

Abstract
Site-specific modification of proteins with functional molecules provides powerful tools for researching and engineering proteins. Here we report a new chemical conjugation method which photocages highly reactive but chemically selective moieties, enabling the use of protein-inert amines for selective protein modification. New amino acids FnbY and FmnbY, bearing photocaged quinone methides (QMs), were genetically incorporated into proteins. Upon light activation, they generated highly reactive QM, which rapidly reacted with amine derivatives. This method features a rare combination of desired properties including fast kinetics, small and stable linkage, compatibility with low temperature, photo-controllability, and widely available reagents. Moreover, labeling via FnbY occurs on the β-carbon, affording the shortest linkage to protein backbone which is essential for advanced studies involving orientation and distance. We installed various functionalities onto proteins, and attached a spin label as close as possible to the protein backbone, achieving high resolution in double electron-electron paramagnetic resonance distance measurements.
INTRODUCTION
Nature uses enzymes to install post-translational modifications onto proteins site-selectively, dramatically increasing the structural and functional diversity of proteins1. Chemists have correspondingly developed a range of chemical reactions for site-specific protein modification, targeting either the canonical amino acids or unnatural amino acids (Uaas) bearing bioorthogonal handles2,3. These site-selective protein conjugation reactions have enabled the generation of protein-based probes, biomaterials, diagnostics, and therapeutics, advancing biological research, material sciences, and medicine4,5. Most useful protein modification chemistries targeting natural residues employ electrophiles or redox reagents to react with nucleophilic groups on proteins, such as the ε-amine of Lys, thiol of Cys, hydroxyl of Tyr, and thiol ether of Met6,7,8,9. However, it is generally challenging to employ nucleophiles to directly modify proteins, because electrophilic groups of proteins do not readily react under biocompatible conditions10. For instance, the carboxyl group of Glu and Asp needs to be activated for reacting with amine. For chemistries targeting Uaas, initial efforts have incorporated Uaas bearing ketone, azide, alkyne, or alkene, but the reaction rates of these functionalities are generally slow under biocompatible conditions11,12,13,14. Fast kinetics have been achieved in recent years through tetrazole-alkene cycloaddition and inverse electron-demand Diels-Alder cycloaddition between tetrazine and strained alkenes or alkynes15,16,17,18, but they generate bulky and long extended linkages keeping the labeled moiety far away from the protein backbone. The size and length of the conjugation linkage is an important yet underappreciated consideration for protein modification. While a long or bulky linkage is sufficient for simple fluorescent tagging of a protein for imaging, advanced studies involving orientation and distance changes prefer a small linkage to minimize potential perturbation and require a short linkage to attach the probe as close as possible to protein backbone so as to minimize errors caused by linker flexibility. These demanding requirements are generally desired for various sophisticated biophysical techniques such as NMR, electron paramagnetic resonance (EPR), Förster resonance energy transfer (FRET), single molecule analysis, and so on19,20,21,22,23. Moreover, a small, short, and stable linkage is also important for biotherapeutics to minimize immunogenicity and to reduce undesired dissociation and degradation24.
Amine derivatives are stable, biocompatible, easy to synthesize, and broadly available. Amine-based nucleophilic reactions can result in small, stable, and short linkages. However, as relatively weak nucleophiles, amines cannot be directly applied to proteins for site-selective modification. In fact, it is a general challenge to efficiently label proteins with chemical functionalities that are either less reactive or inert to proteins. Quinone methides (QM) are excellent Michael acceptors for nucleophiles, with reaction rate constants reaching extremely high 105 to 108 M−1s−1 25. They also have a small size comparable to tyrosine, and their reaction products have small linkages similar to amino acid side chains. QMs have been mainly used in organic synthesis for conjugation reactions generally in organic solvents26,27. Due to its high reactivity, QM cannot be directly introduced into proteins.
Here we report a photo-controlled approach to site-selective protein modification and functionalization with amine reagents. Our strategy is to photocage a highly reactive but also chemically selective species into proteins, release of which in situ would allow selective reaction with the protein-inert labeling reagent for site-specific protein modification. Through expansion of the genetic code28,29, we genetically incorporated into proteins two Uaas FnbY and FmnbY, containing photocaged para-QM (p-QM) and ortho-QM (o-QM), respectively (Fig. 1). We demonstrate that the QM-based Michael addition could be extended on proteins in fully aqueous and biocompatible conditions. Upon light activation, QM was generated in proteins in situ enabling site-specific conjugation of proteins with various amine reagents as well as thiol probes. We report the modification of proteins with diverse bioorthogonal and click functionalities, biophysical probes, and post-translational modification mimics. The labeling reaction features a combination of highly desired properties including fast kinetics (< 10 sec), compatibility with low temperatures (0 °C and 4 °C), widely available reagents, small linkage, and photo-controllability. In addition, FnbY uniquely enabled modifications to be installed on the β-carbon of the amino acid, resulting in the shortest linkage for protein bioconjugation. We further demonstrated that the spin label installed via FnbY provided better resolution in EPR-based distance measurements. Our strategy of caging and releasing highly reactive but chemically selective moieties in proteins may enable the use of a broader range of protein-inert functionalities for selective protein modification.
Fig. 1. Protein labeling through amine-QM Michael addition.
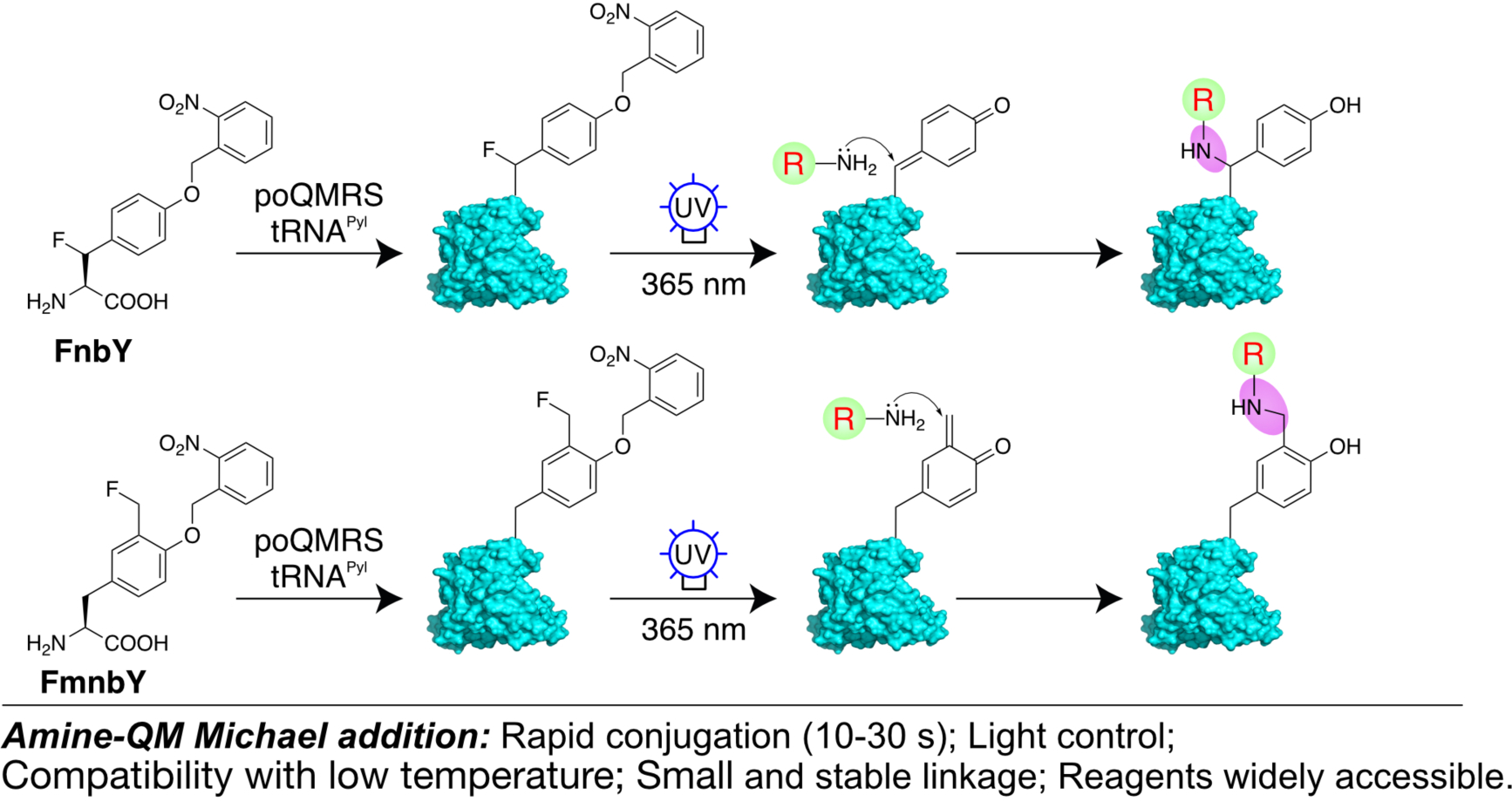
Scheme showing the labeling mechanism. FnbY or FmnbY was genetically incorporated into proteins via genetic code expansion. Upon UV light activation, both amino acids generated the highly reactive QM, which rapidly reacted with amine derivatives.
