

Abstract
Aims:
We sought to determine if some unclassified renal cell carcinomas (RCCs) in children and young adults that are characterized by predominantly eosinophilic cytoplasm are related to the recently described succinate dehydrogenase (SDH)-deficient RCC, fumarate hydratase (FH)-deficient RCC, or eosinophilic solid and cystic (ESC) RCC.
Methods and Results:
We reviewed 33 unclassified RCCs with predominantly eosinophilic cytoplasm in patients age 35 years or younger. Immunohistochemistry (IHC) for SDHB, FH, and CK20 (a marker of ESC) was performed on all cases. IHC for 2-succinocysteine (2SC) was performed on RCC with loss of FH labeling. Four RCC (12%) (median age 18 years) demonstrated loss of FH labeling as well as aberrant 2SC labeling, and thus were classified as FH-deficient RCCs. Importantly, none of these cases demonstrated the characteristic macronucleoli typical of FH-deficient RCC. Eight RCC (24%) (median age 20.5 years) demonstrated loss of SDHB and were reclassified as SDH-deficient RCCs. Importantly, only four of eight SDH-deficient RCC demonstrated the characteristic cytoplasmic vacuoles and inclusions of typical SDH-deficient RCC. Ten RCC (30%) (median age 27 years) were reclassified as ESC RCCs. Four of ten ESC RCC were multifocal (1 bilateral), four of ten ESC RCC occurred in males, and one patient presented with liver and lung metastases, all not previously described in ESC. Eleven RCC (33%) remained unclassified.
Conclusions:
Pathologists should have low threshold for performing FH, SDHB, and CK20 IHC when confronted with unclassified eosinophilic RCC or “oncocytoma” in young patients.
Keywords: pediatric, renal cell carcinoma, fumarate hydratase, succinate dehydratase, eosinophilic solid and cystic RCC
Introduction
Renal cell carcinomas (RCC) are uncommon in children and adolescents, accounting for less than 5% of all pediatric renal neoplasms1–4. Nonetheless, evidence indicates that pediatric RCC are clinically, morphologically, and genetically different from adult RCC5. Unlike adult RCC, the majority of pediatric RCC fall into the category of MiT family translocation RCC, which includes the Xp11 translocation RCC and t(6;11) RCC6–7. There are also two subtypes of RCC that occur almost exclusively in young patients with sickle cell trait. Renal medullary carcinoma was first described in 1995 by Davis et al and termed the seventh sickle cell nephropathy8. More recently, RCC harboring fusion of the vinculin (VCL) and anaplastic lymphoma kinase (ALK) genes have been reported in the same patient population and described as the eighth sickle cell nephropathy9–11. In addition, RCC have been documented to arise in metanephric adenofibroma, a biphasic neoplasm of childhood combining features of metanephric stromal tumor and metanephric adenoma12. RCC has also been described as a second malignancy in children with a history of neuroblastomas (NB), and was initially considered a distinct entity in the 2004 World Health Organization (WHO) classification of renal tumors13. However, the latter are now considered a provisional entity in the 2016 WHO classification as recent studies demonstrate that these neoplasms represent a heterogeneous group of RCC rather than a single entity, with many representing chemotherapy-associated MiT family translocation RCC14–16.
Despite these recent advances, many RCC in children and young adults remain difficult to classify. In particular, a significant percentage of these unclassified RCC in children and young adults have been noted to be characterized by predominantly eosinophilic cytoplasm17,18. During the last few years, three new RCC characterized by eosinophilic cytoplasm have been described. First, succinate dehydrogenase (SDH)-deficient RCC occurs in patients with Carney-Stratakis syndrome which is characterized by germline mutations of one of the SDH subunit genes A, B, C, or D. These SDH-deficient RCC demonstrate loss of immunohistochemical labeling for SDH subunit B (SDHB), which reflects the resulting destabilization of the SDH mitochondrial complex II. SDH-deficient RCC are characterized by cytoplasmic vacuoles and inclusion-like spaces containing eosinophilic fluid or flocculent material19–21. Second, fumarate hydratase (FH)-deficient RCC, associated with hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome and inactivating germline mutations in the FH gene, is characterized by prominent macronucleoli with perinucleolar clearing22–26. Immunohistochemically, these neoplasms demonstrate loss of FH expression and aberrantly overexpress 2-succinocysteine (2SC)26. Third, eosinophilic solid and cystic (ESC) RCC has been reported exclusively in adult female patients and thought to be the sporadic counterpart to one subtype of tuberous sclerosis complex (TSC)-associated RCC27,28. Histologically, the neoplastic cells contain voluminous eosinophilic cytoplasm with coarse cytoplasmic precipitates, and frequently show at least focal expression of cytokeratin 20. Whether these three entities account for a significant proportion of previously unclassified eosinophilic RCC in young patients is unclear.
In this study, we review 33 unclassified RCC from our files that occurred in children and young adults and had predominantly eosinophilic cytoplasm. We assess the morphologic features in combination with immunohistochemical labeling for SDHB, SDHA, FH, and CK20, hypothesizing that a significant number of these previously unclassified neoplasms might be reclassifiable as SDH-deficient RCC, FH-deficient RCC, or ESC RCC.
Material and Methods
Institutional review board approval.
This study was approved by the Institutional Review Board of the Johns Hopkins Hospital and other participating institutions.
