

Abstract
Monoclonal gammopathy of undetermined significance (MGUS) associated to sporadic late onset nemaline myopathy (SLONM) is a rare and severely disabling condition of quickly progressive limb girdle acquired myopathy. It is believed by some authors to be due to myotoxicity of light chain deposits. Two female patients were diagnosed with MGUS associated SLONM. In the first case, diagnosis was delayed by 6 years thus giving time for a severe generalized myopathy and cardiomyopathy to develop. A single anti-myeloma chemotherapy with lenalidomide markedly improved and stabilized the patient’s condition despite respiratory and cardiac insufficiency. In our second patient the condition was identified one year after onset of the first symptom and markedly improved after autologous bone marrow transplantation and lenalidomide. Clinicians should be aware of monoclonal gammopathy associated sporadic late onset nemaline myopathy as this acquired muscle disorder, although extremely rare, may be reversed by adequate management.
Key Words: sporadic late-onset nemaline myopathy, monoclonal gammopathy of undetermined significance, stem cell transplantation, lenalidomide
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Nemaline myopathy was described in 1963,1 as a congenital myopathy with rod inclusions on muscle biopsy. Late (adult) onset rod myopathy was first reported in 1966.2,3 In 1975, WK Engel and Oberc noticed the association of this condition with a monoclonal gammopathy of undetermined significance (MGUS).4 It was only 18 years later that Eymard et al. published the third case of MGUS-associated adult onset rod myopathy, underlining its favourable response to plasma exchanges and suggesting a pathophysiological link between paraproteinemia and myopathy.5 Since then, the number of similar case reports is increasing slowly,6-9 even if larger series and meta-analyses have been appearing since 2014.10-14
The association of SLONM with MGUS (MGUS-SLONM) is often reported (e.g. 50 % of the 14 patients reported by Chahin et al. (2005),15 and 61% of the 28 patients reported by Naddaf et al. (2019).14 SLONM without MGUS may include cases of dermatomyositis,16 HIV-associated myositis,17 paraneoplastic myositis,18 or polymyositis.19 MGUS-SLONM usually presents as an acquired, rapidly progressive limb-girdle or axial myopathy in adults over 40 years old, but onset in younger adults and other clinical features such as camptocormia,20,21 dropped head syndrome, or swallowing troubles have also been described.20,22,23 CK levels are often normal but may be moderately increased.
Deposits of light chains were first reported in the observation of MGUS-SLONM by Eymard et al., (1993) who suggested a pathogenic link through a myotoxic effect.5 By analogy with the light chain deposition disease (LCDD), which usually involves the kidney rather than the muscle, MGUS-SLONM has been successfully treated either with the proteasome inhibitor bortezomib (Velcade®),24 or by high dose melphalan (HDM) followed by stem cell transplantation (SCT), which was recommended to be the reference treatment, based on a retrospective study of 8 cases.10
The exact pathogenic mechanism remains unknown and without treatment the prognosis is poor, due to cardiac and/or respiratory insufficiency following either myocardiopathy and/or chest muscles involvement.13,23,25
The treatment currently recommended is high-dose melphalan (HDM) followed by autologous stem cell transplantation (SCT),10-12,26 but a lenalidomide /dexamethasone protocol has also been reported to be successful.27 When inflammatory features are present, immunotherapy may also be effective.20 However, little is known about long term evolution of the condition. Here we report two additional patients with different particular features and outcomes.
Fig 1.
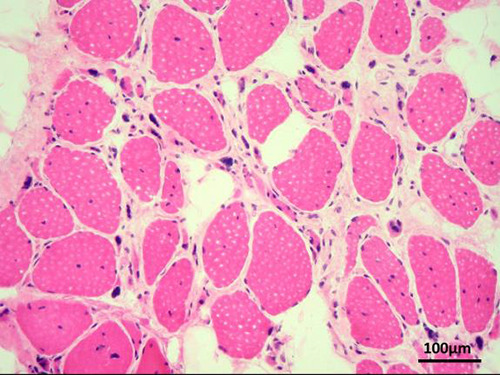
Deltoid muscle biopsy of patient 1, showing many internal nuclei, multiple atrophic fibers, nuclear clumps and fibro-lipomatosis (optic microscopy, HE staining, original magnification 200x).
