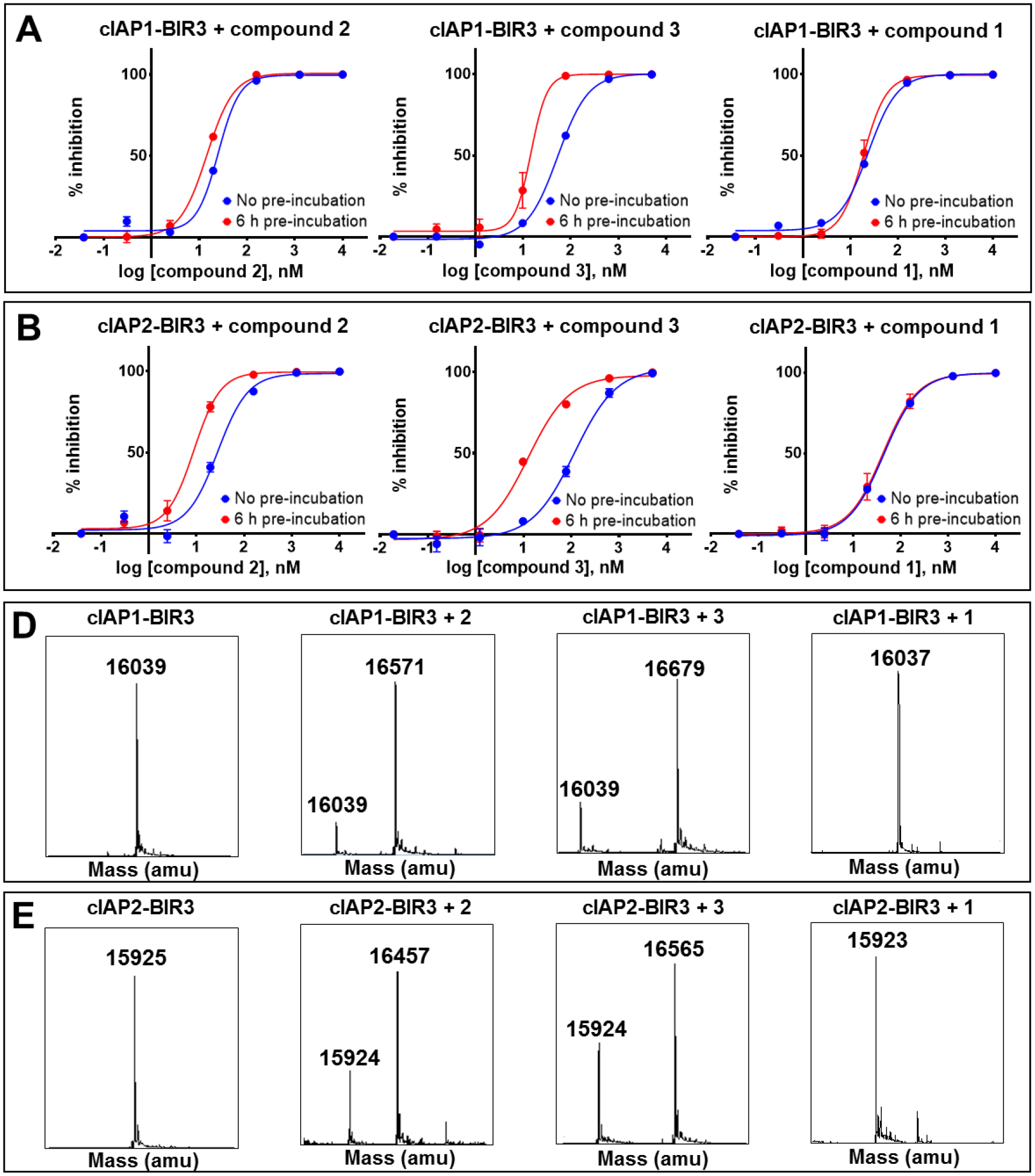
DELFIA displacement curves relative to the BIR3 domain of cIAP1 A) and the BIR3 domain of cIAP2 B) without pre-incubation (blue), and with 6 h pre-incubation (red) with compounds 2 (left), 3 (center), and 1 (right). For compounds 2 and 3, the IC50 values decreased with increasing pre-incubation time, typical for a covalent interaction, while the IC50 of compound 1 is pretty much constant. LC-MS spectra of the BIR3 domain of cIAP1 C) and the BIR3 domain of cIAP2 D) in the absence and in presence of compounds 2, 3, and 1. The mass of the BIR3 domain of cIAP1 is 16,039 Da, while the mass of the BIR3 domain of cIAP2 is 15,925 Da. For both proteins, the mass increases by 533 Da and 641 Da in the presence of compounds 2 and 3, respectively. There is no increase in mass in the presence of compound 1.
Plasma stability, cell permeability, and cellular activity of covalent agents.
To assess the pharmacological properties of compounds 2 and 3 relative to clinical candidates LCL161 or AT406, we compared solubility, aqueous and plasma stability, and cellular permeability of each agent. Solubility and chemical stability were assessed using 1H 1D NMR, measured over time, and at different concentrations (supplementary Figure S3). Compound 2 was long-lived (several hours) and soluble up to 2 mM in aqueous buffer (pH = 7.5, T = 25 °C), similar to what we observed with AT406 and LCL161. Compound 3 was also fairly soluble up to 1 mM. Plasma stability data also revealed that compound 2 was plasma stable with t1/2 > 2 h (again, similar to LCL161 and AT406), while compound 3 appeared less soluble in plasma, but still long-lived.
Most importantly, to assess cell permeability of these agents, and to verify that the covalent agents can reach the target and act covalently in cell, we have obtained a cell line that is stably transfected with HA-BIR3 of XIAP.73 Hence, exposure of cell line to our agents, followed by western blot analyses of cell lysates using an anti-HA antibody was used to monitor whether the test agents enter the cell, and target covalently the BIR3 domain of XIAP, as this could be directly appreciated by a significant shift in molecular weight (band shift) similar to what can be observed in vitro. However, as we reported recently, the unbound BIR3 domain of XIAP is fairly unstable at 37 °C, while the domain gets stabilized by ligand binding, as suggested by our reported thermal shift data (Table 1). Accordingly, exposing the HA-BIR3-expressing HEK293 cell line to cell permeable compounds AT406 or LCL161 stabilized the BIR3 domain that resulted in an increase in band intensity on the western blot (Figure 5A). Similarly, compounds 1, 2, and 3 stabilized HA-BIR3 in this experiment, resulting in increasingly more visible bands. Most importantly, compound 2, and 3 in particular, also induced a clearly appreciable gel shift, both consistent with cell permeability of the agent and covalent adduct formation in cell (Figure 5A).
Figure 5. Covalent bond formation in cell and cellular effect of compounds of IAP levels.