Cases
We reviewed 33 cases originally diagnosed as unclassified renal cell carcinomas with eosinophilic cytoplasm in patients younger than 35 years of age. Where to draw an age cutoff to define “young” is unclear; we chose 35 years of age as our cutoff because it is a full two decades younger than the median age of occurrence of renal cell carcinoma (55 years). Moreover, while our files contained only 13 such cases under the age of 18 years, the cases in the age group of 18–35 were highly similar morphologically. The cases were collected over the years 2008–2017, with one case coming from the institutional files at the Johns Hopkins Hospital and the rest (32 cases) from the consultation files of one of the authors (PA). These 32 cases represented 12% of the 261 renal tumor consultations in patients in this age group reviewed by this author (PA) in this time frame. Thirty one cases were given a final diagnosis of unclassified RCC. Two cases which were reviewed while this study was ongoing were thought to be unclassified based on H and E sections and routine immunohistochemistry, but were reclassified following SDHB and FH immunohistochemistry which was reflexively performed based on our preliminary study results. Formalin-fixed, paraffin-embedded tissue samples and clinical data were obtained from the submitting clinicians (consultation cases) or from the pathology archives and institutional patient database at the Johns Hopkins Hospital.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on the Benchmark XT autostainer (Ventana Medical Systems Inc., Tucson, AZ) for FH (J-13; Santa Cruz Biotechnology, Santa Cruz, CA; 1:200 dilution) and CK20 (ks20.8; Dako, Carpinteria, CA; pre-diluted). IHC for SDHB (catalog #M7195; ABCAM, Cambridge, MA; 1:100 dilution) was performed on the Leica Biosystems autostainer. SDHA IHC was performed as previously described29.Although there is not yet a commercially available antibody for the detection of 2SC, the generation of a polyclonal antibody for experimental purposes has been described by Nagai et al30. IHC for 2SC was performed using this polyclonal antibody and the method described by Bardella et al31. Antigen retrieval was performed by microwaving in pH 6.0 citrate buffer for 15 minutes. The tissues were then blocked for endogenous peroxidase and incubated with 2SC polyclonal antibody (1:5000 dilution). This was followed by washing in PBS (0.1% Tween20) and incubation with anti-rabbit HRP polymer (Envision; Dako) for 30 minutes. The positive control consisted of an RCC from a genetically confirmed HLRCC patient.
For FH, SDHB, and SDHA complete absence of labeling of neoplastic cells in the presence of intact labeling of normal tissue was considered loss of expression. For CK20 and 2SC, any labeling of neoplastic cells was considered as positive result.
Results
Cases
All but one of the cases were sent for consultation of renal tumor classification; the one other case was reviewed from the in-house surgical pathology archives of The Johns Hopkins Hospital. Of note, a total of 13 cases (9 of 10 ESC RCC and 4 of 8 SDH-deficient RCC) were sent for consultation to rule out MiT family translocation RCC. The number of slides reviewed varied, with a mean of 7 slides per case (range, 1–24 slides/case).
FH-deficient RCC
Four cases (12%) demonstrated loss of FH expression as well as diffuse aberrant 2SC overexpression by IHC and were thus reclassified as FH-deficient RCC (Figures 1–2). The four patients had a median age of 18 years (range, 10 to 25 years); three patients were female while one was male. Importantly, none of these cases demonstrated papillary architecture or the characteristic macronucleoli typical of FH-deficient RCC in adults; instead, all demonstrated a nested tubular appearance and inconspicuous nucleoli that suggested oncocytoma. Along these lines, one case was submitted with a diagnosis of oncocytoma. Two of four FH-deficient tumors were partially cystic while one was multifocal. In the surrounding kidney of the latter case, 2SC immunostain highlighted multiple additional small clusters of and single neoplastic cells (Figures 1G–1I). All four FH-deficient RCC demonstrated intact SDHB labeling, and all were negative for cytokeratin 20. Clinical follow-up was available for 1 of 4 patients; she was free of disease at 24 months with no personal or family history of HLRCC.
Figure 1:
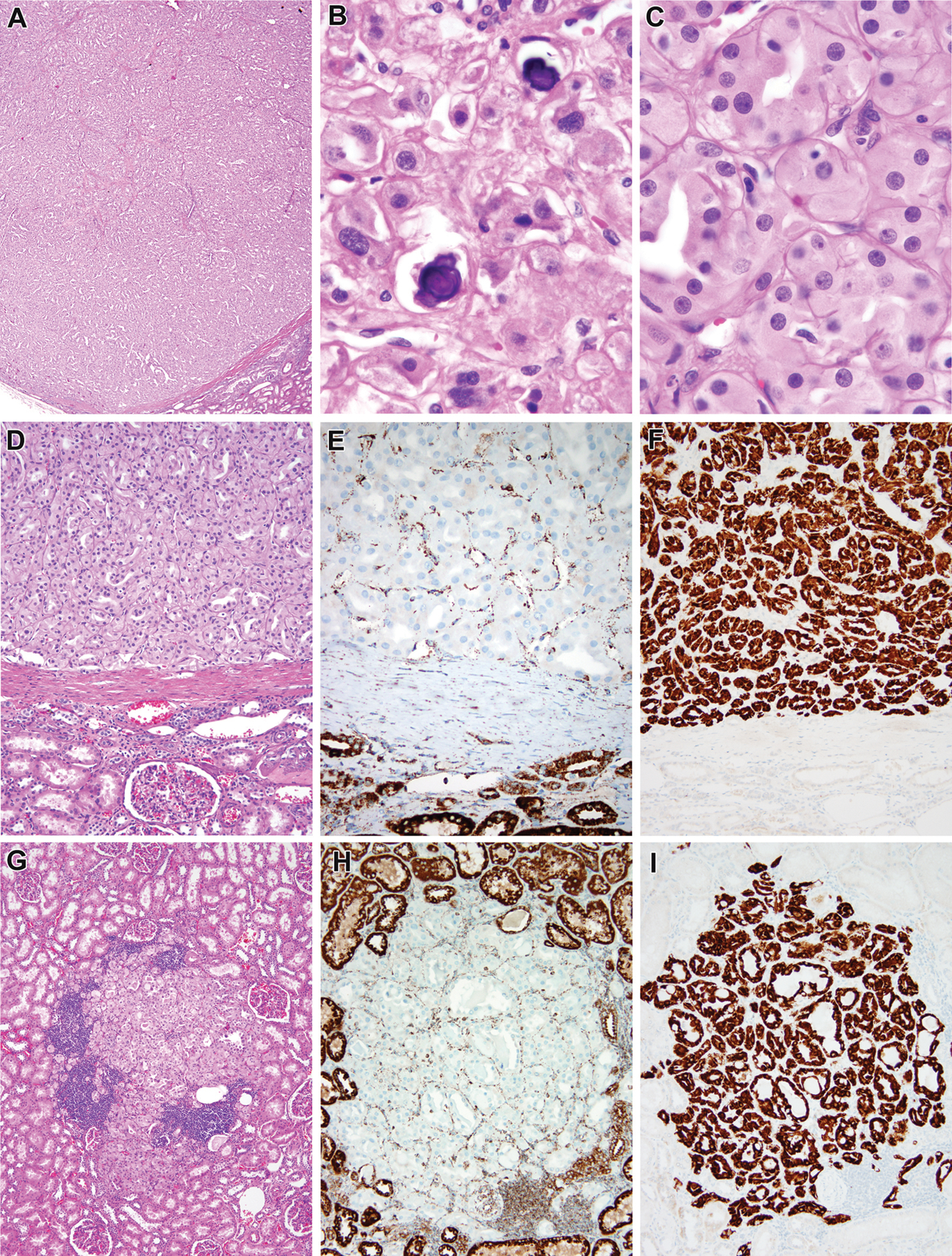
