

Abstract
A number of hormones and growth factors stimulate target cells via the second messenger pathways, which in turn regulate cellular phenotypes. Cyclic adenosine monophosphate (cAMP) is a ubiquitous second messenger that facilitates numerous signal transduction pathways; its production in cells is tightly balanced by ligand-stimulated receptors that activate adenylate cyclases (ACs), i.e., “source” and by phosphodiesterases (PDEs) that hydrolyze it, i.e., “sinks”. Because it regulates various cellular functions, including cell growth and differentiation, gene transcription and protein expression, the cAMP signaling pathway has been exploited for the treatment of numerous human diseases. Reduction in cAMP is achieved by blocking “sources”; however, elevation in cAMP is achieved by either stimulating “source” or blocking “sinks”. Here we discuss an alternative paradigm for the regulation of cellular cAMP via GIV/Girdin, the prototypical member of a family of modulators of trimeric GTPases, Guanine nucleotide Exchange Modulators (GEMs). Cells up- or down-regulate cellular levels of GIV-GEM, which modulates cellular cAMP via spatiotemporal mechanisms distinct from the two most often targeted classes of cAMP modulators, “sources” and “sinks”. A network-based compartmental model for the paradigm of GEM-facilitated cAMP signaling has recently revealed that GEMs such as GIV serve much like a “tunable valve” that cells may employ to finetune cellular levels of cAMP. Because dysregulated signaling via GIV and other GEMs has been implicated in multiple disease states, GEMs constitute a hitherto untapped class of targets that could be exploited for modulating aberrant cAMP signaling in disease states.
Graphical Abstract
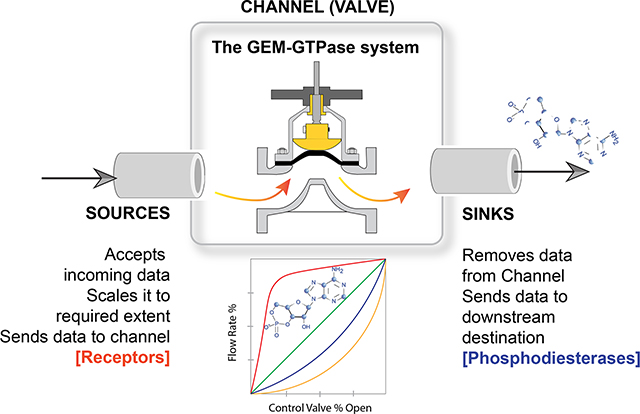
Caption: Schematic displays information transmission in cells, where the data that is carried forward is cyclic AMP flux. Ligand-activated receptors serve as ‘sources’ of such data, whereas phosphodiesterases serve as ‘sinks’ for the same. In this work, the authors describe the presence of a tunable valve (channel) that is poised between the ‘sources’ and the ‘sinks’. Comprised of Guanine nucleotide Exchange Modulators (GEM) and the trimeric GTPase system, this valve adds emergent features that control cAMP flux in cells.
Cellular cyclic AMP: A balancing act between “Sources” and “Sinks”--
For cells to properly react to their environment, cells must constantly sense their external environment and correctly relay them to the intracellular environment. While sensing is mediated by a myriad of cell-surface receptors, relaying such signals depends on protein scaffolding, enzymatic reactions and the production of second messengers, such as cyclic nucleotides (Beavo & Brunton, 2002; Newton, Bootman, & Scott, 2016). Of the various cyclic nucleotides, the first to be identified was cyclic adenosine 3,5-monophosphate (cAMP), a universal second messenger used by diverse forms of life, such as unicellular bacteria, fungi, protozoans, and mammals. cAMP relays signals triggered by hormones, ion channels, and neurotransmitters [reviewed in (Sassone-Corsi, 2012)]. cAMP also binds and regulates other cAMP-binding proteins such as cyclic nucleotide gated channels, PKA, and Epac1. Intracellular levels of cAMP are regulated by the antagonistic action of two classes of enzymes: adenylyl cyclases (ACs) and cyclic nucleotide phosphodiesterases (PDEs). ACs, with the exception of soluble ACs, are membrane-bound enzymes that utilize ATP to generate cAMP transmitting signals from cell-surface receptors to second messengers. In contrast, PDEs are soluble enzymes and catalyze the degradation of the phosphodiester bond resulting in the conversion of cAMP to AMP. Most ACs are activated downstream from G-protein-coupled receptors (GPCRs) by interactions with the α subunit of the Gs protein (Gαs). Gαs is released from heterotrimeric αβγ G-protein complexes following binding of agonist ligands to GPCRs. Gαs is then free to bind to and activate AC (Pierce, Premont, & Lefkowitz, 2002). Alternatively, cAMP activity can be suppressed by ligands that stimulate GPCRs coupled to Gi (which inhibits AC activity) and/or cAMP can be degraded by PDEs. PDE4, for instance, is activated by protein kinase A (PKA), a downstream effector of cAMP, resulting in a negative feedback loop between cAMP and PDE4s (MacKenzie et al., 2002; Murthy, Zhou, & Makhlouf, 2002; Sette & Conti, 1996; Tremblay, Lachance, & Hamet, 1985). It is noteworthy that some, but not all isoforms of PDEs are targets of such cAMP-PKA feedback loops [reviewed in (Bender & Beavo, 2006)].Thus, the level of cAMP in cells is a fine balance between its synthesis by ACs (i.e., source), its degradation by PDEs (i.e., sink) that in some instances may be subject to feedback regulation through the cAMP-PKA-PDE loop [reviewed in (Sassone-Corsi, 2012)]. Existing computational models built within certain contexts such as GPCR or GTPase activation kinetics (Kenakin, 2004; Bornheimer et al., 2004) and compartmentalization (Yang et al., 2016; Agarwal et al., 2016) generally agree that the loss of spatial and temporal control of these “sources” and/or “sinks” can lead to aberrant behavior and loss of cAMP microdomains. In addition, these computational models helped point out what components are crucial for proper spatial and temporal control. For instance, (Yang et al., 2016) and (Agarwal et al., 2016) found that PDEs themselves are not sufficient for the formation of microdomains and secondary mechanisms such as buffering by PKA are needed.
Aberrant circuits that give rise to too much or too little cAMP can be unhealthy; in fact, deregulated signaling events with resultant abnormal levels of cellular cAMP is a key pathophysiologic component in many human diseases (Figure 1; see Table). In the context of cancers, multiple studies across different cancers [breast (Tagliaferri et al., 1988), melanoma (Dumaz et al., 2006), pancreas (Boucher, Duchesne, Lainé, Morisset, & Rivard, 2001), etc.] agree that high levels of cAMP are generally protective, whereas low cAMP levels fuel cancer progression [reviewed in (Fajardo, Piazza, & Tinsley, 2014)]. High cAMP inhibits several harmful phenotypes of tumor cells such as proliferation, invasion, stemness, and chemoresistance, while enhancing differentiation and apoptosis (Figure 1).
Figure 1: An emerging paradigm for modulation of cellular cAMP by growth factors.