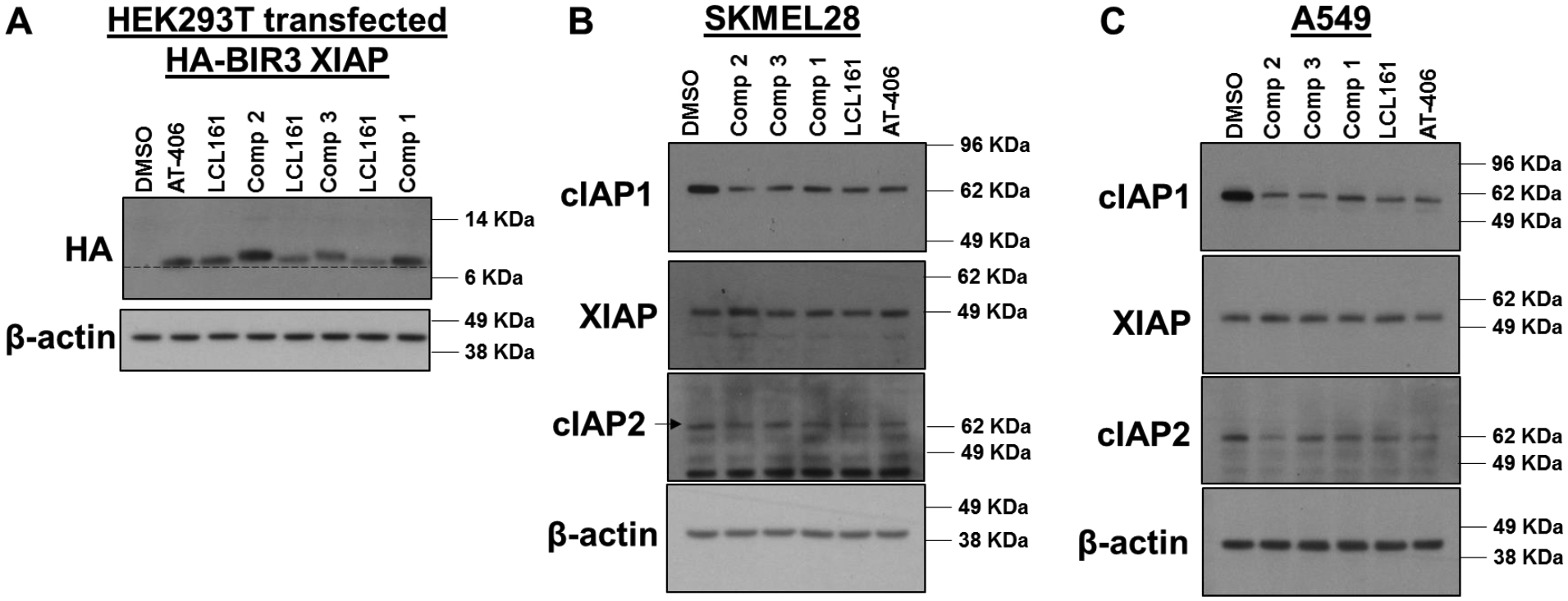
A) WB analysis of HA-BIR3 of XIAP-expressing HEK293T cells after exposure to 10 μM of each of the indicated compounds. Band intensity is qualitatively proportional to compounds’ permeability and induction of protein stability in cell. Covalent adduct formation is clearly appreciable by a band shift due to the slightly increased molecular weight of covalent adducts (see Figure 2) compared to the native protein. This is observed with compounds 2 and 3 in particular, while this is less appreciable with compound 1, indicating an incomplete reaction under these experimental conditions. Effect of each agent on XIAP, cIAP1 and cIAP2 protein levels after exposure of melanoma cell line SK-MEL-28 B) or non-small cell lung cancer cell line A549 C) to the indicated agents (1 μM for 3 h). All agents caused a significant reduction in cIAP1 and cIAP2 (more evident with the A549 cell line) levels, and compound 2 seems overall more effective than the others tested.
Previous studies demonstrated that exposure of cell lines to pan-IAP antagonists can cause degradation of cIAP1 and cIAP2, but not of XIAP. Accordingly, when the melanoma cell line SK-MEL-28 or the non-small cell lung cancer cell line A549 (both high expressors of IAP proteins) were exposed to 1 μM each of our test agents, all compounds caused a significant reduction of cIAP1 and cIAP2 levels (Figure 5B, C), with compound 2 causing the most appreciable effect even compared to clinical agents LCL161 and AT406, in agreement with its in vitro affinity and predicted cell permeability (Figure 5A).
Subsequently, induction of apoptosis was further examined in both SK-MEL-28 and A549 cell lines that, because of the expression of XIAP, cIAP1, and cIAP2 (Figure 5), are resistant to TNFα-induced apoptosis. For this purpose, we used the IncuCyte S3 Caspase-3/7 fluorescence-based apoptosis assay, which consists of a non-fluorescent substrate that freely crosses the cell membrane where it can be cleaved by activated caspase-3/7 to release a green DNA-binding fluorescent label. Hence, apoptotic cells are identified by the appearance of fluorescently-labeled nuclei. After 36-h treatment, the compounds alone at the indicated doses showed similar caspase-3/7 intensity as that of DMSO-treated cells in both cell lines (Figure 6A,B, top panels). In SK-MEL-28 and A549, caspase-3/7 activity was greatly enhanced in a dose-dependent manner when combining test compounds and 1 ng/mL TNFα, demonstrating that the agents can greatly sensitize the cell lines to apoptosis (Figure 6A,B, bottom panels). These data parallel cell viability data (supplementary Figure S2) that clearly indicated that compound 2 and 3 are as efficacious, or more efficacious than tested agents. Collectively, these data suggested that the covalent agents have cellular pharmacological properties that are at the least on par as the clinical candidates LCL161 or AT406.
Figure 6. Induction of apoptosis in the presence of TNFα in cell lines.
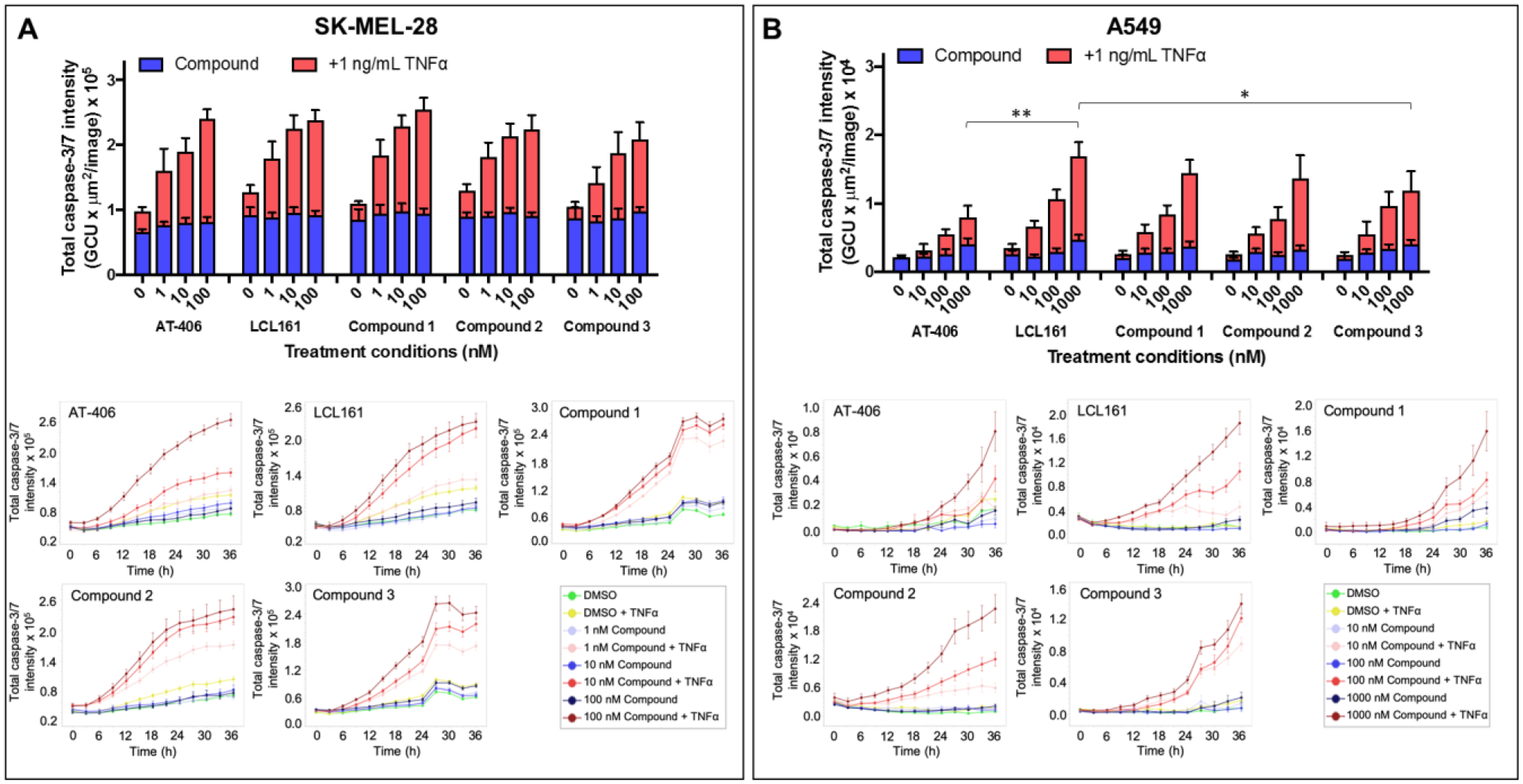
Cells were treated with various compounds and normalized to 1% DMSO (v/v) control in the presence or absence of 1 ng/mL TNFα. Caspase activity was monitored with the IncuCyte S3 live-cell analysis system. Histograms displaying caspase activity for A) human melanoma cell line, SK-MEL-28 and B) lung cancer cell line, A549 were measured at a 36-h time point. Data were presented as mean ± SE of at least 2 independent experiments. *p<0.05, **p<0.005 compared to highest concentration of compound + TNFα used for treatment, as determined by a two-way ANOVA using Bonferroni post-test analysis. Time-response curves for the caspase activity after the indicated treatments for SK-MEL-28 cells and A549 cells were shown on their respective bottom panels.
