

Abstract

Photosystem II (PSII) is a multisubunit pigment–protein complex that uses light-induced charge separation to power oxygenic photosynthesis. Its reaction center chromophores, where the charge transfer cascade is initiated, are arranged symmetrically along the D1 and D2 core polypeptides and comprise four chlorophyll (PD1, PD2, ChlD1, ChlD2) and two pheophytin molecules (PheoD1 and PheoD2). Evolution favored productive electron transfer only via the D1 branch, with the precise nature of primary excitation and the factors that control asymmetric charge transfer remaining under investigation. Here we present a detailed atomistic description for both. We combine large-scale simulations of membrane-embedded PSII with high-level quantum-mechanics/molecular-mechanics (QM/MM) calculations of individual and coupled reaction center chromophores to describe reaction center excited states. We employ both range-separated time-dependent density functional theory and the recently developed domain based local pair natural orbital (DLPNO) implementation of the similarity transformed equation of motion coupled cluster theory with single and double excitations (STEOM-CCSD), the first coupled cluster QM/MM calculations of the reaction center. We find that the protein matrix is exclusively responsible for both transverse (chlorophylls versus pheophytins) and lateral (D1 versus D2 branch) excitation asymmetry, making ChlD1 the chromophore with the lowest site energy. Multipigment calculations show that the protein matrix renders the ChlD1 → PheoD1 charge-transfer the lowest energy excitation globally within the reaction center, lower than any pigment-centered local excitation. Remarkably, no low-energy charge transfer states are located within the “special pair” PD1–PD2, which is therefore excluded as the site of initial charge separation in PSII. Finally, molecular dynamics simulations suggest that modulation of the electrostatic environment due to protein conformational flexibility enables direct excitation of low-lying charge transfer states by far-red light.
1. Introduction
Photosystem II (PSII) is a dimeric multisubunit protein–pigment complex responsible for the light-driven oxidation of water into molecular oxygen and for the supply of reducing equivalents in oxygenic photosynthesis.1−7 Excitation-induced charge separation and the early steps of the electron transfer cascade take place within a cluster of six chlorin molecules known as the reaction center (RC). The RC comprises four chlorophylls (typically chlorophyll a), the central PD1 and PD2 “special” pair flanked by chlorophylls ChlD1 and ChlD2, and two pheophytin molecules, PheoD1 and PheoD2. The RC chromophores are arranged in a symmetric fashion along the D1 and D2 protein subunits of PSII (Figure 1) that are highly conserved across oxygenic photosynthetic organisms. The RC of PSII receives excitation energy from integral chlorophyll-rich polypeptides (CP43 and CP47) and from external light-harvesting antenna systems that vary among different species.
Figure 1.

(a) Arrangement of the photosystem II dimer viewed from the stromal side of the membrane; marked regions are the core-antenna chlorophyll proteins CP43 and CP47, and the reaction center. (b) Reaction center chromophores and selected additional components within the D1 and D2 polypeptides, with a scheme indicating the flow of electrons within PSII.
Following excitation and charge separation within the RC chromophores, the radical anion is localized within 0.3–3 ps8−12 on PheoD1 and the hole is distributed over the PD1/PD2 pair.13−15 The resulting radical cation, known as P680+, is the strongest oxidant in biology. With an estimated reduction potential of 1.1–1.3 V, P680+ is able to drive the oxidation of water at the oxygen-evolving complex (OEC) via a redox active tyrosine residue (Tyr161 or YZ) that interfaces the two sites. A distinctive feature of PSII is the utilization of the D1 branch, which also harbors the OEC, for electron transfer following productive charge separation. On the acceptor side the negative charge proceeds from PheoD1 to plastoquinone QA and finally to the terminal mobile electron acceptor plastoquinone QB.16
Key questions include the nature and localization of initial excited states, the nature and energetics of charge-transfer (CT) excited states that may lead to productive charge separation, and the factors that determine the asymmetry of RC chromophores and directionality of electron transfer.13,17,18
The multimer model19 assumes that the local excitation energies of all pigments are similar, thus favoring delocalized excitation, but other studies supported models where the chromophore energies are distinct.12,20−23 The PD1–PD2 pair is a prime candidate for the initial excitation event in analogy to the “special pair” of bacterial reaction centers.24,25 One of two major charge-transfer pathways26,27 considered includes excitation and charge separation within this pair: [PD1PD2]* → PD1–PD2+, and the negative charge is subsequently transferred to ChlD1, i.e., [PD1PD2]+ChlD1–,28−30 before proceeding to PheoD1. However, the prevailing view is that ChlD1 is the most red-shifted pigment and hence may function as the primary electron donor11,21,23,31,32 (uncertainties remain regarding the low-energy excited states of pheophytins23,33−35). The second pathway is thus described as [ChlD1PheoD1]* → ChlD1+PheoD1–. It has been proposed that room temperature structural perturbation in the protein can induce switching between the different pathways.26 Both would eventually lead to the same PD1+PheoD1– charge-separated state, but the electronic nature of the excitation and all underlying events are fundamentally distinct.
Stark spectroscopy studies suggested the presence of mixed local excitation–charge-transfer excitation in the active D1 branch,36,37 while Styring and co-workers38−40 proposed the presence of low-energy CT states responsible for far-red charge-separation and furthermore that the photochemistry of PSII is wavelength-dependent.39 An important fact is that although the working threshold of PSII is typically considered to be 680 nm, charge separation in the reaction center can be initiated with far-red light (700–780 nm) either by direct excitation of a low-lying charge transfer state in Chl a RCs or by excitation of far-red chlorophylls (Chl d and f).38,39,41−50 These observations highlight the significance of low-energy charge-transfer states for RC function.17 Remarkably, no quantum chemical study has so far identified interpigment CT excited states low enough in energy to be consistent with these observations.
It is useful to keep in mind that experimental studies are typically restricted to nonphysiological and perhaps nonfunctional PSII preparations that may yield varying observations depending on the type of preparation and conditions used. Even disregarding light-harvesting antennae, a PSII monomer comprises more than 20 proteins and dozens of chlorophylls. Core complex preparations (PSII-CC) reduce this complexity by maintaining only the D1, D2, Cytochrome b559, CP43, and CP47 proteins, but the study of RC would still be challenging due to spectral congestion by the core protein chlorophylls,27 therefore most experimental work involves PSII “RC complexes” (PSII-RCC) that maintain D1, D2, and Cytochrome b559.51−55 These are considered as a minimal structural scaffold for studying the RC,26,27,33,56−58 but their actual structure and the extent to which they represent the physiological system are debatable.48,59−61 Computational studies can potentially assist in bridging the gap between observations made on such preparations and the properties of physiological PSII.