Fig 2.
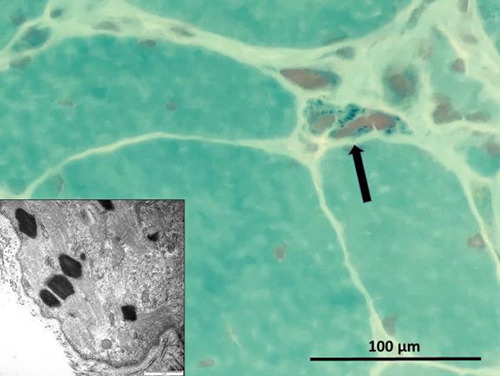
Deltoid muscle biopsy of patient 1, showing small dark particles in an atrophic muscle fiber (arrow, optic microscopy, trichrome staining, original magnification 600x). Inset shows typical nemaline bodies on electron microscopy.
Materials and Methods
This is a retrospective analysis of clinical and histopathological data in two female patients diagnosed with MGUS-SLONM, over prolonged periods of 17 and 8 years. In both patients, muscle biopsy specimens were obtained and processed using hematoylin-eosin (HE), Gomori trichrome and red Congo stainings as well as electron microscopy. In patient 1, a cardiac muscle biopsy specimen could also be performed during the course of her disease
Results
Case report 1.
A 42 years old woman with MGUS started to complain of myalgias and proximal weakness of four limbs in May 2001. Clinical examination showed Trendelenburg gait and severe paresis of upper and lower limb girdle muscles. An EMG examination performed 6 months later showed myopathic changes. CK levels were normal twice at one-year interval: 121 U/l (September 2002) and 89 U/l (September 2003) respectively. In December 2002, a needle muscle biopsy of the quadriceps was normal. The patient’s condition subsequently deteriorated and in September 2003 a second EMG showed a worsening of myopathic abnormalities. A surgical muscle biopsy was performed in November 2005 in the deltoid and showed a severe myopathy, with numerous internal nuclei, atrophic fibers, nuclear clumps and fibrolipomatosis (Fig. 1). In 2006 signs of cardiomyopathy and a respiratory insufficiency appeared, prompting genetic investigations for laminopathies, which were negative. In April 2008, a connection between the pre-existing MGUS and the patient’s muscular condition was made by one of us (AT) after attending a videoconference reporting another patient later published by Novy et al. (2010).28 The pathologist (JAL) was then asked to specifically review the slides of the 2005 biopsy specimen for rods inclusions. On trichrome staining, small dark particles were seen in atrophic fibers (Fig. 2). The sample was reprocessed for electron microscopy and this time (May 2009) revealed characteristic nemaline bodies inside muscle fibers (Fig. 2, inlet), allowing a diagnosis of SLONM to be made, after an 8 years spontaneous evolution since first symptom. HIV serology was negative. A re-staining of the biopsy sample for immunoglobulin light chain deposition, as well as actin immunochemistry could not be done at this time due to lack of material. A myocardial biopsy was performed and showed ultrastructural abnormalities resembling the nemaline bodies observed in the skeletal muscle, although difficult to distinguish from Z-disc material (Fig. 3). Red Congo staining did not disclose amyloid deposits. At this time, a monoclonal light chain IgG kappa was present at a very high level (105 mg/l) and the patient's general condition was considered too poor to undertake a bone marrow transplantation, so that she was merely given an immunosuppressive chemotherapy with lenalidomide. Under this treatment, the monoclonal Ig peak decreased as well as the cardiac and respiratory insufficiency symptoms. Muscle weakness also improved and stabilized but never showed complete restoration; however, the patient was only moderately disabled and accomplished her daily home tasks by herself (last evaluation: December 2017, 16 years after onset of symptoms). At this time, the light chain IgG kappa was still moderately increased but stabilized around 50 mg/l. Retrospective histopathologic studies with complementary immunostainings on myocardial tissue, seeking for light chains deposit, were no more possible due to lack of remaining material. The patient later died from breast carcinoma (November 2018) without having experienced any change of her muscular condition.
