

Abstract
The chemoenzymatic synthesis of heparin, through a multienzyme process, represents a critical challenge in providing a safe and effective substitute for this animal-sourced anticoagulant drug. D-glucuronyl C5-epimerase (C5-epi) is an enzyme acting on a heparin precursor, N-sulfoheparosan, catalyzing the reversible epimerization of D-glucuronic acid (GlcA) to L-iduronic acid (IdoA). The absence of reliable assays for C5-epi has limited elucidation of the enzymatic reaction and kinetic mechanisms. Real time and offline assays are described that rely on 1D 1H NMR to study the activity of C5-epi. Apparent steady-state kinetic parameters for both the forward and the pseudo-reverse reactions of C5-epi are determined for the first time using polysaccharide substrates directly relevant to the chemoenzymatic synthesis and biosynthesis of heparin. The forward reaction shows unusual sigmoidal kinetic behavior, and the pseudo-reverse reaction displays nonsaturating kinetic behavior. The atypical sigmoidal behavior of the forward reaction was probed using a range of buffer additives. Surprisingly, the addition of 25 mM each of CaCl2 and MgCl2 resulted in a forward reaction exhibiting more conventional Michaelis–Menten kinetics. The addition of 2-O-sulfotransferase, the next enzyme involved in heparin synthesis, in the absence of 3′-phosphoadenosine 5′-phosphosulfate, also resulted in C5-epi exhibiting a more conventional Michaelis–Menten kinetic behavior in the forward reaction accompanied by a significant increase in apparent Vmax. This study provides critical information for understanding the reaction kinetics of C5-epi, which may result in improved methods for the chemoenzymatic synthesis of bioengineered heparin.
Keywords: C5-epimerase, divalent cations, enzyme kinetics, N-sulfoheparosan, 2-O-sulfotransferase
Introduction
Heparin is a highly polydisperse (molecular weight 5000–40,000, corresponding to chain lengths of 8–60 disaccharide units) glycosaminoglycan (GAG) composed primarily of the disaccharide repeating unit, 2-O-sulfo-iduronic acid and 6-O-sulfo N-sulfo-glucosamine, and is one of the most widely used anticoagulant drugs (Linhardt 2003). Pharmaceutical grade heparin is currently isolated from mast cells (Feyerabend et al. 2006) present in the inner lining of the small intestine of pigs, and hence, contains small quantities of other GAGs, including heparan sulfate (a less sulfated analog of heparin found in a variety of cell types), chondroitin and dermatan sulfates, as well as other potential tissue-derived impurities such as viruses and prions (Liu et al. 2009). A chemoenzymatically synthesized bioengineered heparin has been proposed as an alternative to animal-sourced heparin (Zhang et al. 2008; Wang 2011). Bioengineered heparin starts with the fermentation of Escherichia coli K5 to obtain the capsular polysaccharide, heparosan, which is then chemically partially N-deacetylated and N-sulfonated to afford N-sulfoheparosan (NSH), of the required molecular weight and polydispersity (PD) to serve as the substrate for a series of enzymatic transformations, based on heparin’s biosynthetic pathway, to afford bioengineered heparin (Feyerabend et al. 2006; Fu et al. 2016; Cress et al. 2019)
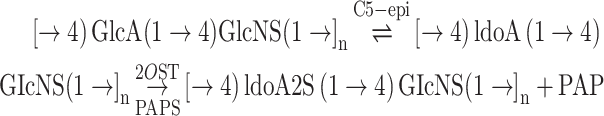 |
(1) |
A key step in heparin synthesis is the epimerization of D-glucuronic acid (GlcA) into L-iduronic acid (IdoA) catalyzed by D-glucuronyl C-5 epimerase (C5-epi) (Eq. 1). Once the IdoA residue is formed in the product, polysaccharide 2-O-sulfotransferase (2OST) preferentially sulfates IdoA using 3′-phosphoadenosine-5′-phosphosulfate (PAPS) as a sulfo donor. The resulting sulfo group at the 2-position of IdoA (IdoA2S) prevents C5-epi from catalyzing reverse epimerization (Vaidyanathan et al. 2016). The action of C5-epi and 2OST on NSH results in the conversion of most of the GlcA residues into IdoA2S residues affording a disaccharide sequence comprised primarily of 2-O-sulfo-iduronic acid (IdoA2S) 1 → 4 (GlcNS) (Bhaskar et al. 2012, 2015). Further, complicating this reaction is the slow conversion of NSH into GlcA2S in the presence of 2OST and absence of C5-epi (Eq. 2)
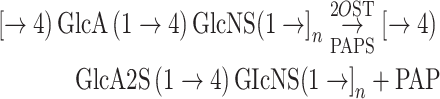 |
(2) |
Measuring the kinetics of C5-epi catalysis is complicated due to the lack of significant physicochemical differences between the substrate and product of the reaction, the reversibility of the reaction and the coupling of the reaction to subsequent action of 2OST. Conventional analysis of heparin (or heparan sulfate) disaccharide composition relies on their treatment with heparin lyases that function as eliminases affording unsaturated ΔUA (deoxy-α-L-threo-hex-4-enopyranosyluronic acid)-residues (Linhardt 2001) followed by liquid chromatography–mass spectrometry (LC-MS). Since action of the heparin lyases results in loss of chirality at the C5 position, such analysis cannot distinguish between disaccharides arising from GlcA and IdoA (Eq. 3)
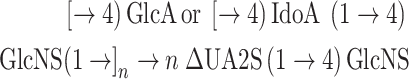 |
(3) |
Kinetic analysis of C5-epi has been performed on NSH using cofactor recycling through a three-enzyme reaction involving C5-epi with excess 2OST and arylsulfotransferase isozyme IV (AST-IV), generating the IdoA2S-containing product (NS2S), converting PAPS to 3′-phosphoadenosine-5′-phosphate (PAP) and converting the sacrificial sulfate donor, p-nitrophenyl sulfate (PNPS) to p-nitrophenol (PNP) (detectable at 405 nm) (Burkart and Wong 1999; Sterner et al. 2014), as PAP is converted to PAPS. Because 2OST can also slowly catalyze formation of GlcA2S from GlcA (Eq. 2), the formation of PNP does not always correlate with conversion of GlcA to IdoA catalyzed by C5-epi. Moreover, this approach requires coupling C5-epi to 2OST and AST-IV, which may lead to changes in rate-determining enzyme step as a function of the reaction conditions, enzyme stability and extent of the reaction. Thus, it is necessary to develop a more direct measure of C5-epi activity.
Recognizing this problem, a radioisotopic assay relying on tritium-labeled NSH oligosaccharide has been used (Sterner et al. 2014). Both substrate and product needed to be removed from the reaction mixture prior to measuring the 3H2O produced. A related method relied on NSH and treating it with C5-epi in D2O and measuring the increase in molecular weight using LC–MS due to deuterium incorporation into the IdoA (Babu et al. 2011). However, mass-spectrometry assays may overestimate the reaction rate and conversion of C5-epi due to the increased likelihood for the reverse reaction to occur (IdoA to GlcA) as the percentage of IdoA residues in the polysaccharide substrate increases, which also results in deuterium incorporation from D2O. Moreover, substantial postreaction processing is required to analyze disaccharide content by LC–MS. Following isolation of polysaccharide product from the reaction, it must be depolymerized using nitrous acid (Conrad 2001) or reactive oxygen species (Li et al. 2014) since heparin lyases cannot be used, followed by desalting steps before disaccharide detection can take place.
