

Abstract

N-Heterocyclic carbene (NHC) gold(I) complexes offer great prospects in medicinal chemistry as antiproliferative, anticancer, and antibacterial agents. However, further development requires a thorough understanding of their reaction behavior in aqueous media. Herein, we report the conversion of the bromido[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(I) ((NHC)AuIBr, 1) complex in acetonitrile/water mixtures to the bis[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(I) ([(NHC)2AuI]+, 7), which is subsequently oxidized to the dibromidobis[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(III) ([(NHC)2AuIIIBr2]+, 9). By combining experimental data from HPLC, NMR, and (LC-)/HR-MS with computational results from DFT calculations, we outline a detailed ligand scrambling reaction mechanism. The key step is the formation of the stacked ((NHC)AuIBr)2 dimer (2) that rearranges to the T-shaped intermediate Br(NHC)2AuI–AuIBr (3). The dissociation of Br– from 3 and recombination lead to (NHC)2AuI–AuIBr2 (5) followed by the separation into [(NHC)2AuI]+ (7) and [AuIBr2]− (8). [AuIBr2]− is not stable in an aqueous environment and degrades in an internal redox reaction to Au0 and Br2. The latter in turn oxidizes 7 to the gold(III) species 9. The reported ligand rearrangement of the (NHC)AuIBr complex differs from that found for related silver(I) analogous. A detailed understanding of this scrambling mechanism is of utmost importance for the interpretation of their biological activity and will help to further optimize them for biomedical and other applications.
Short abstract
By means of experimental data from HPLC and (LC-)MS in combination with DFT calculations, we present a detailed mechanism for the ligand scrambling reaction of (NHC)AuIBr to the corresponding [(NHC)2AuI]+ complex and the oxidation to the [(NHC)2AuIIIBr2]+ species in aqueous solutions.
Introduction
Discovering gold(I) and gold(III) complexes as catalysts,1 luminescence agents,2−4 and more recently as anticancer and antibacterial agents in medicinal chemistry5−9 pushed forward the research in gold chemistry. For example, complexes bearing phosphine and/or thiol ligands with the famous representative auranofin,10,11 cyclometalated gold(III) complexes with C, N donor ligands, and N-heterocyclic carbene (NHC) gold(I) complexes arose in the past as auspicious compounds against abnormal cell growth.7,12−14 In particular, (NHC)AuIX and related complexes came into the focus of medicinal chemists and were examined for their suitability as chemotherapeutic agents.6,9,15−19 The straightforward synthesis of the NHC ligands, the possibility of fine-tuning the physicochemical properties, and the reactivity of the resulting (NHC) gold(I) complexes make the latter to attractive lead structures for the development of novel metal-based drugs.
NHCs are monodentate and electron-rich, σ-donor ligands, whereby their electron donor effect is greater than that of phosphines.20−23 Therefore, they form strong bonds to metals24 and bind to the metal center in a “push–pull” mechanism.25,26
Ligand exchange reactions were investigated for various (NHC)AuIX complexes17,27 indicating a strong trans effect of the NHC and a preferred exchange of X as leaving group.
In biological systems, the complexes frequently bind to cysteine (Cys) or selenocysteine (Sec) in the active site of enzymes, causing their inhibition.28−30 However, the nonselective coordination to biomolecules can also cause unpredictable side effects. It is well accepted that the observed biological response depends on the ligand exchange rate.28−30 Therefore, the suitability of NHCs as leaving groups was investigated, too. Dos Santos et al., for instance, studied the kinetics of the NHC ligand exchange at bis(N,N′-dialkylimidazol-2-ylidene)gold(I) complexes by Cys and demonstrated that the activation enthalpy of the rate-limiting first reaction step depends on both steric and electronic features.31 [(NHC)2AuI]+ complexes can therefore undergo ligand exchange reactions with strong nucleophiles and exert cytotoxic effects, e.g., due to intracellular enzyme inhibition. In this context, the ligand scrambling between two (L)AuIX complexes giving [(L)2AuI]+ must be taken into account.
So far, such a reaction was monitored for several gold complexes containing, phosphine, thiol, selenium, or cyano ligands.32−36 For instance, Hormann-Arendt et al. observed the formation of [(L)2AuI]+ species from (L)AuICN (L = trialkyl-/triarylphosphine).37 Although NHCs are regarded as stronger σ-donor ligands than phosphines and are expected to form stronger bonds to the metal center, ligand scrambling reaction products have already been observed upon crystallization.38,39
From this finding arose the question whether NHC ligands can also interchange between two metal complexes in solution. Indeed, Wang et al. solved the X-ray crystal structure of [(diethylbenzimidazol-2-ylidene)2AgI]+ [AgIBr2]− (Scheme 1-I) as product from the reaction of Ag2O with 1,3-diethylbenzimidazolium bromide. The argentophilic contacts amounted to 2.956 Å.40 After dissolution, the [(NHC)2AgI]+ [AgIBr2]− directly interconverted to (NHC)AgIBr as determined by 13C NMR spectroscopy and led to the proposal of the mechanism depicted in Scheme 1.40
Scheme 1. Intermediates and Transition State of the Rearrangement Reaction of (NHC)AgIX (X = Cl, Br, I) Complexes Adapted from Wang et al.40 and Su et al.41.
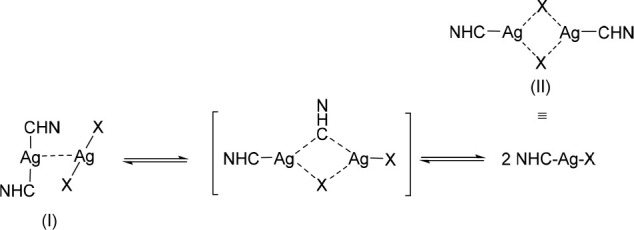
Later, Su et al. confirmed this reaction mechanism by variable temperature NMR measurements and DFT studies of (NHC)AgIX (X = Cl, Br, I).41 In addition, they reported the X-ray crystal structure of the chlorido and bromido complexes, which proved the existence of (NHC)AgIX dimers in the solid state as schematically shown in Scheme 1-II. For example, bromido[N-mesityl-N′-methylimidazol-2-ylidene]silver(I) crystallized from an acetonitrile (ACN)/ether solution in dimeric units of the ((NHC)AgIBr)2 type with strong intermolecular Ag···Br interactions at a distance of 2.928 Å and a disordered NHC–AgI–Br arrangement with an angle of 158° and bridging Br ligands.41
In various crystal structures, the existence of (NHC)AuIX (X = Cl, Br, I) dimers and [(NHC)2AuI]+ [AuIX2]− adducts was confirmed. Guo et al. determined the formation of [(NHC)2AuI]+ [AuII2]− upon crystallization of iodido[4-benzyl-1-methyl-1,2,4-triazolin-5-ylidene]gold(I) with a short AuI–AuI distance of 3.116 Å.39 This distance is shorter than twice the van der Waals radius of gold and suggests strong interactions arising from d10–d10 attractions between two gold(I) centers, which are denoted as aurophilic interactions. Indicative of these interactions are AuI–AuI distances between 2.750 and 3.250 Å,42 which can be considered as exceptionally strong noncovalent bonds due to the relativistic effects of gold(I). Redissolving the crystalline [(NHC)2AuI]+[AuI2]− sample led exclusively to the identification of the (NHC)AuI complex.39 The authors postulated a reversible interchange and a shift of the equilibrium to the mono-NHC species due to loss of AuI–AuI contacts in solution upon elongation of the distance between the metals.
Such ligand scrambling reactions, especially in the presence of water, are of high relevance in medicinal chemistry for the interpretation of biological effects.37,43,44 Ott et al. investigated the chlorido[(4-(4-fluorophenyl)-1,3-diethylimidazol-2-ylidene)]gold(I) complex in 1% DMF/water and 1% DMSO/water mixtures and identified by electrospray ionization (ESI)-MS spectroscopy ligand exchange reactions to NHC–AuI–OH and NHC–AuI–DMF/DMSO species as well as rearrangements reaction to the respective bis(NHC) gold(I) complex.17 However, the formation of these species under MS conditions (gas phase) cannot be excluded.