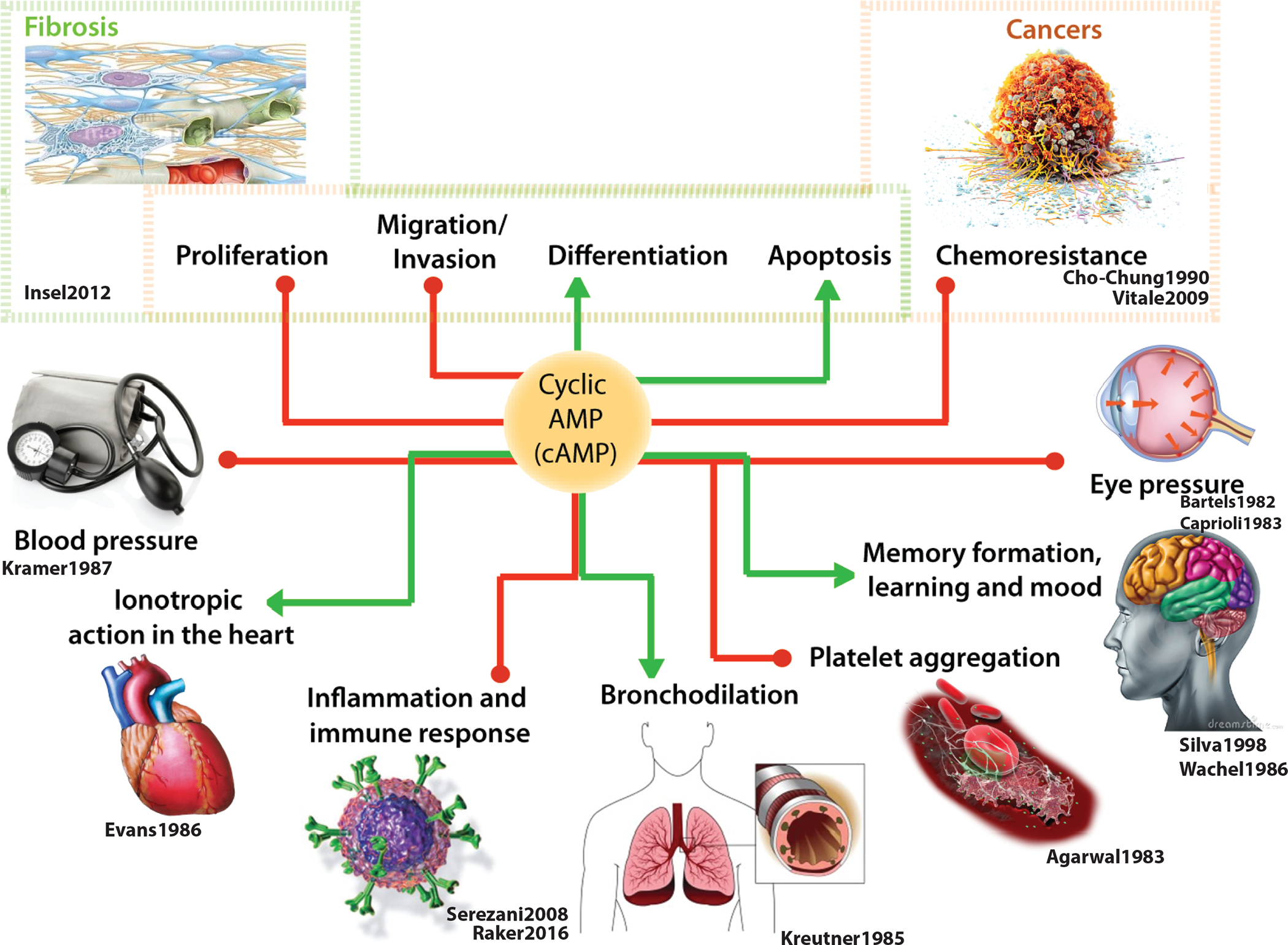
(A) Schematic summarizing the role of cyclic AMP (cAMP) in diverse biological processes. In cancers (top right), cAMP is largely protective as it inhibits proliferation, invasion, chemoresistance, and promotes apoptosis and differentiation of tumor cells. Similarly, in the context of organ fibrosis, cAMP is a potent anti-fibrotic agent because it inhibits proliferation and migration and triggers apoptosis and return to quiescence for myofibroblasts, the major cell type implicated in fibrogenic disorders. Red lines indicate suppression and green lines indicate promotion. Citations for each process can be found under the process images (K. C. Agarwal & Parks Jr, 1983; Bartels et al., 1982; Caprioli & Sears, 1983; Cho-Chung, 1990; Evans, 1986; Insel et al., 2012; Kramer et al., 1987; Kreutner et al., 1985; Raker et al., 2016; Serezani et al., 2008; Silva et al., 1998; Vitale et al., 2009; Wachtel & Löschmann, 1986)
Table:
GIV, cAMP levels and disease states
| Disease state | Tissue | Levels of GIV-GEM/GEM activity, and implicated in disease | Derangement of cAMP (predicted) | Derangement of cAMP pathway (experimentally confirmed and implicated in disease pathogenesis) |
|---|---|---|---|---|
| Cancers [EMT, invasion and metastasis, stemness] | Tumor cells and stroma | Elevated levels of mRNA and protein; GEM function ON [multiple studies, summarized in (Ghosh, 2015)] | Suppressed than normal | Suppressed (pharmacologic elevation inhibits motility, invasion) (Follin-Arbelet et al., 2011; Gooding & Schiemann, 2016; Murata et al., 2000; Ou et al., 2014; Perez et al., 2016; Zimmerman et al., 2015) |
| Cancers (emergence of resistance) | Tumor cells | Elevated levels of mRNA and protein; GEM function ON (Midde et al., 2018; Yu et al., 2017; Zhang, Li, Han, Yin, & Lin, 2014) | Suppressed than normal | Suppressed (pharmacologic elevation overcomes resistance, increase apoptosis) (McEwan et al., 2007; Wang et al., 2016) |
| Nephrotic syndrome | Podocytes | GIV is elevated and activation of its GEF function is required for podocyte recovery (Wang et al., 2015) | Nephritic glomeruli generated less cyclic AMP than normal glomeruli (11 and 26 pmol) (Nagamatsu et al. 2006). Attenuated generation of cyclic AMP in response to ligands is connected to the augmented accumulation of fibronectin in nephritic glomeruli (Nagamatsu, Nishiyama, Goto, Nagao, & Suzuki, 2003) | |
| Liver fibrosis | Hepatic stellate cells, Kupffer cells | Elevated levels of mRNA and protein; GEM function ON (Lopez-Sanchez et al., 2014) | Suppressed than normal | Augmentation of forskolin-induced increase in intracellular cyclic AMP level (inhibitory effect on HSC activation (Shimizu et al., 1999)). Quiescent HSCs have high levels of cAMP-PKA-phospho-CREB signaling, which decreases upon HSC activation; activation of PKA restores phospho-CREB levels and inhibits proliferation of activated HSCs (Houglum, Lee, & Chojkier, 1997; Kawada, Kuroki, Kobayashi, Inoue, & Kaneda, 1996). |
| Type II DM, Insulin resistance | Muscle (skeletal) | Decreased levels of GIV (Hartung et al., 2013; Ma et al., 2015) | Elevated than normal | High levels of cAMP induces insulin resistance (Chai & Fong, 2015; Erion et al., 2009; Kida, Nyomba, Bogardus, & Mott, 1991, p. 1; D. Kirsch, Kemmler, & Häring, 1983; D. M. Kirsch, Bachmann, & Häring, 1984; Tanti, Grémeaux, Rochet, Obberghen, & Marchand-Brustel, 1987); (Ramirez et al., 2015). Insulin triggers PDE activity for cAMP degradation. (Makino et al., 1985). PDE3B is activated by Akt downstream of insulin (Kitamura et al., 1999). Levels of PDE3B are reduced in DM, and restored by TZDs (Tang et al., 1999). |
| Alzheimer’s disease | Neurons | Decreased levels of GIV protein (Liu et al., 2015) | Increased (predicted) | Increased (Martínez et al., 1999) |
| Autism | Cultured peripheral blood lymphocytes | Gain of copy number for GIV (van Daalen et al., 2011). | Suppressed cAMP (predicted) | Low cyclic AMP confirmed in programmed neuronal stem cells. Compensatory high cAMP in CSF and peripheral blood. Addition of cAMP restored defective signaling within the cAMP pathway. |
| Acute myocardial infarction and other vascular endothelial injury | Cardiac and smooth muscle cells | Elevated levels of mRNA and protein (Hayano et al., 2015; Ito et al., 2013; Miyachi, Takahashi, & Komori, 2015) | Suppressed cAMP (predicted) | Suppressed in acute MI in humans: Reviewed in: (Leineweber Kirsten, Böhm Michael, & Heusch Gerd, 2006). Suppression of cAMP after vessel injury is required for neointima repair |
Therapies that target the canonical GPCR/G-protein-cAMP signaling pathway have been successfully translated to the clinics, and account for ~40% of currently marketed drugs that can treat a wide range of ailments (Filmore, 2004), from hypertension to glaucoma. However, such strategies have largely failed to impact cancer care or outcome. Thus, how tumor cells avoid high levels of cAMP, despite the fact that there are is generally hyperactivation of receptors (“sources”), is puzzling. Efforts to elevate cAMP using PDE inhibitors, although perceived as successful at the bench (reviewed in Table 3 in Pantziarka et al., 2018), showed controversial results in well-designed clinical trials (Barone, Giordano, Bonofiglio, Andò, & Catalano, 2017). This would suggest that inhibiting the “sinks” alone may not be enough. Consequently, despite a well-thought out therapeutic goal, i.e., to elevate cellular levels of cAMP, a strategy to accomplish the same in chronic diseases like cancers has not emerged. Here we highlight the importance of an emergent field / paradigm in trimeric GTPase signaling and in the regulation of cellular cAMP; we discuss its importance in the tonic and robust suppression of cAMP, especially in the context of cancers.
Enter GEMs: An emerging paradigm in GPCR-independent G protein and cAMP signaling--
Recent studies by us and others have shown that heterotrimeric G proteins can be activated by integrins and growth factor receptor tyrosine kinases (RTKs) (reviewed in, Ghosh, Rangamani, & Kufareva, 2017). Although these receptor classes are not typically coupled to heterotrimeric G proteins like the G protein-coupled receptors (GPCRs), both classes of receptors have been shown to modulate heterotrimeric G proteins and successfully transduce external stimuli into an intracellular cAMP signal (Alenghat, Tytell, Thodeti, Derrien, & Ingber, 2009; Poppleton, Sun, Fulgham, Bertics, & Patel, 1996). Where these receptors differ from GPCRs is that unlike GPCRs that rapidly perturb cAMP for a finite period of few hundred seconds, growth factor RTKs and Integrins signal over longer periods of time [about 60 minutes after an acute stimulus before a steady state is reached (Burke, Schooler, & Wiley, 2001; Kholodenko, Demin, Moehren, & Hoek, 1999)]. Who or what may allow these receptors to couple to and modulate G proteins remained a puzzle for decades. One mechanism for such coupling that has emerged just within this past decade is non-receptor (i.e., cytosolic) modulators of G proteins that may contextually and dynamically scaffold unlike receptor classes to heterotrimeric G proteins. More specifically, studies focused on GIV (also known as Girdin/HkRP1/APE) the prototypical member of the family of proteins known as Guanine nucleotide Exchange Modulator (GEMs) have exposed the critical roles of a class of cytosolic scaffolds that use their modularity and motifs to trigger G protein signaling downstream of growth factor receptors (Beas et al., 2012; Ghosh et al., 2010; Gupta et al., 2016; Lin et al., 2014, 2011; Ma, Aznar, et al., 2015; Ma, Lopez-Sanchez, et al., 2015; Midde et al., 2015; Parag-Sharma et al., 2016) and integrins (Leyme, Marivin, & Garcia-Marcos, 2016; Leyme, Marivin, Perez-Gutierrez, Nguyen, & Garcia-Marcos, 2015; Lopez-Sanchez et al., 2015; Weng et al., 2014). There have been four GEMs identified thus far, all implicated in diverse signaling paradigms: GIV was independently discovered by four groups (Anai et al., 2005; Enomoto et al., 2005; Le-Niculescu, Niesman, Fischer, DeVries, & Farquhar, 2005; Simpson et al., 2005) in 2005, Calnuc/Nucleobindin 1 and 2 (NUCB1 and NUCB2) in 2011 (Garcia-Marcos, Kietrsunthorn, Wang, Ghosh, & Farquhar, 2011) and Daple in 2015 (Aznar et al., 2015).