Discussion and conclusions
In recent years we have witnessed a resurgence of targeted covalent therapeutics, with several newly covalent agents targeting surface Cys residues entering clinical studies.1–14, 74–76 Arguably, this success is attributable to the proper balance between Cys reactivity, plasma stability, and cell permeability of acrylamides warheads. Alpha-beta unsaturated carbonyls in acrylamides have been particularly attractive for cysteine drug targeting because of their balanced reactivity. Such balance between the rate of reaction, selectivity, and stability, seems ideally suited to target Cys residues, which is at the core of the success of the several FDA approved covalent drugs. Currently, increased drug discovery efforts focus on the Cysteinome,74–76 that is the target space that contains a druggable Cys residue. Based on this success, expanding such relatively limited target space to other more frequently occurring residues in proximity of binding sites such as Lys, Tyr, or His is emerging.15–18, 25 Several recent manuscripts report on covalent targeting of Lys residues in active sites of proteins by introduction of appropriately placed electrophiles on existing ligands.17, 18, 20, 77 Our own recent studies15, 16 and others19, 78 also revealed that it is possible to target Lys residues located at protein-protein interfaces, suggesting that in principle the target space of covalent antagonists of PPIs could be expanded to include Lys. Recent studies are already describing the Lysinome or the Tyrosinome as an ensemble of targets that present a “targetable” Lys or Tyr in proximity of their binding sites for co-factors in enzymes.79, 80 Recently, we further investigated side by side the merits and pitfalls of aryl-sulfonyl fluorides and aryl-fluorosulfates as possible warheads to target Lys, Tyr, and His in protein-protein interactions.16 We found that while sulfonyl fluorides can react quickly with these residues, these are fairly unstable in aqueous buffer and in plasma, in agreement with previous in vitro studies.22 Conversely, while aryl-fluorosulfates seemed very stable in buffer and in plasma,16, 68 their reactivity appeared relatively limited requiring several hours incubation time in vitro for the reaction to go to completion.16 This latter observation is in stark contrast with acrylamides, that, when properly juxtaposed to Cys residues, can react readily in vitro within a few minutes, while in cell they form stable adducts with their targets after relatively short exposure times (1–6 h). Of note is that sulfonyl fluorides and aryl-fluorosulfates also react with Cys residues rapidly but the resulting reaction products are not stable and thus usually not observed.22, 23 Another potential issue with these warheads is that the sulfamate linkage generated upon reaction of fluorosulfates with lysines could be labile towards hydrolytic cleavage.21 Hence, the question we tried to address here was whether aryl-fluorosulfates can approach the proper balance of stability, reactivity and cell permeability, similar to what observed with Cys and acrylamides, to be envisioned as future chemical probes or even therapeutics, in particular when targeting protein-protein interactions.
Targeting the BIR3 domain of XIAP using our recent NMR and enthalpy-based screening approaches,71 resulted in compound 1, as an effective Lys 311-targeting covalent agent.15, 16 However, the reaction rate between compound 1 and the BIR3 domain of XIAP was relatively slow, requiring nearly 6 h incubation at 37 °C to observe a complete adduct formation in vitro, as it can be appreciated by the thermal shift data (Table 1). The reaction of compound 1 with BIR3 was largely incomplete after 2 h incubation as it can be inferred by the relatively small observed thermal shift (ΔTm < 10 °C, Table 1, supplementary Figure S1), while the same measurements after 6 h pre-incubation resulted in a ΔTm > 30 °C, typical of covalent binding.16 These observations made us wondering if such a reactivity is suitable for the development of effective agents that target covalently their targets also in cell. Our hypothesis was that the slow reactivity of compound 1 could be due to the fact that both the Lys 311 and the ligand containing the warhead are predicted to be fairly flexible. Hence, we sought to design novel Lys-targeting inhibitors that more directly juxtaposed a binding site Lys with an aryl-fluorosulfate, in the hope that these could react more promptly, similar to what observed between Cys and acrylamides.
For this purpose, simple structure-based design studies identified Lys residues in proximity of position P4 of the tetrapeptides binding to the BIR3 domain of XIAP (Figure 1B). Lys residues are present in a similar position also in cIAP1 and cIAP2 (Figure 1C,D). Previous HTS by ΔH studies in our laboratory identified a 4-fluoro-Phe in position P4 that later was optimized to a 4fluoro-1-amino indane.15, 16, 81 Likewise we had identified in position P2 a tri-substituted Phe with increased potency with respect to a Val or a cyclohexyl-Gly in that position.15, 81 Hence, we synthesized two novel Lys-targeting ligands in which the 4-fluoro-1-amino indane was replaced by a 4-fluorosulfate-1-amino indane in position P4, while cyclohexyl-Gly (compound 2) or the tri-substituted Phe (compound 3) were placed in position P2 (Table 1).
To assess whether these agents covalently interacted with the BIR3 domains of XIAP, cIAP1, or cIAP2, we performed a series of biochemical and biophysical studies. Incubation-dependent IC50 measurements (Table 1, Figure 3) and thermal shift data (Table 1, supplementary Figure S1) suggested that both compounds 2 and 3 could potentially form a covalent bond with XIAP-BIR3. This was corroborated by mass spectrometry data and SDS gel electrophoresis with XIAP-BIR3 (Figure 3). Intriguingly, however, the time course of the reaction between compound 2 and the BIR3 domain of XIAP indicated a complete reaction within 30 min at 25 °C (Figure 3C). This is in stark contrast with previous data with compound 1, where the warhead is located on a more flexible side chain in position P2, targeting a more distant Lys 311 (Table 1) that required significantly longer incubation times for the covalent adduct to be fully formed.16 Thermal shift measurements with compounds 2 and 3 binding to XIAP-BIR3 also displayed ΔTm values > 30 °C, hence, typical of covalent binding, already after 2 h pre-incubation, again in contrast to compound 1 that required 6 h pre-incubation (Table 1) to display similarly large ΔTm values. Next, we used HEK293 cells transfected with an HA-tagged BIR3 construct. We observed that the isolated BIR3 domain of XIAP, in its unbound form, was particularly unstable at 37 °C as also revealed by our thermal shift studies and previously by 1D 1H NMR.15, 16 Accordingly, western blot analysis using anti-HA antibody of HEK293 cell lysates that express HA-BIR3, revealed only a fainted band (if any) for HA unless the cells were treated with potent, cell permeable BIR3 ligands, including LCL161 or AT406. Hence, exposing the cells to these ligands, resulted in the stabilization of the BIR3 domain as it was manifested in an intense band in the western blot (Figure 5A). Most interestingly, exposing cells to covalent agents 1, 2 and 3, resulted in even more intense bands (especially for compound 2) that are also appreciably shifted (especially for compounds 2 and 3, but much less for compound 1), clearly indicating that compounds 2 and 3 are not only cell permeable, but can also covalently interact with the target in cell (Figure 5A). On the contrary, as mentioned, the gel shift for compound 1 is less appreciable, again likely suggesting that this agent would require longer incubation times for the reaction to take place in cell.
cIAP1 and cIAP2 present only one Lys residue in correspondence to the aryl-fluorosulfate, and modeling studies suggest that these Lys residues could likewise form covalent adduct with compounds 2 and 3 (Figure 1) as corroborated by DELFIA data and MS analyses (Figure 5). The position of these Lys residues is not the structural equivalent to Lys 297 of XIAP but rather of Lys 299. In addition, the proximity of another cationic residue in XIAP (Lys 299) may enhance the nucleophilic character of Lys 297. Both arguments would suggest that the reactivity of compounds 2 and 3 versus cIAP1 and cIAP2 should be less effective compared to XIAP, as it seems to be the case based on our data.
Finally, to further corroborate these data, we preliminarily assessed cellular efficacy of these agents by measuring their ability to induce cell apoptosis in the melanoma cell line SK-MEL-28, and in the non-small cell lung cancer cell line A549, that have been reported to be resistant to TNFα due to overexpression of IAP proteins (Figure 5B,C). Using the IncuCyte® Cytotox Green apoptosis assay, we observed that TNFα (1 μg/ml) alone was not able to significantly induce apoptosis in these cell lines. Time- and dose-dependent apoptosis was restored in both SK-MEL-28 and A549 by co-treatment of TNFα and either LCL161, AT406 or our agents compounds 1–3 in nanomolar concentration (Figure 6), clearly corroborating their cellular activity.
With the resurgence and the success of covalent drugs targeting Cys residues via acrylamides-based Michael acceptors, our studies suggest that equally effective strategies targeting Lys residues could be expected by proper juxtaposition of aryl-fluorosulfates in small molecules or peptide mimetics. These observations should significantly widen the target space from the Cysteinome74–76 to other more abundant residues such as Lys, as shown here, or also Tyr, or His as we and others demonstrated recently.16, 25, 82 In targeting PPIs, in particular, our studies support our vision that aryl-fluorosulfates could be effectively incorporated into drug discovery strategies that have emerged in the past decade, including fragment-, structure-, and/or NMR-based approaches,58, 69, 70, 81, 83–85 phage display, and DNA encoded libraries,80, 86–88 aimed at deriving novel, potent, selective, and cell permeable PPI antagonists for continued lead optimizations and drug development.