The same is true for chlorido[methyl-1-(2-hydroxy-2-methylpropyl)imidazol-2-ylidene]gold(I), which rearranged in aqueous solution to its bis(NHC) gold(I) analogue. In aqueous buffered solutions (20 mM NH4CO3, pH 7.4), however, mainly the [(NHC)AuI–NH3]+ species is built.45 This study was done by TOF-MS analysis.
The conversion of (NHC)AuIX complexes to the corresponding bis(NHC) species, [(NHC)2AuI]+, under physiological conditions has considerable influence on the biological activity. It was already confirmed that [(NHC)2AuI]+ complexes show significantly improved inhibition of cancer cell proliferation compared to the respective (NHC)AuIX complexes.7,9,12,17,46−49
In light of these preliminary findings, we used bromido[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(I) (1), of which the synthesis is described in ref (50), as a model for a detailed investigation of ligand scrambling reactions. In the context of its development as antitumor agent, the HPLC analysis documented impurities of the [(NHC)2AuI]+ complex 7 in an amount of 2–3%.50 Furthermore, high-resolution mass spectrometry (HR-MS) identified further species that are possibly involved in scrambling reactions. Therefore, we combined experimental (HPLC, NMR, (LC-)/HR-MS) and computational investigations with density functional theory (DFT) calculation to get a closer insight into the reaction mechanism. Moreover, in previously performed anticancer investigations, complex 1 was identified as a highly active agent against cisplatin-resistant A2780cis cells.50 Therefore, it is of interest to know how 7 is built, for example, under storage conditions (e.g., in stock solutions) or in aqueous media.
Results and Discussion
Stability Studies
In a first experiment, the stability of 1 in pure ACN (1 mM) at RT was studied by HPLC at 0, 24, 48, and 72 h (for conditions see the Experimental Section). The chromatograms indicate the expected impurity of 7 (2%, t = 0 h, Figure 1A), which remained constant during the incubation time of 72 h.
Figure 1.

Stability of 1 at RT in (A) pure ACN, (B) various ACN/water mixtures after incubation for 72 h, and (C) ACN/water = 50/50 solution, analyzed by HPLC. The numbering of compounds follows their occurrence in the reaction mechanism (vide infra).
Increasing amounts of water (0 to 50%) led to an extensive degradation as visible from Figure 1B.
The ACN/water = 50/50 mixture was additionally investigated in a time dependent manner (analyzed at 0 h and after 24, 48, and 72 h; Figure 1C). After dissolution (0 h), the chromatogram documents 1 as main peak (tret = 6.02 min) with a small amount of 7 (tret = 7.35 min). During the following 72 h, 1 partially degraded to 7 and additionally to 9 (tret = 7.23 min). The latter was assigned to the dibromidobis[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(III) complex ([(NHC)2AuIIIBr2]+) by comparison with a synthesized reference substance (see Synthesis and Characterization section, Supporting Information). Additionally, two further peaks are present in the HPLC chromatogram at tret = 7.68 min (4) and 7.89 min (3), which slightly increased during the incubation time.
The UV–vis spectra taken from the HPLC runs indicate two maxima at 234 and 257 nm for complex 1 (Figure S3, Supporting Information). The maximum at 234 nm is caused by the ligand and that at 256 nm by the characteristic metal-to-ligand charge-transfer transition (MLCT).51 For 7, the MLCT maximum is located at 276 nm. In contrast, 9 as a gold(III) complex induces a strong absorption at 228 nm and a weak band at about 330 nm. Comparable absorption maxima were registered for other [(NHC)2AuIIIX2]+ (X = Cl, Br, I) complexes.52,53
To get further information about the present species, a freshly prepared ACN/water = 50/50 solution was analyzed by HR-MS (Figure 2) and LC-MS (Figures S13–S15).
Figure 2.

Full HR-MS spectrum of a freshly prepared solution of 1 in ACN/water = 50/50 (A). HCD fragmentation spectra of intermediates with m/z = 1175/1177 (B) and 1257 (C).
The HR-MS spectrum (Figure 2A) confirms the presence of species with m/z = 352, 589, 628, 899, 1175 (sometimes also labeled as 1177 due to the isotopic distribution of bromine), and 1257. Complexes 1 and 7 are identified at m/z = 628 and 899, respectively, together with [(NHC)AuIACN]+ at m/z = 589 (labeled as NHC–Au–ACN) and free NHC ligand at m/z = 352 (labeled as NHC). The signals at m/z = 1175 and 1257 show the typical isotopic pattern of bound bromide (Figures S6 and S7) and correspond to the sum formula [(NHC)2AuI2Br]+ (4) and (NHC)2AuI2Br2 (2/3).
To get more insight into the structure of the molecules present in solution, we performed higher energy collisional dissociation (HCD). Compound 4 with m/z = 1175 lost an AuIBr fragment resulting in 7 (m/z = 899, Figure 2B), indicating an [(NHC)2AuI–AuIBr]+ arrangement. Additionally, a low abundant signal with m/z = 576 appeared, caused by the addition of a collision gas molecule N2 to the [(NHC)AuI]+ fragment in the gas phase to form the N2-bound species, labeled as NHC–Au–N2 (Figure 2B).
m/z = 1257 corresponds to the sum formula (NHC)2AuI2Br2, labeled as 2/3 in Figure 2C. The fragmentation to m/z = 628, consistent with the sum formula of (NHC)AuIBr (1), points to a simple dimeric adduct ((NHC)AuIBr)2 (2) and documents the existence of strong AuI–AuI bonds in solution and under MS conditions (gas phase).
Nevertheless, release of Br– from m/z = 1257 and the detection of m/z = 1175 (4) during HCD fragmentation indicate the presence of an (NHC)2AuI2Br2 species (3), however, in very small amounts because of the low intensity.
The proportion of 3 in solution increased during the time of storage as detected by HPLC analysis (tret = 7.89 min, Figure 1). Its UV–vis spectrum (Figure S3) displays a maximum at 235 nm and a low transition at 338 nm very similar to that of tetragonal substituted gold(III) complexes (see e.g. UV–vis spectrum of 9 in Figure S3), indicating a [Br(NHC)2AuI–AuIBr] arrangement.
Interestingly, besides 3 additionally 4 (tret = 7.68 min) can be identified in the chromatograms during the time of incubation.
The UV–vis spectrum of 4 contains a strong absorption maximum at 234 nm and an MLCT at 277 nm, comparable to that of the [(NHC)2AuI]+ complex 7 (Figure S3). These data support the assumption of [(NHC)2AuI–AuIBr]+ as the structure of 4. A possible stabilization by water arises from the theoretical investigations (see below and the Supporting Information). It is worth mentioning that H2O adducts are observed for 1 and 7 as low abundant signals at m/z = 645 and m/z = 917.
Theoretical Investigation of the Ligand Scrambling Reaction
HPLC and HR-MS investigations documented strong aurophilic interactions between gold(I) complexes, but it is very difficult to verify their structures in solution. Therefore, we performed DFT calculations with water as implicit solvent and predict a detailed mechanism of the ligand scrambling from 1 → 7 + 8 in aqueous solution. [AuIBr2]− (8) as cleavage product of 1 → 7 was confirmed by HR-MS in the negative mode (Figure S8).
The theoretical considerations started from the available crystal structure of 1, which confirms a linear arrangement of the (NHC)AuIBr molecules and a stacked dimer formation (2), as visible from the ORTEP plot depicted in Figure 3.50 The AuI–AuI distance amounts to 3.626 Å, somewhat higher than those found for other NHC–AuI complexes.36,39,54 These values are similar to the one obtained from DFT calculations with PBE0/def2-TZVP/BJ, where a distance of 3.716 Å was calculated (Table S3).
Figure 3.

ORTEP plot of 1 as previously published in ref (50). A stacked dimer formation—corresponding to 2—is observed with a distance between the two gold(I) centers of 3.626 Å, which is also likely be formed in solution.