RESULTS
Genetic incorporation of FnbY and FmnbY
We designed and synthesized two Uaas containing photocaged QM in the side chain, o-2-nitrobenzyl-β-fluoro tyrosine (FnbY) and o-2-nitrobenzyl-3-fluoromethyl tyrosine (FmnbY) (Fig. 1). Upon light activation, FnbY will generate p-QM while FmnbY will generate o-QM. The Michael addition reaction occurs at the amino acid β-carbon for p-QM, which is closer to protein backbone than that for o-QM. This distance difference provides flexibility for protein labeling needs.
We previously evolved an orthogonal tRNAPyl/FnbYRS (a mutant aminoacyl-tRNA synthetase specific for the Uaa FnbY) from the Methanosarcina barkeri tRNAPyl/PylRS for genetic incorporation of FnbY into proteins in E. coli and mammalian cells, and demonstrated that FnbY, upon UV activation, cross-links nucleophilic amino acid residues of proteins only if the nucleophilic side chain is in contact with the QM30. In the absence of a contacting nucleophilic side chain, we reasoned that the generated QM could be repurposed for protein modification by adding small nucleophilic molecules. We aimed to explore the genetic incorporation of both FnbY and the new FmnbY into proteins for site-specific protein modification with amine derivatives. However, the tRNAPyl/FnbYRS pair could incorporate FmnbY in very low efficiency only. We then transplanted the mutations of FnbYRS into the PylRS of Methanomethylophilus alvus PylRS31, and discovered that the resultant mutant synthetase was able to incorporate both FnbY and FmnbY in excellent efficiency. We named this synthetase as poQMRS (para- and ortho-quinone methide synthetase, containing mutations L125F/N166A/V168G, Supporting Information). Co-expression in E. coli of the tRNAPyl/poQMRS with the MBP-Z(24TAG) gene, which encodes the maltose binding protein (MBP) fused with the Z protein containing a TAG stop codon at site 24, resulted in the production of full-length MBP-Z fusion protein only when FnbY (Fig. 2a) or FmnbY (Fig. 2b) was added to the media, demonstrating the incorporation of FnbY or FmnbY (Table S2). We also incorporated FnbY or FmnbY into ubiquitin (Ub) by expressing the Ub gene containing a TAG codon at site 6 with tRNAPyl/poQMRS in E. coli (Fig. 2c). The purified Ub proteins were analyzed with electrospray ionization mass spectrometry (ESI-MS), which confirmed that FnbY and FmnbY were incorporated in high fidelity (Fig. 2d, 2e, and S1).
Fig. 2. Genetic incorporation of FnbY and FmnbY into proteins.
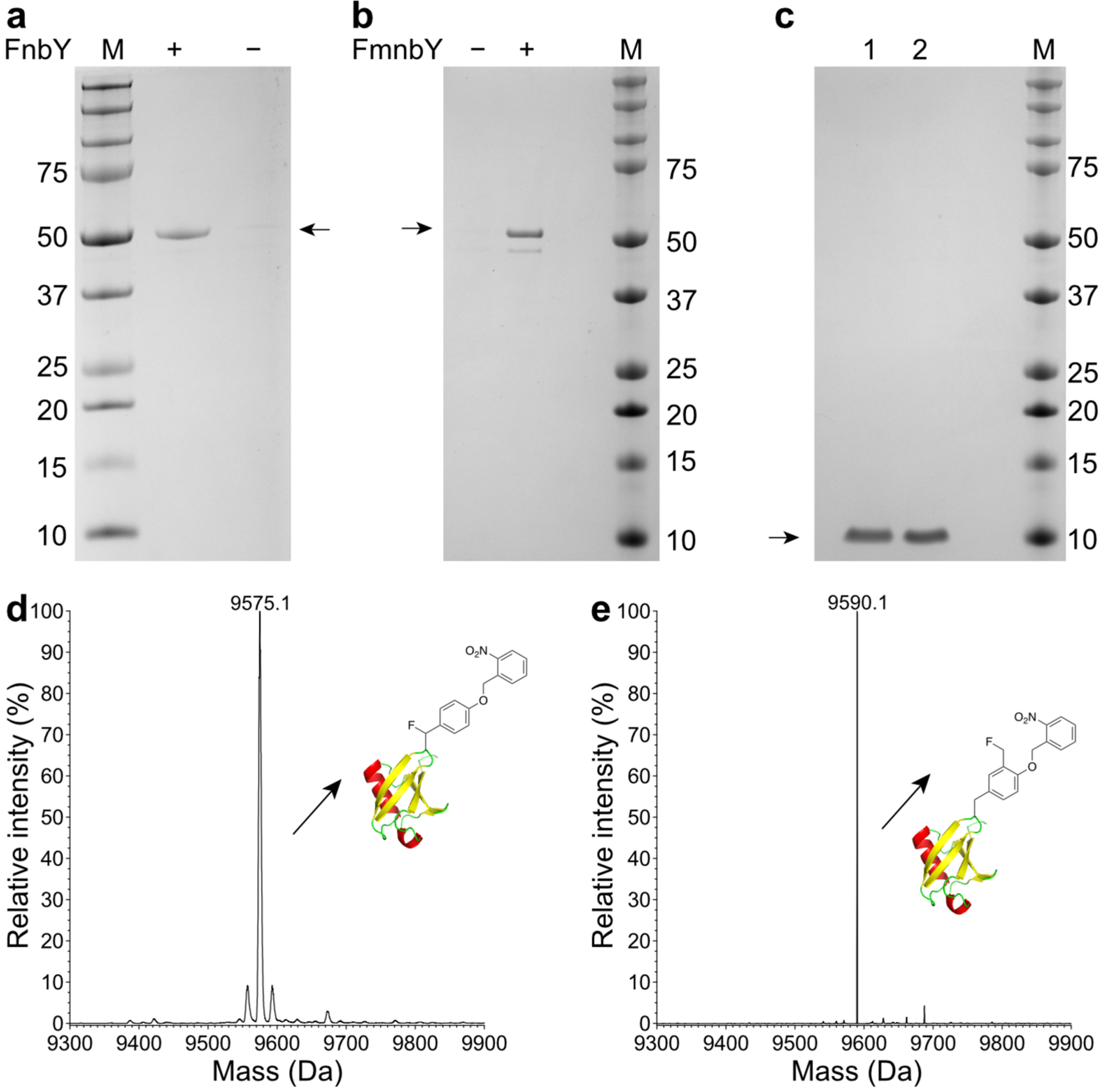
a-b, SDS-PAGE analysis of MBP-Z protein purified from E. coli cells expressing the MBP-Z(24TAG) gene with tRNAPyl/poQMRS in the presence or absence of 1 mM FnbY (a) or 1 mM FmnbY (b). c, SDS-PAGE analysis of purified Ub(6FnbY) (Ub protein with FnbY incorporated at position 6, lane 1) and Ub(6FmnbY) (Ub protein with FmnbY incorporated at position 6, lane 2). d, Deconvoluted ESI-MS spectrum of intact Ub(6FnbY). Expected, 9575.8 Da; measured, 9575.1 Da. e, Deconvoluted ESI-MS spectrum of intact Ub(6FmnbY). Expected, 9589.8 Da; measured, 9590.1 Da.
Protein labeling with widely available amine-derivatized functionalities
Amine derivatives are stable, readily accessible synthetically, and widely available from commercial sources. We therefore focused on investigating amine-QM Michael addition under aqueous condition for protein labeling.
We genetically incorporated FnbY and FmnbY into Ub separately, labeled the Ub with various amine derivatives under aqueous condition (50 mM phosphate, 200 mM NaCl, pH 7.5) at RT, and determined amine-QM conjugation efficiency using mass spectrometry. As shown in Fig. 3, both FnbY and FmnbY mediated efficient amine-QM conjugation of proteins upon UV light activation. Ub was rapidly conjugated with various amine-derivatized functional groups, including alkyne, azide, and alkene (Fig. 3a). The labeling efficiency of FnbY via p-QM was ca. 65% with 50 mM propargylamine, 67% with 50 mM 3-azido-1-propanamine, 67% with 50 mM allylamine (Fig.3a–d, and Fig. S2). FmnbY-mediated protein conjugation via o-QM was more robust. With 50 mM 3-azido-1-propanamine or 50 mM allylamine, the Ub labeling reached almost completion (close to 100%); 90% labeling efficiency was obtained with 50 mM propargylamine (Fig. 3e–g, and Fig. S2). In comparison to p-QM, o-QM has higher reactivity32, and is more exposed and accessible for reaction in proteins, which explains the observed higher conjugation efficiency of FmnbY than FnbY. In addition to alkyl amine derivatives, aryl amine derivatives such as aniline could also be efficiently conjugated with Ub (Fig. S11a).
Fig. 3. Rapid protein labeling with various amine-derivatized functionalities.