Theoretical studies14,15,23,62−82 of photosynthetic reaction centers are challenging due to the size and complexity of the system. Beyond site energies, detailed electronic structure analysis of excited states that may be delocalized among different chromophores necessitates the use of quantum chemical approaches. A landmark study by Frankcombe used time-dependent density functional theory (TD-DFT) for the excited states of all PSII RC chromophores in the absence of the protein environment.62 This and subsequent similar studies find neither asymmetry in local excitations along the D1 and D2 branches, nor low-lying CT states that could be related to the charge-separation function of the RC.62,65 Taking the protein matrix into account with a combined quantum–mechanics/molecular–mechanics approach (QM/MM)83−85 is obviously necessary. However, this approach must not be viewed as a mere technical extension that can automatically deliver good results. Four distinct methodological requirements must be met simultaneously and successfully to ensure a meaningful and reliable outcome. These are (i) explicit atomistic representation of the complete protein matrix, potentially with consideration of conformational dynamics, (ii) high-quality quantum chemical geometric definition of the chromophores86−89 as opposed to the direct use of crystallographic coordinates, (iii) reliable excited state calculations of single and coupled chromophores, because site energies alone are insufficient to address the electronic nature of multichromophore excitations, and (iv) high-level quantum chemical methods that ensure the correct response of excited state energetics to protein electrostatics and, above all, provide a reliable description of the nature and energetics of interpigment charge-transfer excited states. The present study addresses for the first time all of the above requirements in a definitive way, aiming to provide reliable quantitative insights into the excitation profile of the reaction center of PSII.
A large-scale model of an entire membrane-embedded PSII complex is used as the basis for multiscale QM/MM modeling on geometry-optimized pigments to uncover the influence of the protein environment on the excitation profile of reaction center chromophores. Highly accurate quantum chemical descriptions of both local and distributed excitations among pairs of chromophores are obtained by long-range corrected time-dependent DFT as well as by coupled cluster theory at a level employed for the first time in such simulations, namely the similarity transformed equation of motion coupled cluster theory with single and double excitations (STEOM-CCSD). Our results provide a complete view of the nature and energetics of local and, most importantly, of charge-transfer excitations among different chromophore pairs. The results explain the origin of the dual type of asymmetry in the RC and identify the pigment pair responsible for the primary charge transfer excitation. In combination with insights from molecular dynamics simulations we determine the static and dynamic factors that control the excitation profile of the reaction center and enable charge separation to occur with far-red light.
2. Methodology
2.1. Preparation of Models
The lipid-bilayer embedded model of Photosystem II is based on the high-resolution dimeric crystal structure (1.9 Å) of Thermosynechococcus vulcanus (PDB ID: 3WU2).5 In the present work, we have used one of the monomers to build the entire system. Missing structural elements were completed to reproduce the physiological intactness of the complex. All crystallographic water associated with the monomer was retained and additional water molecules were added using the 3D-RISM technique90−94 (three-dimensional reference interaction site model) to achieve a physiological hydration state of the protein complex. The PSII monomer was embedded inside a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayer of dimension 176 × 176 Å2 using Packmol-Memgen.95,96 A total of 784 POPC molecules were added in the upper and lower leaflets of the trans-membrane region. The membrane-embedded protein complex was placed inside a water box. Appropriate amounts of Na+ and Cl– ions were added to neutralize the system and maintain a physiological concentration of 0.15 M. The complete dimensions of the system were 176 × 176 × 160 Å3 and it consists of 512 341 atoms. (Figure 2).
Figure 2.

Side and top (stromal) view of the molecular mechanics model of the PSII monomer embedded in an equilibrated POPC lipid bilayer. The simulation box has dimensions of 176 × 176 × 160 Å3.
The electrostatic charges for all the cofactors were computed based on the MK-RESP (Merz–Kollman Restrained Electrostatic Potential) methodology.97,98 For the organic cofactors, first the hydrogens atoms were optimized at the B3LYP/Def2-SVP level99,100 and then single-point calculations were performed at the HF/6-31G* level of theory97,101,102 using the ORCA program103 and RESP fitting of the charges was performed using the Multiwfn code.104 A bonded model is employed for the computation of the RESP charges on the OEC (Mn4CaO5- Oxygen Evolving Complex) and NHI (nonheme iron) sites, as a first step, a small cluster model is built around metal sites including the side chain of the residues which form a direct coordination with the metal site. The OEC is modeled in its S1 state of the Kok–Joliot cycle, i.e., the oxidation states are Mn1(III)–Mn2(IV)–Mn3(IV)–Mn4(III) and involved ligands are Asp170, Glu354, Ala344, Asp342, Glu189, His332, Glu333, and four H2O molecules. Similarly, the NHI site is modeled as Fe(II) with the ligands HCO3–, His214, His268, His215, and His272. These models were first optimized at B3LYP/Def2-TZVP and then RESP fitting is performed at B3LYP/6-31G* level. More importantly, we restrained the charge of the backbone atoms of the residues according to the original AMBER force field102 as such a procedure is known to produce better backbone dynamics during the simulation.105 The RESP charges of the chlorophylls and the heme iron site were calculated in a similar fashion. The chlorophylls and the heme-iron are ligated axially to amino acids and water molecules, wherever applicable. For example, PD1 and PD2 of the reaction center are axially ligated to histidine residues and ChlD1 and ChlD2 are axially ligated to a single water molecule. Similarly, both heme sites are bound axially with two histidine residues.