A series of studies from our group and others helped understand the unique features and different set of rules of GEM-dependent (and hence, GPCR-independent) G protein signaling (Ghosh et al., 2017; Midde, Aznar, Kalogriopoulos, & Ghosh, 2016). In brief, this signaling pathway has distinctive temporal and spatial features and an unusual profile of receptor engagement: diverse classes of receptors [such as Integrins, RTKs, and LRPs (reviewed in, Ghosh, Rangamani, & Kufareva, 2017)], not just GPCRs can engage with GIV to trigger such activation. Such activation is spatially and temporally unrestricted, that is, can occur both at the plasma membrane (PM) and on internal membranes discontinuous with the PM, and can continue for prolonged periods of time. GEMs act within diverse signaling cascades and couple activation of these cascades to G-protein signaling via an evolutionarily conserved motif of ~30 amino acids that directly binds and modulates Gαi and Gαs proteins. It is via this short motif that GIV-GEM serves as a GEF for Gαi and as a GDI for Gαs in a temporally-spatially segregated manner that is controlled by two kinases CDK5 and PKC-theta (Gupta et al., 2016). Despite this apparent paradox of modulating Gαi and Gαs, both forms of modulation lead to suppression of cellular cAMP (Ghosh et al., 2017). Thus, unlike the canonical G protein/cAMP signaling paradigm, which is rigid (finite, is triggered exclusively by GPCRs and transduced via either Gi or Gs at a time, primarily at the PM), the temporal and spatial features of non-canonical G protein/cAMP signaling via GIV-family of cytosolic GEMs are unusually complex and relaxed. GIV uses this relaxed circuitry to integrate, reinforce and compartmentalize signals downstream of diverse classes of receptors and G proteins in a way that enables it to orchestrate cellular phenotypes in a sustained manner. While the molecular mechanisms that govern the unique spatiotemporal aspects of non-canonical G protein activation by GIV and the relevance of this new paradigm in health and disease has been reviewed elsewhere (Midde et al., 2016), the structural basis for GEM-dependent G protein activation has just begun to emerge. Using the combined synergy of x-ray crystallography, molecular dynamics simulations, and other biophysical and biochemical approaches a recently published study (Kalogriopoulos et al., 2019), has revealed that despite differences in how they independently bind G-proteins, both GPCRs and GEMs share common mechanisms for nucleotide exchange. A part of their allosteric paths converge to disrupt key nucleotide contacts in the hydrophobic core of the nucleotide-binding domain, which ultimately leads to nucleotide exchange (Ghosh & Garcia-Marcos, 2019). Despite these atomic level insights, the impact of the unusual complex and relaxed spatiotemporal aspects of GEM-dependent G protein activation on cAMP production was difficult to deduce intuitively, and hence, required investigations from a systems level.
As a tunable valve between the “sources” and “sinks”, GEMs enable tonic modulation of cAMP, impart robustness--
With the ultimate goal of generating experimentally testable predictions, recently we used a systems biology approach to understand the design principles of the GEM-dependent biological network with a focus on cAMP signaling (Getz, Swanson, Sahoo, Ghosh, & Rangamani, 2019). We constructed the first-ever compartmental network model of growth-factor triggered cAMP signaling and identified two key features of non-canonical G protein signaling via GIV-GEM. We focused the study on the epidermal growth factor (EGF) and its receptor (EGFR) because this pathway had the most experimental evidence (Gupta et al., 2016) to use towards building mathematical models with well-validated tools/readouts/approaches to validate/test model-inspired predictions. This endeavor resulted in two major findings.
First, the model implicated compartmentalized RTK signaling at the PM (where GIV serves as a GEF that triggers activation of Gαi) and on the endosomes (where GIV serves as a GDI that inhibits activation of Gαs) as a key contributor to the delayed and prolonged cAMP dynamics that is observed over an hour. Consequently, RTK-GIV-triggered cAMP dynamics spans 5 to >60 min, which coincides with other RTK-triggered mitogenic signaling pathways, trafficking events, and transcriptional response; this is the major temporal domain of RTK activity, the so-called “window of activity” (Amit, Wides, & Yarden, 2007).
Second, the model predicted and experimentally validated that GIV-GEM may serve as a tunable “valve” for cAMP regulation in cells. When all the known compartmental and reaction kinetics were accounted for, the model indicated that GIV levels (which vary in pathologic states; see Table), in conjunction with EGFR levels, can be thought of as key determinants, and high GIV in the setting of high EGFR may facilitate tonic suppression of cAMP levels regardless of pathway stimulation. In a low-EGFR state, varying GIV concentrations resulted in cAMP changes only within a narrow range; however, in a high-EGFR state, varying GIV concentrations achieved a larger variance in cAMP. In a low-GIV state, varying EGFR concentrations resulted in cAMP changes; however, in high-GIV states, cAMP concentrations remained low regardless of increasing levels of EGFR. GIV levels, in conjunction with EGFR (“source”) levels, emerged as key determinants, and high-GIV state regardless of the levels of activation of EGFR resulted in tonic suppression of cAMP levels, reminiscent of the actions of “valves” in any conduit [Figure 2]. The “valve” appears to be “closed” at high-GIV states, despite high levels of “sources”, and hence, when “sources” and “valve” were compared head to head, the “valve” emerged as the dominant determinant of cellular levels of cAMP.
Figure 2: Schematic summarizing the unique impacts of GIV-GEM on the EGFR→ cAMP pathway, as revealed by systems biology.
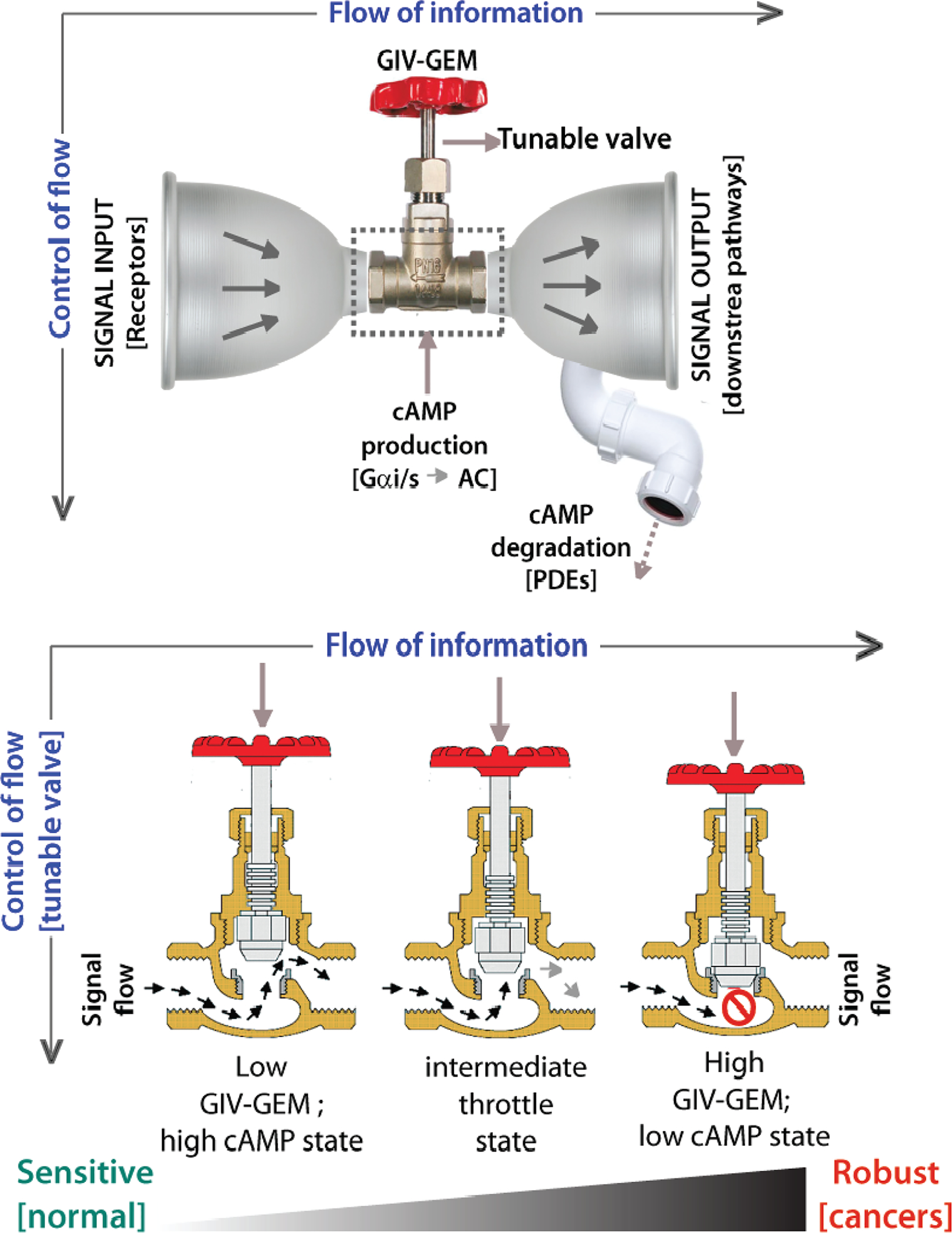
Top: Within the ‘bow-tie’ microarchitecture of layered signal flow in any circuit, incoming signals from RTKs like EGFR [signal input; left] are integrated by core proteins like GIV [center] that activate second messengers like cAMP, which subsequently impacts multiple target proteins such as kinases, phosphatases, and transcription factors [output signals; right]. Prior systems biology work had concluded that cellular concentrations of cAMP is a key determinant of robustness at the core of information (signal) flow (Doyle & Csete, 2011; Friedlander et al., 2015; Kirschner & Gerhart, 1998). While cAMP production is tuned up or down by variable levels of GIV and its compartmentalized action on Gai/Gas and ACs within the RTK-cAMP pathway, cAMP degradation by PDEs serves as a dominant sink [drainpipe]. Bottom: Within the hourglass microarchitecture for vertical flow of ‘control’, up/down-regulation of GIV-GEM in cells serves as a tunable control valve, allowing cells to control cAMP production in cells responding to growth factors. When GIV-GEM expression is low [as seen in the normal epithelium], increasing input signals can trigger some of the highest levels of cellular cAMP, thereby conferring sensitivity (left). Increasing GIV-GEM expression throttles the cAMP response [middle], such that, when GIV-GEM is expressed highly [as seen across all cancers, cAMP levels remain low, regardless of the amount of input signals, thereby conferring robustness [right].