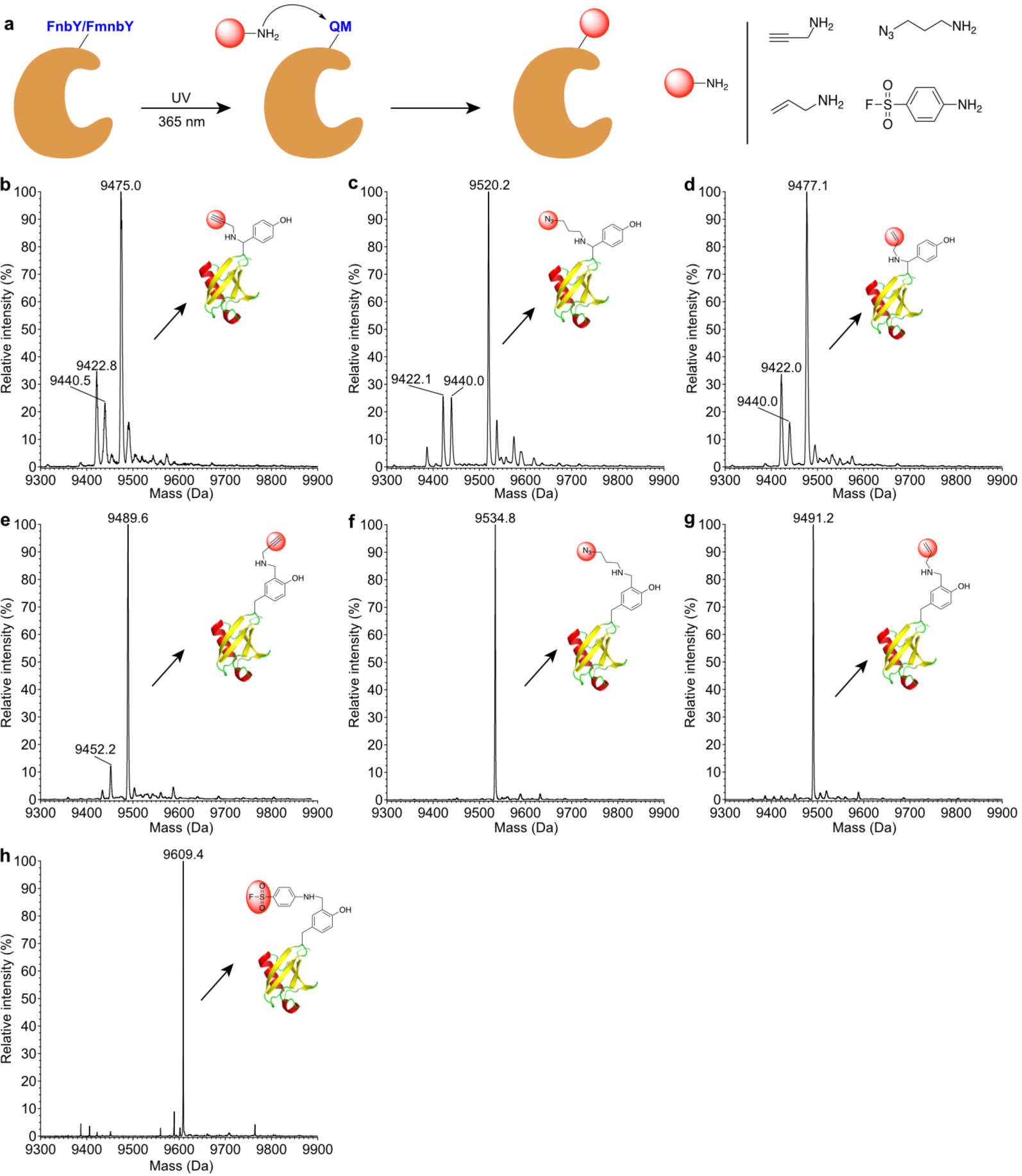
a, Scheme showing the conjugation procedure. b, Mass spectrum of Ub(6FnbY) conjugated with propargylamine. Expected, 9475.8 Da; measured 9475.0 Da. c, Mass spectrum of Ub(6FnbY) conjugated with 3-azido-1-propanamine. Expected, 9520.8 Da; measured 9520.2 Da. d, Mass spectrum of Ub(6FnbY) conjugated with allylamine. Expected, 9477.8 Da; measured 9477.1 Da. In spectra b-d, the peak observed at 9440 Da correspond to water hydrolysis of the photoactivated FnbY; the peak observed at 9422 Da correspond to F loss from the photoactivated FnbY, suggesting possible intramolecular Michael addition by nearby nucleophilic amino acid residue. e, Mass spectrum of Ub(6FmnbY) conjugated with propargylamine. Expected, 9489.8 Da; measured 9489.6 Da. f, Mass spectrum of Ub(6FmnbY) conjugated with 3-azido-1-propanamine. Expected, 9534.8 Da; measured 9534.8 Da. g, Mass spectrum of Ub(6FmnbY) conjugated with allylamine. Expected, 9491.8 Da; measured 9491.2 Da. h, Mass spectrum of Ub(6FmnbY) conjugated with 4-aminobenzene-1-sulfonyl fluoride. Expected, 9609.9 Da; measured 9609.4 Da.
The broad range of amine-derivatized functional molecules readily available together with the rapid amine-QM protein conjugation demonstrated here will allow the facile installation of various functional groups onto proteins via FnbY/FmnbY, facilitating applications requiring different functionalities or reagents. FnbY and FmnbY thus can serve as a general handle of proteins for quick chemical conversion. In addition to bioorthogonal functional groups described above, we also conjugated aryl sulfonyl fluoride onto proteins (25 mM 4-aminobenzene-1-sulfonyl fluoride, 90% efficiency, Fig. 3h), which is a major functionality for Sulfur Fluoride Exchange (SuFEx) reaction, the new generation of click chemistry33, thus enabling SuFEx reaction to be performed on proteins34,35. Therefore, the QM-amine reaction via FnbY/FmnbY can serve as a platform to rapidly install onto proteins various bioorthogonal functionalities and click chemical handles with tuned size, hydrophobicity, and chemical reactivity.
Protein functionalization with probes and post-translational modification mimics
To further investigate the general applicability of amine-QM for protein modification, we conjugated FnbY/FmnbY-incorporated proteins with amino derivatives of biophysical probes and post-translational modification mimics. We first incubated MBP-Z(24FnbY) protein with amine-derivatized fluorophore Cyanine 3 (Cy3-amine, 3 mM) and activated the reaction with UV light (365 nm) (Fig. 4a). The reaction mixture was separated on SDS-PAGE and imaged for Cy3 fluorescence. Strong Cy3 fluorescence was detected on MBP-Z(24FnbY) activated by UV light only, but not on WT MBP-Z or in the absence of UV light (Fig. 4b and Fig. S3), indicating successful Cy3 conjugation to MBP-Z protein through amine-p-QM reaction. Similarly, MBP-Z (24FmnbY) could also undergo Cy3-amine (3 mM) conjugation through amine-o-QM reaction in a photo-triggered fashion (Fig. 4c).
Fig. 4. Site-specific protein conjugation with biophysical probes through amine-QM addition.