The formation of the stacked dimer 2 is energetically favored compared to the monomer 1 by a gain in free energy of ΔG298.15K = −22.6 kJ mol–1 (shown in blue, Figure 4) and an (electronic) energy gain of ΔE = −58.7 kJ mol–1 (shown in red, Figure 4). ΔG298.15K is somewhat higher because of the loss of entropy.
Figure 4.

Detailed proposed reaction mechanism for the ligand scrambling reaction of 1 to 7 + 8 via several intermediates and transition states. Gibbs free energies are listed in blue, whereas relative electronic energies are presented in red. All structures were fully optimized with PBE0/def2-TZVP/BJ, where water was modeled as an implicit solvent. For further details, see the Quantum Chemical Methodology section.
The AuI–AuI distance in the intermediate 3 is reduced to 2.581 Å (dimer 2: Au–Au distance = 3.716 Å) upon rearrangement via the transition state TS(2–3). As illustrated in Figure 5, TS(2–3) possesses a triangular shape constituted by the two AuI centers and one NHC ligand. The length of the AuI–AuI bond amounts to 2.664 Å. This close contact distorts the linear arrangement of the NHC–AuI–Br to angulated units of 61.7° resembling that of (NHC)AgIBr, found in the solid state.40,41
Figure 5.

Structure of TS(2–3), optimized with PBE0/def2-TZVP/BJ and implicit solvent. The dotted lines represent the bonds created and broken during the reaction to form 3.
Formation of the transition state TS(2–3) is accompanied by an energy barrier of ΔG‡298.15K = 105.6 kJ mol–1 (ΔE‡ = 83.0 kJ mol–1). The following NHC shift results in the Br(NHC)2AuI–AuIBr arrangement (3) with ΔG298.15K = 27.7 kJ mol–1 (ΔE = −7.5 kJ mol–1). Several configurations of 3 were tested; however, all of them resulted upon optimization either in regeneration of 1 or 2 or in the proposed structure of complex 3.
In the next step, Br– dissociates from 3, from either the terminal AuI–AuIBr moiety or from the quaternary AuI. DFT calculations indicate for the former one a barrier of ΔG‡298.15K = 125.3 kJ mol–1 and ΔE‡ = 122.5 kJ mol–1 (denoted as TS(3–4′), Figure S19), while for the latter TS(3–4) the barrier is only ΔG‡298.15K = 67.3 kJ mol–1 and ΔE‡ = 70.5 kJ mol–1 (see Figure S18 and also the Quantum Chemical Methodology section). Hence, dissociation from the quarternary AuI is more likely. Upon Br– dissociation, species 4 is formed, which features a trigonal structure (similar to TS(2–3)) with a relative free energy of ΔG298.15K = 48.1 kJ mol–1 (ΔE = 30.2 kJ mol–1) and an AuI–AuI distance of 2.716 Å.
The dissociated free Br– attacks the Br–AuI center to form the transition state TS(4–5) with a barrier height of ΔG‡298.15K = 37.2 kJ mol–1 (ΔE‡ = 58.8 kJ mol–1), resulting in an angular Br–AuI–Br moiety. Subsequently, intermediate 5 is formed (ΔG298.15K = −3.0 kJ mol–1; ΔE = −39.6 kJ mol–1), where the two fragments, [(NHC)2AuI]+ and [AuIBr2]−, are still connected through the Au centers as evident from the AuI–AuI distance of 3.146 Å.
We also tested the possibility of a concerted rearrangement from 2 to 5. However, all attempts failed to localize transition state TS(2–5) for this rearrangement. Investigations on the reaction path always resulted in intermediates that converted in each case to the T-shaped species 3.
A direct transformation of 3 to 5 via an intramolecular transfer of Br– was also computationally examined. On the basis of a calculated transition state energy of ΔE = 93.1 kJ mol–1 and free energy of ΔG298.15K = 152.7 kJ mol–1 as well as a reaction barrier of ΔG‡298.15K = 159.5 kJ mol–1, however, it seems unlikely (compare Table S2 and Figure S23). Because the experimental data confirmed the formation of 4 under aqueous conditions, a reaction via TS(3–5) could probably be a secondary, but not the main, pathway in solution.
Conversion of 5 to 6 takes place via a low-lying transition state TS(5–6) with ΔG‡298.15K = 13.8 kJ mol–1 (ΔE‡ = 14.6 kJ mol–1). For 6 a relative free energy of ΔG298.15K = 4.6 kJ mol–1 (ΔE = −31.6 kJ mol–1) and an AuI–AuI distance of 3.452 Å were calculated. The elongation of the AuI–AuI distance points to the formation of the two charged fragments, [(NHC)2AuI]+ and [AuIBr2]−, that now form an (electrostatic) encounter complex.
Complete dissociation of the two fragments 7 and 8 proceeds through transition state TS(6–7) with a barrier of ΔG‡298.15K = 46.4 kJ mol–1 (ΔE‡ = 68.6 kJ mol–1). Remarkably, as the [AuIBr2]− fragment lost its interaction with [(NHC)2AuI]+, a rotation of the NHCs was observed. In the final structure 7, the two NHC ligands are no longer collinear but adapt a dihedral angle of 111.5°, whereas in 8 the [AuIBr2]− exists in a linear arrangement.
The free energy of 7 + 8 amounts to ΔG298.15K = 25.0 kJ mol–1, while the relative electronic energy is ΔE = 3.6 kJ mol–1. These energy levels show that the driving force for the ligand scrambling is the formation of intermediates 5 and 6. Comparison of their energies with that of 7 + 8 allows an estimation of the energy for the interaction between the [(NHC)2AuI]+and [AuIBr2]− (ΔE = 43.2 kJ mol–1 (5) and 35.2 kJ mol–1 (6)). These stabilizing effects are predominantly aurophilic interactions.
Strikingly, analysis of the ACN/water mixtures by HPLC and HR-MS clearly indicates the existence of 3, 4, and 7 in solution. Intermediates 5 and 6 could not be detected by HPLC. This finding forces the question about the influence of water molecules on the stability of compounds 4–7.
Hence, we investigated the impact of water coordination on the structures and the energy (4WATER to 7WATER, Figure S17; see the Quantum Chemical Methodology section for further details).
In a first step, water was placed at the position of the dissociated Br– ligand in 4, since this intermediate not only is of theoretical nature but also was detected in solution by HPLC analysis. Geometry optimization not only yields the stable complex 4WATER (Figure S17 and Table S4), but also conserves the T-shaped arrangement that is found in 3. This arrangement is even maintained in the case of the presence of a point charge. The distance between the water’s oxygen atom OWATER and AuI is 2.707 Å, pointing to attractive interactions. The energy ΔE = 38.0 kJ mol–1 is very similar to that of 4 (ΔE = 30.2 kJ mol–1), while ΔG298.15K = 57.8 kJ mol–1 is 8–10 kJ mol–1 higher than that of 4.
The linear alignment of the NHC–AuI–NHC unit in 4WATER is also consistent with the UV–vis spectra obtained from HPLC (Figure S3). Thus, we hypothesize that surrounding water plays a role in the stabilization of the structure. Not only for 4 but also for 5 and 6 an explicitly coordinated water molecule was converged (Figure S17 and Table S4). While the electronic energies ΔE = −51.8 kJ mol–1 (5WATER) and ΔE = −40.8 kJ mol–1 (6WATER) are comparable to those of 5 and 6, the free energy ΔG298.15K increases to 8.5 kJ mol–1 (5WATER) and 27.1 kJ mol–1 (6WATER). The AuI–OWATER distances amount to 3.399 and 3.465 Å, respectively, indicating that the oxygen is no longer directly coordinated to Au. Therefore, it can be concluded that explicit water coordination is probably less important in 5WATER and in 6WATER.
The calculated free energies for 5WATER and 6WATER should not be overinterpreted, because they correspond to the change in free energy relative to 1 and an infinitely separated water molecule in an implicit solvent. Hence, the bulk free energy of solvation of a water molecule is not correctly accounted for.
Structure optimization of 7WATER with one explicitly coordinated water molecule showed that the interaction with the Au center is no longer favored as the water molecule moves toward one NHC ligand during optimization (Figure S17).