Herein, we have developed both real-time (continuous) and offline (discontinuous) NMR-based analysis of C5-epi reaction kinetics. Using this assay, we studied C5-epi catalysis on NSH in the forward reaction and on chemically O-desulfated, N-sulfated heparin (CDNSH) in the pseudo-reverse reaction and evaluated the impact of various reaction and substrate parameters on enzyme activity. NMR was used to follow the epimerization reaction with essentially no work-up required. A set of experiments was undertaken to better understand the steady-state kinetics of the epimerization reaction and to elucidate some of the factors that impact C5-epi catalysis using the polymeric, multicomponent NSH substrate that is both relevant in heparin biosynthesis and directly used in bioengineered heparin production. This study provides important mechanistic insight on C5-epi for use in the chemoenzymatic synthesis of bioengineered heparin.
Results
C5-epi catalyzes a key early step in heparin (or heparan sulfate) biosynthesis by reversibly converting GlcA into IdoA (Figure 1A and B). Analysis of C5-epi reaction kinetics has typically employed a coupled multienzyme colorimetric assay relying on C5-epi, 2OST and AST-IV (Figure S1). The NS2S product can be quantified by heparin lyase 1,2 and 3-catalyzed depolymerization of NS2S to disaccharides, ΔUA2S (1 → 4)GlcNS and ΔUA(1 → 4)GlcNS, followed by LC–MS. Unfortunately, this disaccharide analysis cannot distinguish between IdoA2S and GlcA2S and requires multiple steps involving multiple enzymes. As a result, we proceeded to examine whether an NMR assay could be used to reliably follow this reaction.
Fig. 1.

C5-epimerase catalyzes the conversion of GlcA to IdoA. This reaction is reversible and is skewed towards the formation of the more thermodynamically favored GlcA. (A) Forward reaction using NSH and (B) pseudo-reverse reaction using CDNSH. The protons deuterium exchanged at the C5 positions of the GlcA and the IdoA residues are shown in blue. (C) NMR-based assay that is used to detect directly the presence of GlcA and IdoA within the reaction based on differences in chemical shifts of the anomeric proton of GlcA and IdoA. The peak area as a function of the internal standard (DSS) provides quantitative amounts (in mM) of GlcA and IdoA within the reaction. This figure is available in black and white in print and in color at Glycobiology online.
In silico design and expression of C5-epimerase
C5-epi is a eukaryotic membrane-bound enzyme localized on the inner membrane of the Golgi. Like most membrane-associated proteins, human C5-epi has low solubility. We used a previously reported transmembrane domain-truncated, maltose-binding protein (MBP) fusion protein of C5-epi (referred to as “wild-type” since it contains an unmodified sequence in the remaining residues) to improve enzyme solubility (Hagner-McWhirter et al. 2000; Li et al. 2001; Crawford et al. 2001; Liu et al. 2010). In this version, 52 residues at the N-terminus were deleted and the clone was codon optimized. C5-epi wild type was expressed from E. coli Origami B (DE3) using a pMAL expression vector as a variant of the human enzyme. We also designed in silico an engineered version of this truncated, fusion protein using the PROSS algorithm (referred to as the “engineered” enzyme) (Goldenzweig et al. 2016). This computational design relied on the C5-epi zebra fish crystal structure (Qin et al. 2015; Debarnot et al. 2019) to exclude mutations in or near the catalytic and substrate-binding sites of C5-epi. The resulting engineered C5-epi contained 35 amino acid mutations (Figure S2 and Table SI) from the wild-type sequence and gave ~ 8-fold higher soluble expression yield from E. coli than the wild-type enzyme. Both enzyme variants were used in this work. The purity of the cell lysate for wild-type and engineered enzyme was 3 and 4%, respectively (Figure S3). Following purification using an amylose column, the purity of the wild-type and engineered enzyme was 66 and 69%, respectively (Figure S3).
Model system and assay optimization
Full-length polydisperse NSH (Mw 21,000 Da, corresponding to ~ 45 disaccharide residues) primarily → 4) GlcA (1 → 4) GlcNS (1 → was used to assess the forward reaction converting GlcA residues to IdoA residues (Figure 1A). This forward reaction is directly related to the production of bioengineered heparins. CDNSH (~7000 Da, corresponding to ~17 disaccharide residues) primarily → 4) IdoA (1 → 4) GlcNS (1 → was also used to assess the pseudo reverse reaction converting IdoA residues to GlcA residues (Figure 1B). The NSH used in this study contained ~10% GlcNAc (90% GlcNS), which is relevant to heparin biosynthesis.
We used both real-time and off-line NMR spectroscopy in D2O to exploit the different chemical shifts of the H-1 (anomeric proton) of GlcA and IdoA in the 1H-NMR spectrum (Figure 1C) to obtain quantitative information on the concentrations of GlcA and IdoA in their polysaccharide substrates during the course of the enzymatic reaction as opposed to the more commonly used NSH oligosaccharide fragments (Hagner-McWhirter et al. 2000; Sheng et al. 2012; Qin et al. 2015). Experimental parameters were first optimized to ensure that the integral values or peak areas as a function of the internal standard, 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), were accurate. Standard curves for NSH and CDNSH were constructed (Figure S4A and B). The integral signals were linear for both substrates at concentrations up to at least 50 μM (based on the Mw of the substrates).
The 1H-NMR of CDNSH (Figure S4B) afforded a calculated ratio of 77% IdoA to 23% GlcA ([slope IdoA/slope GlcA] × 100) similar to that obtained using disaccharide analysis by nitrous acid followed by HPLC, an orthogonal technique. Due to the proximity of the IdoA (4.96 ppm) and GlcA (4.55 ppm) peaks to the water (HOD) peak (4.7 ppm), tailing was observed in both GlcA and IdoA signals that influenced the integral values. Fitting each of the peaks to a Lorentzian function reduced the error associated with this tailing. Since each of the time points were obtained from separate samples, the instrument was recalibrated between sample runs during the offline experiments (e.g., locking, tuning, matching and shimming). However, these recalibration steps did not impact the analysis. Continuous, real-time, NMR experiments involving a series of online 1D 1H measurements as a function of time were also used to validate the discontinuous offline measurements.
The activity of C5-epi was initially measured using both offline and real-time NMR with 5 μM NSH or CDNSH and 0.5 mg/mL C5-epi using the engineered enzyme. For 5 μM NSH at a weight average molecular weight of 21,000 Da (0.1 mg/mL), this represents 48 disaccharide units (~240 μM). The enzyme concentration was 0.5 mg/mL, which translates into 4.7 μM. Thus, at the lowest NSH concentration in the ratio of substrate:enzyme was 51, assuming all GlcA residues were accessible to C5-epi. Even if all such residues were not accessible to C5-epi, the stoichiometry suggests that a steady-state assumption can be made.