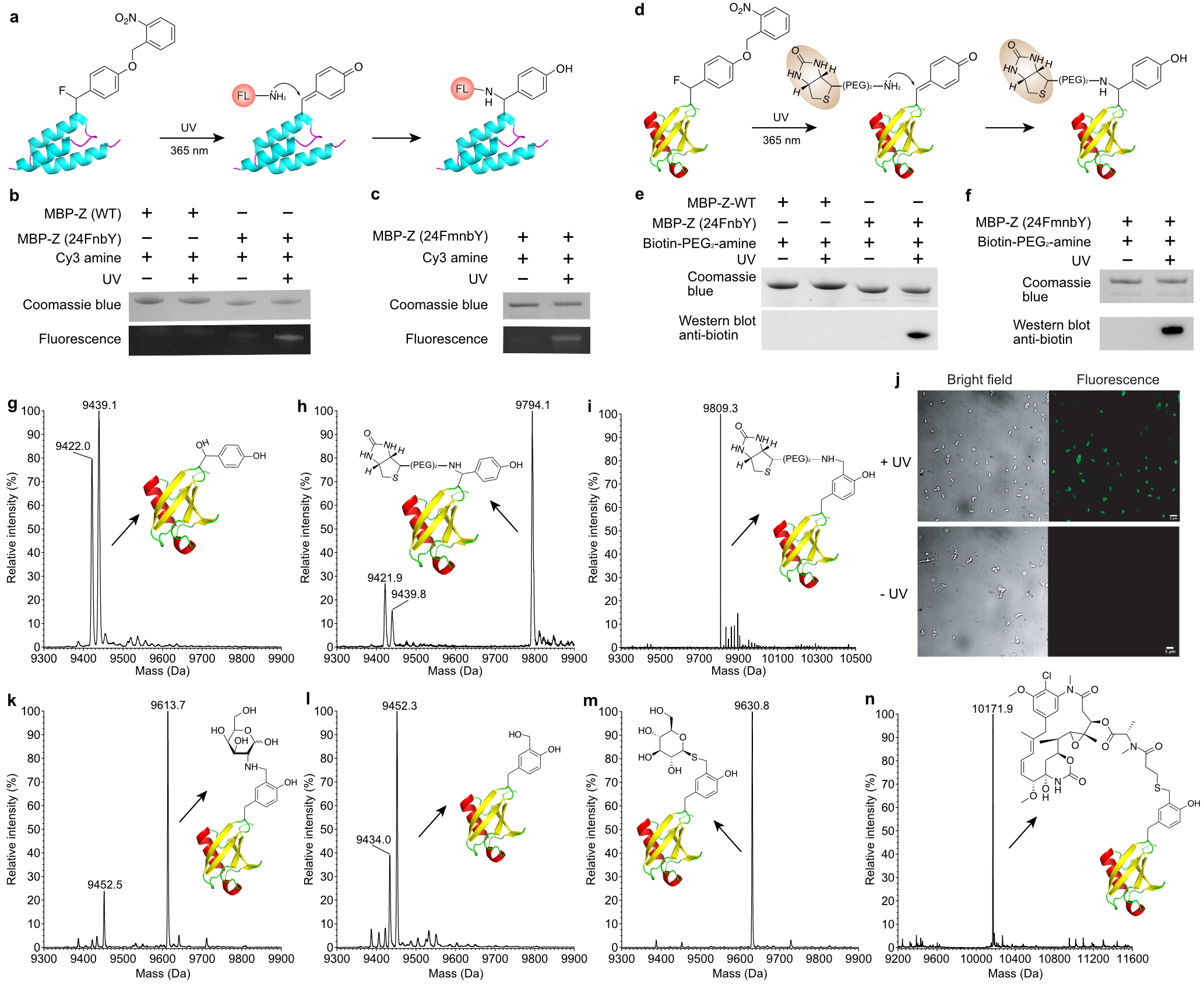
a, Scheme showing the conjugation of MBP-Z (24FnbY) with a fluorescent amine derivative, Cy3-amine. b-c, SDS-PAGE and fluorescent detection of MBP-Z(24FnbY)-Cy3 conjugation (b) and MBP-Z(24FmnbY)-Cy3 conjugation (c). d, Scheme showing the conjugation of Ub(6FnbY) with biotin-PEG2-amine. e-f, SDS-PAGE and Western blot analysis of biotin-PEG2-amine conjugation with MBP-Z(24FnbY) (e) and with MBP-Z(24FmnbY) (f). An anti-biotin antibody was used for detection of biotin conjugation. g, Mass spectrum of Ub(6FnbY) photoactivated in aqueous solution in the absence of added nucleophiles. The calculated mass of Ub(6FnbY) after photoactivation followed by conjugation with H2O was 9438.7 Da, and a peak at 9439.1 Da was observed. The peak at 9422.0 Da corresponds to loss of F, suggesting possible intramolecular Michael addition of nucleophilic residue to QM. h, Mass spectrum of Ub(6FnbY) conjugated with biotin-PEG2-amine. Expected, 9795.2 Da; measured 9794.1 Da. Two small peaks at 9439.8 and 9421.9 were also observed, as explained in (g). i, Mass spectrum of Ub(6FmnbY) conjugated with biotin-PEG2-amine. Expected, 9809.2 Da; measured 9809.3 Da. j, Fluorescence imaging of E. coli cells expressing eCPX(5FmnbY) and labeled with 0.1 mM Alexa Fluor 488 hydrazide with or without UV light (365 nm) activation. k, Mass spectrum of Ub(6FmnbY) conjugated with D-galactosamine. Expected, 9613.9 Da; measured 9613.7 Da. The peak at 9452.5 Da corresponds to water hydrolysis of photoactivated FmnbY. l, Mass spectrum of Ub(6FmnbY) conjugated with D-glucose (500 mM). Expected, 9614.9 Da; Not observed. The peak at 9452.3 Da corresponds to water hydrolysis of photoactivated FmnbY, and the peak at 9434.0 Da corresponds to loss of F. m, Mass spectrum of Ub(6FmnbY) conjugated with 1-thio-β-D-glucose (1 mM). Expected, 9630.9 Da; measured 9630.8 Da. n, Mass spectrum of Ub(6FmnbY) conjugated with Mertansine. Expected, 10173.0 Da; measured, 10171.9 Da.
We next incubated MBP-Z(24FnbY) or WT MBP-Z with 10 mM biotin-(PEG)2-amine with or without UV light (365 nm) activation (Fig. 4d). Again, as shown in Western-blot with anti-biotin antibody (Fig. 4e), MBP-Z(24FnbY) reacted with biotin-(PEG)2-amine only under UV activation. In a similar manner, MBP-Z(24FmnbY) was also conjugated with biotin-(PEG)2-amine (10 mM) through photo-controlled amine-o-QM reaction (Fig. 4f).
To confirm the amine-QM conjugation reaction and to determine the conjugation efficiency, we analyzed the conjugated protein products with mass spectrometry. In the absence of amine nucleophiles, UV activation of Ub(6FnbY) alone in aqueous buffer resulted in hydrolysis of the released p-QM, as confirmed by the intact masses of Ub(6FnbY) after UV exposure (Fig. 4g), consistent with water reacting with QM. In the presence of 10 mM biotin-(PEG)2-amine and UV light, Ub(6FnbY) conjugated with this biotin probe was detected by MS, together with a small portion of hydrolysis product (Fig. 4h). Based on the intensities of these species, the conjugation efficiency was 85%. Under similar conditions, MS analysis of biotin-(PEG)2-amine conjugation with Ub(6FmnbY) also confirmed the reaction of amine probe with the photo-released o-QM, and the labeling efficiency was over 95% (Fig. 4i), which is consistent with the higher reactivity of o-QM over p-QM. These results together indicated that the amine probe was able to compete with H2O for reaction with the photo-released QM on proteins, generating the desired protein conjugation products. In addition, biotin bearing with a stronger nucleophilic alpha effect amine, biotin-hydrazide (1 mM), was also tested for conjugation with Ub(6FnbY) and Ub(6FmnbY), and 47% and 84% labeling efficiency was achieved for Ub(6FnbY) and Ub(6FmnbY), respectively (Fig. S4). The working concentration of the more nucleophilic hydrazide could be lowered to 1 mM without compromising the labeling efficiency for FmnbY.
Since FmnbY mediated amine-QM labeling is more robust, we next tested labeling FmnbY-containing proteins expressed on E. coli cell surface. FmnbY was incorporated into an outer membrane protein eCPX (enhanced circularly permuted outer membrane protein OmpX) in E. coli to display the FmnbY residue on the cell surface. These E. coli cells were incubated with 0.1 mM Alexa Fluor 488 hydrazide, or 5 mM biotin-hydrazide with and without UV light activation for 1 min, followed by a thorough wash to remove labeling molecules. For biotin-hydrazide labeled E. coli cells, Western blot analysis of the cell lysate clearly showed biotin conjugation to the target protein (Fig. S5a) in UV-activated cells. For Alexa Fluor 488 hydrazide labeled E. coli cells, the cell lysate was separated by SDS-PAGE and imaged for Alexa Fluor 488 fluorescence. A fluorescent band corresponding to the target protein was clearly detected in UV-activated cells (Fig. S5b–c). We also imaged Alexa Fluor 488 labeled E. coli cells with a fluorescence microscope (Fig. 4j). Alexa Fluor 488 fluorescence was observed on cells with UV activation. These results together indicate successful labeling of FmnbY-containing protein on E. coli cell surface.
We envisioned that amine-QM conjugation could also be used for facile installation of certain post-translational modification mimics for modulating protein properties. Conjugation of peptide or protein drugs with glycan is a viable strategy to improve the drug’s pharmacological efficacy, such as cell penetration, bio-distribution, and half-life36,37,38,39. To this end, we incubated Ub(6FmnbY) with a commercially available D-galactosamine (50 mM), which contains an amine group. UV light activation resulted in ready conjugation of this sugar onto protein in 80% efficiency, as confirmed by intact MS (Fig. 4k). In contrast, similar treatment of Ub(6FmnbY) with 500 mM D-glucose, which has no amine group, resulted in no conjugation (Fig. 4l), confirming the amine-QM conjugation mechanism. Through FmnbY the sugar was conjugated at the ortho position of tyrosine hydroxyl group. Whether it has similar effect as O-glycosylation of tyrosine awaits additional studies, but further glycan extension through glycosyltransferases should mitigate this difference.
Taken together, the genetically encoded FnbY and FmnbY could both be used for facile and rapid introduction of protein modifications through photo-controlled amine-QM conjugation with high efficiency. Higher concentration and stronger nucleophilicity of the amine derivatives generally lead to higher conjugation efficiency due to more effective competition with water hydrolysis.