Bonded parameters for the chlorophyll a,106 heme,107 and nonheme iron site108 were obtained from the literature. Custom bonded parameters were defined for the OEC based on the study by Guerra et al.109 Parameters for the standard protein residues were described using the Amber14SB force field.110 The TIP3P model111 was chosen for the water. The bonded parameters for the organic cofactors were described with GAFF2.112 The LIPID17 force field113,114 was chosen to describe our POPC bilayer. The nonbonded parameters for the metal ions were based on their respective oxidation states using data sets105,115,116 available for the TIP3P water model. For Na+ and Cl–, we used the Joung–Cheatham parameters compatible with the TIP3P water model.117,118
2.2. Classical Molecular Dynamics Simulations
The complete system was minimized systematically to remove unfavorable geometric clashes. During the equilibration phase, the system is slowly heated from 10 to 100 K during 5 ps in the NVT ensemble. In the next step, the temperature is slowly increased from 100 to 303 K in the NPT ensemble, while maintaining the positional restraints (20 kcal mol–1 Å–2) on the Cα atoms of amino acids. The temperature during this procedure is controlled using the Langevin dynamics119 with a collision frequency of 5 ps–1. We released the restraints on Cα atoms in a controlled fashion (2 kcal mol–1 Å–2/400 ps) and subsequently invoked the Monte Carlo/Molecular Dynamics (MC/MD) module120 for a controlled hydration and dehydration from bulk to protein, and vice versa. The system was then further equilibrated for another 63 ns in the NPT ensemble to properly equilibrate the lipid bilayer. Thereafter, we initiated the production simulation for 12 ns in the NPT ensemble using the collision frequency (Langevin dynamics) of 1 ps–1. The pressure was regulated anisotropically using the Berendsen barostat121 with a relaxation time of 2 ps and maintained at 1 bar. Particle Mesh Ewald (PME)122 approach is used to treat all electrostatic interactions with a 10 Å cutoff. The SHAKE algorithm123 was used to constrain bonds involving hydrogen atoms, which allowed us to use a time step of 2 fs. The frames were saved every 2 ps. Minimizations were performed using the CPU version while equilibration and production simulations were performed using the GPU version of the pmemd engine124−126 of the AMBER18 package.127,128
2.3. QM/MM Protocol
For the purposes of the present work we extracted five snapshots of the complete system from the classical molecular dynamics to be used in the QM/MM calculations. The first snapshot, which represents a structural configuration that is close to the crystal structure of PSII, is derived from the early equilibration procedure, where we clustered129 a series of frames from the trajectory using the CPPTRAJ130,131 module of AmberTools19. The other four snapshots were derived from the production run, that is, from an unbiasedly evolved, completely hydrated, and thoroughly equilibrated system. These four snapshots are equally spaced from each other, i.e., captured with an interval of 4.0 ns, which ensures that these snapshots are structurally uncorrelated. Inspection of the overall protein structure overlay (Figure S1 of the Supporting Information, SI) of these snapshots confirms that the structural configuration of the protein in these snapshots is distinct. For the QM/MM setup we considered the entire PSII monomer and a total of 8000 water molecules, which includes all the waters presents in the protein cavity, various channels, and ∼7 Å bulk-region around the protein. In order to keep the system neutral, we maintained the required amount of Na+ ions at their equilibrated positions. The final QM/MM system contains a total of 76 056 atoms.
The QM/MM calculations were performed with ChemShell 3.7,132−134 where the in-built DL-POLY135 was used for MM computations, whereas ORCA103 was the QM engine. Our calculations are based on the electrostatic embedding technique. Covalent bonds were cut using the hydrogen link atom approach. The charge-shift method implemented in ChemShell is used to avoid overpolarization of the QM region by the MM region. For a given snapshot, we carried individual QM/MM geometry optimization calculations of ChlD1, ChlD2, PheoD1, and PheoD2, while the PD1/PD2 pair is treated as a single QM unit in QM/MM optimizations due to the close proximity of the two chlorophylls. With such a procedure, we retain the symmetry breaking feature of the vinyl moiety in PD1/PD2, which is otherwise poorly represented by the MM force field. Similar concerns were raised by Mennucci and co-workers,136 who found overstabilization of the acetyl group of Bacteriochlorophyll a by the MM force-field. In addition, with this approach both chromophores of the PD1/PD2 pair retain their macrocyclic ring curvature, important for the position of the Q-band,137,138 induced by their spatial proximity. Therefore, the geometry for all the single point calculations on the individual PD1 and PD2 is derived directly from the pair-optimized geometry.
During all QM/MM geometry optimizations the model is divided into two parts, active and static. The active region is defined by a QM subregion and an active MM region, whereas the static region remains fixed in the optimization procedure and can only influence the active region through the electrostatic effect of the point charges. In our cases where single chromophore is studied, we chose all residues and cofactors which are ca. 13 Å around the QM region (approximately 1300–1600 atoms). A larger active MM region (15 Å) was chosen for the optimization of the special pair (ca. 2650 atoms). For all the geometry optimizations, we employed the PBE functional139 and Def2-TVZP basis set,140 along with D3(BJ) dispersion corrections.141,142 To speed up the calculations of Coulomb integrals we employed the resolution of identity (RI) approximation,143,144 in combination with Weigend’s universal Def2/J auxiliary basis set.145 We used tight convergence criteria and a higher than default integration grid (grid6 in ORCA nomenclature) for all optimizations.
2.4. Computation of Excitation Energies
The computation of vertical excitation energies (VEE) in this work is performed on the QM/MM optimized geometries of the chromophores. The explicit effect of the entire PSII monomer environment on the electronic properties of chromophores is included through point-charges, handled by the orca_pc module which adds the contribution of the charges in the one-electron matrices and nuclear repulsion. We elucidated the effect of specific protein components on the excitation energies by switching off the corresponding point charges. Full Time-Dependent Density Functional Theory (TD-DFT), i.e., without the Tamm–Dancoff approximation, was used to compute the excited state properties using the range-separated ωB97X-D3(BJ)146−148 functional along with the Def2-TZVP basis set. The choice of functional was guided by its highly successful application in related studies of biological chromophores149 and its established superior performance for the treatment of charge-transfer states with TD-DFT.150 The appropriateness of the functional is additionally confirmed in the present work via direct comparisons with coupled cluster theory. The RIJCOSX approximation151,152 is used to speed up the calculations. In addition, very tight SCF convergence criteria were set along with higher integration grids (grid6 and gridX7 in ORCA nomenclature) in all cases considered in this work. We have computed 20 roots in all cases. We have also performed calculations with the long-range corrected LC-BLYP153 functional, with the same settings as described above. In addition to individual chromophores we computed the excited states of groups of adjacent RC chromophores. The selection of pairs and trimers is based on proximity and on existing hypotheses about the primary excitation and charge separation.26,29 Independently optimized geometries of individual chromophores were used to set up the oligomeric assemblies.