The integral areas were linear as a function of enzyme concentration (Figure S4C). The intensity of the IdoA H-1 signal increased over time for NSH (Figure 2A) and similarly the intensity of the GlcA H-1 signal increased over time for CDNSH as substrate (Figure S5). Online NMR experiments further confirmed the results of the off-line experiments (Figure 2B). Specifically, an increase in the IdoA signal was observed and was visualized using Topspin 3.2.1 under contour mode in the raw real-time NMR spectra. Good agreement was observed between both the real-time and offline experiments (Figure S6A). We next quantified IdoA by depolymerizing the product using nitrous acid and then measuring GlcA and IdoA content using HPLC to further confirm that the NMR methods provided accurate IdoA and GlcA content during C5-epi catalysis. The results obtained using this approach were similar to those using either of our two NMR methods (Figure S6B and C). We also examined a kinetic isotope effect using nitrous acid depolymerization and HPLC-based separation. The reaction rate in D2O was 1.78 ± 0.03 slower than in H2O for the forward reaction with 5 mg/mL NSH and 1.71 ± 0.04 slower for the reverse reaction with 5 mg/mL CDNSH. This is within the range of the kinetic isotope effects observed for other enzymatic studies performed using hydrogen-deuterium exchange (Cleland 2003).
Fig. 2.

Offline and real-time NMR changes in the H-1 anomeric signal of IdoA. (A) Spectral overlay using the offline experiments using the engineered enzyme with NSH as the substrate, where the IdoA H-1 peak increases as a function of time. The colored lines represent: blue, 15 min; red, 30 min; green, 45 min; purple, 60 min and yellow, 90 min. (B) Real-time 1D 1H-time spectrum showing an increase in signal corresponding to IdoA H-1 as a function of time (Topspin 3.2.7, contour mode). This figure is available in black and white in print and in color at Glycobiology online.
Equilibrium constant determination
The reaction equilibrium was determined using the engineered enzyme by measuring the mole fraction of IdoA in NSH (moles IdoA/(moles GlcA + IdoA)) at 10 h reaction as a function of NSH substrate concentration (from 0 to 60 μM) and at a high C5-epi concentration (3 mg/mL). In all cases, the fraction of IdoA plateaued after 20 min (Figure 3A). Purification of the product and the addition of fresh engineered C5-epi (3 mg/mL) at the 10 h time point did not result in an increase in IdoA content at any of the initial NSH concentrations, suggesting that equilibrium had been reached at 30 ± 5% IdoA. To confirm this equilibrium IdoA concentration, we reacted C5-epi with different concentrations of CDNSH (0–150 μM). This resulted in reduction of IdoA in the pseudo-reverse reaction to an identical percentage as that observed with NSH in the forward reaction (Figure 3B). Similar results were obtained for the wild-type enzyme, which was expected given the independence of equilibrium on the biocatalyst. This measured equilibrium (~30% IdoA and ~ 70% GlcA) agrees with previous reports (Figure 3A and B) (Hagner-McWhirter et al. 2004; Qin et al. 2015; Debarnot et al. 2019).
Fig. 3.

Determination of GlcA/IdoA equilibrium in the forward and pseudo-reverse reactions. (A) Repeated treatment of 3 mg/mL of engineered C5-epi at various concentrations of NSH yielded a plateau in the concentration of IdoA produced, indicating that equilibrium had been reached. (B) Repeated treatment of 3 mg/mL of C5-epi at various concentrations of CDNSH. Error bars are shown on representative points, n = 3.
C5-Epi steady-state reaction kinetics
Steady-state kinetics of C5-epi on NSH using both the wild-type and engineered enzymes constructs followed a nonstandard kinetic profile (Figure 4A and B) with significant sigmoidal behavior. Saturation kinetics were observed at high concentrations of NSH, and the resulting apparent kinetic constants are summarized in Table I. In this table, K50 values are reported, which represents the substrate NSH concentration at half-maximal velocity. Because of non-Michaelis–Menten behavior, we do not use the term Km for the C5-epi reaction. The engineered enzyme had a calculated higher maximal rate but also higher K50 than the wild-type enzyme. The Hill coefficients were roughly similar for both enzyme variants (Table I), and indicated severe sigmoidal behavior. Interestingly, the reverse reaction with CDNSH yielded a nonsaturating linear plot with an apparent Vmax/Km of 5.54 ± 0.23 min−1 for the wild-type and 22.9 ± 0.93 min−1 for the engineered enzyme, respectively (Figure 4C and D).
Fig. 4.

Steady-state kinetic profiles for C5-epi wild type and engineered, MBP-fusion, constructs. (A) Wild-type enzyme acting on NSH; (B) engineered enzyme acting on NSH; (C) wild-type enzyme acting on CDNSH and (D) engineered enzyme acting on CDNSH. Error bars (n = 3) are within the plot symbols.
Table I.
Kinetic parameters of the wild-type and engineered enzymes
| (Vmax)apparent (μmol/(mg enzyme-min)) | K 50 (μM) | Hill coefficient | |
|---|---|---|---|
| Wild-type | 275 ± 12.4 | 10.1 ± 0.14 | 7.62 ± 0.93 |
| Engineered | 360 ± 5.64 | 29.2 ± 0.19 | 7.12 ± 1.41 |
| Engineered +25 mM CaCl2 + 25 mM MgCl2 | 268 ± 15.4 | 5.42 ± 0.12 | ~1 |
| Engineered +2OST | 440 ± 9.80 | 14.2 ± 0.92 | ~1 |
The impact of various additives, including monovalent and divalent metals, surfactant, and the presence of 2OST (without added PAPS), on C5-epi catalysis was examined at 5 μM NSH to further investigate the mechanism underlying the sigmoidal behavior of the forward reaction on NSH (Table II and Figure S7). An NSH concentration of 5 μM was chosen, as it is sufficiently low to be impacted by the sigmoidal regime of the kinetic plot. The engineered enzyme had an initial rate ~3.75-fold faster than the wild-type enzyme, and thus, we focused on the engineered enzyme for this part of the study. Several additives negatively influenced C5-epi activity. For example, Triton X-100 at its critical micelle concentration (CMC) of 0.015% (w/v) reduced C5-epi activity ~3-fold, and this was similar at Triton X-100 concentrations above and below the CMC. Triton X-100 did not alter the sigmoidal kinetic behavior of the forward reaction on NSH resulting in an apparent Vmax of 200 ± 5.88 μmol/(mg enzyme-min), an apparent K50 of 28.7 ± 0.4 μM, and a Hill coefficient of 6.9 ± 0.19 (Figure S7), and therefore, simply reduced enzyme turnover with no impact on apparent substrate affinity. Interestingly, a combination of CaCl2 and MgCl2 at 25 mM each resulted in more conventional saturation kinetics with a Vmax of 268 ± 15.4 μmol/(mg enzyme-min), a K50 of 5.42 ± 0.12 μM, a Hill coefficient of ~1.0, and a nearly 4-fold increase in the initial rate of C5-epi catalysis at 5 μM NSH (Figure 5). Moreover, the presence of 25 mM MgCl2 largely eliminated the sigmoidal behavior, albeit with lower activity.