Experimental Section
General Chemistry.
Solvent and reagents were commercially obtained and used without further purification. NMR spectra used to check concentration were recorded on Bruker Avance III 700MHz. High resolution mass spectral data were acquired on an Agilent LC-TOF instrument. RP-HPLC purifications were performed on a JASCO preparative system equipped with a PDA detector and a fraction collector controlled by a ChromNAV system (JASCO) on a Luna C18 10μ 10 × 250mm (Phenomenex) to > 95% purity. LCL161 and AT406 were obtained from MedChem Express. BAL resin was purchased from Creosalus. Fmoc-amino acids were purchased from Chem-Impex and Novabiochem. The AISF reagent was purchased from Sigma-Aldrich. Peptides were synthesized by using standard solid phase Fmoc peptide synthesis protocols. For each coupling reaction 3 eq. of Fmoc-AA, 3 eq. of HATU and 5 eq. of DIPEA in 1 ml of DMF were used. The coupling reaction was allowed to proceed for 50 min at room temperature, followed by 3 washes with DMF. Kaiser test was employed to monitor reaction completion. Fmoc deprotection was performed in two steps by treating the resin-bound peptide with 20% 4-methylpiperidine in DMF for 5 min then 15 min at room temperature.
Purity of tested compounds was assessed by HPLC using an Atlantis T3 3μm 4.6×150mm column (H2O/ACN gradient from 5% to 100% in 45min). All compounds have a purity >95% (supplementary Figure S4).
Compound 1: 4-((S)-3-((S)-2-(((R)-4-fluoro-2,3-dihydro-1-H-inden-1-yl)carbamoyl)pyrrolidin-1yl)-2-((S)-2-(methylamino)propanamido)-3-oxopropyl)phenyl sulfurofluoridate. The synthesis and purification of compound 1 and the Fmoc protection of the DL-2-amino-3-(2-fluoro-5-methyl-4-(trifluoromethyl)phenyl)propanoic acid were described in our previous publication.16
Compound 2: (R)-1-((S)-1-((S)-2-cyclohexyl-2-((S)-2-(methylamino)propanamido)acetyl)pyrrolidine-2-carboxamido)-2,3-dihydro-1H-inden-4-yl sulfurofluoridate. BAL resin was used as solid-phase support (0.05 mmol scale), and the previously described coupling conditions were used to obtain the peptidic part of the agent. Ary-lfluorosulfate incorporation was performed on resin, using [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF)30 reagent (1.2 eq., 2.2 eq. of DBU in THF, overnight reaction at room temperature). After cleavage, the crude was purified by preparative RP-HPLC using a Luna C18 column (Phenomenex) and water/acetonitrile gradient (5% to 100%) containing 0.1% TFA, obtaining a white powder (15.3 mg, 55.4%). 1D 1H NMR is reported as supplementary Figure S5. HRMS: calcd 552.2418 (M); obs 553.4656 (M+H)+.
Compound 3:(1R)-1-((2S)-1-(3-(2-fluoro-5-methyl-4-(trifluoromethyl)phenyl)-2-((S)-2-(methylamino)propanamido)propanoyl)pyrrolidine-2-carboxamido)-2,3-dihydro-1H-inden-4-yl sulfurofluoridate. BAL resin was used as solid-phase support (0.05 mmol scale), and the previously described coupling conditions were used to obtain the peptidic part of the agent. Aryl-fluorosulfate incorporation was performed on resin, using [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF)30 reagent (1.2 eq., 2.2 eq. of DBU in THF, overnight reaction at room temperature). After cleavage, the crude was purified by preparative RP-HPLC using a Luna C18 column (Phenomenex) and water/acetonitrile gradient (5% to 100%) containing 0.1% TFA, obtaining a white powder (22.0 mg, 66.6%). 1D 1H NMR is reported as supplementary Figure S5. HRMS: calcd 660.2043 (M); obs 661.2114 (M+H)+, 683.1933 (M+Na) +. Attempts to separate the resulting two diastereoisomers of compound 3 were made using three different HPLC columns: Luna 10um C18(2) 100A 250×10mm (Phenomenex); Hypersil GOLD PFP 5μm 250×10mm (Thermo Scientific); XTerra MS C18 OBD 125A 10μm 250×10. However, no appreciable separation of the two epimers was observed after different methods were used. NMR analysis of the purified product showed signals compatible with the presence of a nearly 50:50 mixture of the two species (Supplementary Figure S6).
Protein Expression and Purification.
cDNA fragments encoding the human BIR3 domain of XIAP (residues 253–347) and an N-terminal His tag were used in the expressions of XIAP-BIR3.15 The fragments were transformed into E. coli BL21-Gold(DE3) pLysS cells and grown in LB medium at 37 °C with 100 μg/mL of ampicillin until reaching an OD600 of 0.6–0.7 followed by induction with 1 mM IPTG overnight at 25 °C. Bacteria were then collected by centrifugation and lysed by sonication at 4 °C. Proteins were purified using Ni2+ affinity chromatography, eluted in 25 mM Tris at pH 7.5, 500 mM NaCl, and 500 mM imidazole, and exchanged with a desalting column into an aqueous buffer composed of 25 mM Tris at pH 8, 150 mM NaCl, 50 μM Zn(Ac)2, and 1 mM DTT. The recombinant BIR3 domains of cIAP1 and cIAP2 with N-terminal 6xHis tag were obtained from Reaction Biology Corp. (Malvern, PA).
Dissociation-Enhanced Lanthanide Fluorescence Immunoassay (DELFIA).
Each well of the 96-well streptavidin-coated plates (PerkinElmer) was incubated with 100 μL of 100 nM AVPI-Biotin (AVPIAQKSEK-Biotin) for 1 h followed by three washing steps to remove the unbound AVPI-Biotin. Subsequently, a solution containing 89 μL of Eu-N1-labeled anti-6xHis antibody (PerkinElmer) and a mixture containing 11 μL of the protein and a serial dilution of the test compounds were added to each well and incubated for 2 h with or without pre-incubation (6-h incubation of only the protein and the test compounds). At the end of the incubation period, plates were washed three times with DELFIA wash (PerkinElmer) and 200 μL of the DELFIA enhancement solution (PerkinElmer) was added to each well. Following a 10-min incubation with the enhancement solution, fluorescence was measured using the VICTOR X5 microplate reader (PerkinElmer) with the excitation and emission wavelengths of 340 and 615 nm, respectively. The final antibody concentrations used for XIAP-BIR3 and cIAP1-BIR3 were 22.2 ng/well and 29.7 ng/well for cIAP2-BIR3. The final protein concentrations were 30 nM for XIAP-BIR3 and cIAP1-BIR3 and 15 nM for cIAP2-BIR3. Protein, peptide, and antibody solutions were prepared with DELFIA assay buffer (PerkinElmer) and all the incubations were performed at room temperature. Samples were normalized to 1% DMSO and reported as % inhibition. The IC50 values were calculated from dose-response curves using GraphPad Prism version 7. The reported SE values were obtained from replicate measurements.
Gel Electrophoresis.
10 μM of each protein was incubated at various time points with 100 μM of compounds in a buffer containing 25 mM Tris pH 8, 150 mM NaCl, and 50 μM zinc acetate either at room temperature or 37 μC. Samples were loaded onto the NuPAGE 12% Bis-Tris Protein Gels (Life Technologies) and electrophoresed using MES running buffer (Life Technologies) at 200 V for 35 min. Gels were then stained with SimplyBlue SafeStain (Life Technologies) according to the manufacturer’s protocol.
Cell Culture and Nuclear Labeling.
Non-small cell lung cancer A549 NucLight Red cells were purchased from Essen Bioscience and cultured in Ham’s F-12 nutrient mixture with GlutaMAX-1 (Gibco) supplemented with 10% FBS, 1% PenStrep (100 U/mL penicillin and 100 μg/mL streptomycin) and 0.5 μg/mL puromycin. Human melanoma cell line SK-MEL-28 was obtained from the American Type Culture Collection (ATCC) and the cells were nuclear labeled red (SK-MEL-28 NucLight Red) with the IncuCyte NucLight lentivirus reagent (Essen Bioscience) according to the manufacturer’s protocol. SK-MEL-28 NucLight Red cells were cultured in EMEM (ATCC) supplemented as described above with the exception of 1 μg/mL puromycin. HEK293T cell line was purchased from ATCC and cultured in DMEM supplemented with 10% FBS and 1% PenStrep. All cells were maintained at 37 °C in a humidified incubator with 5% CO2.