Despite using the best available functionals, known problems in the performance of TD-DFT in general and for charge-transfer states in particular,154−162 indicate that independent validation beyond DFT is necessary to build confidence in the results,88,163,164 especially when excited state properties of coupled chromophores are considered. Thus, both in order to obtain results that overcome potential limitations of TD-DFT and to further probe the nature of the excited states of the chromophores with an orthogonal methodology, we employed, for the first time in such a large-scale simulation, the domain based local pair natural orbital (DLPNO) implementation of the similarity transformed equation of motion coupled cluster theory with single and double excitations, STEOM-CCSD.164−170 A recent study showed this method to provide a highly accurate description of all features of the gas-phase absorption spectrum of chlorophyll a,171 while another study172 showed that it performs exceptionally well for charge-transfer states, on par with coupled cluster methods that include triple excitations, in contrast to more approximate approaches such as CC2. For the coupled cluster QM/MM calculations, the first of their kind to be conducted on a photosynthetic reaction center, we computed in total 6 roots for each chromophore using the Def2-TZVP(-f) basis set for all atoms. When applied to pairs of chromophores, to maintain feasibility of the coupled cluster calculations the chlorophyll and pheophytin models were truncated by omitting macrocyclic ring substituents. With this procedure, we keep the number of basis functions within the bounds of computational feasibility while maintaining the chemical information regarding the nature and energetic order of excited states in RC chromophore pairs. For the DLPNO-STEOM-CCSD calculations of chromophore assemblies we have resorted to the Def2-SVP basis set and computed 20 roots in total. The RIJCOSX approximation is used to speed up the calculations throughout. “TightPNO” settings were applied for the DLPNO calculations. The TCutPNOsingles cutoff was set to 6.6 × 10–10 and the active space selection keywords “Othresh” and “Vthresh” were set to 5.0 × 10–3.
3. Results and Discussion
3.1. Structural Aspects of Pigment–Protein Interactions
The intrinsic photophysical properties of the chromophores are engineered in the protein matrix for efficient light harvesting. In Photosystem II various domains of multichromophoric systems exist, such as internal antenna systems (CP43 and CP47) and the reaction center. The chromophores in the RC, i.e., four chlorophyll a and two pheophytin a molecules, are symmetrically placed along the D1 (344 residues) and D2 (342 residues) polypeptide chains. These chromophores are situated deep inside the transmembrane region of PSII, and far away from the solvent exposed stromal and lumenal regions. Due to differences in the amino acid sequence of D1 and D2,14 and overall PSII structural organization, the chromophores may experience distinct strain and electrostatic effects.
The chlorophylls of the PD1/PD2 pair are weakly stacked in the middle of the PSII monomer and both of them are axially ligated with histidine residues, His198 for PD1 and His197 for PD2 (Figure 3). The accessory chlorophylls ChlD1 and ChlD2 are axially ligated with a single water molecule, while another water molecule is present in the PSII crystal structure that hydrogen-bonds simultaneously with the axially ligated water and the C-132–COOCH3 substituent. However, important differences are found in the second coordination sphere of ChlD1 and ChlD2. The axially ligated water forms further hydrogen bond with Thr179 in case of ChlD1, but the axial water of ChlD2 does not participate in any further hydrogen bonding and a hydrophobic Ile178 is found instead in its vicinity. Other important structural differences exist in the peripheral region, where the C-131 keto group of the macrocyclic ring is hydrogen bonded with a single water molecule. This water molecule is also found to be highly conserved across many high-resolution crystal structures. Interestingly, a recent experimental study using absorption spectroscopy and Resonance Raman techniques by Robert and co-workers173 showed that hydrogen bonds to the keto group of Chl a can fine-tune the absorption properties of the LHCII (Light Harvesting Complex) antenna system. Another investigation by Collini and co-workers174 showed how such water-mediated interaction with the peripheral substituents can influence the overall conjugation in Chl a/b found in the water-soluble chlorophyll protein (WSCP). A small number of differences also exist in the immediate environment of the PheoD1 and PheoD2. The C-131 keto group of PheoD1 is hydrogen bonded to the Gln130, whereas, the C-131 keto group of PheoD2 can establish two hydrogen bonds simultaneously with Gln129 and Asn142. The C-132–COOCH3 and C-173–COOR groups of PheoD1 hydrogen-bonds to two tyrosines, Tyr147 and Tyr126. However, C-132–COOCH3 and C-173–COOR groups of PheoD2 are surrounded by hydrophobic residues (Phe255, Phe125, and Phe146).
Figure 3.
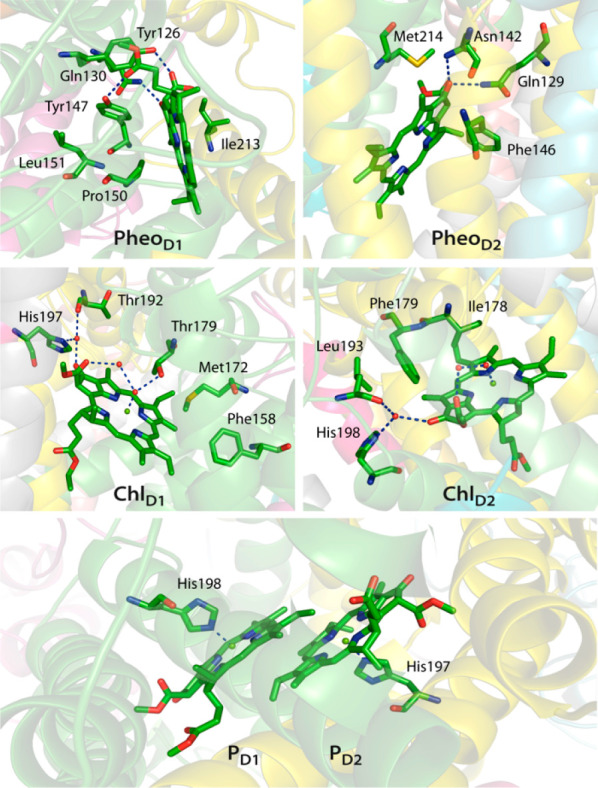
Protein environment around chromophores associated with the reaction center in Photosystem II. Hydrogen bonding interactions are indicated with dashed lines.