Fumarate hydratase (FH)-deficient renal cell carcinoma, multifocal (FH case #1, 25 year old male). The nephrectomy specimen contained multiple oncocytic tumor nodules. The main neoplasm and all of the others demonstrated a solid tubular architecture (A). Occasional psammoma bodies were noted (B). On high power examination, nuclei were round with punctate but non-prominent nucleoli (C). The largest tumor nodule was separated from the surrounding kidney by a thin fibrous capsule (D). These neoplasms demonstrated loss of fumarate hydratase labeling (E, topportion of the figure) and aberrant expression of 2SC (F, topof the figure). The multiple tumor nodules included an incipient unencapsulated oncocytic nodule (G) which similarly demonstrated loss of fumarate hydratase immunoreactivity (H) and aberrant expression of 2SC (I). Interestingly, single 2SC immunoreactive cells were found at the edge of this incipient nodule and elsewhere in the uninvolved kidney.
Figure 2:

Fumarate hydratase (FH)-deficient renal cell carcinoma (FH case #2, 10 year old female). This neoplasm had cystic and solid areas (A). The solid areas demonstrated compact nests and tubules of oncocytic cells (B). The cells in the solid areas and lining cysts demonstrated eosinophilic cytoplasm and punctate, non-prominent nucleoli (C). The neoplasm demonstrated loss of fumarate hydratase immunoreactivity (D) as well as aberrant expression of 2SC (E, F).
SDH-deficient RCC
Eight cases (24%) demonstrated loss of SDHB by IHC and were thus reclassified as SDH-deficient RCC. All eight SDHB IHC-negative cases demonstrated intact SDHA labeling by IHC. All six SDH-deficient RCC tested demonstrated intact FH labeling, and all five cases tested were negative for cytokeratin 20. The patients had a median age of 20.5 years (range, 17 to 34 years); 6 patients were male while 2 were female. Importantly, these cases demonstrated fewer of the cytoplasmic vacuoles and inclusions that are typical of previously reported adult SDH-deficient RCC. Only four of eight SDH-deficient RCC demonstrated cytoplasmic vacuoles and/or inclusions. Three of eight SDH-deficient RCC were submitted with the differential diagnosis of oncocytoma (one of which had been diagnosed as an oncocytoma on intraoperative frozen section), while 2 of 8 SDH-deficient RCCs mimicked the biphasic morphology of the t(6;11) RCC in that it showed clusters of small cells surrounding basement membrane material (Figure 3). These two cases had been submitted in consultation to rule out t(6;11) RCC; however, fluorescence in-situ hybridization (FISH) did not detect a TFEB gene rearrangement, and melanocytic markers were negative. One case demonstrated pleomorphism throughout and extensive necrosis, and presented with bone metastasis. This well-sampled neoplasm (all 13 sections from a 10cm tumor were reviewed) lacked cytoplasmic inclusions and vacuoles; however, a cytokeratin stain highlighted globular, cytoplasmic inclusions (Figure 4). SDHA was diffusely positive in the case but did not highlight the inclusions. Clinical follow-up was available for 2 of 8 patients. The 2 patients showed no evidence of disease at 11 and 61 months; neither had a personal or family history of Carney-Stratakis syndrome.
Figure 3:

Succinate dehydrogenase (SDH)-deficient renal cell carcinoma (SDH case #6, 17 year old male). This oncocytic neoplasm demonstrated solid, nested and acinar architecture (A). The solid nests featured eosinophilic cells with focal cytoplasmic vacuoles but no well-developed cytoplasmic inclusions (B). In multiple areas, micropapillae projected into acinar lumens and surrounded basement membrane material, simulating the appearance of the t(6;11) renal cell carcinoma (C). The neoplasm demonstrated loss of succinate dehydrogenase B immunoreactivity. Note the intact labeling of the renal tubules and stroma at the left of the lesion (D).
Figure 4:

Succinate dehydrogenase (SDH)-deficient renal cell carcinoma (SDH case #8, 34 year old male). The neoplasm has a nested, alveolar appearance (A) and is composed large pleomorphic cells with abundant eosinophilic cytoplasm. Multiple atypical mitotic figures are identified (B). Focally the neoplasm had a lower grade solid nested appearance (C) without diagnostic inclusions or cytoplasmic vacuoles. The neoplasm did demonstrate a peculiar globular staining pattern for cytokeratin (D). The neoplasm demonstrated loss of SDHB labeling (E), and presented with bony metastasis (F).