Table II.
Activity of C5-epimerase as a function of reaction additives. Enzyme and NSH concentrations were 0.5 mg/mL and 5 μM NSH, respectively. Except where indicated, the engineered C5-epi was used
| Additive | Initial rate (μmol/(mg enzyme-min)) |
|---|---|
| None (wild type) | 9.6 ± 1.4 |
| None (engineered) | 36 ± 1.2 |
| Triton X (0.0075%) | 11.6 ± 1.4 |
| Triton X (0.015%) | 9.8 ± 1.0 |
| Triton X (0.03%) | 10.2 ± 1.4 |
| CaCl2 (25 mM) | 18 ± 1.2 |
| CaCl2 (50 mM) | 5.6 ± 3.4 |
| MgCl2 (25 mM) | 6.4 ± 0.4 |
| MgCl2 (50 mM) | 10 ± 1.8 |
| CaCl2 (10 mM) + MgCl2 (40 mM) | 36 ± 3.6 |
| CaCl2 (40 mM) + MgCl2 (10 mM) | 15.4 ± 1.6 |
| CaCl2 (25 mM) + MgCl2 (25 mM) | 136 ± 3.0 |
| 2OST (0.5 mg/mL) | 134 ± 2.0 |
Fig. 5.

Steady-state kinetic profiles for engineered C5-epi (0.5 mg/mL), acting on NSH with no additive (black), with 2OST (0.5 mg/mL) added as an additive without PAPS (red) or with added 25 mM CaCl2 + 25 mM MgCl2 (green). Error bars (n = 3) are within the plot symbols. This figure is available in black and white in print and in color at Glycobiology online.
Finally, because C5-epi acts in concert with 2OST, possibly as a complex with the 2OST (Hagner-McWhirter et al. 2004; Préchoux et al. 2015) in the heparin/heparan sulfate biosynthetic pathway, we tested the addition of 2OST (0.5 mg/mL in the absence of PAPS to prevent 2OST catalysis) to the C5-epi reaction on NSH. In the absence of the PAPS cofactor, 2OST is completely inactive, yet remains structurally intact. Under these conditions, a significant increase in initial rate was observed on 5 μM NSH and the kinetic profile was consistent with a more conventional Michaelis–Menten behavior, yielding a Vmax of 440 ± 9.8 μmol/(mg enzyme-min) and K50 of 14.2 ± 0.92 μM (Figure 5). 2OST, once processed to act as an additive, was also assayed in the presence of PAPS and it generated NS2S indicating that 2OST was active.
We examined enzyme stability at 37°C in the presence of 25 mM each of CaCl2 and MgCl2 or 2OST in relation to the absence of an additive to assess whether enzyme deactivation impacted the observed sigmoidal kinetic behavior. In all cases, C5-epi activity decreased over time (Figure 6A). Interestingly, the no additive incubation was more stable than in the presence of the cation mixture or 2OST; the deactivation rate constants were 0.04, 0.06 and 0.06 h−1 for the no additive, + CaCl2 and MgCl2, and + 2OST, respectively.
Fig. 6.

Stability studies for c5-epi. (A) C5-epi upon incubation at 37°C for 6 h and 24 h was evaluated for kinetic stability in the presence of specific additives. (B) DLS experiments were performed to evaluate the effect of additives on C5-epi and on the NSH substrate after incubation at 37°C for 0 and 30 min. (i) 0.5 mg/mL of C5-epi in 50 mM D-MES, pH 7.0. (ii) 0.5 mg/mL of C5-epi in 50 mM D-MES, pH 7.0 in the presence of 25 mM CaCl2 and 25 mM MgCl2. (iii) 0.5 mg/mL of C5-epi in 50 mM D-MES, pH 7.0 in the presence of 0.5 mg/mL of 2OST. (iv) 5 mg/mL of NSH in 50 mM D-MES, pH 7.0. (v) 5 mg/ml of NSH in 50 mM D-MES, pH 7.0 in the presence of 25 mM CaCl2 and 25 mM MgCl2.
We also examined the physical stability (i.e., aggregation) of the C5-epi and the NSH substrate using dynamic light scattering (DLS). The hydrodynamic diameter of the enzyme was identical in the absence of additive (Figure 6B) or in the presence of CaCl2 and MgCl2 (Figure 6B) at time zero (~5.9 nm). After 30 min, which was the incubation time scale for the enzymatic reaction, the hydrodynamic diameter of the enzyme increased to ~7.7 nm in both cases (Figure 6B); however, there was no difference in the presence or absence of the divalent cation mixture. In the case of the addition of 2OST, the enzyme size at time zero was slightly increased vs. C5-epi alone (Figure 6B), which indicates that the C5-epi and 2OST associate, as has been suggested by Préchoux et al. (2015). However, after 30 min, this size did not change. It is possible that the additives, particularly the divalent cations, can affect the size of the NSH. DLS analysis (Figure 6B) of the NSH in the absence of the C5-epi shows that the NSH size (hydrodynamic diameter of ~11 nm) remained unchanged over time. This size is indicative of an extended polysaccharide chain given the Mw = 21,000 Da. Moreover, the presence of the divalent metal mixture had essentially no effect of NSH size indicating that the presence of the metal ions does not influence the structural features of the polysaccharide.
Discussion
We have developed and optimized quantitative, NMR-based, discontinuous and real-time assays to measure the specific activity and determine the steady-state reaction kinetics of C5-epi on long-chain polymeric and polydisperse substrates. We have used both a wild type, membrane-domain truncated enzyme and an in silico engineered enzyme that was codon and sequence optimized and expressed at high levels in E. coli. While the engineered enzyme is more soluble and has higher activity than the wild-type enzyme, it is important to note that both enzymes show similar kinetic behavior, which suggests that the improved solubility of the engineered variant does not affect significantly the kinetic behavior of the C5-epi. Discontinuous NMR kinetic analysis provided information on reaction equilibrium and steady-state kinetic parameters. The reaction equilibrium position of ~70 ± 5% GlcA and ~30 ± 5% IdoA was the same for both wild type and engineered C5-epi constructs (as expected due to the independence of reaction equilibrium on the catalyst) and these values are consistent with previous reports (Liu et al. 2010; Qin et al. 2015; Debarnot et al. 2019). Interestingly, steady-state kinetic analysis for both wild-type and engineered enzymes yielded sigmoidal kinetic behavior for the forward reaction with NSH. This behavior has not been previously reported. Therefore, it may be possible the higher molecular weight of NSH (21,000 Da) substrate compared to the NSH oligosaccharides of ~1500 Da used in previous kinetic studies (Hagner-McWhirter et al. 2004; Sheng et al. 2012; Qin et al. 2015; Debarnot et al. 2019) played a role in these unusual kinetic properties. Indeed, (Sheng et al. 2012) showed an interesting irreversibility of C5-epi catalysis when acting on NSH-like oligosaccharides; this is distinct from C5-epi catalysis reported (Hagner-McWhirter et al. 2004; Sheng et al. 2012; Qin et al. 2015; Debarnot et al. 2019) previously on NSH substrates that are fully N-sulfated. This is clearly distinct from the current study, which employed both full-length NSH that is ~90% N-sulfated. Nonsaturating kinetic behavior was observed for the pseudo-reverse reaction using CDNSH as substrate for both wild-type and engineered enzymes, suggesting that the conversion of an IdoA residue to a GlcA residue in the polysaccharide is associated with a very low apparent substrate affinity.