Moreover, because 7 is mainly found in the presence of increasing amounts of water, it is very likely that further explicit solvation stabilizes the ions [(NHC)2AuI]+ (7) and [AuIBr2]− (8) in aqueous medium and drives the reaction. Such effects are difficult to model and are only approximately accounted for in our calculations.
Considering the entire mechanism as depicted in Figure 4, the total energy barrier to overcome, that is, the difference between the lowest lying intermediate, here 2, and the highest transition state, here TS(3–4), is 117.6 kJ mol–1, which is quite high given that the reaction is readily observed at RT. However, explicit interaction with a water molecule not only may stabilize 4 but may also lower the reaction barrier (formally, this corresponds to replacement of Br– by water). Similar arguments may hold for the reaction barrier TS(4–5), which may also be lowered by explicit water attachment. Taking these additional interactions into account, our data suggest that the ligand redistribution transition state TS(2–3) is the rate-determining one.
In summary, the experimental and computational data strongly suggest the mechanism for the ligand scrambling reaction of (NHC)AuIBr complexes as illustrated in Figure 4. Aurophilic interactions facilitate the migration of the NHC ligands of the stacked dimer 2, forming 3. According to DFT calculations, the AuI–AuI binding can distinctly be strengthened upon formation of a T-shaped arrangement of the ligands around the AuI–AuI axis. Release of Br– results in a trigonal structure of the AuI centers and the ligands. Subsequently, the attack of the Br– at the AuIBr moiety and linearization of the [(NHC)2AuI]+ [AuIBr2]− fragments increase the AuI–AuI distance, yielding the two charged species 7 and 8. Solvation is likely to play a role in the mechanism as evident from the stabilization of T-shaped intermediate 4 and aids the formation of the degradation products 7 and 8. This mechanism is distinctly different from the one proposed by Su et al. (I),41 where the rearrangement of the ligands is achieved by a simultaneous migration of NHC and Br–, which corresponds to a hypothetical transition state TS(2–5), which despite all efforts has not been found in this study. Our data, however, clearly indicate the formation of 3 followed by Br– dissociation (4) and recombination (5), in a stepwise process.
Formation of the [(NHC)2AuIIIBr2]+ Complex (9)
HPLC analysis of 1 in an ACN/water = 50/50 mixture (Figure 1B) indicated the formation of 9, which corresponds to an [(NHC)2AuIIIBr2]+ species. To the best of our knowledge, such a reaction in situ during the degradation of (NHC)AuIBr complexes in aqueous solution has not yet been reported. Formally, 9 yields from the oxidation of [(NHC)2AuI]+ (7) by Br2—a reaction already described in the literature.12,55−58
In the same straightforward oxidation we transformed 7 to the reference compound 9 and confirmed the structure by X-ray diffraction analysis (Figure 6). The retention time in HPLC of the synthesized [(NHC)2AuIIIBr2]+ complex (tret = 7.35 min, Figure S2) matched that detected during stability studies (Figure 1B).
Figure 6.

ORTEP plot of the gold(III) complex [(NHC)2AuIIIBr2]+ (9) with PF6– as counterion.
To identify the main degradation products of 1 after an incubation time of 72 h in an ACN/water = 50/50 mixture, the solvent was evaporated, and the resulting precipitate was dissolved in CDCl3 and evaluated by 1H NMR (Figure 7) and 13C NMR (Figures S10 and S11) spectroscopy. The use of data from the reference compounds 1, 7, and 9 (see Synthesis and Characterization,e Supporting Information) allowed an unequivocal assignment of the signals in Figure 7.
Figure 7.

Aromatic region of the 1H NMR spectrum obtained from a lyophilized solution (ACN/water = 50/50, incubation time 72 h) of 1. Signals corresponding to 1, 7, and 9 are framed in green, yellow, and blue, respectively.
The positive charge of 7 and 9 led to a deshielding and a downfield shift of the signals (Figure 7: 7, yellow framed, and 9, blue framed) compared to 1 (Figure 7: 1, green framed). Characteristic for the coordination of NHC to gold is the resonance of the metal-bound C, located at 182 ppm (7) and 150 ppm (9) in the 13C NMR spectra (Figure S11).
The oxidation of 7 to 9 was more closely analyzed. Incubation of complex 1 was repeated under an inert atmosphere in a degassed ACN/water = 50/50 mixture to exclude reaction with oxygen. As depicted in Figure 8, the HPLC chromatogram of this solution (line A) shows the same products as that incubated in air for 72 h (Figure 1). Also, the saturation with oxygen did not change the degradation profile (line B).
Figure 8.

HPLC traces of 1 mM solutions of complex 1 after 72 h of incubation: (A) under an inert atmosphere, (B) reference reaction under an oxygen atmosphere, and (C) with addition of 1 equiv of cyclopentene.
To confirm that indeed Br2 is the oxidizing agent converting 7 to 9, a quenching experiment was performed. Therefore, 1 equiv of cyclopentene was added to a freshly prepared solution of 1 to react with in situ formed bromine. The solution was incubated for 72 h and analyzed by HPLC. The chromatogram did not contain the gold(III) species 9 at tret = 7.23 min (Figure 8, line C), but only 7 with tret = 7.35 min. Hence, it can be concluded that any produced bromine is added to the double bond of the cyclopentene and is not available to oxidize 7 to 9.
This finding is confirmed by 1H NMR spectroscopy. The solution of 1 in ACN/water = 50/50 mixture was incubated for 72 h, lyophilized, and dissolved for NMR measurement in CD3CN. Figure 9 depicts the 1H NMR spectra zoomed into the region of aromatic and methoxy signals. The formed 1,2-dibromocyclopentane caused a characteristic resonance at δ = 3.79 ppm (−CH–Br) as depicted in Figure S12.
Figure 9.

1H NMR spectra of 1 after incubation in an ACN/water = 50/50 mixture under various conditions. (A) Spectrum of 1 in the absence of cyclopentene taken at t = 0 h. (B) Spectrum of 1 in the absence of cyclopentene taken at t = 72 h. (C) Spectrum of 1 obtained after incubation in the presence of 1 equiv of cyclopentene for 72 h. For NMR spectroscopy the solvent was removed, and the samples were redissolved in CD3CN.
As visible from Figure 9A (green line), only 1 was present in the mixture at the beginning (t = 0 h). After 72 h, the expected signals of the complexes 7 and 9 appeared (Figure 9B, red line). Incubation with 1 equiv of cyclopentene led exclusively to the formation of 7 (Figure 9C, blue line) and underlines the assumption that bromine is formed in the aqueous solution, which in turn oxidizes complex 7 to the corresponding complex 9.
The (formal) ligand scrambling reaction is sketched in Scheme 2: Complex 1 decomposes in aqueous solution to 7 and 8 (I), followed by oxidation of 7 to the [(NHC)2AuIIIBr2]+ species 9 by Br2 (IV). The latter oxidation step is well-known in the literature to synthesize AuIII complexes.12,55−58
Scheme 2. Overview of the Ligand Scrambling Reaction, Determined for 1.

To realize this reaction sequence, Br2 has to be formed in the reaction mixture. [AuIBr2]−, which was confirmed as a degradation product, has to decompose to AuIBr and Br– in aqueous solution (II). Br– remains as a counterion for either [(NHC)2AuI]+ (7) or [(NHC)2AuIIIBr2]+ (9). AuIBr can undergo an internal redox reaction to form Au0 and Br2 (III). The detection of elemental gold during the time of incubation corroborated this reaction further. Disproportionation of [AuIBr2]− can be excluded, because during (LC-)/HR-MS analyses no further AuxBry species, e.g., [AuIIIBr4]−, was detected.
Up to now, the above-mentioned internal redox reaction (Scheme 2, (III)) has been reported only for high temperatures.59 However, the results of this study point to a water-assisted conversion at lower temperatures.