Fig 3.

Cardiac muscle biopsy of patient 1, electron microscopy. Electro-dense material resembling nemaline bodies, however difficult to distinguish from Z-disc material.
Case report 2.
A 42 years old, previously healthy woman with no familial history started to complain from tremor and blocking sensation in both hands in June 2012. Biological investigation revealed hyperthyroidism, without antibodies, which recovered without any treatment. In October 2012, she was unable to lift her right arm and by the end of December she noticed a slight tendency to limping, due to pelvic limb girdle muscle weakness and Trendelenburg gait. At this time, a monoclonal gammopathy “of undetermined significance” (MGUS) was detected, with an increase of lambda free light chains at 47.3 mg/l, without urinary excretion. At the beginning of the year 2013, the patient noticed her gait to become “like that of a pregnant woman” due to postural disturbances related to axial muscles paresis. Her weakness gradually increased, involving also the left arm. In October 2013, clinical examination revealed a bilateral proximal paresis of upper limbs, and a less pronounced paresis of lower limbs. There was a slight worsening as compared with May 2013. Nerve conduction studies were normal. Needle EMG showed no resting potentials, a myopathic early recruitment and myopathic (low amplitude, short duration, increased polyphasicity) changes of MUAPs in deltoid and infraspinalis muscles. A muscle MRI showed signal abnormality of lumbar paraspinal muscles, whereas iliopsoas and gluteal muscles were normal. Blood laboratory tests showed slight elevation of creatine kinase (CK). In March 2013, a left deltoid muscle biopsy using the Gomori trichrome staining showed only rare atrophic fibers in optic microscopy with no associated pathology (no rods, cores or other myofibrillar alterations, no signs of abnormal autophagy or abnormal mitochondria). Staining with red Congo did not show amyloid deposits, and immunofluorescence for Ig A, Ig G and Ig M light chains was negative. Electron microscopy, however, disclosed some typical nemaline bodies in atrophic fibers (Fig. 4), confirming the suspected diagnosis of MGUS-SLONM. A treatment with lenalidomide + dexamethasone followed by SCT was undertaken at the beginning of 2014, and by August 2014 the patient noticed a slight improvement of her strength. Over the following months, muscle strength partially recovered in parallel with muscular mass gradual restoration. One year later an attempt of lenalidomide removal, even if the MGUS was not completely eliminated, but only stabilized at a low level (11 mg/l), was followed by a relapse of weakness involving proximal lower limb and neck muscles during the summer of 2016, prompting resume of lenalidomide + dexamethasone. However, dexamethasone had to be stopped in 2017 due to iatrogenic Cushing syndrome and lenalidomide was discontinued as well one year later. Up to now (May 2020), the patient currently remains disabled, but her muscular condition stopped to deteriorate and even showed a slight tendency towards improvement on yearly follow-up visits, under supportive care with physiotherapy.
Fig 4.
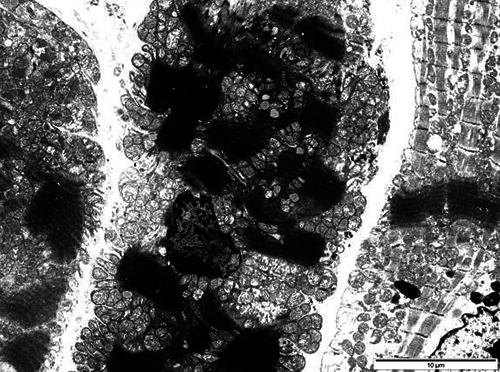
Deltoid muscle biopsy of patient 2, electron microscopy. Typical nemaline bodies are observed in an atrophic muscle fiber.