We hypothesize that the atypical sigmoidal behavior of the forward reaction may be due to one or more factors, including the physicochemical state of the higher molecular weight NSH and/or the structural features of the C5-epi. C5-epi has not been shown previously to be under allosteric control by various reaction medium additives, although the enzyme may form a dimer in solution (Hagner-McWhirter et al. 2004; Qin et al. 2015; Debarnot et al. 2019). Nevertheless, the highly negatively charged state of the polymeric substrates used in this study may play a role in the observed allosteric behavior. Based on homology models, there is a positively charged pocket on the distal end of the catalytic site and this site is more positively charged in the engineered enzyme than in the wild type (Figure 7). It is possible that charge–charge interactions between the positively charged enzyme site and the negatively charged NSH could cause structural changes in the enzyme leading to some degree of allostery. Alternatively, potential nonspecific binding of the NSH to the C5-epi may result in restricted availability of the NSH to the enzyme’s active site (Mehrabi et al. 2019). This is consistent with the higher K50 for the engineered enzyme vs. the wild-type enzyme. The ability of 2OST to ameliorate the allosteric behavior may be consistent with this mechanism. Since these two enzymes appear to form a complex in vitro (Préchoux et al. 2015) and in vivo (Pinhal et al. 2001), it is possible that the 2OST-C5-epi complex blocks this positively charged pocket of C5-epi even at low NSH concentrations overcoming the allosteric behavior (Préchoux et al. 2015).
Fig. 7.

Homology models of both enzyme variants used in this study using the crystal structure of the human C5-epi28 as template. (A) Homology model of the wild-type enzyme with the active site residues in yellow. (B) Homology model of the engineered enzyme. In pink are the residues mutated from the wild type and in yellow are the active site residues. (C) and (D), are catalytic sites of the wild-type and engineered enzymes, respectively, (E) and (F) are the distal end of the catalytic site, a positively charged cleft, of the wild-type and engineered enzymes, respectively. In blue are positively charged and in red are negatively charged residues. This figure is available in black and white in print and in color at Glycobiology online.
The mechanism of how CaCl2 and MgCl2 combined prevents allosteric behavior remains unclear. Enzyme deactivation could result in reduced observed reaction rates at very low substrate concentrations, which appear to be low in the absence of CaCl2 and MgCl2. To further probe this mechanism, enzyme stability studies were performed in the presence and absence of CaCl2 and MgCl2. However, the enzyme in the absence of CaCl2 and MgCl2 was ~50% more stable than in the presence of the divalent metal ion mixture (Figure 6A). Similarly, the presence of 2OST did not further stabilize C5-epi. Thus, enzyme deactivation was not a contributing feature toward the sigmoidal kinetic behavior. In addition to enzyme stability, potential aggregation of the enzyme may result in depressing initial rates at low substrate concentrations. To this end, DLS was performed, yet the apparent size of the enzyme was similar after 30 min in the presence or absence of CaCl2 and MgCl2. Thus, potential enzyme aggregation does not contribute to the sigmoidal kinetic behavior. Finally, the apparently extended chain conformation of NSH, based on hydrodynamic diameter (Figure 6B), is unaffected in the presence of CaCl2 and MgCl2. Thus, substrate reaction sites accessibility, which could influence reactivity differences at low substrate concentrations, is not likely to occur. We considered the potential impact of product inhibition as a cause of the sigmoidal kinetics. While idoA conversion over time was linear in the 30-min reaction period (Figures S8 and S9), this cannot fully rule out the potential for product inhibition. However, the presence of either 25 mM each of CaCl2 and MgCl2, or the 2OST resulted in substantially higher rates and conversion over the 30 min reaction period, which would lead to greater likelihood for product inhibition. Nevertheless, it is not possible to fully rule out the possibility for the presence of these additives to cause a structural change in C5-epi that could reduce the potential deleterious effects of product inhibition.
We also cannot rule out, however, the possibility that the divalent cations interact both with C5-epi and NSH to ensure tighter binding of the high molecular weight substrate to the enzyme, which would be most evident at low NSH concentrations. Indeed, recent crystallographic analysis of C5-epi indicates that the β-sandwich domain has a specific Ca2+ binding site (Debarnot et al. 2019). The K50 for C5-epi in the presence of 2OST was ~ 2-fold and in the case of CaCl2 + MgCl2 was ~ 5-fold lower than in the absence of either additive, respectively. These results are consistent with tighter interaction of NSH with C5-epi when 2OST (Préchoux et al. 2015) or CaCl2 + MgCl2 are present.
In summary, novel online and offline assays have been developed that rely on NMR spectroscopy to determine the specific activity of epimerases. We have used this assay to study comprehensively the steady-state kinetic profile of a wild type (truncated, MBP fusion) human glucuronyl C5-epimerase as well as a soluble and highly expressed protein engineered (truncated, MBP fusion) version. These studies relied for the first time, on high molecular weight polysaccharide substrates, making these relevant to our understanding of heparin and heparan sulfate biosynthesis as well as the production of bioengineered heparin. The reaction kinetics of these two C5-epi constructs were examined using both NSH and CDNSH polysaccharide substrates. The forward reaction on NSH displayed an atypical sigmoidal plot. The nature of these unusual kinetics was further investigated by using various additives. The presence of 25 mM each of CaCl2 and MgCl2, resulted in more typical Michaelis–Menten saturation kinetics and the presence of 2OST in the absence of PAPS resulted in both more typical Michaelis–Menten kinetics and a marked increase in apparent Vmax. Approaches used in this study should prove useful in the kinetic evaluation of other epimerases involved in GAG biosynthesis, such as the dermatan sulfate epimerase or chondroitin C5-epimerase (Tykesson et al. 2018). Key kinetic information obtained herein using the direct polysaccharide substrates should also provide useful insight in the optimization and large-scale production of bioengineered heparin, while providing insight into follow-on experiments to tease out more details of the unusual C5-epi kinetic profiles under specific conditions.