In all, the model directly compared the relative strengths of “valves” and “sinks”. We found that the effect of GIV concentration on cAMP levels in cells is discernible only when PDE activity is low; a high-PDE state overshadowed all effects of changing levels of GIV and virtually abolished GIV-dependent changes in cAMP levels. When PDE activities are high, cAMP levels do not go up even in low-GIV states likely because increased production is balanced by increased degradation. Why would a cell waste energy (ATP) in such a ‘futile cycle’? This situation is reminiscent of the maintenance of steady-state cGMP levels in the sub-μM range in thalamic neurons by concomitant guanylyl cyclase and PDE2 activities (Hepp et al., 2007) and cAMP levels in pyramidal cortical neurons by concomitant AC and PDE4 activities (Castro et al., 2010). Prior studies have suggested that such tonic cAMP production and PKA activity enable signal integration and crosstalk with other cascades (Houslay & Milligan, 1997); unlike an on/off system gated exclusively by Gαs proteins, tonic activity allows both up- and downregulation by activation of Gαi or inhibition of Gαs (via GIVGEM) and by PDEs. Our findings suggest that such up/down tunability is best achieved by changing the cellular concentrations of GIV. Because these predictions were also experimentally validated, PDEs (“sinks”) were determined as a dominant node and GIV (“valve”) as the subordinate node.
Overall, these findings cemented the importance and relevance of GIV-GEM as a tunable “valve” for cellular cAMP within a new network module where there can be many “sources” (EGFR, and other receptors that also engage GIV-GEM) and “sinks” (diverse subtypes of PDEs). The impact of tuning the “valve” up or down (by changing levels of GIV-GEM) was most pronounced in the setting of high “sources” (i.e., ligand activation of multiple receptors) and low “sinks” (PDE activity). While cAMP levels were flexible and responsive to ligand stimuli when GIV levels were maximally tuned down and the “valve” was open, robust suppression of cAMP was seen when GIV levels were maximally tuned up and the “valve” was closed [see Figure 2].
The flow of information in layers within signal transduction circuits in general (Doyle & Csete, 2011; Friedlander, Mayo, Tlusty, & Alon, 2015; Kirschner & Gerhart, 1998), and more specifically for RTKs like EGFR (Amit et al., 2007; Oda, Matsuoka, Funahashi, & Kitano, 2005), is believed to conform to a bow-tie or hourglass macroarchitecture in which diverse functions and diverse components are intertwined via universal carriers. Because cAMP is considered as one of the universal carrier molecules at the knot of such bowties which determines robustness (Friedlander et al., 2015), we conclude that GIV-GEM operates at the knot of the bow-tie as a tunable valve for controlling robustness within the circuit [Figure 3]. GIV’s ability to control the universal carrier, cAMP could explain why GIV has been found to be important for diverse cellular functions and impact diverse components (Aznar, Kalogriopoulos, Midde, & Ghosh, 2016). In an hourglass architecture, the lower and higher layers tend to see frequent evolutionary changes, while the carriers at the waist of the hourglass appears to be constant/invariant and sometimes, virtually ‘ossified’. Of relevance to our model, the importance of cAMP appears to be indeed ossified from unicellular organism to human alike, and GEMs like GIV are expressed ubiquitously in all tissues from fish to man and GIV-like GEMs have so far been identified as early as in C. elegans (Coleman et al., 2016).
Figure 3: Schematic summarizing the diverse pathologic states that feature either too little or too much GIV.
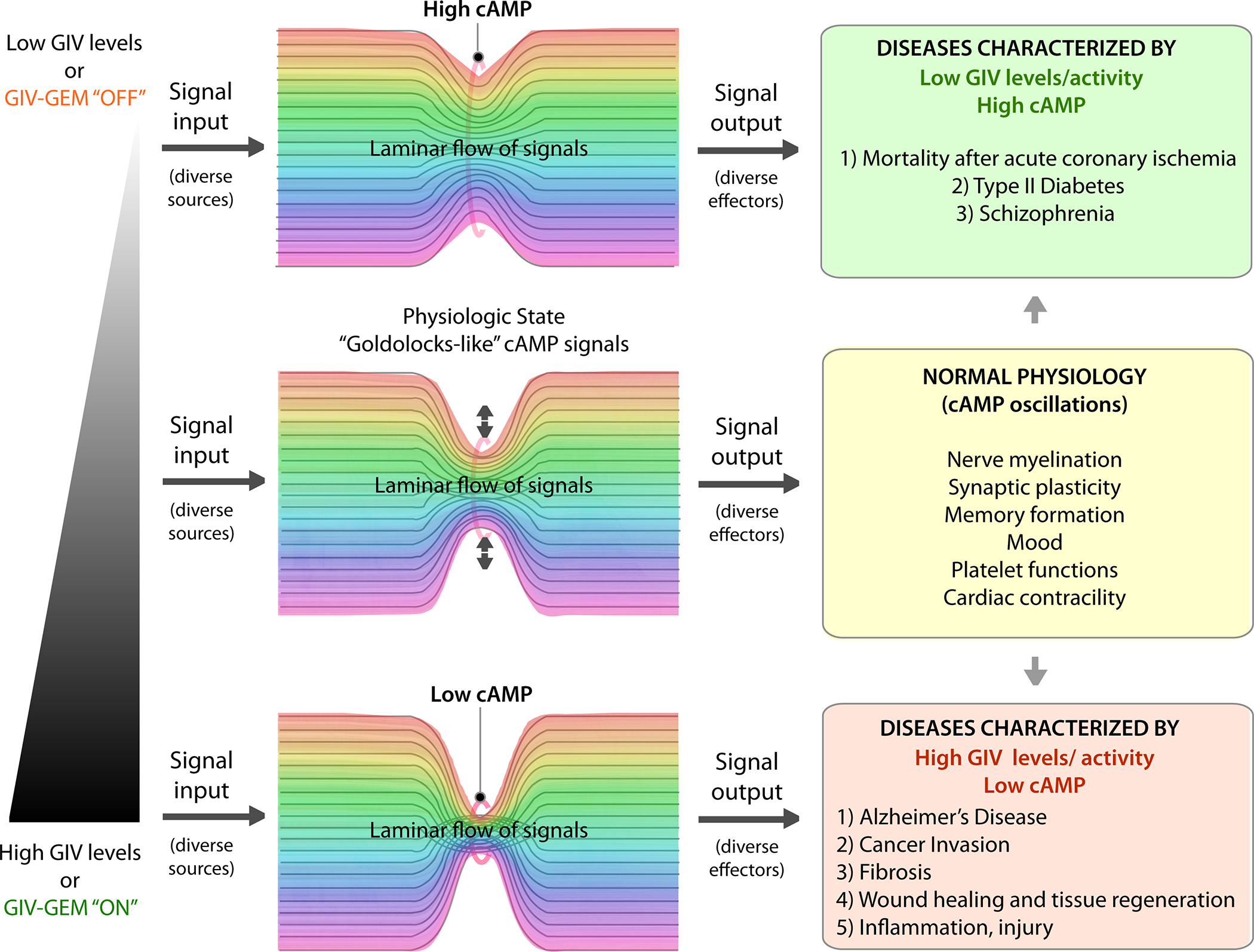
Because of its ability to serve as a tunable valve for cellular cAMP concentrations, too high or too low levels of expression of GIV may robustly regulate the tonic levels of cAMP in cells. Low GIV-states are associated with high cAMP (top), high GIV-states are associated with low cAMP (bottom). Text boxes on the right list pathophysiologic conditions associated with deregulated GIV and cAMP states (see also Table).
