

Abstract
Environmentally sustainable fuel cells with high efficiency have attracted much attention as a promising approach to resolving future energy problems. However, some obstacles must be overcome, such as corrosion, water control, and long-term degradation. Herein, we investigated the improved electrochemical performance and hydrogen oxidation reaction (HOR) mechanism of platinum loaded on carbon nanotube (Pt/CNT) catalyst by conducting experimental and theoretical studies. The Pt/CNT catalyst had a larger active area than the Pt/C (platinum loaded on carbon black) catalyst and also exhibited improved performance due to its long-term stability. In addition, the charge-transfer resistance of Pt/CNT (61.2 Ω cm2) is much smaller than that of Pt/C (90.2 Ω cm2), indicating that the CNT support offers good electron transfer. To further understand the hydrogen dissociation mechanisms of Pt/CNT and Pt/C, we investigated the adsorption characteristics and electron transfer of the catalysts with optimized geometry using the density functional theory (DFT). Pt/CNT exhibited higher adsorption energy and electron transfer than Pt/C, which leads to improved HOR. The integrated experimental and theoretical study conducted here suggests that Pt/CNT is a promising candidate for maintaining the performance of cathode catalysts in the polymer electrolyte membrane fuel cell.
1. Introduction
Efficient and economical future energy systems have been extensively studied to replace the existing hydrocarbon-based power generation because global climate change is partly attributed to conventional energy systems.1,2 Fuel cells hold great potential as a viable nonpolluting power generation option, owing to their high energy densities and a wide range of operating temperatures.3,4 Eco-friendly and highly efficient fuel cell systems, such as proton-exchange membrane fuel cells (PEMFCs), operating at low temperatures (<150 °C) are a promising electrochemical energy device for directly converting hydrogen into power.5−7 In addition, PEMFC is a commercially available candidate for replacing conventional power sources in the field of automotive, marine, and portable applications as they generally operate below 120 °C and have an easy start-up and high energy efficiency.8 However, several aspects of fuel cells could be improved, such as their efficiency, moisture control, and resistance to pollutants. The most commonly used electrode catalyst in fuel cells is Pt loaded on black carbon (Pt/C), which is plated with nanosized platinum on a carbon support.9,10 However, the carbon support material tends to deteriorate under the operating conditions of a PEMFC, resulting in carbon corrosion on the surface and the formation of pores beneath the Pt nanoparticles.11,12 This causes the Ostwald ripening to occur, which condenses and sinters the Pt particles. In short, carbon corrosion results in a reduced electrochemically active surface area (EASA) and inhibits long-term operation.13−16 To reduce the manufacturing cost of the electrode and improve the long-term performance, it is necessary to use catalyst support with excellent electrical conductivity and corrosion resistance to minimize the amount of loading using a Pt alloy and reduce the deterioration of the electrochemical performance due to the growth of Pt particles caused by the long-term operation. Carbon nanotubes (CNTs) have received great interest as nanomaterials in recent decades.17−21 CNTs have been used as catalyst electrodes in various fields and have excellent properties, such as good mechanical strength, high aspect ratio, large specific surface area, high electrical conductivity, high thermal conductivity, high hydrophobicity, and high chemical stability.22−25 Hence, we explored the electrochemical activity of Pt loaded on a CNT catalyst (Pt/CNT) to effectively improve the electrocatalytic performance by virtue of the outstanding properties of CNT as a substrate by conducting integrated experimental and theoretical studies. We performed cyclic voltammetry (CV) to investigate the differences in the electrocatalytic activity of Pt/C and Pt/CNT. In addition to the experiments, we used the density functional theory (DFT) to elucidate the electrochemical adsorption mechanism of the Pt/CNT catalyst in hydrogen oxidation reaction (HOR) catalysis. We also focused on understanding the reaction path of H2 dissociation to determine the effect of H2 and H+ adsorption and the CNT involved in the HOR dependent on the catalyst’s supports (Figure 1).
Figure 1.
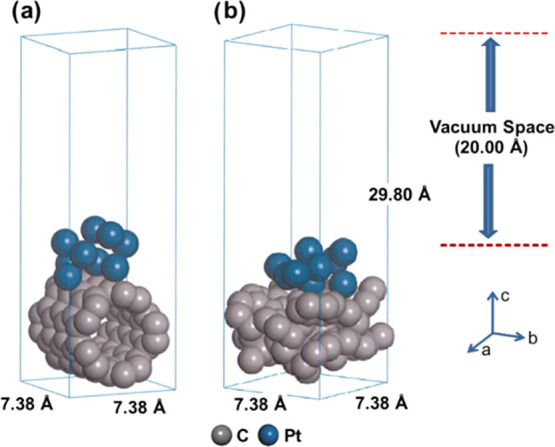
Optimized periodic slab model with 3 × 3 cell crystal structures: (a) Pt(111) loaded on CNT(Pt/CNT) and (b) Pt(111) loaded on black carbon (Pt/C).