Materials and methods
Expression and purification of recombinant C5-epimerases
The human C5-epi (NCBI, NM_015554.3) full gene sequence was previously reported (Sheng et al. 2012) (Table SI). A truncated sequence E53-N617 with the transmembrane domain of the enzyme removed was used as previously described (Qin et al. 2015). The gene was codon-optimized for heterologous expression in E. coli using the iterative particle swarm optimization-based strategy OptimumGene (GenScript, Piscataway, NJ), then was cloned between BamHI–HindIII restriction sites of the pMAL-C3, AmpR vector (NEB) downstream from the malE gene, which encodes MBP (K20-T366), to produce a MBP-C5-epimerase fusion protein. The calculated molecular weight of the resultant fusion protein was 107.3 kDa. E. coli Origami B (DE3), TetR, KanR, (Novagen, Madison, WI) was cotransformed with the recombinant His6-MBP-C5-epi-coding plasmid and with pGro7, CmR (Takara, Kusatsu, Shiga 525-0058, Japan), coding GroES and GroEL chaperonins to facilitate the native folding of the enzyme. A single colony was picked from a selective antibiotics LB agar plate and expanded to prepare glycerol stocks (15%, v/v, glycerol) for subsequent use. The glycerol stocks were stored at –80°C.
C5-epi expression was performed using a standard fermentation protocol. Briefly, overnight bacterial suspension was subcultured at 1:50 dilution into a larger aerated vessel with Terrific Broth and four antibiotics (50 μg/mL ampicillin, 12.5 μg/mL tetracycline, 15 μg/mL kanamycin and 35 μg/mL chloramphenicol). The bacterial culture was propagated at 37°C with aeration using baffled flasks until the cell density reached OD600 of 7–8 (typically 7 h). The temperature was then lowered to 22°C and protein expression was induced by adding 0.5 mM isopropyl-β-D-thiogalactoside (IPTG, for the Ptac promoter) and 1 mM L-arabinose (for the araB promoter). The accumulated biomass was harvested after 16 h and stored frozen.
C5-epi was purified under native conditions by affinity chromatography using amylose resin (NEB, Ipswich, MA) in 25 mM Tris pH 8.0, 500 mM NaCl, and stored at –80°C in the same buffer with 10% (v/v) glycerol. The enzyme and associated chaperonins copurified in one fraction, but separated under reducing conditions, resulting in an SDS-PAGE profile with protein bands at 107 (C5-epi), 57 (GroEL) and 10 (GroES) kDa (Figure S3).
PROSS engineered C5-epimerase
A bioinformatics approach developed by Goldenzweig et al. (Protein Repair One Stop Shop (PROSS) server) was used to design a panel of mutants with iteratively increasing surface polarity. The crystal structure of the human C5-epi, resolved in a complex with heparin hexasaccharide PDB ID 4PXQ, was selected as the scaffold for the PROSS algorithm (Figure 7). The algorithm was set to base possible amino acid substitutions on sequence homologs with at least 75% of the length of the 4PXQ template. This excluded variability among all noneukaryotic epimerase enzymes. We selected a mutant with 35 amino acid substitutions (Table SI and Figure S2). Notably, none of the substitutions was within the active or substrate binding sites of the enzyme. The engineered gene was then cloned into the pMAL-C3 vector (NEB). Origami B (DE3) competent cells were transformed, and the enzyme was expressed as an N-terminal His6 and MBP fusion protein. The expression and purification were performed as described above, omitting chloramphenicol from the growth medium and L-arabinose from the induction step (Figure S3).
Heparosan-based substrates
All heparosan-based substrates were derived either from K5 heparosan or from USP heparin and obtained as previously described (Wang et al. 2011). Briefly, NSH was produced directly from the capsule of E. coli K5 followed by partial (85%) chemical de-N-acetylation using NaOH and complete N-sulfonation using sulfur trioxide triethylamine (Sigma Aldrich, St. Louis, MO) and had a weight average molecular weight (Mw) of ~21,000 and a PD of ~1.1 and contained ~90% GlcNS (Wang et al. 2010, 2011). CDNSH, the NSH analog rich in IdoA content, was prepared from heparin (Raman et al. 2015) and contained ~74% GlcNS with ~10% GlcNAc and ~87% IdoA with ~13% GlcA and a Mw of ~7000 and a PD of ~1.2.
Offline, discontinuous NMR assay
IdoA and GlcA are on either side of the water line (HOD signal), which leads to peak tailing affecting the accuracy of the integral measurements; thus, the C5-epi was buffer-exchanged into D2O (Sigma Aldrich, St. Louis, MO). A 3 K spin membrane was first equilibrated in D2O and centrifuged at 12,000 rpm for 10 min. Then, the enzyme (C5-epi with or without 2OST additive) was loaded onto the washed spin membrane and centrifuged at 12,000 rpm for 20 min. The flow-through was discarded and fresh D2O was added to the spin membrane and centrifuged at 12,000 rpm for 20 min. This step was repeated four times to ensure successful buffer exchange, and then the final volume was adjusted to result in an enzyme concentration of 0.5 mg/mL in D2O. The C5-epi reaction was maintained at pH 7.0 in 50 mM D-MES (2-(N morpholino) ethane sulfonic acid) buffer (Sigma Aldrich). Reactions were performed at 37°C in 300 μL consisting of 100 μL of 50 mM D-MES, 100 μL of substrate solution at different NSH or CDNSH concentrations and 100 μL of buffer-exchanged enzyme at different concentrations.
Aliquots (300 μL) were removed every 15 min and the reactions were terminated by heat shocking the enzyme at 95°C for 10 min. The enzyme was removed by centrifugation at 16,000 rpm for 10 min, discarding the pellet and retaining the supernatant. The supernatant was then lyophilized and resuspended in D2O and placed into an NMR microtube (5 mm outside diameter, (Norell, Morganton, NC) for analysis. NMR spectroscopy was performed on a Brüker Advance II 800 MHz spectrometer (Brüker BioSpin, Billerica, MA) with Topspin 3.2.1 software (Brüker for NMR analysis using a 1D 1H spectrum acquired with 16 scans, acquisition time of 0.991 s, with a relaxation delay of 11.5 s maintained at a temperature of 310 K).
Online, real-time NMR assay
C5-epi was buffer exchanged into D2O as described above. The substrate stock was prepared at the desired concentration in D2O. The reactions were maintained at pH 7.0 in 50 mM D-MES buffer. The reaction volume was prepared at 600 μL by adding 200 μL each of the enzyme, substrate and buffer solutions. The enzyme was added to the reaction mixture right before transferring the NMR tube to an 800 MHz spectrometer ensuring that initial rate of the reaction was measured. To this end, the reaction was transferred to a 5 mM Norell NMR microtube and quickly placed into the Brüker Advance II 800 MHz spectrometer at 310 K and the (free induction decay or time) spectra were acquired, with the size of the FID (free induction decay) set at 32,768 × 128. Data were acquired every 3 min, with 16 dummy scans 64 total scans per FID number with a relaxation delay of 11.5 s and an acquisition time of 2.5 s. Each data point was an average of the 64 scans. For all NMR experiments, 10 μM DSS (Cambridge Isotope Laboratories, Andover, MA) was used as an internal standard for quantification of GlcA and IdoA concentrations. Water suppression was not used since there was a decrease in the signal of the IdoA peak especially at early times. Instead, the real-time spectra were imported into MestreNova v 1.6 to visualize the data as a 2D-stack of a series of 1D 1H spectra. Once stacked, phase and baseline corrections were performed.