ESC RCC
Ten cases (30%) were reclassified as ESC RCCs, with a median age of 27 years (range, 14 to 35 years); 6 patients were female while 4 were male. All ten of these cases were focally immunoreactive for cytokeratin 20, and demonstrated intact FH and SDHB labeling. All 10 neoplasms demonstrated the morphologic features described recently by Trpkov et al27,28: solid and cystic architecture, voluminous eosinophilic cytoplasm and flocculent cytoplasmic precipitates, and focal CK20 expression (Figures 5–6). Four of 10 ESC RCC were multifocal (1 bilateral), while 2 of 10 ESC RCC patients had a history of a prior malignancy (bladder and CNS). One patient presented with metastatic disease involving the lung and liver (Figure 7). Clinical follow-up was available for 2 of 10 patients. The 2 patients showed no evidence of disease at 4.5 and 72 months; neither had a personal or family history of tuberous sclerosis. Of 8 cases tested, 5 (62.5%) showed focal labeling for cathepsin K by IHC. None of these cases demonstrated evidence of MiT family translocations. Three of 5 cases were negative for both TFE3 and TFEB rearrangements by fluorescence in-situ hybridization (FISH), 1 of 5 cases was negative for TFE3 rearrangement by FISH and negative for TFEB by IHC, and FISH could not be performed on 1 case due to technical difficulties.
Figure 5:

Eosinophilic solid and cystic (ESC) renal cell carcinoma (ESC case #3, 28 year old female). The neoplasm demonstrated solid and cystic architecture (A). On higher power magnification one can appreciate the eosinophilic cytoplasm of the neoplastic cells, as well as multinucleate cells lining the cysts (B). At high power, one can appreciate the precipitated, clumped cytoplasm characteristic of this neoplasm (C). The neoplasm demonstrates patchy immunoreactivity for cytokeratin 20, accentuated in the cyst lining (D).
Figure 6:

Eosinophilic solid and cystic (ESC) renal cell carcinoma (ESC case #1, 15 year old male). The neoplasm is well delineated from the surrounded kidney (left), and contains both solid and cystic areas (A). The neoplasm has a solid nested architecture with multinucleate cells associated with the cystic spaces (B). At high power one can appreciate the flocculent, clumped nature of the cytoplasm (C). The neoplasm demonstrates patchy immunoreactivity for cytokeratin 20 (D).
Figure 7:
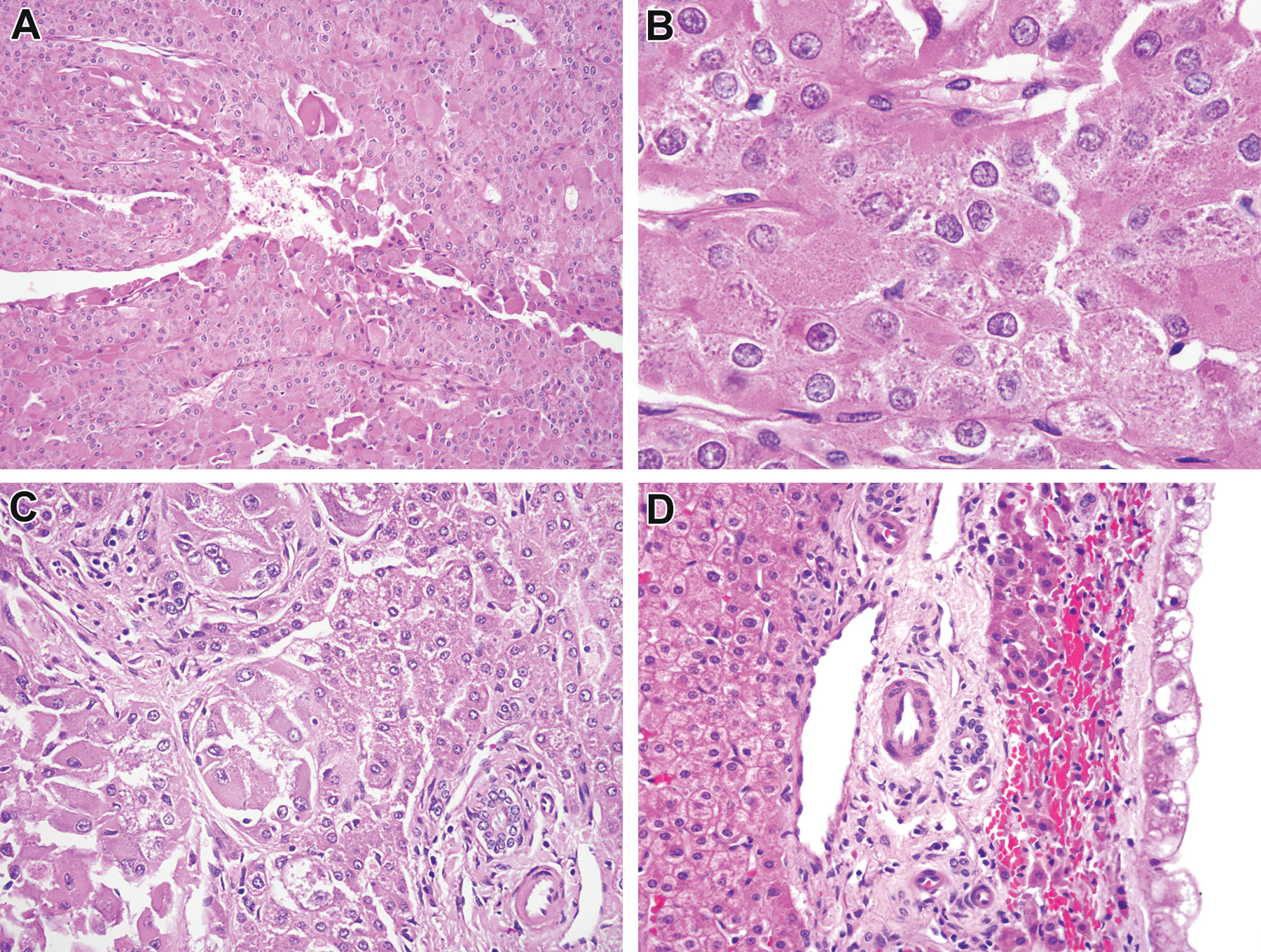
Eosinophilic solid and cystic (ESC) renal cell carcinoma associated with liver metastasis (ESC #5, 15 year old female). The neoplasm demonstrates the typical features of ESC RCC, in that it demonstrates sheets of polygonal cells with eosinophilic cytoplasm focally lining cysts (A). On high power examination, the cytoplasm demonstrates the characteristic flocculent, clumped appearance (B). This patient developed liver metastases (C, note the native liver to the leftof the figure) which were partially cystic (D).