2. Results and Discussion
2.1. Morphology and Structural Characterization
It is necessary to analyze the structural and formation characteristics of catalysts before preparing the electrodes. To investigate the dispersion of Pt/CNT (Figure 2a, b) and Pt/C (Figure 2c, d), we conducted the morphological analysis using a scanning electron microscope (SEM) and a transmission electron microscope (TEM). Metal nanoparticles were uniformly distributed on both carbon supports, amorphous carbon, and CNT. To determine the crystalline structure of the carbon and CNT used as catalyst supports, we measured the X-ray powder diffraction (XRD) patterns, as shown in Figure 3. The (111) planes of both supports were strongly exposed to the reaction; thus, we conducted DFT calculation for the (111) plane. The (111), (200), and (220) planes of Pt/CNT exhibited a higher diffracted ray than the Pt/C catalyst, indicating that Pt/CNT was uniformly distributed and had crystalline characteristics. A significant, wide Pt(111) peak was observed in Figure 3 (blue), while a narrow Pt(111) peak was observed in Figure 3 (red). This suggests that crystal growth was better than that of amorphous carbon when Pt nanoparticles are combined with CNT. Table 1 presents the Pt nanoparticle size and atomic composition determined from the TEM, XRD, and energy-dispersive system (EDS) analyses. Based on the XRD results, the Pt particle sizes determined from the Scherrer formula were 4.9 and 5.0 nm, which is consistent with the values of 5.0 and 5.1 nm measured by TEM. According to the EDS analysis, the atomic composition of the Pt/C and Pt/CNT were Pt (6.6%)/C (93.4%) and Pt (6.5%)/C (93.5%), respectively, thereby indicating the reliability of the chemical performance evaluation of the two electrode catalysts with different supports. The temperature resistance of fuel cell catalysts is an important factor for improving the HOR efficiency and long-term stability, as the current density generally decreases as the operating temperature increases under high overvoltage conditions. Therefore, we performed thermogravimetric analysis (TGA) to assess the mass changes of the electrocatalysts depending on the temperature.
Figure 2.

Morphological analysis using the SEM and TEM images of Pt/CNT (a, b) and Pt/C (c, d).
Figure 3.
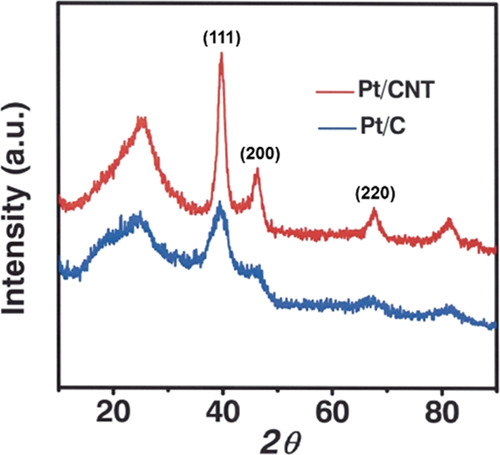
XRD patterns of the Pt/CNT and Pt/CD catalysts.
Table 1. Physical Characteristics of the Catalysts Obtained Using STEM, XRD, and EDS.
| a | loaded
Pt particle size (nm) |
atomic
composition (%) |
|||
|---|---|---|---|---|---|
| catalyst | EASA (m2/g)a | STM | XRD | carbon | platinum |
| Pt/C | 39.5 | 5.0 | 4.9 | 93.4 | 6.6 |
| Pt/CNT | 49.6 | 5.1 | 5.0 | 93.5 | 6.5 |
Electrochemically active surface area is calculated by QH[Pt] × CML (CML = 0.21 mC/cm2) for Pt.
As a result, the rapid mass-loss rates of Pt/C and Pt/CNT were shown as 70 and 50%, respectively, at around 400–450 °C, as shown in Figure 4, which shows high thermal resistance of the CNT support. Thus, the results show that the property of CNTs with high physical and thermal stability led to good resistance to temperature, and they are more suitable as catalysts for PEMFC. It can be considered that the high thermal stability of electrocatalysts can significantly reduce the deterioration of the electrode to enhance the long-term cycling stability.
Figure 4.