Analysis of our molecular dynamics trajectories indicates that the D1 and D2 chains are extremely stable throughout the simulations compared to the dynamic evolution of the complete system (Figure 4). This strongly suggests that the D1 and D2 proteins do not undergo any large-scale conformational change in the PSII core complexes within the time scale of the simulation. The stability of the D1 and D2 poly peptides ensures that the relative orientations of the chromophores stay essentially intact. In terms of individual chromophores (Figures 5 and S2–S7), we observe that the distance between PD1 and PD2 spans only a very narrow range around the average of ca. 3.5 Å along the simulation. Contrasting behavior is seen between ChlD1 and ChlD2 with respect to the hydrogen-bonding interactions of their C-131 keto groups with the nearby water molecule. This hydrogen bond appears quite constrained in the case of ChlD1 but samples a wider range of distances in the case of ChlD2. Finally, we observe that the protein environment around PheoD2 is highly dynamic, with significant degree of rotations for the side-chains of Gln129 and Asn142. In contrast, the hydrogen bonding interaction of PheoD1 with Gln130 remains stable throughout the MD simulation and no large fluctuations in side-chain conformations are observed. Overall, production MD simulations over 12 ns strongly indicate that the environment of the D1 branch chromophores is more rigid and under tighter steric control of the protein matrix than the D2 branch.
Figure 4.

Time evolution of the root-mean-square deviation (RMSD in Å) of the Cα atoms of the complete PSII complex and of the D1 and D2 core polypeptides during the production MD run. Disordered regions of the protein are not considered in the RMSD calculations.
Figure 5.

Time evolution of selected distances involving RC chromophores during the production MD run. Further details are provided in the SI.
3.2. Excitation Profiles of Individual Chromophores
The protein environment is known to influence the excited state properties of the chromophores in various ways, such as geometric strain, hydrogen bonding, and electrostatic effects. To understand the influence of these factors in a bottom-up fashion, first we computed the excited state properties of individual chromophores using their QM/MM optimized geometries but in the absence of any representation of the protein environment. These gas-phase TD-DFT calculations with the ωB97X-D3(BJ) functional lead to approximately the same S0 → S1 (Qy) vertical excitation energies for all chromophores, within the narrow range of 1.920–1.943 eV (see Figure 6 and Tables S1–S4). Similar results were obtained in past studies62,65 where a uniform dielectric medium was used to mimic the protein environment. As the geometries of the pigments already incorporate the structural effects of the protein matrix, we conclude that the protein-induced strain on the macrocyclic rings is not responsible for inducing functional asymmetry in the localization of the lowest excited state in the RC.
Figure 6.

Lowest-energy excitations (site energies), in eV, of PSII reaction center chromophores without (red) and with (green) the protein electrostatic environment. The calculations were performed with the ωB97X-D3(BJ) functional using QM/MM geometries.
Subsequently, excited state calculations using the same QM/MM optimized geometries were performed in a TD-DFT–QM/MM fashion, i.e., in the presence of the protein electrostatic environment (values in green in Figure 6). Two striking observations can be made: (a) all four chlorophyll a chromophores of the RC are red-shifted upon embedding in the protein environment, whereas both pheophytins are blue-shifted (∼0.1 eV), and (b) ChlD1 is the most strongly red-shifted pigment. Interestingly, the “special pair” PD1–PD2 displays negligible internal asymmetry in terms of site energies either with or without the protein matrix. The identification of ChlD1 as the pigment with the lowest energy excited state supports previous interpretations that account for the effects of the protein matrix.21−23,67,76,175
Different quantum chemical methods agree on the absence of noticeable asymmetry in calculations that omit the electrostatic effect of the protein, even though they provide different absolute values for the excitations, which is anticipated (see SI). What is important for the fundamental question of the emergence of excitation asymmetry is the reliable reproduction of the nature and extent of red or blue shift of the lowest excitation when the electrostatic effect of the protein matrix is included in the calculations. Comparison of the shifts produced by different methods (Table 1) shows that ωB97X-D3(BJ) and LC-BLYP produce very similar shifts, in line with those of DLPNO-STEOM-CCSD calculations. The coupled cluster results agree best with ωB97X-D3(BJ). The only significant difference between the two methods is that STEOM-CCSD predicts an even stronger blue shift for PheoD2.
Table 1. Comparison of Electrochromic Shifts (in eV) of Site Energies (Qy) from the Gas-Phase to the Protein Matrix for Individual Reaction Center Chromophores, Computed with TD-DFT Using Two Different Range-Separated Density Functionals and with DLPNO-STEOM-CCSD.
| ωB97X-D3(BJ) | LC-BLYP | DLPNO-STEOM-CCSD | |
|---|---|---|---|
| PD1 | –0.018 | –0.016 | –0.020 |
| PD2 | –0.023 | –0.021 | –0.015 |
| ChlD1 | –0.064 | –0.062 | –0.067 |
| ChlD2 | –0.027 | –0.025 | –0.025 |
| PheoD1 | +0.141 | +0.136 | +0.142 |
| PheoD2 | +0.145 | +0.140 | +0.177 |
It is evident that two types of protein matrix electrostatic asymmetry develop within the transmembrane region: (a) transverse asymmetry, which red-shifts the chlorophylls and blue-shifts the pheophytins, and (b) lateral asymmetry, which differentiates the D1 and D2 branches. This is also reflected in the electrostatic potential as experienced by the chromophores in the protein matrix. The potential is indeed distinct along D1 and D2, and the pheophytins reside in a relatively more positive electrostatic potential pocket (Figure 7). Therefore, the intrinsic protein electrostatic environment is the principal factor in modulating the distribution of excitation energies of RC chromophores and giving rise to both chlorophyll–pheophytin asymmetry (transversely) and D1–D2 branch asymmetry (laterally).
Figure 7.

Electrostatic potential experienced by the reaction center chromophores (in kT/e) inside the protein matrix. The calculations were performed using the APBS (Adaptive Poisson–Boltzmann Solver) program176 on the crystal-structure-like configuration of the protein (snapshot 1).
The asymmetry induced by protein electrostatics can be associated with differences between the D1 and D2 sequences, spatial proximity of charged residues and redox active cofactors, and the overall organization of extrinsic proteins, which differ in cyanobacteria, algae, and higher plants. Each chromophore experiences the intrinsic protein electrostatics differently because of their location and orientation with respect to the transmembrane region. Therefore, the asymmetry in the reaction center is not an intrinsic property related to the spatial arrangement of the chromophores. This explains why past computational studies that used at most a uniform dielectric to mimic the protein environment62,65 reported essentially the same Qy energies for the individual RC chromophores but did not reproduce the excitation asymmetry that is a feature of the real system. It is noted that previous works20,23,33 employing spectral modeling have assigned nearly equal Qy energies of chlorophylls and pheophytins. This is not in line with the present results and further work is required for a confident analysis of the optical spectrum. It cannot be excluded that a more elaborate theoretical treatment of the protein environment, for example with inclusion of polarization effects, may provide a better balance between the spectral fitting and the quantum chemical results.