Unclassified RCC
Eleven cases (33%) remained unclassified, with a median age of 15 years (range, 29 months to 27 years); 8 patients were female while 3 were male. Four of 11 unclassified RCCs showed some features overlapping ESC RCC, including focal CK20 expression, but lacked well-developed cytoplasmic precipitates. The remaining cases demonstrated variable morphology.
Discussion
Pediatric RCC are different from adult RCC in several ways. Most significantly, the majority of pediatric RCC fall into the category of MiT family translocation RCC. Other specific entities that predominate in young patients include renal medullary RCC and VCL-ALK RCC associated with sickle cell trait, along with RCC arising in metanephric adenofibroma. Nonetheless, a high proportion of pediatric RCC have remained unclassified. We and others have previously noted that many of these unclassified pediatric RCC are characterized by predominantly eosinophilic cytoplasm17,18. In this study, we re-evaluated a cohort of previously unclassified RCC in children and young adults of age <35 years with predominant eosinophilic cytoplasm using markers for the recently described entities of FH-deficient RCC, SDH-deficient RCC, and ESC RCC. We find that the majority of the previously unclassified RCC in young patients can now be re-classified into one of these diagnostic categories. Without knowing, one might suspect that these findings occurred simply because our cases date back to 2008, when these entities were not described, and that these were typical examples of these subsequently-described entities. However, this was not the case: our findings in young patients expand the clinical and morphologic spectrum previously described for these RCC entities.
FH-deficient RCC is associated with HLRCC syndrome, an autosomal dominant disorder characterized clinically by an inherited predisposition to aggressive uterine and cutaneous leiomyomas and RCC, and genetically by inactivating germline mutations in the fumarate hydratase gene32–34. Histologically, the neoplastic cells characteristically exhibit prominent macronucleoli with perinucleolar clearing35. Immunohistochemically, these neoplasms demonstrate loss of FH expression and 2SC overexpression. Of note, more recently RCC demonstrating FH loss and 2SC overexpression have been described in patients lacking clinical evidence of HLRCC syndrome. Some of these tumors show areas resembling tubulocystic RCC, but all have demonstrated the prominent macronucleoli that are the hallmark of HLRCC-associated RCC36. These have been referred to as FH-deficient RCC, and some are thought to harbor somatic (not germline) alterations in the fumarate hydratase gene36,37.
Although our four cases of FH-deficient RCC did show loss of FH expression with concomitant 2SC overexpression, they lacked the classic prominent nucleoli typical of previously described FH-deficient RCC. Instead, the neoplasms resembled oncocytomas, which is likely the reason that they were not initially evaluated for FH expression and thus were originally considered unclassified. To our knowledge, this unusual morphology has only been described in one case report of a FH-deficient RCC in a pediatric patient with HLRCC38 as well as recently in a small subset of adult FH-deficient RCC cases39. Chen et al. noted that macronucleoli are not uniformly present throughout HLRCC cases, but did not describe oncocytoma-like areas and noted that large nucleoli were typically seen in scattered cells26. It is unclear as to whether the FH-deficient RCC in our study are HLRCC-associated or sporadic, as germline DNA testing was beyond the scope of this consultation-based study. There were no findings suggestive of syndromic features in the limited clinical follow-up, though the multifocal presentation of FH-deficient case #1 is highly suspicious for a hereditary predisposition, especially given that HLRCC penetrance is variable32,34. Nonetheless, our findings support the routine use of FH IHC in oncocytoma-like or unclassified eosinophilic renal neoplasms in young patients, along with close follow-up and genetic testing, given the aggressive nature of HLRCC-associated RCC.
SDH-deficient RCC occurs in patients with germline mutations in one of the SDH subunit genes SDHA, SDHB, SDHC, or SDHD, though SDHB mutation is most common. Loss of immunohistochemical labeling for SDHB in SDH-deficient RCC reflects the resulting destabilization of the SDH complex. Histologically, the most distinctive feature is the presence of cytoplasmic vacuoles and inclusion-like spaces containing eosinophilic fluid or flocculent material. The 8 cases reclassified as SDH-deficient RCC in our study demonstrated minimal vacuoles and/or inclusions, which is why these cases were not initially tested for SDHB by IHC. The intact SDHA IHC indicates that this gene is likely intact in these cases, and that the mutations or other genetic alterations (such as the methylation of SDHC seen in Carney’s triad) likely affect SDHB, SDHC, or SDHD.