Highlights
• First demonstration of nemaline within the cardiomyocytes in MGUS-SLONM
• Specific abnormalities may not be disclosed by optic microscopy alone
• Lenalidomide alone or in association with autologous SCT is a therapeutic option.
Discussion
The main finding in these 2 patients is long term stabilization of the disease as shown by stopped progression of muscle weakness, using the antitumor and immunomodulatory agent lenalidomide, as also described by Montagnese et al.27 This was observed in our patient 1 even at a late stage of the disease causing severe cardiopulmonary insufficiency and in the absence of STC. In both cases diagnosis was difficult, because no specific abnormalities were disclosed by optic microscopy at the initial stage, even using the more sensitive Gomori trichrome staining,15 as also observed by Doppler et al.24 and Voermans et al.10 of whom some patients had to undergo serial biopsies before the diagnosis was made. In our patients the clue leading to the correct diagnosis was the discovery at laboratory workup of a MGUS which made us review the biopsy specimens with ultrastructural studies. In clinical practice, it may so be recommended that a clinical picture of limb girdle and axial asymmetric, rapidly progressive myopathy in adult subjects should prompt protein electrophoresis for MGUS detection. If this test proves positive, a nearly normal or aspecific muscle biopsy by optic microscopy does not rule out MGUS-SLONM and electron microscopy may be the only way of establishing diagnosis.11, 24
Our first case offers an example of long-term spontaneous evolution of MGUS-SLONM, due to late diagnosis. In Voermans et al. cases series (2014),10 a long course of the disease was associated with a poorer outcome, which was not observed in this patient. Interestingly, the cardiomyopathy that eventually developed displayed ultrastructural abnormalities resembling of the nemaline bodies observed in the skeletal muscle, thus providing the first histopathological evidence of a similar process in skeletal and cardiac muscle cells, which was still lacking up to now12. Doppler at al. (2013) hypothesized a physiopathology similar to light chain deposits disease (LCDD) and light and heavy chains deposits disease (LHCDD) in other organs than muscle.24 However, these authors failed to detect LCD in their patient’s muscle samples. Similarly, in our case 2, we also failed to find any immunoglobulins light or heavy chains deposits in the muscle biopsy. Although removal of serum monoclonal Ig improves muscle condition, there is still no proven direct link between MGUS and occurrence of nemaline myopathy, even if a dysimmune cause seems likely.12 Moreover, there is no evidence that MGUS-SLONM may occur preferentially in individuals with genetic variants
involving proteins expressed in skeletal and cardiac muscle, as no pathogenic mutations could be disclosed in genes known to cause congenital nemaline myopathy in patients with SLONM, either with or without MGUS.11
It is usually reported that treatment of MGUS-SLONM has to be aggressive as in multiple myeloma, to completely eliminate the MGUS. Several possible protocols can be used (bortezomib, HDM-STC). In our experience, lenalidomide and STC also yields good results. If STC is impossible, lenalidomide alone seems a good alternative as it was shown in our first patient to stabilize the myopathy over a prolonged period.
In conclusion, we We report 2 additional cases of MGUS-SLONM with significant but partial recovery, one patient with an unusual 17 years follow up (first 8 years without any treatment and 9 following years under lenalidomide alone), the other patient 6 years after STC followed by 3 years lenalidomide and dexamethasone. This rare condition is important to recognize at an early stage, because without specific haematological care its prognosis is poor, sometimes lethal due to progression of the disease to the myocardium and diaphragm. To prevent this, we suggest that any acquired limb girdle and/or axial weakness picture in an adult should be submitted to active search for MGUS or plasma cell dyscrasia and, in case of negative muscle biopsy search for rods in optic microscopy using Gomori (trichrome) staining, proactive search of nemaline bodies by electron microscopy should be undertaken. Overall, MGUS-SLONM is a good example of a probably under-recognized condition due to lack of knowledge diffusion. In this respect, medical journal reporting clinical cases are essential to help physicians in diagnostic process and for better defining the clinical spectrum of rare/orphan muscle disorders.29, 30
Acknowledgments
None
List of acronyms
- HDM
high dose melphalan
- LCDD
light chain deposition disease
- MGUS
monoclonal gammopathy of undetermined significance
- SCT
stem cell transplantation
- SLONM
sporadic late-onset nemaline myopathy
Funding Statement
Funding This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Contributor Information
Ruxandra Iancu Ferfoglia, Email: ruxandra.iancu@hcuge.ch.