Conclusion
Here we report the ligand scrambling reaction between two bromido[3-ethyl-4-(4-methoxyphenyl)-5-(2-methoxypyridin-5-yl)-1-propylimidazol-2-ylidene]gold(I) complexes (1) in aqueous solutions, yielding [(NHC)2AuI]+ (7) and [AuIBr2]− (8). A detailed reaction mechanism was proposed on the basis of data from HPLC, (LC-)/HR-MS, and NMR experiments in combination with DFT calculations.
The stacked arrangement of the monomeric units 1 in 2 facilitates the migration of an NHC ligand to form intermediates with extraordinary strong aurophilic interactions that allowed the separation of stable intermediates 3 and 4 by HPLC and the determination of their constitution by HR-MS and HCD fragmentation.
DFT calculations provided a detailed structural and energetic picture of the ligand scrambling reaction mechanism. The reaction does not follow that of (NHC)AgIX complexes, where the rearrangement takes place in a concerted associative pathway.40 Our experimental and theoretical data prove for related gold(I) complexes ((NHC)AuIX) the formation of stable (T-shaped) intermediates, followed by Br– dissociation and recombination in a stepwise process forming 7 and 8.
As a surprising side reaction [AuIBr2]− (8) oxidized the initial decomposition product 7 to [(NHC)2AuIIIBr2]+ (9). Thereto, 8 decomposed to Au0 and Br2. The presence of bromine was confirmed by scavenging with cyclopentene and formation of 1,2-dibromopentane as evident from NMR data.
Our findings not only provide a detailed picture of the mechanism of ligand rearrangement reactions of (NHC)AuIBr complexes, but they are also of high relevance for the interpretation of biological results. Scrambling reactions have to be taken into account when preparing aqueous stock solutions for in vitro and in vivo testing. As it is well-known that [(NHC)2AuI]+ complexes are much more active, their formation under physiological conditions could produce false positive results for (NHC)AuIX complexes. In this context, it is of interest to know how the ligand X determines the reaction.
A detailed understanding of these processes is a prerequisite for the successful development of novel biologically active compounds of this class, and stability studies should be conducted with great care.
Experimental Section
Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, Fluka, Alfa Aesar, and Acros) and used without further purification. Column chromatography was performed by using silica gel 60 (0.040–0.063 mm). NMR spectra were recorded on a Bruker Avance 4 Neo, operating at 400 MHz (1H NMR) and 100 MHz (13C NMR) (2 channels, rt BBFO probe) with sample charger and Bruker Avance II+ (3 channels, liquid N2 cooled TCI Prodigy probe) operating at 600 MHz. Deuterated solvents were purchased from Eurisotop. Chemical shifts are given in ppm, and coupling constants (J) are reported in Hz. The center of the solvent signal and the TMS signal served as internal standard.
HPLC Methods
A solution of complex 1 in the appropriate mixture of ACN and water (1 mM in 1.50 mL) was studied over a period of 72 h by HPLC analysis. First, the powdered complexes were dissolved in ACN before water was added. Sample solutions were passed through a 0.20 μm membrane filter and analyzed with a Shimadzu prominence HPLC system with autosampler SIL 20A HT, column oven CT = −10AS VP, degasser DGU-20A, detector SPD-M20A, pump LC 20AD, and a KNAUER 250 × 4 nm2 Eurospher 100-5 C18 column. The mobile phase consists of ACN and water with 0.1% TFA. To achieve a separation of the compounds, the gradient elution from 90 to 70% ACN/water was used with a flow rate of 1 mL/min at an oven temperature of 35 °C. All solvents have been degassed before use. The injection volume was 20 μL, and the detection wavelength was set at 254 nm. Each measurement was performed in triplicates and displayed in 3D graphics by Origin Pro 2016 (Origin LabCorporation, Northampton, MA).
Mass Spectrometry
Mass spectra were recorded on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA) using direct infusion and heated electrospray ionization. HCD fragmentations were performed on isolated ions (isolation width typically 5 Da) with stepwise increase of energy to obtain optimum results.
For LC-MS measurement, an Agilent 1100 series LC coupled to an Orbitrap Elite mass spectrometer was used. Separation was achieved on a Zorbax Eclipse Plus C18 column (2.1 × 150 mm2, 3.5 μm) by using a eluation gradient from 90 to 70% ACN/water (0.1% formic acid) at a flow rate of 0.1 mL/min over 25 min. Mass spectra were recorded with a resolution of 240000.
Crystallography
A Bruker D8 Quest Kappa diffractometer equipped with a Photon 100 detector was used to collect the single-crystal intensity data. Monochromatized Mo Kα radiation was generated by an Incoatec microfocus X-ray tube (50 kV/1 mA power settings) in combination with a multilayer optic. The supplementary crystallographic data were deposited as CCDC 1923122 (9). The crystal structure of 1 (illustrated as dimer 2) has previously been published and has the number CCDC 1923124.50 Copies of the data can be obtained, free of charge, at the Cambridge Crystallographic Data Centre.
Quantum Chemical Methodology
The initial geometry was taken from the provided X-ray crystal structure of 1. Subsequently, structures were modified according to the mass information obtained from the LC-MS experiments. As no structural information except the mass of intermediate 3 was known, several configurations were tested and subjected to density functional theory (DFT) structure optimizations. Based on the obtained structural information, the intermediate structures 3–7 were set up and optimized.
Structure optimizations were performed with Turbomole 7.2.1,60 using the PBE0 functional,61 which is known to provide good results when investigating transition metals such as gold.62−64 The def2-SV(P)65 basis set was utilized for preoptimization, while the def2-TZVP65 basis sets employing Becke–Johnson dispersion corrections66 (BJ) were used to refine the structures. Relativistic effects of gold were taken into account by effective core potentials (ECPs).67 Frequency calculations were performed on the optimized structures to verify that they are indeed energy minima or first-order saddle points. Implicit solvent corrections were accounted for by using the Conductor-Like Screening Model (COSMO)68,69 as implemented in Turbomole, where a dielectric constant of ε = 80 was used to model the aqueous environment. The GoodVibes tool70 was utilized to obtain the zero-point energy and thermal corrections for the selected structures by calculating an approximate partition function using the harmonic oscillator model. The frequency cutoff was set to 50 cm–1, and the chosen scaling factor was 0.9944.71 The entropic contributions of frequencies below the cutoff value was calculated by a free rotator model,72 whereas those above the cutoff were treated with the standard rigid-rotator quasi-harmonic oscillator approximation. As it is known that the entropy is significantly quenched in solution73 compared to the gas phase (where it is calculated), values for the entropy were scaled by 0.5.74 Reported energies are either relative electronic energies ΔE (PBE0/def2-TZVP/BJ in implicit solvent) or relative free energies ΔG at 298.15 K, denoted as ΔG298.15K.
The transition states (TS) were obtained by using the eigenvector following method implemented in Turbomole. A successful transition state was confirmed by the presence of exactly one imaginary frequency matching the reaction coordinate.
Transition states for dissociation reactions could not be localized using reaction path optimization methods, such as the nudged elastic band, as implemented in ORCA,75,76 or eigenvector following methods, because dissociations were found to be energetically uphill in the electronic energy. However, as the reactants separate, they gradually gain rotational and translational entropy until they can freely rotate and translate when infinitely separated. This gain in entropy is not accounted for in the standard approach, where upon dissociation the two fragments were still considered as one supermolecule. In this supermolecule, the two fragments cannot move individually, which is of course a poor description of reality. To take this gradual increase in rotational and translational entropy upon dissociation into account and to localize an approximate transition state, a method put forward by Baik77 was applied. Here, the optimized supermolecule was modified by gradually separating it into the two components and optimizing each structure along this pathway, while restraining the reaction coordinate. In addition, the two infinitely separated fragments were optimized, too. The free energy was computed for every structure along this path as well as for the two infinitely separated fragments. A sigmoid function was fitted along the −TΔS term. The two fragments were considered separated and only noncovalently bound, when the natural bond orbital partial charges did no longer change upon further separation. This point was taken as the last of the supermolecule. The sigmodal fit allows for an estimation of the onset of rotational and translational entropy upon dissociation and allows for an approximate determination of the reaction free energy barrier.