Two of our SDH-deficient cases exhibited oncocytoma-like features, though with higher cellularity and less edematous stroma. Two cases mimicked the t(6;11) RCC, as they demonstrated biphasic morphology and clusters of small cells surrounded by basement membrane material. One case showed prominent pleomorphism with no identifiable vacuoles or inclusions and H and E sections; however, the dot-like cytokeratin IHC labeling suggested incipient, inclusion-like cytoplasmic filament condensation, though SHDA IHC labeling did not highlight these inclusions. The significance of this pattern is not clear. In the largest study published to date, all 17 SDH-deficient RCC patients who underwent genetic testing demonstrated germline mutations in the SDHB subunit19. Although somatic SDH loss is not well described, syndromic manifestations of SDH deficiency have variable penetrance. For example, a significant subset of SDH-deficient syndromic paragangliomas (7–24%) clinically mimic sporadic paraganglioma40. As such, the absence of a family history does not exclude the presence of a hereditary syndrome. Therefore, while there were no findings suggestive of Carney-Stratakis syndrome in our cases, caution is warranted as clinical follow-up is limited. Nonetheless, our results support the routine use of SDHB IHC in evaluating unclassified eosinophilic RCC, including oncocytoma-like lesions, in young patients as well as clinical follow-up for stigmata of Carney-Stratakis syndrome.
ESC RCC was recently described by Trpkov et al. as a novel and indolent lesion occurring exclusively in adult females. It is thought to be the sporadic counterpart to one subtype of tuberous sclerosis complex (TSC)-associated RCC41,42. Histologically, the neoplastic cells contain voluminous eosinophilic cytoplasm with flocculent cytoplasmic precipitates. Immunohistochemistry usually reveals at least focal expression of CK20. No metastases or multifocal lesions were initially reported. Our cases meet the morphologic and immunohistochemical criteria for ESC RCC; however, they differ in several aspects from previously reported cases. First, these tumors occurred in young patients (median age 27 years), including 3 teenagers. Second, four of our cases occurred in males. Third, one patient presented with metastatic disease, in this case lung and liver metastases. Fourth, 4 cases were multifocal and one was bilateral, suggesting a hereditary predisposition. It should be noted that an updated study from Trpkov et al. recently published online reported one multifocal tumor out of 19 total cases28. Finally, 5 of the cases demonstrated focal labeling for cathepsin K, typically a marker of MiT family translocation RCC and PEComas. The latter is intriguing given that ESC is proposed to be a sporadic counterpart to one subtype of tuberous-sclerosis associated RCC, and the classic TSC-associated neoplasm PEComa consistently expresses cathepsin K43,44.
With regards to the cases that remained unclassified following our review, some demonstrated ESC-like morphology but lacked other important features, mainly cytoplasmic precipitates, despite focal CK20 expression. We excluded these cases, but it is possible that they are related to the ESC RCC. Similarly, given the lack of proven specificity of CK20 labeling and the somewhat subjective nature of the morphologic criteria for ESC, it is possible that some of our cases (and those in the original descriptions) are not true ESC. Such diagnostic challenges highlight the need for more specific genetic markers of this proposed tumor type.
In summary, we report that renal neoplasms with FH or SDHB loss in young patients often lack the previously described typical morphologic features, and often mimic oncocytoma. In addition, we find that ESC RCC occur in children, can affect males, are often multifocal and can metastasize. Thus, pathologists should have a low threshold for performing FH, SDHB and CK20 IHC when confronted with unclassified eosinophilic RCC or oncocytoma-like lesions in young patients. Indeed, it is now our practice to perform such IHC stains (especially FH and SHDB) when evaluating otherwise unclassifiable eosinophilic RCC in patients of any age. We think that loss of FH or SDHB in an otherwise unclassifiable RCC establishes a working diagnosis. Whether any of these cases (particularly the FH and SDH-deficient RCC) reflect germline alterations remains unclear. We did not have adequate material to address this possibility directly: there was typically only minimal normal tissue present in the blocks or unstained slides available to us, and peripheral blood was not available. Moreover, not all alterations (such as FH deletions) may be detected by many sequencing techniques. While we found no clinical evidence for a hereditary syndrome in any of our cases, our clinical follow-up was extremely limited despite maximal efforts. We attempted to directly contact the patients’ treating physicians from these consult cases (some from almost 10 years ago), but this met with little success. Hence, our findings do not exclude the possibility of familial implications. At this time, we suggest that young patients whose RCC show FH or SDH deficiency, like their adult counterparts, undergo in depth clinical assessment to evaluate for a hereditary syndrome and be offered the possibility of germline FH and SDH gene mutation analysis.