The raw data from Mestrenova were then exported into a MatLab code, designed to measure the area under GlcA and IdoA peaks from the raw data. This MatLab code was used to integrate the peaks, subtract the noise and also avoid the noise from the water line. This code was also used to fit the peaks to a Lorentzian to ensure that there was true signal and also to ensure that there was no error during manual integration. The signal-to-noise ratio was calculated to be three at the lowest NSH concentration used, and this ratio increased as NSH concentration increased. Three biological replicate experiments were performed to incorporate error. Data were acquired for a total of 410 min, but initial rate was calculated from 0 to 120 min (see Figures S8 and S9). The data were exported into Mestrenova as mentioned above and the in house Matlab code was used to determine integral values as a function of time. Areas under the peaks thus measured as a function of the internal standard, to provide real-time measurements of product formed and substrate consumed as a function of time. A linear regression model was used to determine the slope, which was reported for initial rate, the r2 value for the model was above 0.9 in all cases. Three biological replicate experiments were run in order to incorporate error.
Online, real-time NMR assay to assess the effect of additives
Engineered C5-epi was buffer exchanged in D2O as described above. The reaction was maintained at pH 7.0 using D-MES. The various additives were added to 200 μL of buffer at the desired concentrations. The total reaction volume was maintained at 600 μL as described above with 200 μL each of the components of the reaction. Online NMR experiments were performed as described above. In the case of 2OST as an additive, 2OST was also buffer exchanged in D2O just as in the case of C5-epi using a spin membrane. C5-epi and 2OST were mixed at a final concentration of 0.5 mg/mL each. 2OST was also examined using disaccharide analysis, separately in the presence of PAPS to ensure that the activity and stability of the enzyme was not compromised from the buffer exchange. The reaction volume was maintained at 600 μL with 200 μL each of the enzyme mixture, buffer and substrate. Online NMR experiments were also performed a described above.
Nitrous acid depolymerization and HPLC-based detection
NSH samples were prepared to a final stock concentration of 1 mg/mL. The samples (30 μL) were mixed with 60 μL of 2 M citric acid solution in wells of a 96-well plate, then 30 μL of 700 mM sodium nitrite solution was added to the wells and mixed. The 96-well plate was sealed and incubated at 65°C at 600 rpm for 2 h using a plate shaker. Then, 60 μL of 2,4-dinitrophenylhydrazine (Spectrum Laboratories, Rancho Dominguez, CA) (100 mM in acetonitrile) was added and mixed well. Incubation was performed at 45°C, 600 rpm for 2 h on the plate shaker. When the reactions were terminated, the solutions were transferred into HPLC vials. HPLC was performed on an Agilent 1200 HPLC (Agilent, Santa Clara, CA) with a 365 nm detector with mobile phase A consisting of 50 mM ammonium formate, pH 4.5, and mobile phase B being acetonitrile. Separation was performed at a flow rate 650 μL/min at room temperature on 5 μm hydrosphere C18 120 Å, 250 mm column (YMC, Yawata City, Kyoto, Japan). The sample injection volume was 8 μL, and a gradient was run between 10 and 70% MPB in 24 min.
Enzyme stability experiments
The stability of C5-epi was evaluated by incubating the enzyme in the presence and absence of additives of interest at 37°C for 0, 6 and 24 h. Once incubated at the given times, 5 mg/mL of NSH was added and formation of IdoA determined over time, as described above. The conditions tested were C5-epi in the presence or absence of 25 mM CaCl2 and 25 mM MgCl2, and C5-epi in the presence of 0.5 mg/mL of 2OST (in the absence of PAPS). The experiment was performed with three biological replicates.
Dynamic light scattering measurements
The effect of the additives of interest was evaluated on C5-epi in solution as well as the substrate (NSH) in solution using DLS (Litesizer 500, Anton Paar, Graaz, Austria) To test the effect of the additives, C5-epi or NSH was incubated at 37°C in the presence and absence of with 25 mM CaCl2 and 25 mM MgCl2 or 0.5 mg/mL of 2OST for 30 min. DLS measurements were acquired using the low-volume disposable cuvette (Uvette, Anton Paar, Graaz Austria) for three technical and three biological replicates.
Evaluation of kinetic isotope effect
C5-epi was buffer exchanged in the same way as described above into H2O and D2O. NSH (5 mg/mL) and CDNSH (5 mg/mL) were resuspended in H2O and D2O. In triplicate, NSH and CDNSH were resuspended in H2O and D2O and was treated with C5-epi buffer exchanged in H2O and D2O, respectively, by using 50 mM H-MES or D-MES at pH 7.0, at 37°C for 30 min. Reactions were terminated by heat shocking the protein at 95°C for 10 min. Centrifugation at 16,000 rpm for 10 min was performed to separate the aggregated protein. The supernatant containing the product was freeze-dried and depolymerized using nitrous acid. HPLC was used to evaluate the IdoA and GlcA obtained in the reaction as described above. Initial rates were calculated in a protonated environment, as well as in a deuterated environment, and the kinetic isotope effect was determined based on the initial rates obtained.
Supplementary Material
Acknowledgments
This work was supported by the Heparin Applied Research Center (J.S.D. and R.J.L.) and National Institutes of Health DC111958 and CA231074 (R.J.L.). The authors acknowledge Dr. Daisuke Mori who provided the CDNSH substrate used in this work and Dr. Marc Douaisi for helpful discussions. The authors would like to acknowledge Dr. Runye Zha for use of the DLS instrument and Tanner Fink for DLS training. Use of and assistance by the CBIS NMR and Microbiology Core Facilities are acknowledged, particularly Dr. Joel Morgan in the Microbiology Core.
Abbreviations
- CDNSH
chemically desulfated N-sulfated heparin
- C5-epi
glucuronyl C5-epimerase
- D-MES
deuterated 2-(N-morpholino) ethanesulfonic acid
- GlcA
glucuronic acid
- GA
glycosaminoglycan
- HPLC
high-performance liquid chromatography
- IdoA
iduronic acid
- NSH
N-sulfoheparosan
- NMR
nuclear magnetic resonance
- OD600
optical density at 600 nm
- 2OST
2-O-sulfotransferase.
Conflict of interest statement
None declared.