Protein conjugation with thiol reagents
Due to its lower pKa and higher nucleophilicity, thiol reacts with QM 102 to 103 times faster than primary amine25. However, thiol derivatives are prone to oxidation, have short shelf life, and are usually expensive40. Treating proteins with thiols may oxidize exposed cysteines to form scrambled disulfide bonds during long incubation. Since our QM-based conjugation is fast and efficient (usually completed within a few minutes or 30 s, see the following section), such side reactions of thiol reagents can be minimized. Also due to the stronger reactivity of thiol than amine, a lower concentration of labeling molecule can be employed. Therefore, when thiol derivative is preferred, we thought that thiol-QM addition could also be used for protein conjugation. We incubated Ub(6FmnbY) with 1 mM or 10 mM 1-thio-β-D-glucose. Mass spectrometric analysis of the reaction products indicate that 1-thio-β-D-glucose was conjugated to Ub almost quantitatively, nearly 100% conversion efficiency under both conditions (Fig. 4m and S6). As mentioned above (Fig. 4l), no conjugation was detected when 500 mM D-glucose was similarly used. We also incubated an thiol derivative of cytotoxic Mertansine (DM1, 0.75 mM) with Ub(6FmnbY), and found it was conjugated to Ub in quantitative conversion in 30 s (Fig. 4n), which can be useful for preparing antibody-drug conjugates41. To test if concentrations of the protein and the labeling reagent could be further lowered, we incubated 10 μM Ub(6FmnbY) with 0.1 mM 2-Mercaptoethanol (10 × equivalents), at elevated pH 9. Mass spectrometric analysis of the reaction product also showed nearly 100% labeling efficiency (Fig. S7).
Fast reaction controllable by light intensity
QMs are highly reactive. The half-life of p-QM in water is only a few seconds42,32,43, and o-QM is reported more reactive than p-QM in aqueous solution32. The nucleophile thiourea reacts with p-QM at a second order rate constant of 3.95 × 107 M−1s−1, thiolate reacts with o-QM at 2.8 × 108 M−1s−1, and amine reacts with o-QM on the order of 105 M−1s−1 25. Therefore, the rate limiting step for amine-QM protein conjugation should be photo-releasing of QM from FnbY or FmnbY.
We first used a UV lamp commonly found in chemistry or molecular biology labs, the 3UV-38 UV lamp made by UVP, which has a low irradiance output of 1.5 mW/cm2 at 365 nm. Biotin labeling on MBP-Z (24FnbY) was assayed using this lamp to activate the reaction for different time duration (Fig. 5a). A clear band that correlated with biotin conjugation was detected within 2 min, and the labeling reaction reached saturation after 10 min. We next increased the light intensity by using a UV lamp (Omnicure S1000) with an irradiance output of 5.3 W/cm2 at 365 nm. Activated by this lamp, the reaction of Ub(FmnbY) with 5 mM biotin-hydrazide was completed in 10 s, while WT Ub incubated with 5 mM biotin-hydrazide did not show any labeling even when exposed to the same light wavelength and intensity for 2 min, as demonstrated by Western blot analysis using a biotin-specific antibody (Fig. 5b). We further characterized the Ub labeling at different time duration using mass spectrometry. Intact MS revealed that 36% of Ub(FmnbY) was labeled with biotin in 1 s, and the labeling was completed in 10 s, while WT Ub was not labeled at all and remained intact after UV illumination for 1 min (Fig. 5c, Fig. S8), consistent with Western blot analysis. These data indicate that light-mediated amine-QM protein conjugation is specific and extremely fast.
Fig. 5. Effect of light intensity and temperature on amine-QM conjugation reaction.
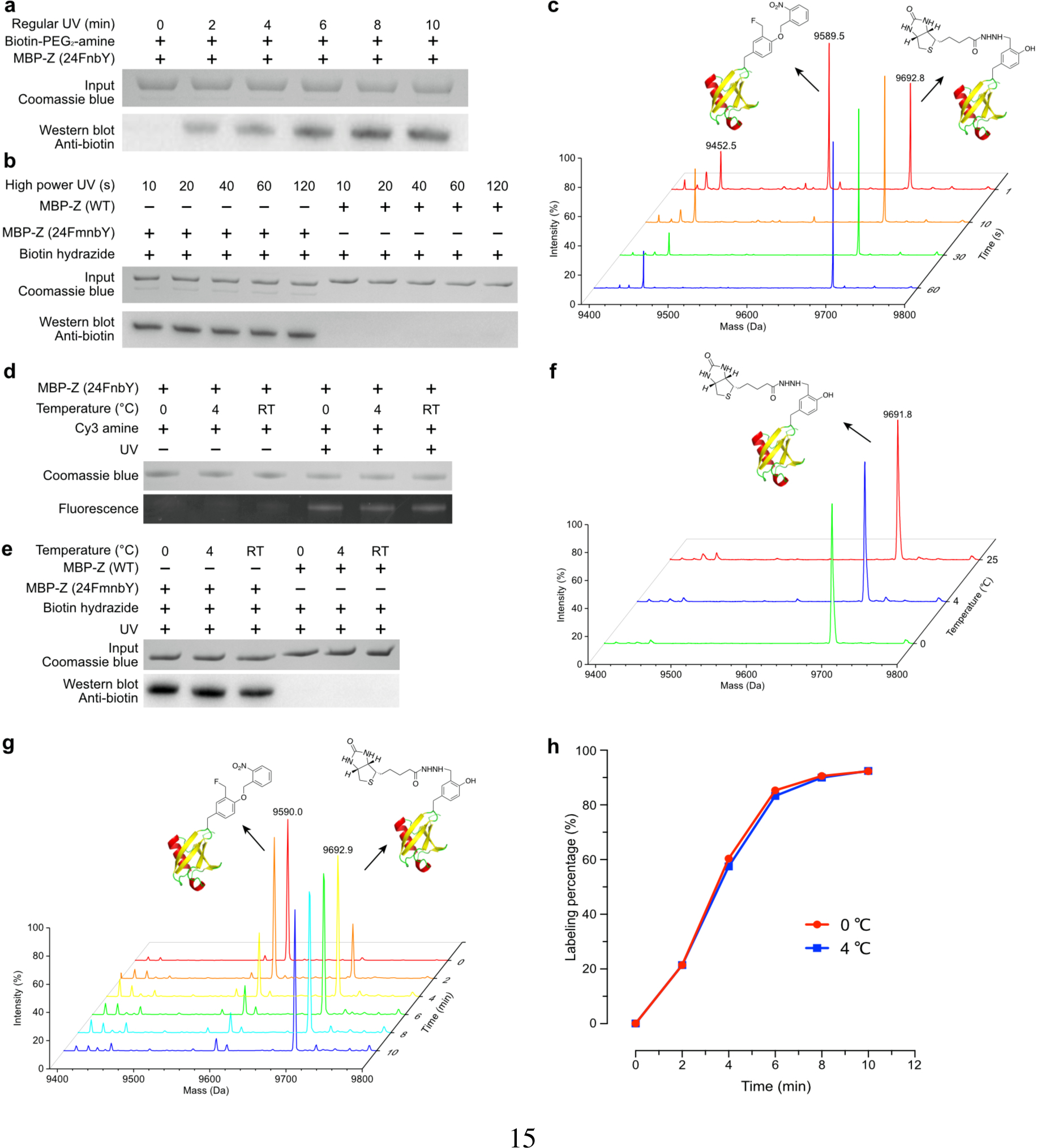
a, Western blot analysis of MBP-Z(24FnbY) conjugation with 10 mM biotin-PEG2-amine, activated by low power UV lamp for indicated time duration. b, Western blot analysis of MBP-Z (24FmnbY) conjugation with 1 mM hydrazide-biotin, activated by high power UV lamp for indicated time duration. c, Mass spectra of Ub(6FmnbY) conjugated with 1 mM biotin-hydrazide with the reaction activated by the high-power UV lamp for 1 s, 10 s, 30 s, and 60 s, respectively. Expected 9693.0 Da; measured 9692.8 Da. d, Fluorescent SDS-PAGE analysis of MBP-Z(24FnbY) conjugation with Cy3-amine (3 mM) under different temperatures. Reaction was activated by low power UV lamp for 10 min. e, Western blot analysis of MBP-Z(24FmnbY) conjugation with biotin-hydrazide (5 mM) under different temperatures. Reaction was activated by high power UV lamp for 30 s. f, Mass spectra of Ub(6FmnbY) conjugation with biotin-hydrazide (5 mM) at 0 °C, 4 °C, and room temperature, respectively. Reaction was activated by high power UV lamp for 30 s. g, Mass spectra of Ub(6FmnbY) conjugation with biotin-hydrazide (5 mM) at 0 °C with the reaction activated by low power UV lamp for indicated time duration. h, Labeling efficiency of samples in g and comparison to those of aliquot samples labeled at the same conditions except at 4 °C.
We evaluated the effect of UV exposure under the labeling conditions on E. coli survival and growth rate. Labeling with the low power UV light for 6 min did not result in significant change of E. coli survival rate, while labeling with the high power UV light for 10 s led to 9% decrease in colony forming unit. UV exposure of both labeling conditions did not significantly affect E. coli growth in liquid media (Fig. S9).