Apoptosis Assay.
A549 NucLight Red or SK-MEL-28 NucLight Red cells were seeded at 5 × 103 cell/well in 96-well plates and allowed to attach overnight. The media were removed, and the cells were treated with various concentrations of compounds in the presence or absence of 1 ng/mL TNFα (R&D Systems) and at a final concentration of 2.5 μM of the IncuCyte Caspase-3/7 Green Apoptosis Reagent (Essen Bioscience) for 4 days. Live images were taken every 3 h with the IncuCyte S3 live-cell analysis system and the data at a 36-h time point were analyzed. The Top-Hat method was used to subtract background noise from the red and green channels.
Immunoblots.
The HA-XIAP-BIR3 plasmid was a gift from Dr. Colin Duckett (Addgene plasmid #25689). One million HEK293T cells were plated in 6-well plates and left to attach overnight. The following day, cells were transfected with 0.5 μg of the HA-XIAP-BIR3 plasmid using Lipofectamine 2000 (Thermo Fisher) in complete DMEM media supplemented with 10% FBS and 1% PenStrep. 18 h post-transfection, the media was replaced with serum-free DMEM containing 10 μM of compounds [1% of DMSO] and incubated for an additional 6 h. Finally, the cells were lysed with lysis buffer [20 mM Tris, pH 7.4, 120 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1% IGEPAL, 5 mM EDTA] supplemented with EDTA-free protease inhibitor cocktail and PhosSTOP (SigmaAldrich) for 10 min on cold ice.
One million cells of A549 NucLight Red cells or SK-MEL-28 cells were plated in 6-well plates. The following day, media was removed, and cells were incubated with media serum-free containing DMSO or 1 μM of each compound. After 3 h of incubation, cells were washed and lysed as mentioned earlier. Lysates were centrifuged and supernatants were collected. The protein content was quantified, and the samples were prepared using a NuPAGE antioxidant and LDS sample buffer (Thermo Fisher) and heated for 10 min at 70 °C. Each sample containing 10 μg of proteins were loaded into 4–12% or 12% NuPAGE Bis–Tris precast gels and transferred to PVDF membranes. The membranes were blocked with 5% milk in TBS and 0.1% Tween (TBST) and incubated with anti-HA (Santa Cruz Biotechnology, Y-11, sc-805), anti-cIAP1 (Cell Signaling 7065), XIAP (Cell Signaling 2045) or cIAP2 (Cell Signaling 3130) overnight at 4 °C. Next day, the membrane was washed with TBST and incubated with goat anti-rabbit HRP secondary antibodies. The antigen–antibody complexes were visualized using a Clarity Western ECL Max kit (BIO-RAD). The membrane was stripped, and the western blot was repeated using a β-actin primary antibody (Santa Cruz Biotechnology, sc-69879) to check for loading.
Thermal Shift Assay.
Thermal shift assays for BIR3 construct/inhibitor complexes were obtained with a BioRad CFX Connect Real-Time PCR Detection System. Each data point was collected in triplicate. Incubation of BIR3 protein with inhibitor followed one of two parameters, either 37 °C for 6 h or 25 °C for 2 h. Protein/inhibitor complexes and 5000x SYPRO Orange dye (Sigma) were diluted using reaction buffer, 50 mM Tris pH 8.0, 150 mM NaCl, 50 μM Zinc acetate, to obtain final concentrations of 5 μM BIR3, 10 μM inhibitor, and 60x SYPRO Orange. Sample plates were heated from 10 °C to 95 °C with heating increments of 0.05 °C, over 30 min. Fluorescence intensity was measured within the excitation/emission ranges 470–505/540–700 nm.
Plasma stability.
To test the stability of the agents reported in Table 1, mouse plasma at 37 °C (GenTex: GTX73236) was diluted to 80% with 0.05 M PBS (pH 7.4). Test compounds were added to a 1 ml plasma solution to yield a final concentration of 200 μM (37 °C in triplicate). Samples (50 μl) were taken at various time points (0, 15, 30, 45, 60, 90 and 120 min) and dissolved in 200 μl acetonitrile (4 °C) in order to deproteinize the plasma. After vortexing for 1 min followed by centrifugation (4 °C for 15 min at 14,000 rpm), the clear supernatants were analyzed by Mass Spec Analysis.
Nuclear Magnetic Resonance.
Solution Nuclear Magnetic Resonance (NMR) experiments were conducted on a 700 MHz Bruker Avance spectrometer equipped with a TCI cryoprobe. Each protein sample was dissolved into an NMR tube at a final concentration of 20 μM (1% d6-dmso) in the presence of 100 μM of compound 2 in a buffer containing 25 mM TRIS (2-amino-2-(hydroxymethyl)propane-1,3-diol), pH = 8, 150 mM NaCl, 50 μM of zinc acetate, and 1 mM of DTT. 2D [15N,1H]-sofast HMQC and 1D 1H-aliphatic experiments were acquired.84 NMR data were processed and analyzed using TopSpin 3.6.1 (Bruker).
Molecular modeling.
Covalent docking of compounds in Figure 1, was obtained using Gold (Cambridge Crystallographic Data Center; www.ccdc.cam.ac.uk) (compound 1) or by non-covalent docking followed by manual bond formation and energy minimization of the covalent adduct (SYBYL-X 2.1.1; Certara, Princeton, NJ; compound 2) using Protein Data Bank entries 3HL5, 3UW4, and 2UVL for XIAP, cIAP1 and cIAP2, respectively. The docking preparation for both protein and ligands was performed using SYBYL-X 2.1.1 (Certara, Princeton, NJ) and MOE 2019.0101 (Chemical Computing Group). The figures were generated using MOE 2019.0101 (Chemical Computing Group). The coordinates for models of compound 2 in complex with the BIR3 domains are provided as supplementary information.
Supplementary Material
Acknowledgements
Financial support was obtained in part by the NIH, with grants CA168517, CA242620, and NS107479 (to MP), a City of Hope – UC Riverside Biomedical Research Initiative (CUBRI) grant (to MP), and UC CRCC CRN-18-524906 and UCOP LFR-17-476732 grants (to JJPP). MP holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR. PU is a recipient of the 2017–2018 Pease Cancer Fellowship through the Division of Biomedical Sciences, School of Medicine at UCR. We thank Dr. J. Momper and his associates at the University of California San Diego, Drug Development Pipeline core facility for plasma stability data. HA-XIAP-BIR3 plasmid was a gift from Dr. Colin Duckett (Addgene plasmid # 25689). Our agents can be distributed in small amounts (1–5 mg) for research purposes upon request and signing of a standard material transfer agreement.
Abbreviations used
- XIAP
X-linked Inhibitor of Apoptosis Protein
- BIR
baculovirus IAP repeat domains
- cIAP1
cellular Inhibitor of Apoptosis Protein 1
- cIAP2
cellular Inhibitor of Apoptosis Protein 2
- DELFIA
Dissociation-Enhanced Lanthanide Fluorescent Immunoassay
- DMF
Dimethylformamide
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- AISF
[4-(Acetylamino)phenyl]imidodisulfuryl difluoride
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- THF
Tetrahydrofuran
Footnotes
Ancillary information
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website. Figure S1 contains the individual thermal shift data plots that generated the date reported in Table 1. Figure S2 reports time-dependent cell viability data for two cell lines and each of the compounds reported in Table 1. Figure S3 reports solubility data for the compounds. Figure S4 reports the HPLC traces for compounds 1, 2, and 3. Figures S5 and S6 report 1D 1H NMR spectra of compound 2 and compound 3. PDB files relative to the models prepared for compound 2 in complex with XIAP-BIR3, cIAP1-BIR3, cIAP2-BIR3 are also provided. A Molecular Formula Strings file is also available.
Agents reported in this article have been subjected to a patent application by the University of California, Riverside (UCR). Sunshine Bio Inc., of San Diego (CA) has signed an option to license these agents from UCR to be part their drug development pipeline. MP, CB, LG, and PU are listed as co-inventors in the patent application and may receive a share of eventual future royalties that such license may trigger, according to the University of California policies.