Investigating the discrepancies between experiment and theory further, we evaluated the explicit interaction of water molecules with the gold(I) complexes. While the inclusion of explicit solvent molecules may improve the model, it imposes several additional challenges: the number, position(s), and orientation(s) of the water molecules need to be carefully evaluated. Potential interactions of one or several water molecules with the solute (the gold(I) complex) need to be sampled to find the energetically most favorable position. However, assuming at most one coordinated water molecule, which we assign to the gold(I) complexes 4–7 based on chemical intuition, we obtained the stable water coordinated structures 4WATER–7WATER (Figure S16 and Table S4).
Structural parameter measurements and visualizations were done with PyMol.78
Acknowledgments
The Austrian Research Promotion Agency FFG [West Austrian BioNMR 858017] and the Austrian Science Fund (M-2005) (postdoctoral fellowship to M.P.) are kindly acknowledged for financial support. The computational results presented in this work have been achieved using the HPC infrastructure of the University of Innsbruck (leo3e) and the Vienna Scientific Cluster VSC3.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c02298.
Synthesis and characterization of the complexes 7 and 9; UV–vis spectra of 1, 3, 4, 7, and 9; mass spectra, 1H and 13C NMR spectra of the reaction mixture, LC-MS spectra, crystal structure data, thermodynamic, kinetic and structural data from DFT calculations, and XYZ coordinates of the calculated structures (PDF)
Accession Codes
CCDC 1923122 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
S.K.G. and C.M.G. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Gaillard S.; Cazin C. S. J.; Nolan S. P. N-heterocyclic carbene gold(I) and copper(I) complexes in C-H bond activation. Acc. Chem. Res. 2012, 45, 778–787. 10.1021/ar200188f. [DOI] [PubMed] [Google Scholar]
- Pal S.; Kathewad N.; Pant R.; Khan S. Synthesis, characterization, and luminescence studies of gold(I) complexes with PNP- and PNB-based ligand systems. Inorg. Chem. 2015, 54, 10172–10183. 10.1021/acs.inorgchem.5b01046. [DOI] [PubMed] [Google Scholar]
- Balch A. L. Polymorphism and luminescent behaviour of linear, two-coordinate gold(I) complexes. Gold Bull. 2004, 37, 45–50. 10.1007/BF03215516. [DOI] [Google Scholar]
- Penney A. A.; Starova G. L.; Grachova E. V.; Sizov V. V.; Kinzhalov M. A.; Tunik S. P. Gold(I) alkynyls supported by mono- and bidentate NHC ligands: Luminescence and isolation of unprecendented ionic complexes. Inorg. Chem. 2017, 56, 14771–14787. 10.1021/acs.inorgchem.7b01508. [DOI] [PubMed] [Google Scholar]
- Bertrand B.; Casini A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219. 10.1039/C3DT52524D. [DOI] [PubMed] [Google Scholar]
- Mora M.; Gimeno M. C.; Visbal R. Recent advances in gold–NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. 10.1039/C8CS00570B. [DOI] [PubMed] [Google Scholar]
- Liu W.; Bensdorf K.; Proetto M.; Abram U.; Hagenbach A.; Gust R. NHC gold halide complexes derived from 4,5-diarylimidazoles: Synthesis, structural analysis, and pharmacological investigations as potential antitumor agents. J. Med. Chem. 2011, 54, 8605–8615. 10.1021/jm201156x. [DOI] [PubMed] [Google Scholar]
- Oberkofler J.; Aikman B.; Bonsignore R.; Pöthig A.; Platts J.; Casini A.; Kühn F. E. Exploring the reactivity and biological effects of heteroleptic N-heterocyclic carbene gold(I)-alkynyl complexes. Eur. J. Inorg. Chem. 2020, 2020, 1040–1051. 10.1002/ejic.201901043. [DOI] [Google Scholar]
- Schmidt C.; Karge B.; Misgeld R.; Prokop A.; Franke R.; Brönstrup M.; Ott I. Gold(I) NHC complexes: Antiproliferative activity, cellular uptake, inhibition of mammalian and bacterial thioredoxin reductases, and gram-positive directed antibacterial effects. Chem. - Eur. J. 2017, 23, 1869–1880. 10.1002/chem.201604512. [DOI] [PubMed] [Google Scholar]
- Blodgett R. C. Auranofin: Experience to date. Am. J. Med. 1983, 75, 86–89. 10.1016/0002-9343(83)90480-1. [DOI] [PubMed] [Google Scholar]
- Snyder R. M.; Mirabelli C. K.; Crooke S. T. The cellular pharmacology of Auranofin. Semin. Arthritis Rheum. 1987, 17, 71–80. 10.1016/0049-0172(87)90017-5. [DOI] [PubMed] [Google Scholar]
- Liu W.; Bensdorf K.; Proetto M.; Hagenbach A.; Abram U.; Gust R. Synthesis, characterization, and in vitro studies of bis[1,3-diethyl-4,5-diarylimidazol-2-ylidene]gold(I/III) complexes. J. Med. Chem. 2012, 55, 3713–3724. 10.1021/jm3000196. [DOI] [PubMed] [Google Scholar]
- Citta A.; Schuh E.; Mohr F.; Folda A.; Massimino M. L.; Bindoli A.; Casini A.; Rigobello M. P. Fluorescent silver(I) and gold(I)-N-heterocyclic carbene complexes with cytotoxic properties: Mechanistic insights. Metallomics 2013, 5, 1006–1015. 10.1039/c3mt20260g. [DOI] [PubMed] [Google Scholar]
- Baker M. V.; Barnard P. J.; Berners-Price S. J.; Brayshaw S. K.; Hickey J. L.; Skelton B. W.; White A. H. Synthesis and structural characterisation of linear Au(I) N-heterocyclic carbene complexes: New analogues of the Au(I) phosphine drug Auranofin. J. Organomet. Chem. 2005, 690, 5625–5635. 10.1016/j.jorganchem.2005.07.013. [DOI] [Google Scholar]
- Dada O.; Curran D.; O’Beirne C.; Müller-Bunz H.; Zhu X.; Tacke M. Synthesis and cytotoxicity studies of novel NHC–Gold(I) pseudohalides and thiolates. J. Organomet. Chem. 2017, 840, 30–37. 10.1016/j.jorganchem.2017.03.050. [DOI] [Google Scholar]
- Rubbiani R.; Kitanovic L.; Alborzinia H.; Can S.; Kitanovic A.; Onambele L. A.; Stefanopoulou M.; Geldmacher Y.; Sheldrick W. S.; Wolber G.; Prokop A.; Wölfl S.; Ott I. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. 10.1021/jm100801e. [DOI] [PubMed] [Google Scholar]
- Schmidt C.; Albrecht L.; Balasupramaniam S.; Misgeld R.; Karge B.; Brönstrup M.; Prokop A.; Baumann K.; Reichl S.; Ott I. A gold(I) biscarbene complex with improved activity as a TrxR inhibitor and cytotoxic drug: Comparative studies with different gold metallodrugs. Metallomics 2019, 11, 533–545. 10.1039/C8MT00306H. [DOI] [PubMed] [Google Scholar]
- Kızrak Ü.; Çiftçi O.; Özdemir İ.; Gürbüz N.; Düşünceli S. D.; Kaloğlu M.; Mansour L.; Zaghrouba F.; Hamdi N.; Özdemir İ. Amine-functionalized silver and gold N-heterocyclic carbene complexes: Synthesis, characterization and antitumor properties. J. Organomet. Chem. 2019, 882, 26–32. 10.1016/j.jorganchem.2018.12.018. [DOI] [Google Scholar]
- Guarra F.; Marzo T.; Ferraroni M.; Papi F.; Bazzicalupi C.; Gratteri P.; Pescitelli G.; Messori L.; Biver T.; Gabbiani C. Interaction of a gold(I) dicarbene anticancer drug with human telomeric DNA G-quadruplex: Solution and computationally aided X-ray diffraction analysis. Dalton Trans. 2018, 47, 16132–16138. 10.1039/C8DT03607A. [DOI] [PubMed] [Google Scholar]