Implications of the new network for cAMP signaling in disease pathogenesis--
There are several implications of the newly built network model, which we summarize below. First, our work provides valuable clues into the impact of increased robustness at high-GIV states in cancers. Robustness in signaling is an organizing principle in biology, not only for the maintenance of homeostasis but also in the development and progression of chronic debilitating diseases like cancers; it is widely accepted that tumor cells hijack such robustness to gain growth and survival advantage during the development of cancer (Amit et al., 2007; Iadevaia, Nakhleh, Azencott, & Ram, 2014; Kitano, 2004). Consistently, GIV mRNA levels and DNA copy numbers are invariably higher across multiple cancers when compared to their respective normal tissue of origin [summarized in (Getz et al., 2019). Because GIV has been found to regulate several sinister properties of tumor cells across a variety of cancers (multiple studies, reviewed in (Ghosh, 2015)), it is possible that the high-GIV driven robustness maintains cAMP at low constant levels despite increasing input signals as a tumor evolves when targeted by biologics or chemotherapy agents. Such a phenomenon could be a part of a higher order organizing principle in most aggressive cancers, and therefore, justify GIV as a potential target for network-based anti-cancer therapy.
Second, the findings from network modeling impacts biomarker development. Multiple biomarker studies in bona fide EGFR-driven cancers (lung cancer (Sigismund et al., 2018), colon cancer (Lo Nigro et al., 2016), and GBMs (Westphal et al., 2017)) that are currently treated with anti-EGFR agents have tried to harness the ability to measure EGFR protein or mRNA as predictive or prognostic biomarkers to tell us which patients will do better or worse. However, none have panned out. Our model predicted that changing levels of EGFR may be overshadowed in high-GIV states that can robustly suppress cellular cAMP. This prediction from our model, and the decades-old body of experiments showing that the impact of such tonic cAMP suppressive state on tumors is expected to be an increase in aggressive traits (Table) prompted us to hypothesize that levels of expression of EGFR may provide meaningful information about tumor aggressiveness only if it is evaluated in tumors with low GIV. Our findings on patients validated these predictions using one of the most important readouts of cancer aggressiveness, i.e., patient survival-- i.e., high vs low EGFR levels correlated with poor vs good outcomes only when tumors had low GIV. By contrast, EGFR levels were irrelevant when tumors had high GIV. Moreover, consistent with the mathematical model which showed that high-GIV/high-EGFR states were accompanied by a robust inhibition of cAMP despite high levels of stimuli, Kaplan–Meier curves for a high GIV/high EGFR signature carried the worst prognosis compared to all other patients combined. Thus, high levels of EGFR signaling does not, by itself, fuel aggressive traits or carry a poor prognosis, but does so when GIV levels are concurrently elevated. In addition for tumors with low GIV, the high EGFR signaling state may be beneficial for maintaining high cAMP levels and therefore, critical for dampening several aggressive tumor traits (Table). Because cAMP levels in tumor cells and GIV levels have been previously implicated in anti-apoptotic signaling (McEwan et al., 2007) and the development of chemoresistance (Waugh, 2012), it is possible that the GIV-EGFR crosstalk we modeled recently also determines how well patients may respond to anti-EGFR therapies and who may be at highest risk for developing drug resistance. Whether such is the case, remains to be evaluated.
Third, the network model may also guide the development of anti-cancer therapeutics. For example, in the context of PDEs, it has been demonstrated that overexpression of PDE isoforms in various cancers leads to impaired cAMP and/or cGMP generation (Bender & Beavo, 2006). PDE inhibitors in tumour models in vitro and in vivo have been shown to induce apoptosis and cell cycle arrest in a broad spectrum of tumour cells (Savai et al., 2010). Despite the vast amount of preclinical evidence, there have been conflicting reports on its efficacy in the clinic (Barone et al., 2017). Our finding that low PDE levels in the setting of high GIV carries a poor prognosis predicts that the benefits of PDE inhibitors may be limited to patients who have low GIV expression in their tumors. Similarly, in the context of anti-EGFR therapies, researchers have come to realize that anti-EGFR therapeutics may unpredictably lead to two flavors of outcome that are attributed to kinase-independent functions of EGFR [reviewed in (Thomas & Weihua, 2019)] on the one hand, they may achieve the desirable therapeutic benefits, but on the other hand, their use may trigger the acquisition of resistance during treatment or may cause more harm in tumors that are innately resistant. Our model predicts that those with low GIV/high EGFR [high cAMP state] are likely to respond well to anti-EGFR therapy inducing tumor cell apoptosis, whereas those with high GIV/high EGFR [low cAMP state] may be at highest risk for developing drug resistance. Whether such predictions hold true, and whether these insights may impact patient outcomes remains to be seen.
Fourth, the network model helps extrapolate findings to other disease states beyond cancers that are fueled by aberrant cAMP signaling, where GIV levels are also concomitantly altered. Because our network model revealed how the ‘tunability’ of the “valve” impacts tonic levels of cellular cAMP over long time spans reaching steady-state kinetics, and valve-like GEMs are indeed found to be persistently dysregulated (either up- or downregulated) in diverse chronic disease states beyond cancers (cataloged in Table), it is possible that GIV levels may need to be maintained only within a narrow range in the healthy state [middle; Figure 3]. Because GIV is expressed at very high levels in the brain and reproductive organs (testes and ovary) and only in low levels in epithelial cells (Enomoto et al., 2005; Le-Niculescu et al., 2005), it is likely that the optimal physiologic range of expression varies between cell types. What is clear is that in each disease state, the level of GIV expression and its predicted impact on cellular cAMP (based on our model) is consistent with the observed impact of cAMP in disease pathogenesis [see Table]. It is noteworthy that each of these disease states have multiple different classes of receptors (“sources”) and, in some cases also PDEs (“sinks”) implicated in pathogenesis, further supporting the previously drawn conclusion that GIV-GEM may be operating as a tunable valve es at the knot of the bow-tie shaped network, controlling robustness within the circuit; persistent “open” or “closed”-states of the valve may contribute to disease pathogenesis perhaps via its ability to control cellular concentrations of second messengers such as cAMP [Figure 3].
Conclusions and Perspective
Cellular levels of cAMP impact a wide range of signals in diverse pathways, and a cell’s ability to maintain these levels within a physiologic range is critical for health. Too much or too little cAMP is often encountered in disease states. Although diverse “sources” (receptors) and “sinks” within the cAMP network may be contributing to these diseases, it is possible that a tunable “valve” such as GEMs that is stuck persistently in either “open” or “closed” state may be a common (i.e., an invariant) contributor or driver in the disease network. Because GIV and GEMs like GIV have been implicated in multiple disease states, GEMs constitute a hitherto untapped class of targets that could be exploited for reinstating physiologic cAMP signaling in multiple diseases.
Funding information
This work was supported by ARO W911NF-16-1-0411, AFOSR FA9550-15-1-0124, and National Science Foundation (NSF) PHY1505017 Grants to P.R. and National Institutes of Health (NIH) Grants CA238042, AI141630, CA100768, CA160911, and DK099226 to P.G. M.G. was supported by NIH Grant T32EB009380.