Johannes Alexander Lobrinus, Email: johannes.a.lobrinus@hcuge.ch.
Kaveh Samii, Email: kaveh.samii@hcuge.ch.
André Kohler, Email: a.kohler@hin.ch.
References
- 1.Shy GM, Engel WK, Somers JE, et al. Nemaline myopathy: a new congenital myopathy. Brain 1963; 86: 793-810. [DOI] [PubMed] [Google Scholar]
- 2.Engel AG. Late onset rod myopathy. Mayo Clin Proc 1966; 41: 713-41. [PubMed] [Google Scholar]
- 3.Engel WK, Resnick JS. Late onset rod myopathy: a newly recognized acquired and progressive disease. Neurology 1966; 16: 308-9. [Google Scholar]
- 4.Engel WK, Oberk MA. Abundant nuclear rods in adult onset rod disease. J Neuropathol Exp Neurol 1975; 34: 119-32. [DOI] [PubMed] [Google Scholar]
- 5.Eymard B, Brouet JC, Collin H, et al. Late-onset rod myopathy associated with monoclonal gammopathy. Neuromusc Disord 1993; 3: 557-60. [DOI] [PubMed] [Google Scholar]
- 6.Deconinck N, Laterre EC, Van den Bergh PYK. Adult-onset nemaline myopathy and monoclonal gammopathy. Acta Neurol Belg 2000; 100: 34-40. [PubMed] [Google Scholar]
- 7.Nakagawa M, Hirata K. Adult onset nemaline myopathy and monoclonal gammopathy [in Japanese]. Ryoikibetsu Shokogun Shirizu. 2001; 406-13. [PubMed] [Google Scholar]
- 8.Keller CE, Hays AP, Rowland LP, et al. Adult-onset nemaline myopathy and monoclonal gammopathy. Arch Neurol 2006; 63: 132-4. [DOI] [PubMed] [Google Scholar]
- 9.Milone M, Katz A, Amato AA, et al. Sporadic late onset nemaline myopathy responsive to IVIG and immunotherapy. Muscle Nerve 2010; 41: 272-6. [DOI] [PubMed] [Google Scholar]
- 10.Voermans NC, Benveniste O, Minnema MC, et al. Sporadic late-onset nemaline myopathy with MGUS. Long term follow-up after melphalan and SCT. Neurology 2014; 83: 1-7. [DOI] [PubMed] [Google Scholar]
- 11.Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, et al. Sporadic late-onset nemaline myopathy: clinico-pathological characteristics and review of 76 cases. Orphanet J Rare Dis 2017; 12; 86: 1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uruha A, Benveniste O. Sporadic-late-onset nemaline myopathy with monoclonal gammopathy of undetermined significance. Curr Opin Neurol 2017; 30: 1-7. [DOI] [PubMed] [Google Scholar]
- 13.Monforte M, Primiano G, Silvestri G, et al. Sporadic late-onset nemaline myopathy: clinical, pathology and imaging findings in a single center cohort. J Neurol 2018; 265: 542-51. [DOI] [PubMed] [Google Scholar]
- 14.Naddaf E, Milone M, Kansagra A, et al. Sporadic late-onset nemaline myopathy: clinical spectrum, survival and treatment outcomes. Neurology 2019; 93: 298-305. [DOI] [PubMed] [Google Scholar]
- 15.Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology 2005; 65: 1158-64. [DOI] [PubMed] [Google Scholar]
- 16.Danon MJ, Giometti CS, Manaligod JR, et al. Adult-onset nemaline rods in a patient treated for suspected dermatomyositis. Arch Neurol 1981; 38: 761-6. [DOI] [PubMed] [Google Scholar]