References
- 1.Akher FB; Farrokhzadeh A; Soliman MES Covalent Vs. Non-Covalent Inhibition: Tackling Drug Resistance in Egfr - a Thorough Dynamic Perspective. Chem Biodivers 2019, 16, e1800518. [DOI] [PubMed] [Google Scholar]
- 2.Singh J; Petter RC; Baillie TA; Whitty A The Resurgence of Covalent Drugs. Nat Rev Drug Discov 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- 3.Kalgutkar AS; Dalvie DK Drug Discovery for a New Generation of Covalent Drugs. Expert Opin Drug Discov 2012, 7, 561–581. [DOI] [PubMed] [Google Scholar]
- 4.Mah R; Thomas JR; Shafer CM Drug Discovery Considerations in the Development of Covalent Inhibitors. Bioorg Med Chem Lett 2014, 24, 33–39. [DOI] [PubMed] [Google Scholar]
- 5.Baillie TA Targeted Covalent Inhibitors for Drug Design. Angew Chem Int Ed Engl 2016, 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- 6.Basu D; Richters A; Rauh D Structure-Based Design and Synthesis of Covalent-Reversible Inhibitors to Overcome Drug Resistance in Egfr. Bioorg Med Chem 2015, 23, 2767–2780. [DOI] [PubMed] [Google Scholar]
- 7.Bauer RA Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discov Today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 8.Engel J; Richters A; Getlik M; Tomassi S; Keul M; Termathe M; Lategahn J; Becker C; Mayer-Wrangowski S; Grutter C; Uhlenbrock N; Krull J; Schaumann N; Eppmann S; Kibies P; Hoffgaard F; Heil J; Menninger S; Ortiz-Cuaran S; Heuckmann JM; Tinnefeld V; Zahedi RP; Sos ML; Schultz-Fademrecht C; Thomas RK; Kast SM; Rauh D Targeting Drug Resistance in Egfr with Covalent Inhibitors: A Structure-Based Design Approach. J Med Chem 2015, 58, 6844–6863. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK; Samanta I; Mondal A; Liu WR Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lonsdale R; Ward RA Structure-Based Design of Targeted Covalent Inhibitors. Chem Soc Rev 2018, 47, 3816–3830. [DOI] [PubMed] [Google Scholar]
- 11.Vasudevan A; Argiriadi MA; Baranczak A; Friedman MM; Gavrilyuk J; Hobson AD; Hulce JJ; Osman S; Wilson NS Covalent Binders in Drug Discovery. Prog Med Chem 2019, 58, 1–62. [DOI] [PubMed] [Google Scholar]
- 12.Nussinov R; Tsai CJ The Design of Covalent Allosteric Drugs. Annu Rev Pharmacol Toxicol 2015, 55, 249–267. [DOI] [PubMed] [Google Scholar]
- 13.Adeniyi AA; Muthusamy R; Soliman ME New Drug Design with Covalent Modifiers. Expert Opin Drug Discov 2016, 11, 79–90. [DOI] [PubMed] [Google Scholar]
- 14.Bjij I; Olotu FA; Agoni C; Adeniji E; Khan S; El Rashedy A; Cherqaoui D; Soliman MES Covalent Inhibition in Drug Discovery: Filling the Void in Literature. Curr Top Med Chem 2018, 18, 1135–1145. [DOI] [PubMed] [Google Scholar]
- 15.Baggio C; Gambini L; Udompholkul P; Salem AF; Aronson A; Dona A; Troadec E; Pichiorri F; Pellecchia M Design of Potent Pan-Iap and Lys-Covalent Xiap Selective Inhibitors Using a Thermodynamics Driven Approach. J Med Chem 2018, 26, 6350–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gambini L; Baggio C; Udompholkul P; Jossart J; Salem AF; Perry JJP; Pellecchia M Covalent Inhibitors of Protein-Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J Med Chem 2019, 62, 5616–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mortenson DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET; Sharpless KB; Wilson IA; Kelly JW “Inverse Drug Discovery” Strategy to Identify Proteins That Are Targeted by Latent Electrophiles as Exemplified by Aryl Fluorosulfates. J Am Chem Soc 2018, 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pettinger J; Jones K; Cheeseman MD Lysine-Targeting Covalent Inhibitors. Angew Chem Int Ed Engl 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- 19.Akcay G; Belmonte MA; Aquila B; Chuaqui C; Hird AW; Lamb ML; Rawlins PB; Su N; Tentarelli S; Grimster NP; Su Q Inhibition of Mcl-1 through Covalent Modification of a Noncatalytic Lysine Side Chain. Nat Chem Biol 2016, 12, 931–936. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J Am Chem Soc 2017, 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baranczak A; Liu Y; Connelly S; Du WG; Greiner ER; Genereux JC; Wiseman RL; Eisele YS; Bradbury NC; Dong J; Noodleman L; Sharpless KB; Wilson IA; Encalada SE; Kelly JW A Fluorogenic Aryl Fluorosulfate for Intraorganellar Transthyretin Imaging in Living Cells and in Caenorhabditis Elegans. J Am Chem Soc 2015, 137, 7404–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukherjee H; Debreczeni J; Breed J; Tentarelli S; Aquila B; Dowling JE; Whitty A; Grimster NP A Study of the Reactivity of S((Vi))-F Containing Warheads with Nucleophilic Amino-Acid Side Chains under Physiological Conditions. Org Biomol Chem 2017, 15, 9685–9695. [DOI] [PubMed] [Google Scholar]
- 23.Gehringer M; Laufer SA Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J Med Chem 2019, 62, 5673–5724. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee H; Grimster NP Beyond Cysteine: Recent Developments in the Area of Targeted Covalent Inhibition. Curr Opin Chem Biol 2018, 44, 30–38. [DOI] [PubMed] [Google Scholar]
- 25.Chen W; Dong J; Plate L; Mortenson DE; Brighty GJ; Li S; Liu Y; Galmozzi A; Lee PS; Hulce JJ; Cravatt BF; Saez E; Powers ET; Wilson IA; Sharpless KB; Kelly JW Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(S) through Chemoselective Sufex Reaction with a Binding Site Tyr Residue. J Am Chem Soc 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corti A; Milani M; Lecis D; Seneci P; de Rosa M; Mastrangelo E; Cossu F Structure-Based Design and Molecular Profiling of Smac-Mimetics Selective for Cellular Iaps. FEBS J 2018, 285, 3286–3298. [DOI] [PubMed] [Google Scholar]
- 27.Barile E; Marconi GD; De SK; Baggio C; Gambini L; Salem AF; Kashyap MK; Castro JE; Kipps TJ; Pellecchia M Hbfl-1/Hnoxa Interaction Studies Provide New Insights on the Role of Bfl-1 in Cancer Cell Resistance and for the Design of Novel Anticancer Agents. ACS Chem Biol 2017, 12, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stebbins JL; Santelli E; Feng Y; De SK; Purves A; Motamedchaboki K; Wu B; Ronai ZA; Liddington RC; Pellecchia M Structure-Based Design of Covalent Siah Inhibitors. Chem Biol 2013, 20, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narayanan A; Jones LH Sulfonyl Fluorides as Privileged Warheads in Chemical Biology. Chem Sci 2015, 6, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H; Mukherjee P; Liu R; Evrard E; Wang D; Humphrey JM; Butler TW; Hoth LR; Sperry JB; Sakata SK; Helal CJ; Am Ende CW Introduction of a Crystalline, Shelf-Stable Reagent for the Synthesis of Sulfur(Vi) Fluorides. Org Lett 2018, 20, 812–815. [DOI] [PubMed] [Google Scholar]
- 31.Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(Vi) Fluoride Exchange (Sufex): Another Good Reaction for Click Chemistry. Angew Chem Int Ed Engl 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- 32.Deveraux QL; Reed JC Iap Family Proteins--Suppressors of Apoptosis. Genes Dev 1999, 13, 239–252. [DOI] [PubMed] [Google Scholar]
- 33.Salvesen GS; Duckett CS Iap Proteins: Blocking the Road to Death’s Door. Nat Rev Mol Cell Biol 2002, 3, 401–410. [DOI] [PubMed] [Google Scholar]