REFERENCES
- Agarwal KC, & Parks RE Jr (1983). Forskolin: A potential antimetastatic agent. International Journal of Cancer, 32(6), 801–804. 10.1002/ijc.2910320622 [DOI] [PubMed] [Google Scholar]
- Agarwal SR, Clancy CE, & Harvey RD (2016). Mechanisms Restricting Diffusion of Intracellular cAMP. Scientific Reports, 6, 19577 10.1038/srep19577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat FJ, Tytell JD, Thodeti CK, Derrien A, & Ingber DE (2009). Mechanical control of cAMP signaling through integrins is mediated by the heterotrimeric Gαs protein. Journal of Cellular Biochemistry, 106(4), 529–538. 10.1002/jcb.22001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Wides R, & Yarden Y (2007). Evolvable signaling networks of receptor tyrosine kinases: Relevance of robustness to malignancy and to cancer therapy. Molecular Systems Biology, 3 10.1038/msb4100195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anai M, Shojima N, Katagiri H, Ogihara T, Sakoda H, Onishi Y, … Asano T (2005). A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. The Journal of Biological Chemistry, 280(18), 18525–18535. 10.1074/jbc.M500586200 [DOI] [PubMed] [Google Scholar]
- Aznar N, Kalogriopoulos N, Midde KK, & Ghosh P (2016). Heterotrimeric G protein signaling via GIV/Girdin: Breaking the rules of engagement, space, and time. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 38(4), 379–393. 10.1002/bies.201500133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, … Ghosh P (2015). Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. ELife, 4, e07091 10.7554/eLife.07091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone I, Giordano C, Bonofiglio D, Andò S, & Catalano S (2017). Phosphodiesterase type 5 and cancers: Progress and challenges. Oncotarget, 8(58), 99179–99202. 10.18632/oncotarget.21837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels SP, Lee SR, & Neufeld AH (1982). Forskolin stimulates cyclic AMP synthesis, lowers intraocular pressure and increases outflow facility in rabbits. Current Eye Research, 2(10), 673–681. 10.3109/02713688209019996 [DOI] [PubMed] [Google Scholar]
- Beas AO, Taupin V, Teodorof C, Nguyen LT, Garcia-Marcos M, & Farquhar MG (2012). Gαs promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Molecular Biology of the Cell, 23(23), 4623–4634. 10.1091/mbc.E12-02-0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, & Brunton LL (2002). Cyclic nucleotide research—Still expanding after half a century. Nature Reviews. Molecular Cell Biology, 3(9), 710–718. 10.1038/nrm911 [DOI] [PubMed] [Google Scholar]
- Bender AT, & Beavo JA (2006). Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacological Reviews, 58(3), 488–520. 10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- Bornheimer SJ, Maurya MR, Farquhar MG, & Subramaniam S (2004). Computational modeling reveals how interplay between components of a GTPase-cycle module regulates signal transduction. Proceedings of the National Academy of Sciences, 101(45), 15899–15904. 10.1073/pnas.0407009101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher MJ, Duchesne C, Lainé J, Morisset J, & Rivard N (2001). CAMP protection of pancreatic cancer cells against apoptosis induced by ERK inhibition. Biochemical and Biophysical Research Communications, 285(2), 207–216. 10.1006/bbrc.2001.5147 [DOI] [PubMed] [Google Scholar]
- Burke P, Schooler K, & Wiley HS (2001). Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis and Intracellular Trafficking. Molecular Biology of the Cell, 12(6), 1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli J, & Sears M (1983). Forskolin lowers intraocular pressure in rabbits, monkeys, and man. Lancet (London, England), 1(8331), 958–960. 10.1016/s0140-6736(83)92084-6 [DOI] [PubMed] [Google Scholar]
- Castro LRV, Gervasi N, Guiot E, Cavellini L, Nikolaev VO, Paupardin-Tritsch D, & Vincent P (2010). Type 4 Phosphodiesterase Plays Different Integrating Roles in Different Cellular Domains in Pyramidal Cortical Neurons. The Journal of Neuroscience, 30(17), 6143–6151. 10.1523/JNEUROSCI.5851-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai S-P, & Fong JC (2015). Synergistic induction of insulin resistance by endothelin-1 and cAMP in 3T3-L1 adipocytes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1852(10, Part A), 2048–2055. 10.1016/j.bbadis.2015.06.026 [DOI] [PubMed] [Google Scholar]
- Chiefari E, Paonessa F, Iiritano S, Le Pera I, Palmieri D, Brunetti G, … Brunetti A (2009). The cAMP-HMGA1-RBP4 system: A novel biochemical pathway for modulating glucose homeostasis. BMC Biology, 7(1), 24 10.1186/1741-7007-7-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho-Chung YS (1990). Role of cyclic AMP receptor proteins in growth, differentiation, and suppression of malignancy: New approaches to therapy. Cancer Research, 50(22), 7093–7100. [PubMed] [Google Scholar]
- Coleman BD, Marivin A, Parag-Sharma K, DiGiacomo V, Kim S, Pepper JS, … Garcia-Marcos M (2016). Evolutionary Conservation of a GPCR-Independent Mechanism of Trimeric G Protein Activation. Molecular Biology and Evolution, 33(3), 820–837. 10.1093/molbev/msv336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JC, & Csete M (2011). Architecture, constraints, and behavior. Proceedings of the National Academy of Sciences, 108(Supplement 3), 15624–15630. 10.1073/pnas.1103557108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, … Marais R (2006). In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Research, 66(19), 9483–9491. 10.1158/0008-5472.CAN-05-4227 [DOI] [PubMed] [Google Scholar]
- Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, … Takahashi M (2005). Akt/PKB regulates actin organization and cell motility via Girdin/APE. Developmental Cell, 9(3), 389–402. 10.1016/j.devcel.2005.08.001 [DOI] [PubMed] [Google Scholar]
- Erion DM, Ignatova ID, Yonemitsu S, Nagai Y, Chatterjee P, Weismann D, … Shulman GI (2009). Prevention of Hepatic Steatosis and Hepatic Insulin Resistance by Knockdown of cAMP Response Element-Binding Protein. Cell Metabolism, 10(6), 499–506. 10.1016/j.cmet.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DB (1986). Modulation of cAMP: Mechanism for positive inotropic action. Journal of Cardiovascular Pharmacology, 8 Suppl 9, S22–29. [PubMed] [Google Scholar]
- Fajardo AM, Piazza GA, & Tinsley HN (2014). The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers, 6(1), 436–458. 10.3390/cancers6010436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmore D (2004). It’s a GPCR world. Modern Drug Discovery, 7(11), 24–28. [Google Scholar]
- Follin-Arbelet V, Hofgaard PO, Hauglin H, Naderi S, Sundan A, Blomhoff R, … Blomhoff HK (2011). Cyclic AMP induces apoptosis in multiple myeloma cells and inhibits tumor development in a mouse myeloma model. BMC Cancer, 11(1), 301 10.1186/1471-2407-11-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander T, Mayo AE, Tlusty T, & Alon U (2015). Evolution of bow-tie architectures in biology. PLoS Computational Biology, 11(3), e1004055 10.1371/journal.pcbi.1004055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M, Kietrsunthorn PS, Wang H, Ghosh P, & Farquhar MG (2011). G Protein Binding Sites on Calnuc (Nucleobindin 1) and NUCB2 (Nucleobindin 2) Define a New Class of Gαi-regulatory Motifs. The Journal of Biological Chemistry, 286(32), 28138–28149. 10.1074/jbc.M110.204099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz M, Swanson L, Sahoo D, Ghosh P, & Rangamani P (2019). A predictive computational model reveals that GIV/Girdin serves as a tunable valve for EGFR-stimulated Cyclic AMP Signals. Molecular Biology of the Cell, mbc.E18-10-0630. 10.1091/mbc.E18-10-0630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P (2015). Heterotrimeric G proteins as emerging targets for network based therapy in cancer: End of a long futile campaign striking heads of a Hydra. Aging, 7(7), 469–474. 10.18632/aging.100781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, … Farquhar MG (2010). A Gαi–GIV Molecular Complex Binds Epidermal Growth Factor Receptor and Determines Whether Cells Migrate or Proliferate. Molecular Biology of the Cell, 21(13), 2338–2354. 10.1091/mbc.E10-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, & Garcia-Marcos M (2019). Do All Roads Lead to Rome in G-Protein Activation? Trends in Biochemical Sciences. 10.1016/j.tibs.2019.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Rangamani P, & Kufareva I (2017). The GAPs, GEFs, GDIs and…now, GEMs: New kids on the heterotrimeric G protein signaling block. Cell Cycle, 16(7), 607–612. 10.1080/15384101.2017.1282584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding AJ, & Schiemann WP (2016). Harnessing protein kinase A activation to induce mesenchymal-epithelial programs to eliminate chemoresistant, tumor-initiating breast cancer cells. Translational Cancer Research, 5(Suppl 2), S226–S232. 10.21037/tcr.2016.08.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, & Czech MP (2008). Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nature Reviews. Molecular Cell Biology, 9(5), 367–377. 10.1038/nrm2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Bhandari D, Leyme A, Aznar N, Midde KK, Lo I-C, … Ghosh P (2016). GIV/Girdin activates Gαi and inhibits Gαs via the same motif. Proceedings of the National Academy of Sciences, 113(39), E5721–E5730. 10.1073/pnas.1609502113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung A, Ordelheide A-M, Staiger H, Melzer M, Häring H-U, & Lammers R (2013). The Akt substrate Girdin is a regulator of insulin signaling in myoblast cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1833(12), 2803–2811. 10.1016/j.bbamcr.2013.07.012 [DOI] [PubMed] [Google Scholar]
- Hayano S, Takefuji M, Maeda K, Noda T, Ichimiya H, Kobayashi K, … Murohara T (2015). Akt-dependent Girdin phosphorylation regulates repair processes after acute myocardial infarction. Journal of Molecular and Cellular Cardiology, 88, 55–63. 10.1016/j.yjmcc.2015.09.012 [DOI] [PubMed] [Google Scholar]