Compatibility with low temperature
We tested the temperature effect on the amine-QM labeling reaction under low and high intensity UV light. MBP-Z(24FnbY) was labeled with Cy3-amine and generated the QM using the low power UV lamp at 0 °C, 4 °C, and room temperature (RT). Fluorescence imaging showed that the labeling reaction was equally robust at these three temperatures (Fig. 5d). We then labeled MBP-Z(24FmnbY) with biotin-hydrazide and generated the QM using the high-power UV lamp at these temperatures. Again, Western blot detection of biotin showed that the labeling reached similar level for three temperatures (Fig. 5e). MS analysis of the biotin-hydrazide labeled Ub(6FmnbY) at three temperatures further revealed that the labeling reached almost completion for each temperature when activated by the high power UV lamp for 10 or 30 s (Fig. 5f, Fig. S10). We further characterized the labeling kinetics of Ub(6FmnbY) with biotin-hydrazide at 0 and 4 °C activated by the low power UV lamp for different time duration (Fig. 5g, Fig. S11). Mass spectra showed that labeling reached 85% in 6 min, with no significant difference between 0 and 4 °C (Fig. 5h). The excellent compatibility of the amine-QM labeling reaction at low temperature will be beneficial for preserving unstable proteins during protein conjugation.
Linkage stability
A stable protein conjugation linkage is essential for downstream biological applications. To evaluate the stability of amine-QM linkage, we incubated the aniline or allylamine conjugated Ub at RT for 24 h and 48 h in aqueous solution at pH 7.5, followed by MS characterization. The mass spectra of the conjugated Ub did not change along the time of incubation, indicating that the linkage was stable in aqueous solution (Fig. S12a–12f). We also incubated the allylamine conjugated Ub in pH 5, pH 9, or pH 7.5 but with strong reducing agent (50 mM DTT) for 24 h. MS analyses of these samples again showed no change (Fig. S12g–12i). These data demonstrate that the N-C bond resulting from amine-QM conjugation is stable under aqueous buffer condition, resisting to acidic or basic change as well as strong reducing condition common in protein processing.
pH, salt, and solvent effect
The nucleophilicity of amine is pH dependent, and more deprotonated amine will be available for amine-QM conjugation at higher pH. We thus labeled MBP-Z(24FmnbY) with biotin-amine at pH 5.0, 7.5, and 9.0. Western blot analysis using anti-biotin antibody showed that, in comparison to pH 7.5, pH 5.0 prevented biotin labeling, while pH 9.0 greatly accelerated the labeling (Fig. S13). We also evaluated the labeling in the presence of 2.5 M NaCl, 10% methanol, 10% DMSO, and 10% DMF. Western blot results showed that all these did not significantly affect the labeling, indicating excellent compatibility of amine-QM protein conjugation with conditions of high salt and organic solvents commonly present in labeling reactions and protein research.
Narrow inter-spin distance distribution enabled by the shortest linkage between spin label and protein
Site-directed spin labeling (SDSL) of proteins in combination with EPR/DEER measurement affords a powerful tool to study the structure, dynamics, and interaction of proteins. SDSL can be achieved through labeling of natural amino acids, bioorthogonal labeling of incorporated Uaas, or direct incorporation of spin labels as Uaas into proteins44,21,45,46. A major challenge of existing labeling methods is the high flexibility of the traditional nitroxide side chain, which leads to diffuse electron spin densities and thus DEER-based distance distributions with relatively broad peaks47. A unique feature of FnbY is that the reaction point is at the β-carbon of tyrosine, which enables the conjugation of a spin label to be as close as possible to the protein backbone (Fig. 6a). Such a short linkage will greatly reduce the flexibility of the spin label and thus simplify the resulting inter-spin distance distributions.
Fig. 6. Site-specific spin labeling via FnbY.
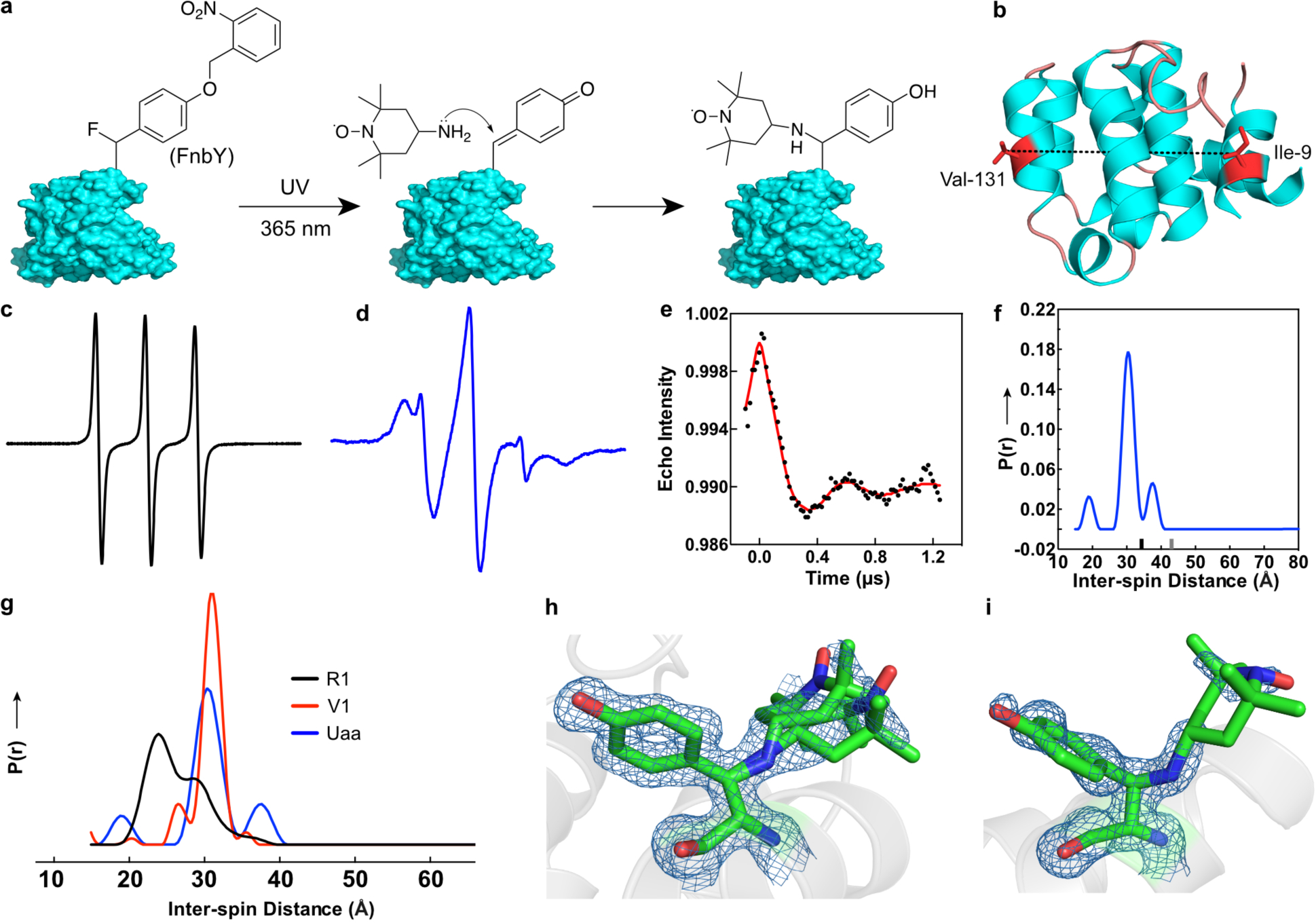
a, Scheme showing photo-controlled site specific labeling of 4-amine-TEMPO to proteins via FnbY. Note the labeling site is on the β-carbon. b, Ribbon diagram of T4 lysozyme (PDB 3LZM) showing sites Ile9 and Val131 for FnbY incorporation and spin labeling. Residues 12–67 of the structure are not shown for clarity. c, CW-EPR spectrum of 4-amine-TEMPO. d, CW-EPR spectrum of T4L with sites 9 and 131 both labeled with 4-amine-TEMPO through reacting with FnbY. e, Background corrected dipolar evolution function of the double spin labeled T4 lysozyme (black dotted trace) with a fit to the data shown in red. f, Derived interspin distance distribution of the double spin-labeled T4L (blue). The main peak in the distribution was fit with a gaussian function with a full width at half maximum (FWHM) of 3.9 Å. Ticks on the x-axis of the plot show the upper distance limits for reliability of peak shapes (black tick) and peak position (gray tick) as obtained by the program LongDistances (see methods). g, For comparison, distance distributions normalized to the area under the curves are shown for T4L containing spin label side chains R1 (black) or V1 (red) at positions 9 and 131 (data replotted from Ref. 47). h,i, 2Fo – Fc electron density map contoured at 1.0σ of spin-labeled FnbY-incorporated T4L at residue 9 and 131, respectively, as determined by x-ray crystallography. Two rotamers are modeled for position 9.