Identification of the key protein components that give rise to asymmetry in the reaction center is important for our overall understanding of unidirectional electron transfer. Protein electrostatics are responsible for red-shifting ChlD1 by 0.064 eV (516 cm–1), which is by far the most drastic protein matrix effect on a single chromophore. The major contributors to this red-shift include Met172 (76 cm–1) and Phe158 (48 cm–1). Interestingly, the pseudosymmetric partners of these ChlD1 “red-shifters” are different on the ChlD2 side, which suggests that localized factors control the asymmetry around these chlorophylls. Additional contributions arise from PD1 (79 cm–1), and from the chloride ions in the vicinity of the OEC (61 cm–1). However, major contributors toward the 0.141 eV (1137 cm–1) blue shift of PheoD1 were found to be mostly the closely lying amino acids and cofactors, including Tyr147 (115 cm–1), Pro150 (89 cm–1), ChlD1 (66 cm–1), Leu151 (52 cm–1), and Ile213 (28 cm–1).
It has been suggested that the OEC or residues that coordinate the Mn4CaO5 cluster contribute to the red shift of ChlD1.67 Our calculations show that this is not the case and we attribute this to incorrect MM setup. Specifically, the use of integer charges for the Mn ions of the OEC in accordance to formal oxidation states results in exaggerated concentration of charge on the inorganic core and individually on its ligands. The physically motivated approach is to use distributed charges as in the present work, deducing them from RESP calculations that treat the Mn4CaO5 cluster and all its covalently bonded ligands as a single chemical entity. This eliminates long-range Coulombic artifacts. Beyond the fundamental technical aspect, attribution of a major site energy determining role to the OEC is conceptually problematic because normal function of the reaction center is required for photoassembly of the OEC177−179 and hence must be independent of its presence.
Although we pinpointed certain major contributors to the observed shifts, the total shifts in each case are not completely produced by a limited list of contributions, but instead an ever increasing number of residues and cofactors with ever smaller contributions can be found. Therefore, not only localized but also global electrostatics23 play a key role to produce the total shift compared to the gas-phase result, which shows that the evolutionary optimization of the enzyme operates on all scales. The present description has a parallel in the photosynthetic bacterial reaction center, where the chromophores were also found to be embedded in a dielectrically asymmetric environment.180,181
Some comments on methodological issues must be made at this point. First of all, the above observations underline the necessity of the QM/MM approach for obtaining meaningful results. We confirm that enlarging a gas-phase QM model by including several selected amino acid residues around each chromophore does not even begin to approximate the full QM/MM results. Another critical methodological issue relevant to studies that employ QM or QM/MM methods is the use of experimental (crystallographic) structures for the pigments. This choice is detrimental for quantum chemical approaches (alternative approaches23,175,182 for studying site energies are largely unaffected) because experimental structures typically do not exhibit correct bond length alternation of conjugated systems,86,87 often not even qualitatively. This is, however, the key geometric parameter that governs the electronic structure of the ground and excited states because it directly determines the nature and energetics of frontier orbitals. Therefore, the use of experimental geometries introduces a fundamental inconsistency between the quantum chemical approach and the structural model on which it operates, leading to randomization of results through uncontrolled errors. When this is coupled with the neglect of protein matrix electrostatics, the combined methodological deficiencies practically guarantee that the quantum chemical results are of little relevance to the real system (for example, a study183 that satisfies none of these conditions finds the lowest-energy excitation of the RC to be localized on the PheoD1). The same holds for the direct use of force-field (MM) geometries.67 These considerations apply equally to the quantum chemical treatment of multiple RC chromophores, an even more delicate case because of the sensitivity of interpigment charge-transfer states on both the geometries of interacting chromophores and on protein matrix electrostatics.
3.3. Excitation Profiles of Chromophore Pairs
Intermolecular charge-transfer (CT) excitations, i.e., those where the electron donor and acceptor are two different chromophores, are central in the function of reaction centers across photosynthetic organisms.36,37,39 Although site energies already reveal a lot about the RC of PSII, understanding the initiating events of productive primary photoexcitation requires direct insight into the excitation profiles of multiple chromophores. To obtain this information we have systematically studied the excited states of pairs of chromophores in the D1 and D2 branches, i.e., PD1–PD2, PD1–ChlD1, PD2–ChlD2, ChlD1–PheoD1, and ChlD2-PheoD2.
The “special pair” holds a special status both in bacterial reaction centers13,184 and in Photosystem II,13,26,29 therefore we focus on this one first. TD-DFT calculations performed in the gas phase (using the QM/MM optimized geometry of the pair) reveal that the nature of the lowest excitation, at 1.917 eV, is a linear combination of local excitations (LE) on PD1 and PD2 individually. Analysis of the excitations in terms of natural transition orbitals (NTOs) is provided in Figure 8. The first excitation with CT character (root 5), PD1–PD2+, is situated considerably higher in energy, at 3.091 eV. In comparison to the gas-phase results, TD-DFT QM/MM computations performed with full account of protein electrostatics result in an overall red-shift of the lowest excited state, which is now predicted at 1.884 eV, however the nature of the excited state remains the same, i.e., superposition of local excitations. We note that this is what one would expect in a Frenkel excitation picture and that the second excited state (S2, at 1.911 eV, see also Table S5) is also a linear combination of the two local excited states. Protein electrostatics affect the “directionality” of the lowest charge transfer state (PD1+PD2–) but do not stabilize it significantly compared to the gas-phase result (2.999 eV).
Figure 8.

A detailed description of the identity and nature of the lowest excited state (S1) and of the first root with significant charge transfer character for the PD1–PD2 and ChlD1–PheoD1 pairs in terms of Natural Transition Orbitals (NTOs) and relative contributions to a given excitation. In the case of ChlD1–PheoD1 both states depicted have significant CT character. Vertical excitation energies (in eV) and oscillator strengths (f) are provided for each state depicted (from ωB97X-D3(BJ) TD-DFT calculations), comparing the results in the absence (left) and in the presence (right) of the electrostatic effect of the complete PSII monomer.