- 17.Dalakas MC, Pezeshkpour GH, Flaherty M. Progressive nemaline (rod) myopathy associated with HIV infection. N Engl J Med 1987; 317: 1602-3. [DOI] [PubMed] [Google Scholar]
- 18.Kimura M, Furuta M, Hiruma S, et al. Nemaline bodies of skeletal muscle fibers of the neck in a case of pharyngeal cancer. Pathol Int 1997; 47:256-9. [DOI] [PubMed] [Google Scholar]
- 19.Sun AP, Ohtsuki Y, Yano T, et al. Typical nemaline bodies presenting in a patient with polymyositis. Med Electron Microsc 2002; 35: 167-72. [DOI] [PubMed] [Google Scholar]
- 20.Mizuno Y, Mori-Yoshimura M, Okamoto T., et al. Two cases of sporadic late onset nemaline myopathy effectively treated with immunotherapy. Rinsho Shinkeigaku (Clin Neurol) 2016; 56: 605-11. [DOI] [PubMed] [Google Scholar]
- 21.Türk M, Nagel AM, Roemer F, et al. Camptocormia as the presenting symptom in sporadic late onset nemaline myopathy: a case report. BMC Musculoskeletal Disorders 2019; 20: 553-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voermans NC, Minnema N, Lammens M, et al. Sporadic late-onset nemaline myopathy effectively treated by melphalan and stem cell transplant. Neurology 2008; 71: 532-4. [DOI] [PubMed] [Google Scholar]
- 23.Maeda HM, Ohta H, Izutsu K, et al. Sporadic late-onset nemaline myopathy as a rare cause of slowly progressive muscle weakness with young adult onset. Muscle Nerve 2015; 51: 772-4. [DOI] [PubMed] [Google Scholar]
- 24.Doppler K, Knop S, Einsele H, Sommer C. Sporadic late onset nemaline myopathy and immunoglobulin deposition disease. Muscle Nerve 2013; 48: 983-8. [DOI] [PubMed] [Google Scholar]
- 25.Belhomme N, Maamar A, Le Gallou T, et al. Rare myopathy associated to MGUS causing heart failure and responding to chemotherapy. Ann Hematol 2017; 96: 695-6. [DOI] [PubMed] [Google Scholar]
- 26.Benveniste O, Laforet P, Dubourg O, et al. Stem cell transplantation in a patient with late-onset nemaline myopathy and gammopathy. Neurology 2008; 71: 531-2. [DOI] [PubMed] [Google Scholar]
- 27.Montagnese F, Portaro S, Musumeci O, et al. Sporadic late-onset nemaline myopathy in a woman with multiple myeloma successfully treated with lenalidomide/dexamethasone. Muscle Nerve 2015; 51: 934-5. [DOI] [PubMed] [Google Scholar]
- 28.Novy J, Rosselet A, Spertini O, et al. Chemotherapy is successful in sporadic late onset nemaline myopathy (SLONM) with monoclonal gammopathy. Muscle Nerve 2010; 41: 286-7. [DOI] [PubMed] [Google Scholar]
- 29.Pennisi EM, Arca M, Bertini E, Bruno C., et al. ; Italian NLSD Group. Neutral Lipid Storage Diseases: clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J Rare Dis 2017;12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Missaglia S, Pegoraro V, Marozzo R, Tavian D, Angelini C. Correlation between ETFDH mutations and dysregulation of serum myomiRs in MADD patients. Eur J Transl Myol 2020;30:8880. doi: 10.4081/ejtm.2019.8880 [DOI] [PMC free article] [PubMed] [Google Scholar]