- Hepp R, Tricoire L, Hu E, Gervasi N, Paupardin-Tritsch D, Lambolez B, & Vincent P (2007). Phosphodiesterase type 2 and the homeostasis of cyclic GMP in living thalamic neurons. Journal of Neurochemistry, 102(6), 1875–1886. 10.1111/j.1471-4159.2007.04657.x [DOI] [PubMed] [Google Scholar]
- Houglum K, Lee KS, & Chojkier M (1997). Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. The Journal of Clinical Investigation, 99(6), 1322–1328. 10.1172/JCI119291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, & Milligan G (1997). Tailoring cAMP-signalling responses through isoform multiplicity. Trends in Biochemical Sciences, 22(6), 217–224. 10.1016/S0968-0004(97)01050-5 [DOI] [PubMed] [Google Scholar]
- Iadevaia S, Nakhleh LK, Azencott R, & Ram PT (2014). Mapping network motif tunability and robustness in the design of synthetic signaling circuits. PloS One, 9(3), e91743 10.1371/journal.pone.0091743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L, Snead A, Lu D, & Aroonsakool N (2012). CAMP and Epac in the regulation of tissue fibrosis. British Journal of Pharmacology, 166(2), 447–456. 10.1111/j.1476-5381.2012.01847.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Komeima K, Yasuma T, Enomoto A, Asai N, Asai M, … Terasaki H (2013). Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. The American Journal of Pathology, 182(2), 586–596. 10.1016/j.ajpath.2012.10.012 [DOI] [PubMed] [Google Scholar]
- Kalogriopoulos NA, Rees SD, Ngo T, Kopcho NJ, Ilatovskiy AV, Sun N, … Kufareva I (2019). Structural basis for GPCR-independent activation of heterotrimeric Gi proteins. Proceedings of the National Academy of Sciences, 116(33), 16394–16403. 10.1073/pnas.1906658116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada N, Kuroki T, Kobayashi K, Inoue M, & Kaneda K (1996). Inhibition of myofibroblastic transformation of cultured rat hepatic stellate cells by methylxanthines and dibutyryl cAMP. Digestive Diseases and Sciences, 41(5), 1022–1029. 10.1007/bf02091547 [DOI] [PubMed] [Google Scholar]
- Kenakin T (2004). Principles: Receptor theory in pharmacology. Trends in Pharmacological Sciences, 25(4), 186–192. 10.1016/j.tips.2004.02.012 [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, Demin OV, Moehren G, & Hoek JB (1999). Quantification of Short Term Signaling by the Epidermal Growth Factor Receptor. Journal of Biological Chemistry, 274(42), 30169–30181. 10.1074/jbc.274.42.30169 [DOI] [PubMed] [Google Scholar]
- Kida Y, Nyomba BL, Bogardus C, & Mott DM (1991). Defective insulin response of cyclic adenosine monophosphate-dependent protein kinase in insulin-resistant humans. The Journal of Clinical Investigation, 87(2), 673–679. 10.1172/JCI115045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch D, Kemmler W, & Häring HU (1983). Cyclic AMP modulates insulin binding and induces post-receptor insulin resistance of glucose transport in isolated rat adipocytes. Biochemical and Biophysical Research Communications, 115(1), 398–405. 10.1016/0006-291x(83)91017-3 [DOI] [PubMed] [Google Scholar]
- Kirsch DM, Bachmann W, & Häring HU (1984). Ciglitazone reverses cAMP-induced post-insulin receptor resistance in rat adipocytes in vitro. FEBS Letters, 176(1), 49–54. 10.1016/0014-5793(84)80909-6 [DOI] [PubMed] [Google Scholar]
- Kirschner M, & Gerhart J (1998). Evolvability. Proceedings of the National Academy of Sciences, 95(15), 8420–8427. 10.1073/pnas.95.15.8420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, … Kasuga M (1999). Insulin-Induced Phosphorylation and Activation of Cyclic Nucleotide Phosphodiesterase 3B by the Serine-Threonine Kinase Akt. Molecular and Cellular Biology, 19(9), 6286–6296. 10.1128/MCB.19.9.6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H (2004). Cancer as a robust system: Implications for anticancer therapy. Nature Reviews. Cancer, 4(3), 227–235. 10.1038/nrc1300 [DOI] [PubMed] [Google Scholar]
- Kirsten Leineweber, Michael Böhm, & Gerd Heusch. (2006). Cyclic Adenosine Monophosphate in Acute Myocardial Infarction With Heart Failure. Circulation, 114(5), 365–367. 10.1161/CIRCULATIONAHA.106.642132 [DOI] [PubMed] [Google Scholar]
- Kramer W, Thormann J, Kindler M, & Schlepper M (1987). Effects of forskolin on left ventricular function in dilated cardiomyopathy. Arzneimittel-Forschung, 37(3), 364–367. [PubMed] [Google Scholar]
- Kreutner W, Chapman RW, Gulbenkian A, & Tozzi S (1985). Bronchodilator and antiallergy activity of forskolin. European Journal of Pharmacology, 111(1), 1–8. 10.1016/0014-2999(85)90106-2 [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H, Niesman I, Fischer T, DeVries L, & Farquhar MG (2005). Identification and characterization of GIV, a novel Galpha i/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. The Journal of Biological Chemistry, 280(23), 22012–22020. 10.1074/jbc.M501833200 [DOI] [PubMed] [Google Scholar]
- Leyme A, Marivin A, & Garcia-Marcos M (2016). GIV/Girdin (Gα-interacting, Vesicle-associated Protein/Girdin) Creates a Positive Feedback Loop That Potentiates Outside-in Integrin Signaling in Cancer Cells. The Journal of Biological Chemistry, 291(15), 8269–8282. 10.1074/jbc.M115.691550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyme A, Marivin A, Perez-Gutierrez L, Nguyen LT, & Garcia-Marcos M (2015). Integrins activate trimeric G proteins via the nonreceptor protein GIV/Girdin. The Journal of Cell Biology, 210(7), 1165–1184. 10.1083/jcb.201506041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Ear J, Midde K, Lopez-Sanchez I, Aznar N, Garcia-Marcos M, … Ghosh P (2014). Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Molecular Biology of the Cell, 25(22), 3654–3671. 10.1091/mbc.E14-05-0978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Ear J, Pavlova Y, Mittal Y, Kufareva I, Ghassemian M, … Ghosh P (2011). Tyrosine Phosphorylation of the Guanine Nucleotide Exchange Factor GIV Promotes Activation of PI3K During Cell Migration. Science Signaling, 4(192), ra64 10.1126/scisignal.2002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Jiang P, Cui D, Du J, He L, Yao J, & Liu D (2015). [Expression of Girdin in brain tissues of Alzheimer’s disease]. Zhonghua Bing Li Xue Za Zhi = Chinese Journal of Pathology, 44(5), 301–304. [PubMed] [Google Scholar]
- Lo Nigro C, Ricci V, Vivenza D, Granetto C, Fabozzi T, Miraglio E, & Merlano MC (2016). Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World Journal of Gastroenterology, 22(30), 6944–6954. 10.3748/wjg.v22.i30.6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez I, Dunkel Y, Roh Y-S, Mittal Y, De Minicis S, Muranyi A, … Ghosh P (2014). GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nature Communications, 5, 4451 10.1038/ncomms5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez I, Kalogriopoulos N, Lo I-C, Kabir F, Midde KK, Wang H, & Ghosh P (2015). Focal adhesions are foci for tyrosine-based signal transduction via GIV/Girdin and G proteins. Molecular Biology of the Cell, 26(24), 4313–4324. 10.1091/mbc.E15-07-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma GS, Aznar N, Kalogriopoulos N, Midde KK, Lopez-Sanchez I, Sato E, … Ghosh P (2015). Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proceedings of the National Academy of Sciences of the United States of America, 112(20), E2602–E2610. 10.1073/pnas.1505543112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma GS, Lopez-Sanchez I, Aznar N, Kalogriopoulos N, Pedram S, Midde K, … Ghosh P (2015). Activation of G proteins by GIV-GEF is a pivot point for insulin resistance and sensitivity. Molecular Biology of the Cell, 26(23), 4209–4223. 10.1091/mbc.E15-08-0553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, … Houslay MD (2002). Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). British Journal of Pharmacology, 136(3), 421–433. 10.1038/sj.bjp.0704743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Kanatsuka A, Suzuki T, Kuribayashi S, Hashimoto N, Yoshida S, & Nishimura M (1985). Insulin Resistance of Fat Cells from Spontaneously Diabetic KK Mice: Analysis of Insulin-sensitive Phosphodiesterase. Diabetes, 34(9), 844–849. 10.2337/diab.34.9.844 [DOI] [PubMed] [Google Scholar]
- Martínez M, Fernández E, Frank A, Guaza C, de la Fuente M, & Hernanz A (1999). Increased cerebrospinal fluid cAMP levels in Alzheimer’s disease. Brain Research, 846(2), 265–267. 10.1016/s0006-8993(99)01981-2 [DOI] [PubMed] [Google Scholar]
- McEwan DG, Brunton VG, Baillie GS, Leslie NR, Houslay MD, & Frame MC (2007). Chemoresistant KM12C colon cancer cells are addicted to low cyclic AMP levels in a phosphodiesterase 4-regulated compartment via effects on phosphoinositide 3-kinase. Cancer Research, 67(11), 5248–5257. 10.1158/0008-5472.CAN-07-0097 [DOI] [PubMed] [Google Scholar]
- Midde KK, Aznar N, Kalogriopoulos N, & Ghosh P (2016). Heterotrimeric G Proteins: Breaking the rules of engagement, space and time. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology, 38(4), 379–393. 10.1002/bies.201500133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midde KK, Aznar N, Laederich MB, Ma GS, Kunkel MT, Newton AC, & Ghosh P (2015). Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proceedings of the National Academy of Sciences of the United States of America, 112(9), E937–946. 10.1073/pnas.1420140112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midde K, Sun N, Rohena C, Joosen L, Dhillon H, & Ghosh P (2018). Single-Cell Imaging of Metastatic Potential of Cancer Cells. IScience, 10, 53–65. 10.1016/j.isci.2018.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyachi H, Takahashi M, & Komori K (2015). A Novel Approach against Vascular Intimal Hyperplasia Through the Suppression of Girdin. Annals of Vascular Diseases, 8(2), 69–73. 10.3400/avd.ra.14-00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Sudo T, Kameyama M, Fukuoka H, Mukai M, Doki Y, … Imaoka S (2000). Cyclic AMP specific phosphodiesterase activity and colon cancer cell motility. Clinical & Experimental Metastasis, 18(7), 599–604. 10.1023/A:1011926116777 [DOI] [PubMed] [Google Scholar]