Table 1:
Fumarate Hydratase Deficient RCC
| Case | Age/Sex | Size/Stage | Notable pathologic features | Clinical history |
|---|---|---|---|---|
| FH #1 | 25/M | Multifocal. 4 nodules (4–62mm) pT1Nx |
Nested, solid and cystic, oncocytoma-like | No f/u available |
| FH #2 | 10/F | 5.5 cm, extensively cystic grossly pT1N0Mx |
Nested tubular and cystic | Presented with hypertension No h/o neoplasia or genetic syndrome NED x 24 months |
| FH #3 | 19/F | 3.0 cm pT1Nx (partial nephrectomy) |
Nested tubular; originally diagnosed as oncocytoma | No f/u available |
| FH #4 | 17/F | 16cm pT3aNX |
Nested tubular | No f/u available |
f/u= follow-up; h/o= history of; NED= no evidence of disease
Table 2:
SDH-Deficient RCC
| Case | Age/Sex | Size/Stage | Notable pathologic features | Clinical history |
|---|---|---|---|---|
| SDH #1 | 21/F | 6 cm pT1Nx |
Nested, vacuolated cytoplasm, rare inclusions | NED x 61 months, no h/o neoplasia or genetic syndrome |
| SDH #2 | 17/M | 9.1 cm pT2Nx |
Tubular with focal papillary architecture. Called oncocytoma on frozen section. |
Presented with hypertension. No f/u available |
| SDH #3 | 20/M | 7.4 cm pT2Nx |
Vacuolated with inclusions | No f/u available |
| SDH #4 | 19/M | Size unknown | Nested with vacuolated cytoplasm and rare inclusions | NED x 11 months, no h/o neoplasia or genetic syndrome |
| SDH #5 | 31/M | 15 cm pT2N0 |
Pleomorphic, vacuolated, t(6:11)-like | No f/u available |
| SDH #6 | 17/M | 6.5 cm pT1Nx |
t(6;11)-like | Presented with hematuria. No f/u available |
| SDH #7 | 22/F | 9.0 cm pT2Nx |
Vacuolated cytoplasm | No f/u available |
| SDH #8 | 34/M | 10 cm pT2NxMx |
Pleomorphic with necrosis | T12 bone metastasis at diagnosis |
f/u= follow-up; h/o= history of; NED= no evidence of disease
Table 3:
Eosinophilic Solid and Cystic Renal Cell Carcinoma
| Case | Age/Sex | Size/Stage | Notable pathologic features | Clinical history |
|---|---|---|---|---|
| ESC #1 | 15/M | Multifocal: 3.8 cm, 1.2 cm. pT1Nx (two left partial nephrectomies). Clinically bilateral. |
Focal Cathepsin K labeling | h/o brain tumor. No f/u available |
| ESC #2 | 30/F | 3 cm pT1Nx (partial nephrectomy) |
Cathepsin K negative | Presented with flank pain during pregnancy. No f/u available |
| ESC #3 | 28/F | 2.4 cm pT1Nx (partial nephrectomy) |
No h/o neoplasia or chemotherapy. No f/u available | |
| ESC #4 | 25/M | 5 cm pT1Nx (partial nephrectomy) |
Rare Cathepsin K positive cells | h/o” bladder carcinoma” at 16 months. No f/u available |
| ESC #5 | 15/F | Multifocal: 9 cm, 4cm pT4NxM1 |
Necrosis | Presented with inferior vena cava involvement and pulmonary emboli. Developed liver metastases 2 years later s/p chemotherapy. AWD x 72 months. No h/o neoplasia or genetic syndromes |
| ESC #6 | 26/F | Multifocal and cystic: 3.3 cm, 1 cm, 1 cm pT1cNxMx |
Cathepsin K positive | No f/u available |
| ESC #7 | 35/M | 1.3 cm pT1Nx |
Focally Cathepsin K positive | No f/u available |
| ESC #8 | 34/M | 2.2 cm pT1Nx |
Cathepsin K negative | No f/u available |
| ESC #9 | 32/F | 2.3 cm pT1Nx |
Cathepsin K negative | No f/u available |
| ESC #10 | 14/F | Multifocal and cystic: 8 cm, 4 cm, 2 cm, 0.5 cm. pT2NxMx |
Focally Cathepsin K positive | History of sickle cell trait NED x 4.5 months. No history of neoplasia or genetic syndrome. |
f/u= follow-up; h/o= history of; NED= no evidence of disease; AWD= alive with disease
Acknowledgement:
Supported in part by the Gary Hill Award (YL), Joey’s Wings (PA), and Dahan Translocation Carcinoma Fund (PA).
Footnotes
Disclosures: None
References:
- 1.Spreafico F, Collini P, Terenziani M, Marchianò A, Piva L. Renal cell carcinoma in children and adolescents. Expert Rev Anticancer Ther 2010;10:1967–78. [DOI] [PubMed] [Google Scholar]
- 2.Silberstein J, Grabowski J, Saltzstein SL, Kane CJ. Renal Cell Carcinoma in the Pediatric Population: results from the California Cancer Registry. Pediatr Blood Cancer 2009;52:237–41. [DOI] [PubMed] [Google Scholar]
- 3.Indolfi P Renal cell carcinoma in children: a clinicopathologic study. J Clin Oncol 2003;21:530–5. [DOI] [PubMed] [Google Scholar]
- 4.Estrada CR, Suthar AM, Eaton SH, Cilento BG. Renal cell carcinoma: Children’s Hospital Boston experience. Urology 2005;66: 1296–300. [DOI] [PubMed] [Google Scholar]
- 5.Selle B, Furtwängler R, Graf N, Kaatsch P, Bruder E, Leuschner I. Population-based study of renal cell carcinoma in children in Germany, 1980–2005. More frequently localized tumors and underlying disorders compared with adult counterparts. Cancer 2006;107:2906–14. [DOI] [PubMed] [Google Scholar]
- 6.Argani P, Ladanyi M. Distinctive neoplasms characterised by specific chromosomal translocations comprise a significant proportion of paediatric renal cell carcinomas. Pathology 2003;35:492–8. [DOI] [PubMed] [Google Scholar]
- 7.Argani P MiT family translocation renal cell carcinoma. Semin Diagn Pathol. 2015;32:103–13. [DOI] [PubMed] [Google Scholar]
- 8.Davis CJ Jr, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19:1–11. [DOI] [PubMed] [Google Scholar]
- 9.Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol. 2011;24:430–442. [DOI] [PubMed] [Google Scholar]
- 10.Mariño-Eñríquez A, Ou WB, Weldon CB, et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. 2011;50:146–153. [DOI] [PubMed] [Google Scholar]
- 11.Smith NE, Deyrup AT, Marino-Enriquez A, Fletcher JA, Bridge JA, Illei PB, Netto GJ, Argani P. 2014. VCL-ALK renal cell carcinoma in children with sickle-cell trait: The eight sickle cell nephropathy? Am J Surg Pathol 38:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arroyo MR, Green D, Perlman EJ, Beckwith JB, Argani P. Metanephric adenofibroma and related lesions. Clinicopathologic study of 25 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 2001;25: 433–444.. [DOI] [PubMed] [Google Scholar]