Next, we have investigated the symmetry-related PD1-ChlD1 and PD2-ChlD2 pairs along the active and inactive chains. The lowest excited state (1.905 eV) computed from gas-phase calculations on PD1-ChlD1 is a linear combination of local excitations on ChlD1 and PD1, whereas the first CT state (root 9, PD1– ChlD1+) was found much higher in energy (3.639 eV). Protein electrostatics induce an overall red shift in the lowest excited state (1.843 eV) and character alteration of the lowest excited state, which becomes a local excitation on ChlD1. In addition, the lowest CT state (root 5) changes in directionality (PD1+ ChlD1–) and is found much lower in energy, at 3.130 eV. Similar observations are made for the PD2–ChlD2 pair, where in relation to the gas-phase results the protein electrostatics induces a red-shift of the lowest excited state (1.895 eV, LE on PD2), lowers the energy and alters the directionality of the first CT state (PD2+ ChlD2–, root 5, 3.232 eV, see also Table S7).
In the case of the ChlD2-PheoD2 pair (see Figure S8), the gas-phase calculations indicate that the lowest energy state (1.926 eV) is a local excitation on ChlD2, whereas the lowest CT state (ChlD2+PheoD2–, root 9) is located at 3.726 eV. Within the protein matrix we computed a slight red-shift of the lowest excited state (1.903 eV, LE on ChlD2) and a significant stabilization of the CT state (ChlD2+PheoD2–, root 3) at 2.092 eV.
The most profound demonstration of protein matrix control is observed in the case of the ChlD1–PheoD1 pair. The nature of the lowest excited state at 1.905 eV obtained from the gas-phase calculations on the ChlD1–PheoD1 is primarily a local excitation on the ChlD1. The first CT state (root 9, ChlD1+PheoD1–) is much higher in energy (3.728 eV). In this case, however, protein electrostatics drastically reorganize the excitation profile of the pair. The lowest excited state is computed at 1.828 eV and has significant ChlD1+PheoD1– charge transfer character (Figure 8). Crucially, this particular CT state marks the lowest-energy excited state among all chromophore pairs of the PSII RC. In fact, not only the first but also the second excited state computed for this pair display significant or dominant CT character favoring excitation from ChlD1 to PheoD1. The picture obtained from the NTOs is consistent with the difference densities computed for these roots, as will also be shown for the additional MD snapshots discussed in the present work and which have pure ChlD1+PheoD1– charge-transfer character (vide infra).
The above results are in line with observations from Stark spectroscopy,36,37 where the ChlD1+PheoD1– CT state was found to be mixed with LE on ChlD1. This mixing was proposed to be crucial for initiating charge separation in the RC. A similar observation was made by Valkunas and co-workers,66 where a good fit of the Stark spectrum (using complex time-dependent Redfield theory) was obtained with inclusion of the ChlD1+PheoD1– CT state, which was found to be more important in reproducing the Stark spectrum than the CT state originating from PD1–PD2+.
Calculation of the excited states of the ChlD1–PheoD1 pair using the conductor-like polarizable continuum model (CPCM)185,186 with a constant dielectric (ε = 4, appropriate for the transmembrane region) does not produce significant differences in the excitation spectrum of the chromophore pair compared to the gas-phase results. The lowest gas-phase CT state (S9, 3.728 eV) is only marginally stabilized by CPCM (S8, 3.592 eV). Overall the nature of all excited states remains essentially indistinguishable from the gas-phase TD-DFT spectrum, and hence the continuum dielectric approach is no substitute for the electrostatically asymmetric protein matrix.
Even by using one of the best available DFT methods, the fact that we predict the lowest excited state to have dominant CT character creates the need for further confirmation, because the correct prediction of CT character has been historically a challenge for TD-DFT. The only definitive way to achieve this is to go beyond DFT. Excited states of truncated pigment pairs computed by DLPNO-STEOM-CCSD (see discussion in the SI and Tables S15 and S16) confirm the nature of excited states obtained with ωB97X-D3(BJ), and therefore fully support the above conclusions beyond any conceivable uncertainty arising from the level of theory.
In conclusion, our QM/MM results on monomer and pair excitation energies confirm ChlD1 as the pigment with the lowest site energy and furthermore identify the lowest excitation of the RC as associated with a CT state of the ChlD1–PheoD1 pair.
3.4. Excited States of the PD1–PD2–ChlD1 Trimer
The results presented above firmly support a CT excited state of the type ChlD1+PheoD1– as the lowest energy excited state among pairs of RC chromophores, and hence suggest that actual charge separation would occur accordingly within this pair. An alternative pathway mentioned in the introduction as one of the possibilities under discussion involves excitation of PD1 or the PD1–PD2 pair with charge transfer to ChlD1. To investigate this possibility we conducted the same type of QM/MM calculations with simultaneous inclusion of all three relevant chromophores in the QM region (PD1, PD2, and ChlD1). The TD-DFT QM/MM results presented and analyzed in terms of NTO compositions in Table S17 show that the lowest excited state of the trimer (S1 at 1.836 eV) is fully localized on ChlD1, which is consistent with the attribution of the lowest site energy of the RC to this pigment. The second and third (S2 at 1.880 eV and S3 at 1.910 eV) are localized excitations on PD1 and PD2. Charge transfer states begin to appear above 3 eV. The lowest is a CT state within the PD1–PD2 pair (PD1+PD2–, S7 at 3.050 eV), while the first CT state with PD1+ChlD1– CT character is S8 at 3.093 eV. These results are in line with those obtained for monomers and dimers. Therefore, the present data on the trimer exclude the possibility of an energetically accessible CT excited state within the RC that involves delocalization of negative charge onto ChlD1, and strongly disfavor the hypothetical participation of an anionic ChlD1 species in native PSII charge separation.
3.5. Dynamic Control of Low-Energy Charge-Transfer States
In view of the key role of the electrostatic environment described above, it is interesting to investigate if the dynamic evolution of the protein conformation influences the excited state properties of the RC established so far within a single structural configuration of PSII. For this purpose, we performed the same set of excited state calculations, each with individually optimized QM/MM geometries, on structurally independent snapshots obtained from the molecular dynamics simulations. These snapshots were obtained from unbiased production simulations of the PSII–membrane complex, i.e., with no restraints or constraints. We chose four distinct structural configurations of the PSII–membrane complex with a consecutive interval of 4 ns. This has the advantage that the excited state properties are computed on uncorrelated protein configurations that are removed from the crystal structure minima and are properly hydrated and equilibrated with the surrounding environment.