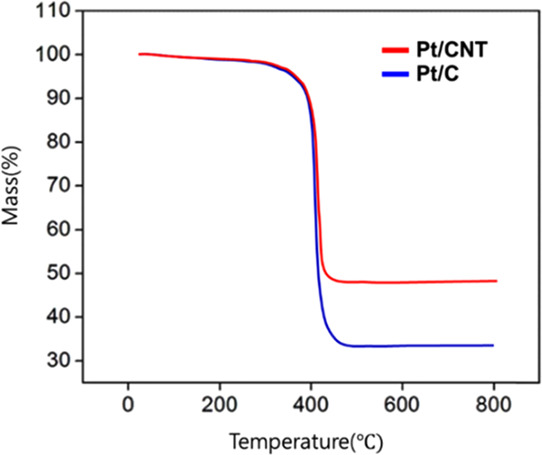
TGA of the Pt/CNT and Pt/CD catalysts.
2.2. Electrochemical Performance of Pt/CNT and Pt/C
To investigate the feasibility of the proposed electrocatalyst, we evaluated the electrochemical performance of both the Pt/CNT and Pt/C electrocatalysts for HOR applications, as shown in Figure 3. We measured the CV profiles using both electrocatalysts as the working electrodes in an acidic medium (0.5 M H2SO4) to determine the role of the carbon structure in enhancing the electrochemical characteristics of Pt/CNT (Figure 5a) for HOR and compared their characteristics to those of Pt/C (Figure 5b). We found that the peak current density in the forward scan of Pt/CNT (0.33 mA) is significantly higher than that of Pt/C (0.17 mA). Meanwhile, the integrated peak intensity of Pt/CNT was higher than that of Pt/C, confirming that Pt/CNT is more active than Pt/C toward the hydrogen adsorption and desorption processes, as Pt/CNT has more available electrochemical sites for HOR, stemming from the increased EASA of Pt/CNT (49.6 m2/g) compared to that of Pt/C (39.5 m2/g; Table 1 cycle show that the reduced current density of Pt/CNT is higher than that of Pt/C, which can be attributed to the use of the nanotube structure as a support. The CNT plays an important role in stably anchoring the Pt nanoparticles and improving the resistance to Pt agglomeration during HOR, leading to high electrocatalytic activity and high stability. The high electrical conductivity and specific surface area of the CNT as catalyst support resulted in excellent electrical activity and improved long-term durability, owing to its high physical strength and thermal stability. To evaluate the long-term stability of both electrocatalysts using the HOR profiles, the change in the EASA during multiple cycles was measured using a CV instrument, as shown in Figure 5c. The EASA of Pt/CNT and Pt/C decreased to 68 and 57% after 300 repeated cycles, respectively, which clearly represents the improved long-term stability of Pt/CNT to provide a sufficiently active surface area for HOR. Low resistance plays an important role in the durability of electrocatalysts, as sufficient conductivity is important to allow the free transport of electrons between electrodes. Since CNT has relatively low corrosiveness and excellent durability with the large specific surface area, well-distributed Pt particles over the pore of the CNT support lead to the improved performance with a low agglomeration of Pt particles. We conducted electrochemical impedance spectroscopy (EIS) analysis to measure the charge-transfer resistance (Rct), which influences the electrocatalytic activity of the electrocatalyst in HOR. Figure 5d shows the Nyquist plots of the electrocatalysts in an acidic medium. In the Nyquist plots, the charge-transfer resistance of Pt/CNT (61.2 Ω cm2) is much smaller than that of Pt/C (90.2 Ω cm2), which illustrates that CNT readily paves the way for superior electron transport between the electrodes. The carbon nanotube structure provides a more stable electro-oxidation activity through a continuous conduction pathway in the PEMFC system than the conventional black carbon.
Figure 5.
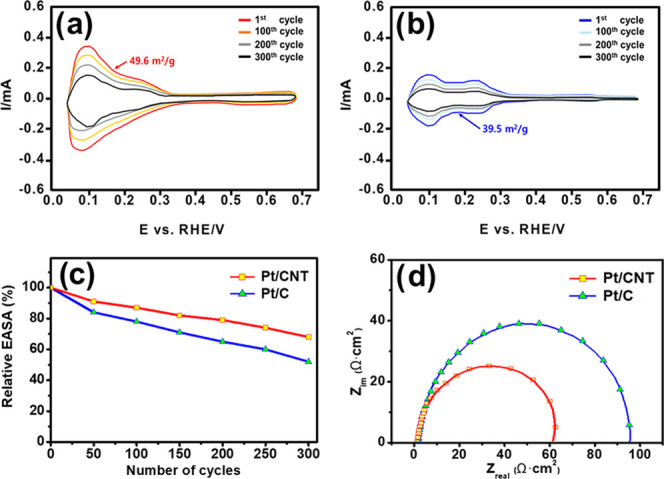
Electrochemical performance of the electrocatalysts: (a) cyclic voltammograms of Pt/CNT and (b) Pt/C in an acidic medium (0.5 M H2SO4), (c) relationship between the normalized EASA as a function of the cycle number in an acidic medium (at +0.75 V vs reversible hydrogen electrode (RHE)) and (d) Nyquist plots of the electrocatalysts.