- Pyykkö P.; Li J.; Runeberg N. Predicted ligand dependance of the Au(I)-Au(I) attraction in (XAuPH3)2. Chem. Phys. Lett. 1994, 218, 133–138. 10.1016/0009-2614(93)E1447-O. [DOI] [Google Scholar]
- Crudden C. M.; Allen D. P. Stability and reactivity of N-heterocyclic carbene complexes. Coord. Chem. Rev. 2004, 248, 2247–2273. 10.1016/j.ccr.2004.05.013. [DOI] [Google Scholar]
- Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Slattery J.; Thatcher R. J.; Shi Q.; Douthwaite R. E. Comparison of donor properties of N-heterocyclic carbenes and N-donors containing the 1H-pyridin-(2E)-ylidene motif. Pure Appl. Chem. 2010, 82, 1663–1671. 10.1351/PAC-CON-09-11-10. [DOI] [Google Scholar]
- Mata J. A.; Chianese A. R.; Miecznikowski J. R.; Poyatos M.; Peris E.; Faller J. W.; Crabtree R. H. Reactivity differences in the syntheses of chelating N-heterocyclic carbene complexes of rhodium are ascribed to ligand anisotropy. Organometallics 2004, 23, 1253–1263. 10.1021/om034240+. [DOI] [Google Scholar]
- Phillips E. M.; Chan A.; Scheidt K. A. Discovering new reactions with N-heterocyclic carbene catalysis. Aldrichim. Acta 2009, 42, 55–65. [PMC free article] [PubMed] [Google Scholar]
- Bourissou D.; Guerret O.; Gabbai F. P.; Bertrand G. Stable carbenes. Chem. Rev. 2000, 100, 39–92. 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]
- Głodek M.; Makal A.; Paluch P.; Kadziołka-Gaweł M.; Kobayashi Y.; Zakrzewski J.; Plażuk D. (Ar–CO–C≡C)(PEt3)Au and (Ar–C≡C)(PEt3)Au complexes bearing pyrenyl and ferrocenyl groups: Synthesis, structure, and luminescence properties. Dalton Trans. 2018, 47, 6702–6712. 10.1039/C8DT01061G. [DOI] [PubMed] [Google Scholar]
- Deponte M.; Urig S.; Arscott L. D.; Fritz-Wolf K.; Réau R.; Herold-Mende C.; Koncarevic S.; Meyer M.; Davioud-Charvet E.; Ballou D. P.; Williams C. H.; Becker K. Mechanistic studies on a novel, highly potent gold-phosphole inhibitor of human glutathione reductase. J. Biol. Chem. 2005, 280, 20628–20637. 10.1074/jbc.M412519200. [DOI] [PubMed] [Google Scholar]
- Urig S.; Fritz-Wolf K.; Réau R.; Herold-Mende C.; Tóth K.; Davioud-Charvet E.; Becker K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew. Chem., Int. Ed. 2006, 45, 1881–1886. 10.1002/anie.200502756. [DOI] [PubMed] [Google Scholar]
- Bindoli A.; Rigobello M. P.; Scutari G.; Gabbiani C.; Casini A.; Messori L. Thioredoxin reductase: A target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. 10.1016/j.ccr.2009.02.026. [DOI] [Google Scholar]
- Dos Santos H. F.; Vieira M. A.; Delgado G. Y. S.; Paschoal D. Ligand exchange reaction of Au(I) R-N-heterocyclic carbene complexes with cysteine. J. Phys. Chem. A 2016, 120, 2250–2259. 10.1021/acs.jpca.6b01052. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Isab A. A.; Perzanowski H. P. Ligand scrambling reactions of cyano (thione) gold (I) complexes and determination of their equilibrium constants. Can. J. Chem. 2002, 80, 1279–1284. 10.1139/v02-165. [DOI] [Google Scholar]
- Hormann A.; Shaw C. III; Bennett D.; Reiff W. Solid-state structure and solution equilibria of cyano (triethylphosphine) gold(I). Inorg. Chem. 1986, 25, 3953–3957. 10.1021/ic00242a025. [DOI] [Google Scholar]
- Ahmad S.; Isab A. A.; Perzanowski H. P.; Hussain M. S.; Akhtar M. N. Gold(I) complexes with tertiary phosphine sulfide ligands. Transition Met. Chem. 2002, 27, 177–183. 10.1023/A:1013920208168. [DOI] [Google Scholar]
- Albert A.; Brauckmann C.; Blaske F.; Sperling M.; Engelhard C.; Karst U. Speciation analysis of the antirheumatic agent Auranofin and its thiol adducts by LC/ESI-MS and LC/ICP-MS. J. Anal. At. Spectrom. 2012, 27, 975–981. 10.1039/c2ja30109a. [DOI] [Google Scholar]
- Onaka S.; Katsukawa Y.; Shiotsuka M.; Kanegawa O.; Yamashita M. Synthesis, X-ray molecular structure analysis, and study on ligand scrambling reactions of new thiolatogold(I) complexes with various phosphines. Inorg. Chim. Acta 2001, 312, 100–110. 10.1016/S0020-1693(00)00353-4. [DOI] [Google Scholar]
- Hormann-Arendt A. L.; Shaw C. F. Ligand-scrambling reactions of cyano(trialkyl/triaryl-phosphine)gold(I) complexes: Examination of factors influencing the equilibrium constant. Inorg. Chem. 1990, 29, 4683–4687. 10.1021/ic00348a019. [DOI] [Google Scholar]
- Sivaram H.; Jothibasu R.; Huynh H. V. Gold complexes of an alicyclic indazole-derived N-heterocyclic carbene: Syntheses, characterizations, and ligand disproportionation. Organometallics 2012, 31, 1195–1203. 10.1021/om201268m. [DOI] [Google Scholar]
- Guo S.; Bernhammer J. C.; Huynh H. V. 1,2,4-triazole-derived carbene complexes of gold: Characterization, solid-state aggregation and ligand disproportionation. Dalton Trans. 2015, 44, 15157–15165. 10.1039/C4DT03201B. [DOI] [PubMed] [Google Scholar]
- Wang H. M. J.; Lin I. J. B. Facile synthesis of silver(I)–carbene complexes. Useful carbene transfer agents. Organometallics 1998, 17, 972–975. 10.1021/om9709704. [DOI] [Google Scholar]
- Su H.-L.; Pérez L. M.; Lee S.-J.; Reibenspies J. H.; Bazzi H. S.; Bergbreiter D. E. Studies of ligand exchange in N-heterocyclic carbene silver(I) complexes. Organometallics 2012, 31, 4063–4071. 10.1021/om300340w. [DOI] [Google Scholar]
- Schmidbaur H. The fascinating implications of new results in gold chemistry. Gold Bull. 1990, 23, 11–21. 10.1007/BF03214710. [DOI] [Google Scholar]
- Pan P.; Wood S. A Gold-chloride complexes in very acidic aqueous solutions and at temperatures 25–300°C: A laser Raman spectroscopic study. Geochim. Cosmochim. Acta 1991, 55, 2365–2371. 10.1016/0016-7037(91)90112-I. [DOI] [Google Scholar]
- Theilacker K.; Schlegel H. B.; Kaupp M.; Schwerdtfeger P. Relativistic and solvation effects on the stability of gold(III) halides in aqueous solution. Inorg. Chem. 2015, 54, 9869–75. 10.1021/acs.inorgchem.5b01632. [DOI] [PubMed] [Google Scholar]
- Karaca Ö.; Scalcon V.; Meier-Menches S. M.; Bonsignore R.; Brouwer J. M. J. L.; Tonolo F.; Folda A.; Rigobello M. P.; Kühn F. E.; Casini A. Characterization of hydrophilic gold(I) N-heterocyclic carbene (NHC) complexes as potent TrxR inhibitors using biochemical and mass spectrometric approaches. Inorg. Chem. 2017, 56, 14237–14250. 10.1021/acs.inorgchem.7b02345. [DOI] [PubMed] [Google Scholar]
- Kaps L.; Biersack B.; Müller-Bunz H.; Mahal K.; Münzner J.; Tacke M.; Mueller T.; Schobert R. Gold(I)–NHC complexes of antitumoral diarylimidazoles: Structures, cellular uptake routes and anticancer activities. J. Inorg. Biochem. 2012, 106, 52–58. 10.1016/j.jinorgbio.2011.08.026. [DOI] [PubMed] [Google Scholar]