References
- Babu P, Victor XV, Nelsen E, Nguyen TK, Raman NK, Kuberan B. 2011. Hydrogen/deuterium exchange-LC-MS approach to characterize the action of heparan sulfate C5-epimerase. Anal Bioanal Chem. 401:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar U, Sterner E, Hickey AM, Onishi A, Zhang F, Dordick JS, Linhardt RJ. 2012. Engineering of routes to heparin and related polysaccharides. Appl Microbiol Biotechnol. 93:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar U, Li G, Fu L, Onishi A, Suflita M, Dordick JS, Linhardt RJ. 2015. Combinatorial one-pot chemoenzymatic synthesis of heparin. Carbohydr Polym. 122:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart MD, Wong CH. 1999. A continuous assay for the spectrophotometric analysis of sulfotransferases using aryl sulfotransferase IV. Anal Biochem. 274:131–137. [DOI] [PubMed] [Google Scholar]
- Conrad HE. 2001. Nitrous acid degradation of glycosaminoglycans. Curr Protoc Mol Biol. 17:17–22A. [DOI] [PubMed] [Google Scholar]
- Crawford BE, Olson SK, Esko JD, Pinhal MAS. 2001. Cloning, golgi localization, and enzyme activity of the full-length heparin/heparan sulfate-glucuronic acid C5-epimerase. J Biol Chem. 276:21538–21543. [DOI] [PubMed] [Google Scholar]
- Cress BF, Bhaskar U, Vaidyanathan D, Williams A, Cai C, Liu X, Fu L, M-Chari V, Zhang F, Mousa SA et al. 2019. Heavy heparin: A stable isotope-enriched, chemoenzymatically-synthesized, poly-component drug. Angew Chemie. 58:5962–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarnot C, Monneau YR, Roig-Zamboni V, Delauzun V, Le Narvor C, Richard E, Hénault J, Goulet A, Fadel F, Vivès RR et al. 2019. Substrate binding mode and catalytic mechanism of human heparan sulfate d-glucuronyl C5 epimerase. Proc Natl Acad Sci USA. 11:6760–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyerabend TB, Li J-P, Lindahl U, Rodewald H-R. 2006. Heparan sulfate C5-epimerase is essential for heparin biosynthesis in mast cells. Nat Chem Biol. 2:195–196. [DOI] [PubMed] [Google Scholar]
- Fu L, Suflita M, Linhardt RJ. 2016. Bioengineered heparins and heparan sulfates. Adv Drug Deliv Rev. 97:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, Dym O, Unger T, Albeck S, Prilusky J et al. 2016. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol Cell. 63:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagner-McWhirter Å, Hannesson H, Campbell H, Westley P, Rodén L, Lindahl U, Li J-P. 2000. Biosynthesis of heparin/heparan sulfate: Kinetic studies of the glucuronyl C5-epimerase with N-sulfated derivatives of the Escherichia coli K5 capsular polysaccharide as substrates. Glycobiology. 10:159–171. [DOI] [PubMed] [Google Scholar]
- Hagner-McWhirter Å, Li J-P, Oscarson S, Lindahl U. 2004. Irreversible glucuronyl C5-epimerization in the biosynthesis of heparan sulfate. J Biol Chem. 279:14631–14638. [DOI] [PubMed] [Google Scholar]
- Li G, Cai C, Li L, Fu L, Chang Y, Zhang F, Toida T, Xue C, Linhardt RJ. 2014. New contaminant in heparin and determination by radical depolymerization and liquid chromatography-mass spectrometry. Anal Chem. 86:326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-P, Gong F, Darwish KE, Jalkanen M, Lindahl U. 2001. Characterization of the D-glucuronyl C5-epimerase involved in the biosynthesis of heparin and heparan sulfate. J Biol Chem. 276:20069–20077. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ. 2001. Analysis of glycosaminoglycans with polysaccharide lyases. Curr Protoc Mol Biol. 17:17-13.17–17-13.32. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ. 2003. Heparin: Structure and activity. J Med Chem. 46:2551–2554. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang Z, Linhardt RJ. 2009. Lessons learned from the contamination of heparin. Nat Prod Rep. 26:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Xu Y, Chen M, Weïwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. 2010. Chemoenzymatic design of heparan sulfate oligosaccharides. J Biol Chem. 285:34240–34249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabi P, Di Pietrantonio C, Kim TH, Sljoka A, Taverner K, Ing C, Kruglyak N, Pomès R, Pai EF, Prosser RS. 2019. Substrate-based allosteric regulation of a homodimeric enzyme. J Am Chem Soc. 141:11540–11556. [DOI] [PubMed] [Google Scholar]
- Préchoux A, Halimi C, Simorre J-P, Lortat-Jacob H, Laguri C. 2015. C5-Epimerase and 2-O-sulfotransferase associate in vitro to generate contiguous epimerized and 2-O-sulfated heparan sulfate domains. ACS Chem Biol. 10:1064–1071. [DOI] [PubMed] [Google Scholar]
- Pinhal MA, Smith B, Olson S, Aikawa J, Kimata K, Esko JD. 2001. Enzyme interactions in heparan sulfate biosynthesis: Uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc Natl Acad Sci USA. 98:12984–12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Ke J, Gu X, Fang J, Wang W, Cong Q, Li J, Tan J, Brunzelle JS, Zhang C et al. 2015. Structural and functional study of D-glucuronyl C5-epimerase. J Biol Chem. 290:4620–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W. 2003. The use of isotope effects to determine enzyme mechanisms. J Biol Chem. 278:51975–51984. [DOI] [PubMed] [Google Scholar]
- Raman K, Kuberan B, Arungundram S. 2015. Chemical modification of heparin and heparosan In: Balagurunathan K, Nakato H, Desai UR, Editors. Glycosaminoglycans: Chemistry and biology. Methods in Molecular Biology, Chapter 4. Humana Press: Springer Protocols p. 31–36. ISBN 978-1-4939-1714-3. [DOI] [PubMed]
- Sheng J, Xu Y, Dulaney SB, Huang X, Liu J. 2012. Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J Biol Chem. 287:20996–21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner E, Li L, Paul P, Beaudet JM, Liu J, Linhardt RJ, Dordick JS. 2014. Assays for determining heparan sulfate and heparin O-sulfotransferase activity and specificity. Anal Bioanal Chem. 406:525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykesson E, Hassinen A, Zielinska K, Thelin MA, Frati G, Ellervik U, Westergren-Thorsson GL, Malmström A, Kellokumpu S, Maccarana M. 2018. Dermatan sulfate epimerase 1 and dermatan 4-O-sulfotransferase 1 form complexes that generate long epimerized 4-O-sulfated blocks. J Biol Chem. 293:13725–13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan D, Williams A, Dordick JS, Koffas MAG, Linhardt RJ. 2016. Engineered heparins as new anticoagulant drugs. Bioeng Translat Med. 2:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang Z, McCallum SA, Linhardt RJ. 2010. NMR quantification for monitoring heparosan K5 capsular polysaccharide production. Anal Biochem. 398:275–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yang B, Zhang Z, Ly M, Takieddin M, Mousa S, Liu J, Dordick JS, Linhardt RJ. 2011. Control of the heparosan N-deacetylation leads to an improved bioengineered heparin. Appl Microbiol Biotechnol. 91:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, McCallum SA, Xie J, Nieto L, Corzana F, Jiménez-Barbero J, Chen M, Liu J, Linhardt RJ. 2008. Solution structures of chemoenzymatically synthesized heparin and its precursors. J Amer Chem Soc. 130:12998–13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.