2.3. Proton Adsorption Characterization Using DFT Calculation
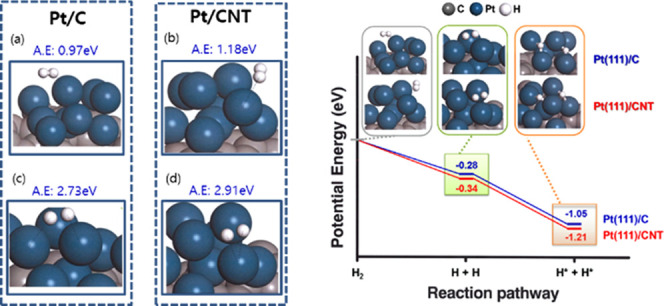
The final product, H+, is adsorbed on the surface of Pt catalyst through the hydrogen oxidation reaction in the anode of the PEMFC. Therefore, we investigated the hydrogen–catalyst adsorption behavior by geometry optimization between the electrocatalysts (Pt/CNT and Pt/C) and H2, H+, as shown in Figure 6 and Table 2. The adsorption energy was Pt/CNT (−2.91 eV) > Pt/C (−2.73 eV) for the H+ catalysts and Pt/CNT (−1.18 eV) > Pt/C (−0.97 eV) for the H2 catalysts, which elucidates that the Pt/CNT catalyst supports H+ formation to have a favorable structure for the HOR. The adsorption calculation between the catalyst and H+ is in good agreement with the other theories. To explore these results, we investigated the charge-transfer characteristics dependent on the interactions between the catalysts and H2 through Mulliken analysis. Figure 7 represents the charge transfer in Pt(111)/CNT and Pt(111)/C. The proton has a bidentate formation with a short bond length, as it approaches the catalyst’s surface during the chemical adsorption process. The analytical results show that the charge-transfer capacity of Pt/CNT (2.64 × 1019 e–/gcatalyst) is approximately double that of Pt/C (1.37 × 1019 e–/gcatalyst), which is attributed to the better electrical conductivities of carbon nanotubes than that of amorphous carbon.
Figure 6.
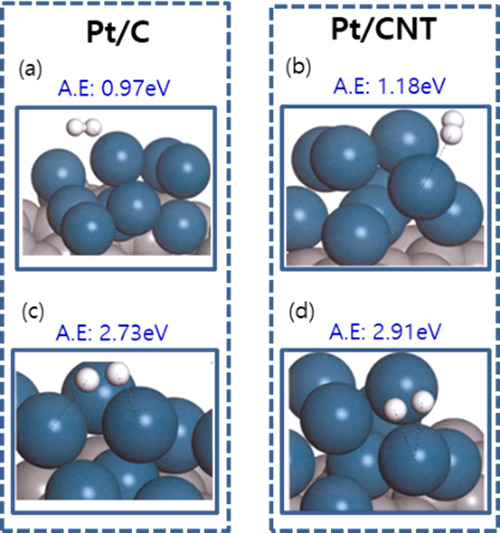
Adsorption energy (AE, ΔEa) of H+ and H2 on catalysts: (a) H2 + Pt/C, (b) H2 + Pt/CNT, (c) H+ + Pt/C, and (d) H+ + Pt/CNT.
Table 2. Characteristics of H+ and H2 Adsorption on Catalysts Determined by DFT Calculations.
Figure 7.
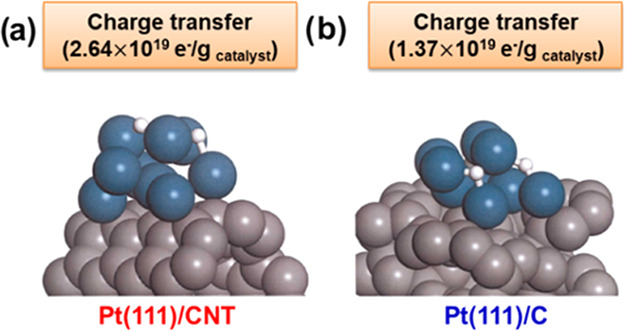
Charge transfer in (a) Pt(111)/CNT and (b) Pt(111)/C catalysts.