- 34.Cong H; Xu L; Wu Y; Qu Z; Bian T; Zhang W; Xing C; Zhuang C Inhibitor of Apoptosis Protein (Iap) Antagonists in Anticancer Agent Discovery: Current Status and Perspectives. J Med Chem 2019, 62, 5750–5772. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y; Rich RL; Myszka DG; Wu H Requirement of Both the Second and Third Bir Domains for the Relief of X-Linked Inhibitor of Apoptosis Protein (Xiap)-Mediated Caspase Inhibition by Smac. J Biol Chem 2003, 278, 49517–49522. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z; Sun C; Olejniczak ET; Meadows RP; Betz SF; Oost T; Herrmann J; Wu JC; Fesik SW Structural Basis for Binding of Smac/Diablo to the Xiap Bir3 Domain. Nature 2000, 408, 1004–1008. [DOI] [PubMed] [Google Scholar]
- 37.Wu G; Chai J; Suber TL; Wu JW; Du C; Wang X; Shi Y Structural Basis of Iap Recognition by Smac/Diablo. Nature 2000, 408, 1008–1012. [DOI] [PubMed] [Google Scholar]
- 38.Samuel T; Welsh K; Lober T; Togo SH; Zapata JM; Reed JC Distinct Bir Domains of Ciap1 Mediate Binding to and Ubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 2 and Second Mitochondrial Activator of Caspases. J Biol Chem 2006, 281, 1080–1090. [DOI] [PubMed] [Google Scholar]
- 39.Cai Q; Sun H; Peng Y; Lu J; Nikolovska-Coleska Z; McEachern D; Liu L; Qiu S; Yang CY; Miller R; Yi H; Zhang T; Sun D; Kang S; Guo M; Leopold L; Yang D; Wang S A Potent and Orally Active Antagonist (Sm-406/at-406) of Multiple Inhibitor of Apoptosis Proteins (Iaps) in Clinical Development for Cancer Treatment. J Med Chem 2011, 54, 2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen F; Alicke B; Elliott LO; Flygare JA; Goncharov T; Keteltas SF; Franklin MC; Frankovitz S; Stephan JP; Tsui V; Vucic D; Wong H; Fairbrother WJ Orally Bioavailable Antagonists of Inhibitor of Apoptosis Proteins Based on an Azabicyclooctane Scaffold. J Med Chem 2009, 52, 1723–1730. [DOI] [PubMed] [Google Scholar]
- 41.Flygare JA; Beresini M; Budha N; Chan H; Chan IT; Cheeti S; Cohen F; Deshayes K; Doerner K; Eckhardt SG; Elliott LO; Feng B; Franklin MC; Reisner SF; Gazzard L; Halladay J; Hymowitz SG; La H; LoRusso P; Maurer B; Murray L; Plise E; Quan C; Stephan JP; Young SG; Tom J; Tsui V; Um J; Varfolomeev E; Vucic D; Wagner AJ; Wallweber HJ; Wang L; Ware J; Wen Z; Wong H; Wong JM; Wong M; Wong S; Yu R; Zobel K; Fairbrother WJ Discovery of a Potent Small-Molecule Antagonist of Inhibitor of Apoptosis (Iap) Proteins and Clinical Candidate for the Treatment of Cancer (Gdc-0152). J Med Chem 2012, 55, 4101–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaither A; Porter D; Yao Y; Borawski J; Yang G; Donovan J; Sage D; Slisz J; Tran M; Straub C; Ramsey T; Iourgenko V; Huang A; Chen Y; Schlegel R; Labow M; Fawell S; Sellers WR; Zawel L A Smac Mimetic Rescue Screen Reveals Roles for Inhibitor of Apoptosis Proteins in Tumor Necrosis Factor-Alpha Signaling. Cancer Res 2007, 67, 11493–11498. [DOI] [PubMed] [Google Scholar]
- 43.Hashimoto K; Saito B; Miyamoto N; Oguro Y; Tomita D; Shiokawa Z; Asano M; Kakei H; Taya N; Kawasaki M; Sumi H; Yabuki M; Iwai K; Yoshida S; Yoshimatsu M; Aoyama K; Kosugi Y; Kojima T; Morishita N; Dougan DR; Snell GP; Imamura S; Ishikawa T Design and Synthesis of Potent Inhibitor of Apoptosis (Iap) Proteins Antagonists Bearing an Octahydropyrrolo[1,2-a]Pyrazine Scaffold as a Novel Proline Mimetic. J Med Chem 2013, 56, 1228–1246. [DOI] [PubMed] [Google Scholar]
- 44.Li L; Thomas RM; Suzuki H; De Brabander JK; Wang X; Harran PG A Small Molecule Smac Mimic Potentiates Trail- and Tnfalpha-Mediated Cell Death. Science 2004, 305, 1471–1474. [DOI] [PubMed] [Google Scholar]
- 45.Ndubaku C; Varfolomeev E; Wang L; Zobel K; Lau K; Elliott LO; Maurer B; Fedorova AV; Dynek JN; Koehler M; Hymowitz SG; Tsui V; Deshayes K; Fairbrother WJ; Flygare JA; Vucic D Antagonism of C-Iap and Xiap Proteins Is Required for Efficient Induction of Cell Death by Small-Molecule Iap Antagonists. ACS Chem Biol 2009, 4, 557–566. [DOI] [PubMed] [Google Scholar]
- 46.Oost TK; Sun C; Armstrong RC; Al-Assaad AS; Betz SF; Deckwerth TL; Ding H; Elmore SW; Meadows RP; Olejniczak ET; Oleksijew A; Oltersdorf T; Rosenberg SH; Shoemaker AR; Tomaselli KJ; Zou H; Fesik SW Discovery of Potent Antagonists of the Antiapoptotic Protein Xiap for the Treatment of Cancer. J Med Chem 2004, 47, 4417–4426. [DOI] [PubMed] [Google Scholar]
- 47.Peng Y; Sun H; Nikolovska-Coleska Z; Qiu S; Yang CY; Lu J; Cai Q; Yi H; Kang S; Yang D; Wang S Potent, Orally Bioavailable Diazabicyclic Small-Molecule Mimetics of Second Mitochondria-Derived Activator of Caspases. 8 J Med Chem 2008, 51, 8158–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheng R; Sun H; Liu L; Lu J; McEachern D; Wang G; Wen J; Min P; Du Z; Lu H; Kang S; Guo M; Yang D; Wang S A Potent Bivalent Smac Mimetic (Sm-1200) Achieving Rapid, Complete, and Durable Tumor Regression in Mice. J Med Chem 2013, 56, 3969–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H; Lu J; Liu L; Yi H; Qiu S; Yang CY; Deschamps JR; Wang S Nonpeptidic and Potent Small-Molecule Inhibitors of Ciap-1/2 and Xiap Proteins. J Med Chem 2010, 53, 6361–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun H; Nikolovska-Coleska Z; Lu J; Meagher JL; Yang CY; Qiu S; Tomita Y; Ueda Y; Jiang S; Krajewski K; Roller PP; Stuckey JA; Wang S Design, Synthesis, and Characterization of a Potent, Nonpeptide, Cell-Permeable, Bivalent Smac Mimetic That Concurrently Targets Both the Bir2 and Bir3 Domains in Xiap. J Am Chem Soc 2007, 129, 15279–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun H; Nikolovska-Coleska Z; Lu J; Qiu S; Yang CY; Gao W; Meagher J; Stuckey J; Wang S Design, Synthesis, and Evaluation of a Potent, Cell-Permeable, Conformationally Constrained Second Mitochondria Derived Activator of Caspase (Smac) Mimetic. J Med Chem 2006, 49, 7916–7920. [DOI] [PubMed] [Google Scholar]
- 52.Sun H; Nikolovska-Coleska Z; Yang CY; Xu L; Tomita Y; Krajewski K; Roller PP; Wang S Structure-Based Design, Synthesis, and Evaluation of Conformationally Constrained Mimetics of the Second Mitochondria-Derived Activator of Caspase That Target the X-Linked Inhibitor of Apoptosis Protein/Caspase-9 Interaction Site. J Med Chem 2004, 47, 4147–4150. [DOI] [PubMed] [Google Scholar]
- 53.Sun W; Nikolovska-Coleska Z; Qin D; Sun H; Yang CY; Bai L; Qiu S; Wang Y; Ma D; Wang S Design, Synthesis, and Evaluation of Potent, Nonpeptidic Mimetics of Second Mitochondria-Derived Activator of Caspases. J Med Chem 2009, 52, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang S Design of Small-Molecule Smac Mimetics as Iap Antagonists. Curr Top Microbiol Immunol 2011, 348, 89–113. [DOI] [PubMed] [Google Scholar]
- 55.Tse C; Shoemaker AR; Adickes J; Anderson MG; Chen J; Jin S; Johnson EF; Marsh KC; Mitten MJ; Nimmer P; Roberts L; Tahir SK; Xiao Y; Yang X; Zhang H; Fesik S; Rosenberg SH; Elmore SW Abt-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res 2008, 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- 56.Zobel K; Wang L; Varfolomeev E; Franklin MC; Elliott LO; Wallweber HJ; Okawa DC; Flygare JA; Vucic D; Fairbrother WJ; Deshayes K Design, Synthesis, and Biological Activity of a Potent Smac Mimetic That Sensitizes Cancer Cells to Apoptosis by Antagonizing Iaps. ACS Chem Biol 2006, 1, 525–533. [DOI] [PubMed] [Google Scholar]
- 57.Hennessy EJ; Saeh JC; Sha L; MacIntyre T; Wang H; Larsen NA; Aquila BM; Ferguson AD; Laing NM; Omer CA Discovery of Aminopiperidine-Based Smac Mimetics as Iap Antagonists. Bioorg Med Chem Lett 2012, 22, 1690–1694. [DOI] [PubMed] [Google Scholar]