- 13.Medeiros LJ, Palmedo G, Krigman HR, Kovacs G, Beckwith JB. Oncocytoid renal cell carcinoma after neuroblastoma: a report of four cases of a distinct clinicopathologic entity. Am J Surg Pathol. 1999;23:772–780. [DOI] [PubMed] [Google Scholar]
- 14.Falzarano SM, McKenney JK, Montironi R, et al. Renal cell carcinoma occurring in patients with prior neuroblastoma: a heterogenous group of neoplasms. Am J Surg Pathol. 2016;40:989–997. [DOI] [PubMed] [Google Scholar]
- 15.Moch H, Humphrey PA, Ulbright TM, Reuter VE.WHO classification of tumours of the urinary system and male genital organs. (4th ed). IARC; Lyon, 2016. [DOI] [PubMed] [Google Scholar]
- 16.Argani P, Laé M, Ballard ET, Amin M, Manivel C, Hutchinson B, Reuter VE, Ladanyi M. Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol. 2006;1;24:1529–34. [DOI] [PubMed] [Google Scholar]
- 17.Bruder E, Passera O, Harms D, Leuschner I, Ladanyi M, Argani P, Eble JN, Struckmann K, Schraml P, Moch H. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol. 2004;28:1117–32. [DOI] [PubMed] [Google Scholar]
- 18.Renshaw AA, Granter SR, Fletcher JA, Kozakewich HP, Corless CL, Perez-Atayde AR. Renal cell carcinomas in children and young adults: increased incidence of papillary architecture and unique subtypes. Am J Surg Pathol. 1999;23:795–802. [DOI] [PubMed] [Google Scholar]
- 19.Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol. 2014;38:1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Housley SL, Lindsay RS, Young B, et al. Renal carcinoma with giant mitochondria associated with germ-line mutation and somatic loss of the succinate dehydrogenase B gene. Histopathology. 2010;56:405–408. [DOI] [PubMed] [Google Scholar]
- 21.Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase- deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28:80–94. [DOI] [PubMed] [Google Scholar]
- 22.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA. 2001;98:3387–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159: 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30: 406–410. [DOI] [PubMed] [Google Scholar]
- 25.Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trpkov K, Hes O, Bonert M, et al. Eosinophilic, solid, and cystic renal cell carcinoma: clinicopathologic study of 16 unique, sporadic neoplasms occurring in women. Am J Surg Pathol. 2016;40:60–71. [DOI] [PubMed] [Google Scholar]
- 28.Trpkov K, Abou-Ouf H, Hes O, et al. Eosinophilic solid and cystic renal cell carcinoma (ESC RCC): further morphologic and molecular characterization of ESC RCC as a distinct entity. Am J Surg Pathol. August 2017. Epub ahead of print (doi: 10.1097/PAS.0000000000000838). [DOI] [PubMed]
- 29.Ozluk Y, Taheri D, Matoso A, Sanli O, Berker NK, Yakirevich E, Balasubramanian S, Ross JS, Ali SM, Netto GJ. Renal carcinoma associated with a novel succinate dehydrogenase A mutation: a case report and review of literature of a rare subtype of renal carcinoma. Hum Pathol. 2015. 46:1951–5 [DOI] [PubMed] [Google Scholar]
- 30.Nagai R, Brock JW, Blatnik M, et al. Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem. 2007;282:34219–34228. [DOI] [PubMed] [Google Scholar]
- 31.Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011; 225:4–11. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trpkov K, Hes O, Agaimy A, Bonert M, et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am J Surg Pathol. 2016;40(7):865–75. [DOI] [PubMed] [Google Scholar]
- 34.Menko FH, Maher ER, Schmidt LS, Middelton LA, Aittomäki K, Tomlinson I, Richard S, Linehan WM. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merino MJ1, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–85. [DOI] [PubMed] [Google Scholar]
- 36.Smith SC, Trpkov K, Chen YB, Mehra R, et al. Tubulocystic Carcinoma of the Kidney With Poorly Differentiated Foci: A Frequent Morphologic Pattern of Fumarate Hydratase-deficient Renal Cell Carcinoma. Am J Surg Pathol. 2016;40:1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alrashdi I, Levine S, Paterson J et al. Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Familial Cancer. 2010; 9: 239. [DOI] [PubMed] [Google Scholar]
- 39.Smith SC, Sirohi D, Ohe C, McHugh JB, et al. A Distinctive, Low Grade Oncocytic Fumarate Hydratase-Deficient Renal Cell Carcinoma, Morphologically Reminiscent of SDH-deficient Renal Cell Carcinoma. Histopathology 2017;71: 42–52 [DOI] [PubMed] [Google Scholar]
- 40.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44:285–92 [DOI] [PubMed] [Google Scholar]
- 41.Guo J, Tretiakova MS, Troxell ML, Osunkoya AO, et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014;38:1457–67. [DOI] [PubMed] [Google Scholar]
- 42.Yang P, Cornejo KM, Sadow PM, Cheng L, et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014;38:895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martignoni G, Pea M, Gobbo S, Brunelli M, Bonetti F, Segala D, Pan CC, Netto G, Doglioni C, Hes O, Argani P, Chilosi M. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol 2009; 22: 1016–1022. [DOI] [PubMed] [Google Scholar]
- 44.Martignoni G, Gobbo S, Camparo P, Brunelli M, Munari E, Segala D, Pea M, Bonetti F, Illei PB, Netto G, Ladanyi M, Chilosi M, Argani P.Differential expression of cathepsin-K in neoplasms harbouring TFE3 gene fusions. Mod Pathol 2011:24: 1313–1319. [DOI] [PubMed] [Google Scholar]