2.4. Reaction Kinetic of H2 Dissociative Chemisorption
To deeply understand the effects of changes in the carbon support on the HOR, we investigated the H2 dissociation chemisorption pathway on the surfaces of Pt/CNT and Pt/C. Figure 8 illustrates the calculation of the potential energy for each step in the HOR reaction pathway. In step 1, the H2 molecule is physically adsorbed to the electrocatalysts due to the low adsorption energy and long bond length. Afterward, H2 easily dissociates to produce the intermediate product of hydrogen atoms, 2H+, which is the most stable state for H2 adsorption.
Figure 8.
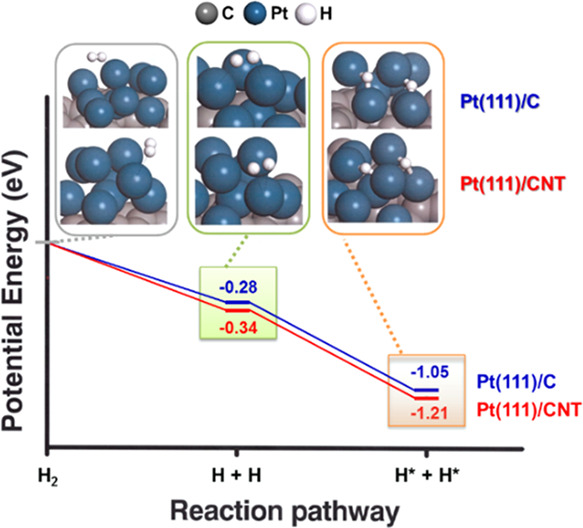
Potential energy profile for H2 dissociation on various catalysts with the optimized geometry of the intermediates on Pt(111)/CNT and Pt(111)/C at stationary points.
3. Conclusions
We investigated the property effects of different Pt-loaded carbon substrates (Pt/CNT and Pt/C) on the improvement of HOR of a PEMFC anode. The experimental and DFT calculation results provide insight into the basic and systematic characteristics of adsorption/dissociation on the electrode catalysts. The Pt/CNT electrocatalyst exhibited excellent durability under severe anodic conditions. Pt/CNT also had a wider electrochemical active area than Pt/C, which leads to enhanced current density and electrochemical stability in the HOR. Along with experimental studies, theoretical properties investigated by DFT calculation also support that Pt/CNT exhibited better activity than Pt/C in the HOR. Pt/CNT has excellent adsorption and electron exchange abilities for H2 and H+, suggesting that it could be a highly electroactive catalyst. In conclusion, the experiment and first-principle calculation using the density functional theory can be used to develop materials and elucidate the adsorption mechanisms in the field of materials, which allows us to utilize basic data to play an important role in fusing theory and experiments.
4. Materials and Methods
4.1. Sample Preparation
The catalyst was synthesized following the chemical reduction method using a reducing agent (NaBH4, Aldrich). We each dispersed 1 g of carbon powder (Vulcan XC-72) and CNT in 100 mL of deionized water and ultrasonicated the sample for 2 h. We then added a carbon solution to 2.5 mL of a chloroplatinic acid solution (8 wt % H2PtCl6 in H2O, Sigma-Aldrich) with 10 wt % Pt (based on the amount of the carbon support) of Pt/C and Pt/CNT. We then agitated the mixture of carbon and metal precursor at room temperature (298 K) for 1 h. The reducing agent was prepared from 3 g of NaBH4 and 100 mL of distilled water and added to the carbon solution and metal salts to prevent the rapid reduction reaction. After stirring the solution for 1 h, we precipitated and centrifuged the electrode catalyst solution. Finally, we freeze-dried the catalyst to preserve its pore structure.
4.2. Characterizations of Catalysis
To determine the Pt distribution dependent on catalyst supports, we analyzed the catalyst samples using an ultrahigh-resolution field emission scanning transmission electron microscope (UHR-FE-STEM; Hitachi SU2900, resolution 0.4 nm). We conducted X-ray powder diffraction (XRD, Bruker, D4, Endeavor) to confirm the loading and changes in the crystalline structure of the catalyst. The XRD patterns were measured with 0.02° intervals at 4°/min using a Philips X’pert Pro X-ray diffractometer (tube voltage: 40 kV; tube current: 40 mA).26 We measured the full width at half-maximum (FWHM) from the analysis results and confirmed the size of the Pt particles using the Scherrer formula. Thermogravimetric analysis was conducted, in which the mass of the catalysts was measured over time as the temperature changed using TA Instruments DSC Q2000 at a heating rate of 5 °C min–1 under N2 purging.