- 58.Huang JW; Zhang Z; Wu B; Cellitti JF; Zhang X; Dahl R; Shiau CW; Welsh K; Emdadi A; Stebbins JL; Reed JC; Pellecchia M Fragment-Based Design of Small Molecule X-Linked Inhibitor of Apoptosis Protein Inhibitors. J Med Chem 2008, 51, 7111–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brunckhorst MK; Lerner D; Wang S; Yu Q At-406, an Orally Active Antagonist of Multiple Inhibitor of Apoptosis Proteins, Inhibits Progression of Human Ovarian Cancer. Cancer Biol Ther 2012, 13, 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang T; Li Y; Zou P; Yu JY; McEachern D; Wang S; Sun D Physiologically Based Pharmacokinetic and Pharmacodynamic Modeling of an Antagonist (Sm-406/at-406) of Multiple Inhibitor of Apoptosis Proteins (Iaps) in a Mouse Xenograft Model of Human Breast Cancer. Biopharm Drug Dispos 2013, 34, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hurwitz HI; Smith DC; Pitot HC; Brill JM; Chugh R; Rouits E; Rubin J; Strickler J; Vuagniaux G; Sorensen JM; Zanna C Safety, Pharmacokinetics, and Pharmacodynamic Properties of Oral Debio1143 (at-406) in Patients with Advanced Cancer: Results of a First-in-Man Study. Cancer Chemother Pharmacol 2015, 75, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perimenis P; Galaris A; Voulgari A; Prassa M; Pintzas A Iap Antagonists Birinapant and at-406 Efficiently Synergise with Either Trail, Braf, or Bcl-2 Inhibitors to Sensitise Brafv600e Colorectal Tumour Cells to Apoptosis. BMC Cancer 2016, 16, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen KF; Lin JP; Shiau CW; Tai WT; Liu CY; Yu HC; Chen PJ; Cheng AL Inhibition of Bcl-2 Improves Effect of Lcl161, a Smac Mimetic, in Hepatocellular Carcinoma Cells. Biochem Pharmacol 2012, 84, 268–277. [DOI] [PubMed] [Google Scholar]
- 64.Houghton PJ; Kang MH; Reynolds CP; Morton CL; Kolb EA; Gorlick R; Keir ST; Carol H; Lock R; Maris JM; Billups CA; Smith MA Initial Testing (Stage 1) of Lcl161, a Smac Mimetic, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2012, 58, 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Infante JR; Dees EC; Olszanski AJ; Dhuria SV; Sen S; Cameron S; Cohen RB Phase I Dose-Escalation Study of Lcl161, an Oral Inhibitor of Apoptosis Proteins Inhibitor, in Patients with Advanced Solid Tumors. J Clin Oncol 2014, 32, 3103–3110. [DOI] [PubMed] [Google Scholar]
- 66.Qin Q; Zuo Y; Yang X; Lu J; Zhan L; Xu L; Zhang C; Zhu H; Liu J; Liu Z; Tao G; Dai S; Zhang X; Ma J; Cai J; Sun X Smac Mimetic Compound Lcl161 Sensitizes Esophageal Carcinoma Cells to Radiotherapy by Inhibiting the Expression of Inhibitor of Apoptosis Protein. Tumour Biol 2014, 35, 2565–2574. [DOI] [PubMed] [Google Scholar]
- 67.Tian A; Wilson GS; Lie S; Wu G; Hu Z; Hebbard L; Duan W; George J; Qiao L Synergistic Effects of Iap Inhibitor Lcl161 and Paclitaxel on Hepatocellular Carcinoma Cells. Cancer Lett 2014, 351, 232–241. [DOI] [PubMed] [Google Scholar]
- 68.Fadeyi OO; Hoth LR; Choi C; Feng X; Gopalsamy A; Hett EC; Kyne RE Jr.; Robinson RP; Jones LH Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem Biol 2017, 12, 2015–2020. [DOI] [PubMed] [Google Scholar]
- 69.Wu B; Zhang Z; Noberini R; Barile E; Giulianotti M; Pinilla C; Houghten RA; Pasquale EB; Pellecchia M Hts by Nmr of Combinatorial Libraries: A Fragment-Based Approach to Ligand Discovery. Chem Biol 2013, 20, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu B; Barile E; De SK; Wei J; Purves A; Pellecchia M High-Throughput Screening by Nuclear Magnetic Resonance (Hts by Nmr) for the Identification of Ppis Antagonists. Curr Top Med Chem 2015, 15, 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baggio C; Udompholkul P; Barile E; Pellecchia M Enthalpy-Based Screening of Focused Combinatorial Libraries for the Identification of Potent and Selective Ligands. ACS Chem Biol 2017, 12, 2981–2989. [DOI] [PubMed] [Google Scholar]
- 72.Martin JS; MacKenzie CJ; Fletcher D; Gilbert IH Characterising Covalent Warhead Reactivity. Bioorg Med Chem 2019, 27, 2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis J; Burstein E; Reffey SB; Bratton SB; Roberts AB; Duckett CS Uncoupling of the Signaling and Caspase-Inhibitory Properties of X-Linked Inhibitor of Apoptosis. J Biol Chem 2004, 279, 9023–9029. [DOI] [PubMed] [Google Scholar]
- 74.Chaikuad A; Koch P; Laufer SA; Knapp S The Cysteinome of Protein Kinases as a Target in Drug Development. Angew Chem Int Ed Engl 2018, 57, 4372–4385. [DOI] [PubMed] [Google Scholar]
- 75.Wu S; Luo Howard H; Wang H; Zhao W; Hu Q; Yang Y Cysteinome: The First Comprehensive Database for Proteins with Targetable Cysteine and Their Covalent Inhibitors. Biochem Biophys Res Commun 2016, 478, 1268–1273. [DOI] [PubMed] [Google Scholar]
- 76.Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anscombe E; Meschini E; Mora-Vidal R; Martin MP; Staunton D; Geitmann M; Danielson UH; Stanley WA; Wang LZ; Reuillon T; Golding BT; Cano C; Newell DR; Noble ME; Wedge SR; Endicott JA; Griffin RJ Identification and Characterization of an Irreversible Inhibitor of Cdk2. Chem Biol 2015, 22, 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoppmann C; Wang L Proximity-Enabled Bioreactivity to Generate Covalent Peptide Inhibitors of P53-Mdm4. Chem Commun (Camb) 2016, 52, 5140–5143. [DOI] [PubMed] [Google Scholar]
- 79.Zhao Z; Bourne PE Progress with Covalent Small-Molecule Kinase Inhibitors. Drug Discov Today 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- 80.Liu R; Yue Z; Tsai CC; Shen J Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J Am Chem Soc 2019, 141, 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baggio C; Cerofolini L; Fragai M; Luchinat C; Pellecchia M Hts by Nmr for the Identification of Potent and Selective Inhibitors of Metalloenzymes. ACS Med Chem Lett 2018, 9, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tamura T; Ueda T; Goto T; Tsukidate T; Shapira Y; Nishikawa Y; Fujisawa A; Hamachi I Rapid Labelling and Covalent Inhibition of Intracellular Native Proteins Using Ligand-Directed N-Acyl-N-Alkyl Sulfonamide. Nat Commun 2018, 9, 1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bottini A; Wu B; Barile E; De SK; Leone M; Pellecchia M High-Throughput Screening (Hts) by Nmr Guided Identification of Novel Agents Targeting the Protein Docking Domain of Yoph. ChemMedChem 2016, 11, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barile E; Pellecchia M Nmr-Based Approaches for the Identification and Optimization of Inhibitors of Protein-Protein Interactions. Chem Rev 2014, 114, 4749–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pellecchia M; Sem DS; Wuthrich K Nmr in Drug Discovery. Nat Rev Drug Discov 2002, 1, 211–219. [DOI] [PubMed] [Google Scholar]
- 86.Favalli N; Bassi G; Scheuermann J; Neri D DNA-Encoded Chemical Libraries - Achievements and Remaining Challenges. FEBS Lett 2018, 592, 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Favalli N; Biendl S; Hartmann M; Piazzi J; Sladojevich F; Graslund S; Brown PJ; Nareoja K; Schuler H; Scheuermann J; Franzini R; Neri D A DNA-Encoded Library of Chemical Compounds Based on Common Scaffolding Structures Reveals the Impact of Ligand Geometry on Protein Recognition. ChemMedChem 2018, 13, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neri D; Lerner RA DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu Rev Biochem 2018, 87, 479–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.