To test this idea, we first conjugated the spin label 4-amino-TEMPO (30 mM) to Ub(6FnbY). The conjugation was verified with MS, and a labeling efficiency of 81% was determined (Fig. S14). We also conjugated this spin label to Ub(6FmnbY) and confirmed the conjugation with MS, yielding 87% labeling with 1.5 mM and 100% labeling with 50 mM of 4-amino-TEMPO (Fig. S15–S16). We next moved onto FnbY-mediated spin labeling to take advantage of its shorter linkage and to achieve less rotamer conformations of the spin label. T4 lysozyme (T4L), a model protein extensively characterized by EPR, was chosen for evaluation. For DEER measurements on site-specifically labeled T4L with two TEMPO spin labels, we genetically incorporated FnbY into sites 9 and 131 of T4L simultaneously, followed by conjugation with 4-amino-TEMPO (Fig. 6b). The continuous-wave (CW) EPR spectrum of the double-labeled T4L showed restricted nitroxide motion, suggesting successful labeling of TEMPO onto the protein (Fig. 6c and 6d). The DEER experiment was performed to measure the resulting distance distribution between the two spin labels (Fig. 6e). This distance distribution revealed a sharp peak at a distance of 30.4 angstroms between the two nitroxides (Fig. 6f). The full width at half maximum (FWHM) was 3.9 angstroms. Additional peaks can arise from different rotamers or most likely represent artifacts from data processing. To the best of our knowledge, this result provides the narrowest distance distribution among Uaa-based SDSL44,45,48, highlighting the advantage of using FnbY to afford a short linkage. For comparison with distance distributions obtained with other common spin labels, distributions for T4L with either R1 or V1 side chains at positions 9 and 131 are shown in Fig. 6g, indicating that the FnbY–TEMPO approach is close to the narrow distribution obtained with the V1 label. To further corroborate the labeling and EPR/DEER measurements, we crystallized the double-labeled T4L protein and solved its structure at the resolution of 1.5 Å (PDB entry: 6V51). The crystal structure showed a tyrosine residue with the spin label conjugated at the β-carbon position of residue 9 and 131, respectively (Fig. 6h, 6i), confirming FnbY incorporation and 4-amino-TEMPO labeling via the amine-QM mechanism. The electron density for 4-amino-TEMPO is not as well-defined as the Tyr side chain, most likely due to flexibility of the TEMPO moiety and incomplete labeling with TEMPO, which may explain the low modulation depth of the DEER signal. Despite rotation and various conformers of the 6-membered ring, the nitroxide remains in a relatively small volume element, which corresponds with a narrow DEER distance distribution. The distance between the two spin labels measured by DEER fits with the distance observed in the crystal structure, i.e. the distance between the average location of the free electrons sitting on two N–O bonds in both TEMPO nitroxides. These results demonstrate that FnbY-mediated SDSL with 4-amino-TEMPO provides a relatively rigid linkage to the protein backbone making it a valuable approach for further DEER applications.
DISCUSSION
We developed a general and versatile protein conjugation method, which reversed the common paradigm to directly use weak electrophilic amine reagents for protein modification and functionalization. Although conventional photo-crosslinkers (such as aryl azides and benzophenone) are photocaged highly reactive species, the released radicals or nitrenes are chemically nonspecific and thus cannot be used for selective protein conjugation. Our strategy is to photocage the highly reactive QM species that is also chemically selective; this double requirement uniquely enabled the photo-controlled selective protein modification with a class of most common chemical reagent amine, which cannot be directly applied to protein modification hitherto. Our strategy should inspire future development of photo- or chemical-caged species in proteins for selective modification with a broader range of protein-inert and biocompatible functionalities.
The FnbY/FmnbY-based protein modification features a rare combination of desired properties addressing unmet challenges: (1) Genetically encoding of FnbY and FmnbY ensures site-specificity of the labeling and compatibility with various proteins. No harsh chemical components or reaction conditions are involved, ensuring biocompatibility. (2) The labeling reaction has fast kinetics and is usually completed within 10 s, which will be valuable for labeling low abundant proteins and pulse-tracing dynamic protein changes with high temporal resolution. (3) The reaction is controlled by UV light (365 nm), offering spatiotemporal resolution for future studies. Conventional photo-crosslinking Uaas such as p-azido-phenylalanine and p-benzoyl-phenylalanine can be used for crosslinking proteins only but not for protein labeling. FnbY and FmnbY are unique in that they could be used for both protein crosslinking and protein labeling. For protein crosslinking, FnbY/FmnbY should be incorporated at sites that have direct contact with the target nucleophilic residue30; at sites where there is no such direct contact, FnbY/FmnbY is suitable for protein labeling. (4) Amine derivatized probes and functional molecules are widely available from commercial sources and readily synthesizable, making this method easily accessible to non-specialists. The ready synthesis of amine probes is especially important for the conjugation of radioactive probes with short half-lives such as 18F (< 2 h) onto proteins for in vivo biomedical imaging. (5) The labeling is equally efficient at low temperatures (0 and 4 °C), which is beneficial for preserving proteins which are unstable at room temperature after purification. (6) The labeling moiety (amine or thiol) and resultant linkage are both small in size, minimizing the perturbation to target protein and the interference with the biological process under study. The stability of the amine reagents and label linkage also afford robust conjugation, reducing complications of label dissociation or degradation for downstream applications. (7) Conjugation via FnbY occurs on the β-carbon, which uniquely places the labeled molecule as close as possible to the protein backbone. Such a short linkage is especially valuable for advanced studies demanding distance and orientation, as demonstrated here in the SDSL and DEER experiments. It would also be highly valuable for NMR, dynamic nuclear polarization (DNP), FRET, single molecule analysis, and so on. Lastly, due to the presence of nucleophiles inside cells, this QM-based labeling method is more suitable for in vitro and cell surface labeling instead of intracellular labeling.
In summary, we expect that this labeling strategy with advantageous characteristics of site-specificity, fast kinetics, photo-controllability, compatibility with low temperature, minimal perturbation, shortest linkage, and widely available reagents will be an accessible and valuable tool for protein research as well as biological and biomedical studies.
Supplementary Material
Acknowledgments
L.W. acknowledges the support of NIH (R01GM118384). S.R. acknowledges the support of NIH (R01GM121607). O.P.E. acknowledges the support of NSERC (RGPIN-2017-06862) and the Canada Excellence Research Chairs (CERC) program. O.P.E. is a fellow of the Canadian Institute for Advanced Research (CIFAR) and holds the Anne and Max Tanenbaum Chair in Neuroscience at the University of Toronto. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. We specifically thank the staff at the GM/CA beamlines 23-ID. We thank Drs Shaoqing Zhang, Lizhi Tao and Joerg Reichenwallner for helpful discussions on EPR experiments. J.L. and L.W. thank Dr. Sinan Wang, Lingchao Cai, Shanshan Li and Robert Flavell for helpful discussions.