4.3. Electrochemical Characterization
We conducted a CV analysis using a three-electrode system to investigate the hydrogen adsorption–desorption characteristics dependent on the changes in the carbon supports. The working electrode was a solution of Nafion and distilled water in a ratio of 9:1 (vol %). The catalyst ink consisting of 5 wt % of the Nafion solution, the electrode catalyst (Pt/C, Pt/CNT), and distilled water was dropped on glassy carbon and dried. For fixation with a binder, we coated and dried the electrode with 5 wt % of the Nafion solution. The reference electrode was Ag/AgCl, and the CV profile and EASA (electrochemically active surface area) were calculated in a N2-purged environment with 0.5 M H2SO4 using a platinum wire as the counter electrode.
4.4. Computational Details
We analyzed the adsorption behavior of Pt/C and Pt/CNT on H+ at an atomic level using quantum mechanical modeling. The quantum mechanical geometry optimization was performed by density functional theory (DFT) calculation using the Dmol3 module, which evaluates the electronic structure and properties of materials.27 DFT has been used to investigate condensate materials, including their surface. Figure 1 shows the geometry-optimized model of Pt/CNT and Pt/C. After the initial atomic arrangement, we optimized the geometries to refine the model structure of the model. To determine the adsorption properties on electrocatalysts, we built 3 × 3 unit cell structures for loading adsorbate molecules and cleaved the (111) faces of the Pt surface using the surface with cleaving process to expose the reaction sites, subsequently. Note that the (111) faces of the Pt are the most available face for reaction activity, as confirmed by XRD patterns. During calculation, the 20 Å of vacuum thickness, defined by the layer of vacuum over the slab surface, was applied along the z-direction to prohibit self-interactions with the periodic image of the bottom layer of atoms on the Pt surface. Finally, the geometry of the prepared cell was optimized. The exchange–correlation function was applied with the Perdew–Burke–Ernzerhof correlation (PBE) and the generalized gradient approximation (GGA) functional in the Dmol3 module.28−32 We investigated the adsorption behaviors between atoms by applying the Kohn–Sham wave function and a double numerical basis set with dynamic nuclear polarization (DNP) for all electrons.33 To analyze the molecular interactions caused by Van der Waals forces, we performed the Grimme DFT-D dispersion correction.34 We designed the vacuum separation by setting 20 Å along the z-axis direction to prevent interactions occurring beyond the periodic boundary conditions. The k-point sampling mesh for the Brillouin zone was established following the 4 × 4 × 1 Monkhorst–Pack method. The k-point was set to 4 × 4 × 1 to improve the accuracy of calculation and the actual space was set to less than 0.03Å–1. The self-consistent field (SCF) convergence was set to 1 × 10–5 Ha, and the charge-transfer rates were determined following the Mulliken charge method.
The adsorption energy (Ea) of Pt/C and Pt/CNT catalysts with H+ was determined using the following independent calculation. We classified three cases and calculated the (1) geometric optimization energy of the catalysts, (2) geometric optimization energy of H2 or H+, and (3) geometric optimization of the catalyst + (H2 or H+) using eq 1.
| 1 |
where ΔEa is the adsorption energy between the catalyst and H2 or H+, Ecatalyst+H2,H+ is the energy of the catalyst with hydrogen, EH2,H+ is the energy of hydrogen, and Ecatalyst is the energy of the catalyst. The adsorption energy is stable when the value is negative (−), and the negative value of ΔEa is proportional to the hydrogen adsorption capacity. Furthermore, we analyzed the charge by conducting the Mulliken analysis to evaluate the electrochemical stability of Pt/C and Pt/CNT with H+.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2019R1A2C2008733).
The authors declare no competing financial interest.