- Muenzner J. K.; Biersack B.; Kalie H.; Andronache I. C.; Kaps L.; Schuppan D.; Sasse F.; Schobert R. Gold(I) biscarbene complexes derived from vascular-disrupting combretastatin A-4 address different targets and show antimetastatic potential. ChemMedChem. 2014, 9, 1195–1204. 10.1002/cmdc.201400049. [DOI] [PubMed] [Google Scholar]
- Bertrand B.; Stefan L.; Pirrotta M.; Monchaud D.; Bodio E.; Richard P.; Le Gendre P.; Warmerdam E.; de Jager M. H.; Groothuis G. M. M.; Picquet M.; Casini A. Caffeine-based gold(I) N-heterocyclic carbenes as possible anticancer agents: synthesis and biological properties. Inorg. Chem. 2014, 53 (4), 2296–2303. 10.1021/ic403011h. [DOI] [PubMed] [Google Scholar]
- Bazzicalupi C.; Ferraroni M.; Papi F.; Massai L.; Bertrand B.; Messori L.; Gratteri P.; Casini A. Determinants for tight and selective binding of a medicinal dicarbene gold(I) complex to a telomeric DNA G-Quadruplex: A joint ESI MS and XRD investigation. Angew. Chem., Int. Ed. 2016, 55, 4256–9. 10.1002/anie.201511999. [DOI] [PubMed] [Google Scholar]
- Gallati C. M.; Goetzfried S. K.; Ausserer M.; Sagasser J.; Plangger M.; Wurst K.; Hermann M.; Baecker D.; Kircher B.; Gust R. Synthesis, characterization and biological activity of bromido[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes. Dalton Trans. 2020, 49, 5471–5481. 10.1039/C9DT04824C. [DOI] [PubMed] [Google Scholar]
- Messori L.; Marchetti L.; Massai L.; Scaletti F.; Guerri A.; Landini I.; Nobili S.; Perrone G.; Mini E.; Leoni O.; Pasquali M.; Gabbiani C. Chemistry and biology of two novel gold(I) carbene complexes as prospective anticancer agents. Inorg. Chem. 2014, 53, 2396–2403. 10.1021/ic401731a. [DOI] [PubMed] [Google Scholar]
- Mageed A. H.; Skelton B. W.; Baker M. V. Stable Au(III) complexes with four N-heterocyclic carbene groups can be prepared in high yield directly from KAuCl4. Dalton Trans. 2017, 46, 7844–7856. 10.1039/C7DT01272A. [DOI] [PubMed] [Google Scholar]
- Huynh H. V.; Guo S.; Wu W. Detailed structural, spectroscopic, and electrochemical trends of halido mono- and bis(NHC) complexes of Au(I) and Au(III). Organometallics 2013, 32, 4591–4600. 10.1021/om400563e. [DOI] [Google Scholar]
- Stocker F.; Britton D. 1,2-Dicyano-1,2-bis(imidazolidine-2-thione)digold(I) and 2,2-dicyano-1,1-bis(dimethylthiourea)digold(I). Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2000, 56, 798–800. 10.1107/S0108270100005783. [DOI] [PubMed] [Google Scholar]
- Schneider D.; Schuster O.; Schmidbaur H. Bromination of (phosphine)gold(I) bromide complexes: Stiochiometry and structure of products. Dalton Trans. 2005, 1940–1947. 10.1039/b502861b. [DOI] [PubMed] [Google Scholar]
- Samantaray M. K.; Dash C.; Shaikh M. M.; Pang K.; Butcher R. J.; Ghosh P. Gold(III) N-heterocyclic carbene complexes mediated synthesis of β-enaminones from 1,3-dicarbonyl compounds and aliphatic amines. Inorg. Chem. 2011, 50, 1840–1848. 10.1021/ic102268n. [DOI] [PubMed] [Google Scholar]
- Kriechbaum M.; List M.; Berger R. J. F.; Patzschke M.; Monkowius U. Silver and gold complexes with a new 1,10-phenanthroline analogue N-heterocyclic carbene: A combined structural, theoretical, and photophysical study. Chem. - Eur. J. 2012, 18, 5506–5509. 10.1002/chem.201200465. [DOI] [PubMed] [Google Scholar]
- Hirtenlehner C.; Krims C.; Hölbling J.; List M.; Zabel M.; Fleck M.; Berger R. J. F.; Schoefberger W.; Monkowius U. Syntheses, crystal structures, reactivity, and photochemistry of gold(III) bromides bearing N-heterocyclic carbenes. Dalton Trans. 2011, 40, 9899–9910. 10.1039/c1dt11175b. [DOI] [PubMed] [Google Scholar]
- Thomsen J. Darstellung und Eigenschaften der Chlor- und Bromverbindungen und des Oxydes des Goldes. Arch. Pharm. 1877, 210, 266–268. 10.1002/ardp.18772100334. [DOI] [Google Scholar]
- TURBOMOLE V7.2 2017, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH: since 2007; available from http://www.turbomole.com/. [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Langseth E.; Scheuermann M. L.; Balcells D.; Kaminsky W.; Goldberg K. I.; Eisenstein O.; Heyn R. H.; Tilset M. Generation and structural characterization of a gold(III) alkene complex. Angew. Chem., Int. Ed. 2013, 52, 1660–1663. 10.1002/anie.201209140. [DOI] [PubMed] [Google Scholar]
- York J. T. Determining the impact of ligand and alkenes substituents on bonding in gold(I)–alkene complexes supported by N-heterocyclic carbenes: A computational study. J. Phys. Chem. A 2016, 120, 6064–6075. 10.1021/acs.jpca.6b03819. [DOI] [PubMed] [Google Scholar]
- Kang R.; Chen H.; Shaik S.; Yao J. Assessment of theoretical methods for complexes of gold(I) and gold(III) with unsaturated aliphatic hydrocarbon: Which density functional should we choose?. J. Chem. Theory Comput. 2011, 7, 4002–4011. 10.1021/ct200656p. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Andrae D.; Häußermann U.; Dolg M.; Stoll H.; Preuß H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Schäfer A.; Klamt A.; Sattel D.; Lohrenz J. C. W.; Eckert F. COSMO implementation in TURBOMOLE: Extension of an efficient quantum chemical code towards liquid systems. Phys. Chem. Chem. Phys. 2000, 2, 2187–2193. 10.1039/b000184h. [DOI] [Google Scholar]
- Luchini G.; Alegre-Requena J.; Funes-Ardoiz I.; Paton R. GoodVibes: Automated thermochemistry for heterogeneous computational chemistry data. F1000Research 2020, 9, 291–303. 10.12688/f1000research.22758.1. [DOI] [Google Scholar]
- Kesharwani M. K.; Brauer B.; Martin J. M. L. Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): Can anharmonic force fields be avoided?. J. Phys. Chem. A 2015, 119, 1701–1714. 10.1021/jp508422u. [DOI] [PubMed] [Google Scholar]
- Grimme S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. - Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Falivene L.; Barone V.; Talarico G. Unraveling the role of entropy in tuning unimolecular vs. bimolecular reaction rates: The case of olefin polymerization catalyzed by transition metals. Mol. Catal. 2018, 452, 138–144. 10.1016/j.mcat.2018.04.012. [DOI] [Google Scholar]
- Dewyer A. L.; Zimmerman P. M. Simulated mechanism for palladium-catalyzed, directed γ-arylation of piperidine. ACS Catal. 2017, 7, 5466–5477. 10.1021/acscatal.7b01390. [DOI] [Google Scholar]
- Neese F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Neese F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar]
- Ryu H.; Park J.; Kim H. K.; Park J. Y.; Kim S.-T.; Baik M.-H. Pitfalls in computational modeling of chemical reactions and how to avoid them. Organometallics 2018, 37, 3228–3239. 10.1021/acs.organomet.8b00456. [DOI] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.