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- (1).Walsh CT; Garneau-Tsodikova S; Gatto GJJ Protein Posttranslational Modifications: the Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. Engl 2005, 44 (45), 7342–7372. [DOI] [PubMed] [Google Scholar]
- (2).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5 (1), 4740–14. [DOI] [PubMed] [Google Scholar]
- (3).Tamura T; Hamachi I Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems. J. Am. Chem. Soc 2019, 141 (7), 2782–2799. [DOI] [PubMed] [Google Scholar]
- (4).Steen Redeker E; Ta DT; Cortens D; Billen B; Guedens W; Adriaensens P Protein Engineering for Directed Immobilization. Bioconjug. Chem 2013, 24 (11), 1761–1777. [DOI] [PubMed] [Google Scholar]
- (5).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and Challenges for the Next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov 2017, 16 (5), 315–337. [DOI] [PubMed] [Google Scholar]
- (6).Vinogradova EV; Zhang C; Spokoyny AM; Pentelute BL; Buchwald SL Organometallic Palladium Reagents for Cysteine Bioconjugation. Nature 2015, 526 (7575), 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Taylor MT; Nelson JE; Suero MG; Gaunt MJ A Protein Functionalization Platform Based on Selective Reactions at Methionine Residues. Nature 2018, 562 (7728), 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang C; Welborn M; Zhu T; Yang NJ; Santos MS; Van Voorhis T; Pentelute BL Π-Clamp-Mediated Cysteine Conjugation. Nat. Chem 2016, 8 (2), 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-Based Reagents for Chemoselective Methionine Bioconjugation. Science 2017, 355 (6325), 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martín-Gago P; Fansa EK; Winzker M; Murarka S; Janning P; Schultz-Fademrecht C; Baumann M; Wittinghofer A; Waldmann H Covalent Protein Labeling at Glutamic Acids. Cell Chem. Biol 2017, 24 (5), 589–597.e5. [DOI] [PubMed] [Google Scholar]
- (11).Wang L; Zhang Z; Brock A; Schultz PG Addition of the Keto Functional Group to the Genetic Code of Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (1), 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Z; Smith Brian AC; Wang L; Brock A; Cho C; Schultz PG A New Strategy for the Site-Specific Modification of Proteins in Vivo. Biochemistry 2003, 42 (22), 6735–6746. [DOI] [PubMed] [Google Scholar]
- (13).Kim CH; Axup JY; Schultz PG Protein Conjugation with Genetically Encoded Unnatural Amino Acids. Curr. Opin. Chem. Biol 2013, 17 (3), 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Freedy AM; Matos MJ; Boutureira O; Corzana F; Guerreiro A; Akkapeddi P; Somovilla VJ; Rodrigues T; Nicholls K; Xie B; Jiménez-Osés G; Brindle KM; Neves AA; Bernardes GJL Chemoselective Installation of Amine Bonds on Proteins Through Aza-Michael Ligation. J. Am. Chem. Soc 2017, 139 (50), 18365–18375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Song W; Wang Y; Qu J; Lin Q Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells. J. Am. Chem. Soc 2008, 130 (30), 9654–9655. [DOI] [PubMed] [Google Scholar]
- (16).Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc 2008, 130 (41), 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kaya E; Vrabel M; Deiml C; Prill S; Fluxa VS; Carell T A Genetically Encoded Norbornene Amino Acid for the Mild and Selective Modification of Proteins in a Copper-Free Click Reaction. Angew. Chem. Int. Ed 2012, 51 (18), 4466–4469. [DOI] [PubMed] [Google Scholar]
- (18).Plass T; Milles S; Koehler C; Szymański J; Mueller R; Wießler M; Schultz C; Lemke EA Amino Acids for Diels-Alder Reactions in Living Cells. Angew. Chem. Int. Ed. Engl 2012, 51(17), 4166–4170. [DOI] [PubMed] [Google Scholar]
- (19).Taraska JW; Puljung MC; Zagotta WN Short-Distance Probes for Protein Backbone Structure Based on Energy Transfer Between Bimane and Transition Metal Ions. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (38), 16227–16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sindbert S; Kalinin S; Nguyen H; Kienzler A; Clima L; Bannwarth W; Appel B; Mueller S; Seidel CAM Accurate Distance Determination of Nucleic Acids via Forster Resonance Energy Transfer: Implications of Dye Linker Length and Rigidity. J. Am. Chem. Soc 2011, 133 (8), 2463–2480. [DOI] [PubMed] [Google Scholar]
- (21).Hubbell WL; López CJ; Altenbach C; Yang Z Technological Advances in Site-Directed Spin Labeling of Proteins. Curr. Opin. Struct. Biol 2013, 23 (5), 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lilly Thankamony AS; Wittmann JJ; Kaushik M; Corzilius B Dynamic Nuclear Polarization for Sensitivity Enhancement in Modern Solid-State NMR. Prog. Nucl. Mag. Res. Spectr 2017, 102–103, 120–195. [DOI] [PubMed] [Google Scholar]
- (23).Forns N; de Lorenzo S; Manosas M; Hayashi K; Huguet JM; Ritort F Improving Signal/Noise Resolution in Single-Molecule Experiments Using Molecular Constructs with Short Handles. Biophys. J 2011, 100 (7), 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hall MP Biotransformation and in Vivo Stability of Protein Biotherapeutics: Impact on Candidate Selection and Pharmacokinetic Profiling. Drug Metab. Dispos 2014, 42 (11), 1873–1880. [DOI] [PubMed] [Google Scholar]
- (25).Modica E; Zanaletti R; Freccero M; Mella M Alkylation of Amino Acids and Glutathione in Water by O-Quinone Methide. Reactivity and Selectivity. J. Org. Chem 2001, 66 (1), 41–52. [DOI] [PubMed] [Google Scholar]
- (26).Caruana L; Fochi M; Bernardi L The Emergence of Quinone Methides in Asymmetric Organocatalysis. Molecules 2015, 20 (7), 11733–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Doria F; Lena A; Bargiggia R; Freccero M Conjugation, Substituent, and Solvent Effects on the Photogeneration of Quinone Methides. J. Org. Chem 2016, 81 (9), 3665–3673. [DOI] [PubMed] [Google Scholar]
- (28).Wang L; Brock A; Herberich B; Schultz PG Expanding the Genetic Code of Escherichia Coli. Science 2001, 292 (5516), 498–500. [DOI] [PubMed] [Google Scholar]
- (29).Wang L Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc. Chem. Res 2017, 50 (11), 2767–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu J; Li S; Aslam NA; Zheng F; Yang B; Cheng R; Wang N; Rozovsky S; Wang PG; Wang Q; Wang L Genetically Encoding Photocaged Quinone Methide to Multitarget Protein Residues Covalently in Vivo. J. Am. Chem. Soc 2019, 141 (24), 9458–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Borrel G; Parisot N; Harris HMB; Peyretaillade E; Gaci N; Tottey W; Bardot O; Raymann K; Gribaldo S; Peyret P; O’Toole PW; Brugère J-F Comparative Genomics Highlights the Unique Biology of Methanomassiliicoccales, a Thermoplasmatales-Related Seventh Order of Methanogenic Archaea That Encodes Pyrrolysine. BMC genomics 2014, 15, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chiang Y; Kresge AJ; Zhu Y Flash Photolytic Generation and Study of P-Quinone Methide in Aqueous Solution. an Estimate of Rate and Equilibrium Constants for Heterolysis of the Carbon-Bromine Bond in P-Hydroxybenzyl Bromide. J. Am. Chem. Soc 2002, 124 (22), 6349–6356. [DOI] [PubMed] [Google Scholar]
- (33).Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. Engl 2014, 53 (36), 9430–9448. [DOI] [PubMed] [Google Scholar]
- (34).Wang N; Yang B; Fu C; Zhu H; Zheng F; Kobayashi T; Liu J; Li S; Ma C; Wang PG; Wang Q; Wang L Genetically Encoding Fluorosulfate-L-Tyrosine to React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J. Am. Chem. Soc 2018, 140 (15), 4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yang B; Wu H; Schnier PD; Liu Y; Liu J; Wang N; DeGrado WF; Wang L Proximity-Enhanced SuFEx Chemical Cross-Linker for Specific and Multitargeting Cross-Linking Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (44), 11162–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu H; Wang L; Brock A; Wong C-H; Schultz PG A Method for the Generation of Glycoprotein Mimetics. J. Am. Chem. Soc 2003, 125 (7), 1702–1703. [DOI] [PubMed] [Google Scholar]
- (37).Moradi SV; Hussein WM; Varamini P; Simerska P; Toth I Glycosylation, an Effective Synthetic Strategy to Improve the Bioavailability of Therapeutic Peptides. Chem. Sci 2016, 7 (4), 2492–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cai L; Gu Z; Zhong J; Wen D; Chen G; He L; Wu J; Gu Z Advances in Glycosylation-Mediated Cancer-Targeted Drug Delivery. Drug Discovery Today 2018, 23 (5), 1126–1138. [DOI] [PubMed] [Google Scholar]
- (39).Alavi SE; Cabot PJ; Moyle PM Glucagon-Like Peptide-1 Receptor Agonists and Strategies to Improve Their Efficiency. Mol. Pharm 2019, 16 (6), 2278–2295. [DOI] [PubMed] [Google Scholar]
- (40).Alcock LJ; Perkins MV; Chalker JM Chemical Methods for Mapping Cysteine Oxidation. Chem. Soc. Rev 2018, 47 (1), 231–268. [DOI] [PubMed] [Google Scholar]
- (41).Girish S; Gupta M; Wang B; Lu D; Krop IE; Vogel CL; Burris Iii HA; LoRusso PM; Yi J-H; Saad O; Tong B; Chu Y-W; Holden S; Joshi A Clinical Pharmacology of Trastuzumab Emtansine (T-DM1): an Antibody-Drug Conjugate in Development for the Treatment of HER2-Positive Cancer. Cancer Chemother. Pharmacol 2012, 69 (5), 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Toteva MM; Richard JP The Generation and Reactions of Quinone Methides. Adv. Phys. Org. Chem 2011, 45, 39–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhou Q; Turnbull KD Quinone Methide Phosphodiester Alkylations Under Aqueous Conditions. J. Org. Chem 2001, 66 (21), 7072–7077. [DOI] [PubMed] [Google Scholar]
- (44).Fleissner MR; Brustad EM; Kalai T; Altenbach C; Cascio D; Peters FB; Hideg K; Peuker S; Schultz PG; Hubbell WL Site-Directed Spin Labeling of a Genetically Encoded Unnatural Amino Acid. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (51), 21637–21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Schmidt MJ; Borbas J; Drescher M; Summerer D A Genetically Encoded Spin Label for Electron Paramagnetic Resonance Distance Measurements. J. Am. Chem. Soc 2014, 136 (4), 1238–1241. [DOI] [PubMed] [Google Scholar]
- (46).Roser P; Schmidt MJ; Drescher M; Summerer D Site-Directed Spin Labeling of Proteins for Distance Measurements in Vitro and in Cells. Org. Biomol. Chem 2016, 14 (24), 5468–5476. [DOI] [PubMed] [Google Scholar]
- (47).Balo AR; Feyrer H; Ernst OP Toward Precise Interpretation of DEER-Based Distance Distributions: Insights From Structural Characterization of V1 Spin-Labeled Side Chains. Biochemistry 2016, 55 (37), 5256–5263. [DOI] [PubMed] [Google Scholar]
- (48).Kugele A; Braun TS; Widder P; Williams L; Schmidt MJ; Summerer D; Drescher M Site-Directed Spin Labelling of Proteins by Suzuki-Miyaura Coupling via a Genetically Encoded Aryliodide Amino Acid. Chem. Commun 2019, 55 (13), 1923–1926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.