References
- Xiao Y. H.; Zhan G. H.; Fu Z. G.; Pan Z. C.; Xiao C. M.; Wu S. K.; Chen C.; Hu G. H.; Wei Z. G. Titanium cobalt nitride supported platinum catalyst with high activity and stability for oxygen reduction reaction. J. Power Sources 2015, 284, 296–304. 10.1016/j.jpowsour.2015.03.001. [DOI] [Google Scholar]
- Steele B. C. H. Material science and engineering: The enabling technology for the commercialisation of fuel cell systems. J. Mater. Sci. 2001, 36, 1053–1068. 10.1023/A:1004853019349. [DOI] [Google Scholar]
- Sharma S.; Pollet B. G. Support materials for PEMFC and DMFC electrocatalysts – A review. J. Power Sources 2012, 208, 96–119. 10.1016/j.jpowsour.2012.02.011. [DOI] [Google Scholar]
- Kwon S.; Lee S. G. Influence of defective sites in Pt/C catalysts on the anode of direct methanol fuel cell and their role in CO poisoning: a first-principles study. Carbon Lett. 2015, 16, 198–202. 10.5714/CL.2015.16.3.198. [DOI] [Google Scholar]
- Ganesan R.; Ham D. J.; Lee J. S. Platinized mesoporous tungsten carbide for electrochemical methanol oxidation. Electrochem. Commun. 2007, 9, 2576–2579. 10.1016/j.elecom.2007.08.002. [DOI] [Google Scholar]
- Ham D. J.; Han S.; Pak C.; Ji S. M.; Jin S. A.; Chang H.; Lee J. S. High electrochemical performance and stability of co-deposited pd-au on phase-pure tungsten carbide for hydrogen oxidation. Top. Catal. 2012, 55, 922–930. 10.1007/s11244-012-9875-2. [DOI] [Google Scholar]
- Sharma S.; Pollet B. G. Support materials for PEMFC and DMFC electrocatalysts – a review. J. Power Sources 2012, 208, 96–119. 10.1016/j.jpowsour.2012.02.011. [DOI] [Google Scholar]
- Chalkova E.; Fedkin M.; Komarneni S.; Lvov S. N. Nafion/Zirconium Phosphate Composite Membranes for PEMFC Operating at up to 120 °C and down to 13% RH. J. Electrochem. Soc. 2007, 154, B288–B295. 10.1149/1.2405731. [DOI] [Google Scholar]
- Kim M.; Park J. N.; Kim H.; Song S.; Lee W. H. The preparation of Pt/C catalysts using various carbon materials for the cathode of PEMFC. J. Power Sources 2006, 163, 93–97. 10.1016/j.jpowsour.2006.05.057. [DOI] [Google Scholar]
- Yu X.; Ye S. Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC: Part I. Physico-chemical and electronic interaction between Pt and carbon support, and activity enhancement of Pt/C catalyst. J. Power Sources 2007, 172, 134–144. 10.1016/j.jpowsour.2007.07.049. [DOI] [Google Scholar]
- Reiser C. A.; Bregoli L.; Patterson T. W.; Yi J. S.; Yang J. D.; Perry M. L.; Jarvi T. D. A Reverse-Current Decay Mechanism for Fuel Cells. Electrochem. Solid-State Lett. 2005, 8, A273. 10.1149/1.1896466. [DOI] [Google Scholar]
- Knights S. D.; Colbow K. M.; St-Pierre J.; Wikinson D. P. Aging mechanisms 0and lifetime of PEFC and DMFC. J. Power Sources 2004, 127, 127–134. 10.1016/j.jpowsour.2003.09.033. [DOI] [Google Scholar]
- Ferreira P. J.; La O G. J.; Horn Y. S.; Morgan D.; Makharia R.; Kocha S.; Gasteiger H. A. Instability of Pt/C Electrocatalysts in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2005, 152, A2256–A2271. 10.1149/1.2050347. [DOI] [Google Scholar]
- Wang X. P.; Kumar R.; Myers D. J. Effect of Voltage on Platinum Dissolution: Relevance to Polymer Electrolyte Fuel Cells. Electrochem. Solid-State Lett. 2006, 9, A225–A227. 10.1149/1.2180536. [DOI] [Google Scholar]
- Guilminot E.; Corcella A.; Charlot F.; Maillard F.; Chatenet M.; et al. Membrane and Active Layer Degradation upon PEMFC Steady-State Operation. J. Electrochem. Soc. 2007, 154, B1106–B1114. 10.1149/1.2775218. [DOI] [Google Scholar]
- Virkar A. V.; Zhou Y. Mechanism of Catalyst Degradation in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2007, 154, B540. 10.1149/1.2722563. [DOI] [Google Scholar]
- Odom T. W.; Huang J. L.; Kim P.; Lieber C. M. Atomic structure and electronic properties of single-walled carbon nanotubes. Nature 1998, 391, 62–64. 10.1038/34145. [DOI] [Google Scholar]