- Murthy KS, Zhou H, & Makhlouf GM (2002). PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. American Journal of Physiology. Cell Physiology, 282(3), C508–517. 10.1152/ajpcell.00373.2001 [DOI] [PubMed] [Google Scholar]
- Nagamatsu T, Nishiyama T, Goto I, Nagao T, & Suzuki Y (2003). Adenosine 3’, 5’ cyclic monophosphate attenuates the production of fibronectin in the glomeruli of anti-glomerular basement membrane antibody-associated nephritic rats. British Journal of Pharmacology, 140(7), 1245–1251. 10.1038/sj.bjp.0705564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC, Bootman MD, & Scott JD (2016). Second Messengers. Cold Spring Harbor Perspectives in Biology, 8(8). 10.1101/cshperspect.a005926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Matsuoka Y, Funahashi A, & Kitano H (2005). A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular Systems Biology, 1, 2005.0010. 10.1038/msb4100014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Zheng X, Gao Y, Shu M, Leng T, Li Y, … Hu H (2014). Activation of cyclic AMP/PKA pathway inhibits bladder cancer cell invasion by targeting MAP4-dependent microtubule dynamics. Urologic Oncology, 32(1), 47.e21–28. 10.1016/j.urolonc.2013.06.017 [DOI] [PubMed] [Google Scholar]
- Pantziarka P, Sukhatme V, Crispino S, Bouche G, Meheus L, & Sukhatme VP (2018). Repurposing drugs in oncology (ReDO)—Selective PDE5 inhibitors as anti-cancer agents. Ecancermedicalscience, 12 10.3332/ecancer.2018.824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parag-Sharma K, Leyme A, DiGiacomo V, Marivin A, Broselid S, & Garcia-Marcos M (2016). Membrane Recruitment of the Non-receptor Protein GIV/Girdin (Gα-interacting, Vesicle-associated Protein/Girdin) Is Sufficient for Activating Heterotrimeric G Protein Signaling. Journal of Biological Chemistry, 291(53), 27098–27111. 10.1074/jbc.M116.764431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DR, Smagley Y, Garcia M, Carter MB, Evangelisti A, Matlawska-Wasowska K, … Chigaev A (2016). Cyclic AMP efflux inhibitors as potential therapeutic agents for leukemia. Oncotarget, 7(23), 33960–33982. 10.18632/oncotarget.8986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, & Lefkowitz RJ (2002). Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology, 3(9), 639–650. 10.1038/nrm908 [DOI] [PubMed] [Google Scholar]
- Poppleton H, Sun H, Fulgham D, Bertics P, & Patel TB (1996). Activation of G by the Epidermal Growth Factor Receptor Involves Phosphorylation. Journal of Biological Chemistry, 271(12), 6947–6951. 10.1074/jbc.271.12.6947 [DOI] [PubMed] [Google Scholar]
- Raker VK, Becker C, & Steinbrink K (2016). The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Frontiers in Immunology, 7 10.3389/fimmu.2016.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CE, Nian H, Yu C, Gamboa JL, Luther JM, Brown NJ, & Shibao CA (2015). Treatment with Sildenafil Improves Insulin Sensitivity in Prediabetes: A Randomized, Controlled Trial. The Journal of Clinical Endocrinology and Metabolism, 100(12), 4533–4540. 10.1210/jc.2015-3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Bedossa P, Francque SM, Larrey D, Aithal GP, Serfaty L, … Traudtner K (2014). Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis. Clinical Gastroenterology and Hepatology: The Official Clinical Practice Journal of the American Gastroenterological Association, 12(10), 1724–1730.e5. 10.1016/j.cgh.2014.01.040 [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P (2012). The cyclic AMP pathway. Cold Spring Harbor Perspectives in Biology, 4(12). 10.1101/cshperspect.a011148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savai R, Pullamsetti SS, Banat G-A, Weissmann N, Ghofrani HA, Grimminger F, & Schermuly RT (2010). Targeting cancer with phosphodiesterase inhibitors. Expert Opinion on Investigational Drugs, 19(1), 117–131. 10.1517/13543780903485642 [DOI] [PubMed] [Google Scholar]
- Serezani CH, Ballinger MN, Aronoff DM, & Peters-Golden M (2008). Cyclic AMP. American Journal of Respiratory Cell and Molecular Biology, 39(2), 127–132. 10.1165/rcmb.2008-0091TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette C, & Conti M (1996). Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. The Journal of Biological Chemistry, 271(28), 16526–16534. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Kobayashi Y, Oki Y, Kawasaki T, Yoshimi T, & Nakamura H (1999). OPC-13013, a cyclic nucleotide phosphodiesterase type III, inhibitor, inhibits cell proliferation and transdifferentiation of cultured rat hepatic stellate cells. Life Sciences, 64(23), 2081–2088. 10.1016/s0024-3205(99)00157-5 [DOI] [PubMed] [Google Scholar]
- Sigismund S, Avanzato D, & Lanzetti L (2018). Emerging functions of the EGFR in cancer. Molecular Oncology, 12(1), 3–20. 10.1002/1878-0261.12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, & Kida S (1998). CREB and memory. Annual Review of Neuroscience, 21, 127–148. 10.1146/annurev.neuro.21.1.127 [DOI] [PubMed] [Google Scholar]
- Simpson F, Martin S, Evans TM, Kerr M, James DE, Parton RG, … Wicking C (2005). A Novel Hook-Related Protein Family and the Characterization of Hook-Related Protein 1. Traffic, 6(6), 442–458. 10.1111/j.1600-0854.2005.00289.x [DOI] [PubMed] [Google Scholar]
- Tagliaferri P, Katsaros D, Clair T, Ally S, Tortora G, Neckers L, … Cho-Chung YS (1988). Synergistic Inhibition of Growth of Breast and Colon Human Cancer Cell Lines by Site-selective Cyclic AMP Analogues. Cancer Research, 48(6), 1642–1650. [PubMed] [Google Scholar]
- Tang Y, Osawa H, Onuma H, Nishimiya T, Ochi M, & Makino H (1999). Improvement in insulin resistance and the restoration of reduced phosphodiesterase 3B gene expression by pioglitazone in adipose tissue of obese diabetic KKAy mice. Diabetes, 48(9), 1830–1835. 10.2337/diabetes.48.9.1830 [DOI] [PubMed] [Google Scholar]
- Tanti JF, Grémeaux T, Rochet N, Obberghen EV, & Marchand-Brustel YL (1987). Effect of cyclic AMP-dependent protein kinase on insulin receptor tyrosine kinase activity. Biochemical Journal, 245(1), 19–26. 10.1042/bj2450019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, & Weihua Z (2019). Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Frontiers in Oncology, 9 10.3389/fonc.2019.00800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay J, Lachance B, & Hamet P (1985). Activation of cyclic GMP-binding and cyclic AMP-specific phosphodiesterases of rat platelets by a mechanism involving cyclic AMP-dependent phosphorylation. Journal of Cyclic Nucleotide and Protein Phosphorylation Research, 10(4), 397–411. [PubMed] [Google Scholar]
- Van Daalen E, Kemner C, Verbeek NE, van der Zwaag B, Dijkhuizen T, Rump P, … Poot M (2011). Social responsiveness scale-aided analysis of the clinical impact of copy number variations in autism. Neurogenetics, 12(4), 315–323. 10.1007/s10048-011-0297-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Dicitore A, Mari D, & Cavagnini F (2009). A new therapeutic strategy against cancer: CAMP elevating drugs and leptin. Cancer Biology & Therapy, 8(12), 1191–1193. 10.4161/cbt.8.12.8937 [DOI] [PubMed] [Google Scholar]
- Wachtel H, & Löschmann PA (1986). Effects of forskolin and cyclic nucleotides in animal models predictive of antidepressant activity: Interactions with rolipram. Psychopharmacology, 90(4), 430–435. 10.1007/bf00174056 [DOI] [PubMed] [Google Scholar]
- Wahlang B, McClain C, Barve S, & Gobejishvili L (2018). Role of cAMP and phosphodiesterase signaling in liver health and disease. Cellular Signalling, 49, 105–115. 10.1016/j.cellsig.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Misaki T, Taupin V, Eguchi A, Ghosh P, & Farquhar MG (2015). GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. Journal of the American Society of Nephrology: JASN, 26(2), 314–327. 10.1681/ASN.2013090985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li Y, Zhu JY, Fang D, Ding H-F, Dong Z, … Huang S (2016). Triple negative breast cancer development can be selectively suppressed by sustaining an elevated level of cellular cyclic AMP through simultaneously blocking its efflux and decomposition. Oncotarget, 7(52), 87232–87245. 10.18632/oncotarget.13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh MG (2012). Phosphatidylinositol 4-kinases, phosphatidylinositol 4-phosphate and cancer. Cancer Letters, 325(2), 125–131. 10.1016/j.canlet.2012.06.009 [DOI] [PubMed] [Google Scholar]
- Weng L, Enomoto A, Miyoshi H, Takahashi K, Asai N, Morone N, … Takahashi M (2014). Regulation of cargo-selective endocytosis by dynamin 2 GTPase-activating protein girdin. The EMBO Journal, 33(18), 2098–2112. 10.15252/embj.201488289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M, Maire CL, & Lamszus K (2017). EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs, 31(9), 723–735. 10.1007/s40263-017-0456-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P-C, Boras BW, Jeng M-T, Docken SS, Lewis TJ, McCulloch AD, Harvey RD, & Clancy CE (2016). A Computational Modeling and Simulation Approach to Investigate Mechanisms of Subcellular cAMP Compartmentation. PLOS Computational Biology, 12(7), e1005005 10.1371/journal.pcbi.1005005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Sun Y, Li J, Wang Y, Zhu Y, Shi Y, … Cao P (2017). Silencing the Girdin gene enhances radio-sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. Journal of Experimental & Clinical Cancer Research, 36(1), 110 10.1186/s13046-017-0580-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-J, Li A-J, Han Y, Yin L, & Lin M-B (2014). Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World Journal of Gastroenterology, 20(25), 8229–8236. 10.3748/wjg.v20.i25.8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman NP, Roy I, Hauser AD, Wilson JM, Williams CL, & Dwinell MB (2015). Cyclic AMP regulates the migration and invasion potential of human pancreatic cancer cells. Molecular Carcinogenesis, 54(3), 203–215. 10.1002/mc.22091 [DOI] [PMC free article] [PubMed] [Google Scholar]