- Falvo M. R.; Clary G. J.; Taylor R. M.; Chi V.; Brooks J. F. P.; Washburn S.; et al. Bending and buckling of carbon nanotubes under large strain. Nature 1997, 389, 582–584. 10.1038/39282. [DOI] [PubMed] [Google Scholar]
- Serp P.; Corrias M.; Kalck P. Carbon nanotubes and nanofibers in catalysis. Appl. Catal., A 2003, 253, 337–358. 10.1016/S0926-860X(03)00549-0. [DOI] [Google Scholar]
- Wang C.; Waje M.; Wang X.; Tang J. M.; Haddon R. C.; Yan Y. Proton exchange membrane fuel cells with carbon nanotube based electrodes. Nano Lett. 2004, 4, 345–348. 10.1021/nl034952p. [DOI] [Google Scholar]
- Zhang W.; Chen J.; Swiegers G. F.; Ma Z.; Wallace G. Microwaveassisted synthesis of Pt/CNT nanocomposite electrocatalysts for PEM fuel cells. Nanoscale 2010, 2, 282–286. 10.1039/B9NR00140A. [DOI] [PubMed] [Google Scholar]
- Lee T. K.; Jung J. H.; Kim J. B.; Hur S. H. Improved durability of Pt/CNT catalysts by the low temperature self-catalyzed reduction for the PEM fuel cells. Int. J. Hydrogen Energy 2012, 37, 17992–18000. 10.1016/j.ijhydene.2012.09.088. [DOI] [Google Scholar]
- Leela Mohana Reddy A.; Ramaprabhu S. Pt/SWNT–Pt/C Nanocomposite Electrocatalysts for Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. C 2007, 111, 16138–16146. 10.1021/jp066985+. [DOI] [Google Scholar]
- Rajalakshmi N.; Ryu H.; Shaijumon M. M.; Ramaprabhu S. Performance of polymer electrolyte membrane fuel cells with carbon nanotubes as oxygen reduction catalyst support material. J. Power Sources 2005, 140, 250–257. 10.1016/j.jpowsour.2004.08.042. [DOI] [Google Scholar]
- Niu J. J.; Wang J. N. Activated carbon nanotubes-supported catalyst in fuel cells. Electrochim. Acta 2008, 53, 8058–8063. 10.1016/j.electacta.2008.06.026. [DOI] [Google Scholar]
- Kwon S. C.; Ham D. J.; Kim T. Y.; Kwon Y.; Lee S. G.; Cho M. Active Methanol Oxidation Reaction by Enhanced CO Tolerance on Bimetallic Pt/Ir Electrocatalysts Using Electronic and Bifunctional Effects. ACS Appl. Mater. Interfaces 2018, 10, 39581–39589. 10.1021/acsami.8b09053. [DOI] [PubMed] [Google Scholar]
- Dmol3 Program of Materials Studio Software; Dassault Systemes ` BIOVIA Corp.: San Diego, CA, 2018. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Local and gradient-corrected density functionals. ACS Symp. Ser. 1996, 629, 453–462. 10.1021/bk-1996-0629.ch030. [DOI] [Google Scholar]
- Kwon S.; Hwang J.; Lee H.; Lee W. R. Interactive CO2 adsorption on the BaO (100) surface: A density functional theory (DFT) study. Bull. Korean Chem. Soc. 2010, 31, 2219–2222. 10.5012/bkcs.2010.31.8.2219. [DOI] [Google Scholar]
- Koh W.; Choi J. I.; Donaher K.; Lee S. G.; Jang S. S. Mechanism of Li adsorption on carbon nanotube-fullerene hybrid system: a first-principles study. ACS Appl. Mater. Interfaces 2011, 3, 1186–1194. 10.1021/am200018w. [DOI] [PubMed] [Google Scholar]
- Kwon S. C.; Fan M.; Dacosta H. F. M.; Russell A. G.; Tsouris C. Reaction kinetic of CO2 carbonation with Mg-rich minerals. J. Phys. Chem. A 2011, 115, 7638–7644. 10.1021/jp2040899. [DOI] [PubMed] [Google Scholar]
- Kim T. Y.; Kwon Y.; Lee J. B.; Lee D. W.; Shin H. S.; Cho M.; Kwon S. C. Development of hydrophilicity on the proton exchange using sulfonic acid on PEEK in the presence of water: A Density functional theory study. Theor. Chem. Acc. 2017, 136, 130. 10.1007/s00214-017-2153-4. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Kreig H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Desai S. K.; Neurock M.; Kourtakis K. A periodic density functional theory study of the dehydrogenation of methanol over Pt(111). J. Phys. Chem. B 2002, 106, 2559–2568. 10.1021/jp0132984. [DOI] [Google Scholar]
- Wang H.; He C. Z.; Huai L. Y.; Liu J. Y. Decomposition and oxidation of methanol on Ir(111): A first-principles study. J. Phys. Chem. C 2013, 117, 4574–4584. 10.1021/jp311227f. [DOI] [PubMed] [Google Scholar]
- Park Y. J.; Kim G.; Lee Y. H. Adsorption and dissociation of hydrogen molecules on a Pt atom on defective carbon nanotubes. Appl. Phys. Lett. 2008, 92, 083108 10.1063/1.2838732. [DOI] [Google Scholar]