The overall trend in the respective blue and red-shift of the individual RC chromophores and the relative ranking of the Qy excitation energies remains the same, i.e., ChlD1 has the lowest site energy (Figure 9). Focusing on chromophore pairs, we find that the lowest-lying excited state of the RC remains on the ChlD1–PheoD1 pair and retains its CT character (ChlD1+PheoD1–) irrespective of the dynamics of the protein. The above observations suggest that the nature of the intrinsic electric field of the protein matrix, and the resulting excitation asymmetry are essentially unperturbed by the conformational dynamics of the protein. Figure 10 depicts difference densities for the lowest excitation of the ChlD1–PheoD1 pair, which also demonstrate that the effect of the protein matrix is the same both in the crystallographic conformation of the protein and in the selected MD snapshots. Overall, we conclude that asymmetry does not arise as a result of the random conformational fluctuations. However, our findings indicate that the conformational flexibility of the PSII complex plays another important role.
Figure 9.

Effect of protein conformational flexibility on the first excited state (in eV) of the individual chromophores (a) and on the two lowest excited states of the PD1–PD2 and ChlD1–PheoD1 pairs of chromophores (b) from ωB97X-D3(BJ) TD-DFT QM/MM calculations on QM/MM geometries optimized individually for each snapshot. Snapshot 1 represents the “crystal-like-conformation” of the protein, whereas snapshots 2–5 were derived from the production molecular dynamics simulations with regular intervals of 4 ns.
Figure 10.

Difference densities describing the lowest excitation of the ChlD1–PheoD1 pair with exclusion (top row) and inclusion (bottom row) of protein electrostatics. With explicit consideration of the protein matrix the lowest excited state of the RC has dominant ChlD1+PheoD1– charge transfer character in the “crystal-like-conformation” of snapshot 1 and exclusive charge transfer character in the production MD snapshots 2–5.
Protein dynamics are seen to affect chromophore pairs in different ways. The conformational flexibility of the protein has little impact on the excited state properties of the inactive branch or of the PD1–PD2 pair (Figures 9 and S9), whereas there is a significant impact on the ChlD1–PheoD1 pair of the active branch, where we observe high sensitivity in the energy of the first excited state. For the conformations of the protein studied here, we find the S1 states with dominant (snapshot 1) to pure (all snapshots along the MD) ChlD1+PheoD1– CT character in the range 1.828 eV (678 nm) to 1.595 (777 nm). Although the absolute computed values are not suggested to be “exact”, since there is a dependence of the absolute values on the choice of QM method, it is interesting to note that this is beyond the nominal threshold for oxygenic photosynthesis of 680 nm (1.82 eV).
We stress that the number of snapshots used here is very small and a considerably more extensive sampling of the MD trajectory would be needed for quantitative analysis. Nevertheless, the present observations serve adequately as demonstration of principle, namely that the intrinsic electrostatic environment and flexibility of the protein are responsible for enabling access to low-energy charge-transfer states. This implies that protein matrix dynamics can push the red limit of oxygenic photosynthesis even in species that do not benefit from alternative types of chlorophyll (d or f). In this sense, specific variants of core PSII proteins available to different organisms might be utilized to adjust the red limit in response to environmental conditions not only by presenting alternate localized electrostatic contributors to critical pigments but also by favoring different distribution of global protein conformations. Therefore, electrostatic control by the dynamically evolving protein matrix must be considered equally important to the intrinsic absorption properties of participating chromophores in determining the red limit of photosynthesis.
3.6. Implications for Charge Separation Pathways
The computational results presented above form a solid basis for exploring the physiological function of the reaction center in PSII and connecting with various experimental observations. Studies on sunflower and bean leaves,43 spinach,38 green algae,46 and cyanobacteria47 showed that the known threshold for charge-separation in oxygenic photosynthesis can be pushed to the far-red region.38,40,45 Pettai et al.43,44 reported that higher plants can evolve oxygen using wavelengths as long as 780 nm. Hughes et al.45 showed that charge separation in PSII can be induced with light of 690–730 nm wavelength (1.7–1.8 eV) at 1.7 K. Similarly, Styring and co-workers38,40 documented generation of the cation radical in RC using far-red light (up to 750 nm), however a decrease in the overall charge-separated centers was observed with increasing wavelength. Furthermore, it was suggested that a significant population of the ChlD1 cation radical is trapped at cryogenic conditions (5 K) and that Tyr161 (YZ) is the preferred electron donor in this case over the Cyt-b559/ChlZ/CarD2 pathway (see Figure 1) in far-red light at 5 K. This is fully consistent with our results, which show the “dark” low-lying CT states of the ChlD1–PheoD1 pair to be in the red and far-red region. This implies that after charge separation within this pair takes place, the cation radical would initially be formed on ChlD1. The hole would subsequently migrate to PD1 under physiological conditions. Our results are also in very good agreement with the suggestion that YZ becomes the preferred electron donor under far-red light at 5 K. The migration of the ChlD1 hole to the special pair would require a reorganization of the protein environment that is probably inhibited at cryogenic temperature, resulting in YZ becoming an electron donor to ChlD1. Further EPR experiments39 showed that charge separation upon light excitation is wavelength-dependent, leading to the hypothesis that PD1 is excited with visible light (532 nm), whereas ChlD1 is excited with far-red light.39 Our results on the PD1–PD2–ChlD1 trimer do not support the presence of any energetically accessible CT state that could lead directly to productive charge separation along the D1 branch, but the conditions under which the special pair may function as the primary donor will need further studies to be clarified.
The present results are also of relevance to understanding RC function in organisms that employ alternative types of chlorophyll. Specifically, some cyanobacteria acclimatized to far-red light synthesize Chl d (Acaryochloris marina)187 or Chl f (Chroococcidiopsis thermalis)49,188 (absorption maxima at 710 and 750 nm, respectively) with alterations of chlorophyll pigments in the light harvesting antennae and possibly in the reaction centers themselves. The existence and location of Chl d pigments in the RC is a subject of debate.189,190 In the case of C. thermalis Nürnberg et al.49 assigned Chl f to be the ChlD1 position, while recent work by Judd et al.191 demonstrated the likelihood of PD2 being occupied by Chl f. Future work will evaluate these possibilities using the QM/MM approach presented in this study.
Finally, our results indicate that protein conformational flexibility would play a critical role in charge separation and subsequent oxygen evolution with far-red light. Due to the high dependence of the far-red absorption capability of ChlD1–PheoD1 on conformational dynamics, only a fraction of PSII centers38 could lead to formation of the charge-separated state in far-red. Since four “productive” charge separation events are required to make one O2 molecule, there is reduced likelihood that the OEC can advance regularly under these conditions, i.e., that subsequent far-red charge separations can occur on-time to advance the catalytic cycle outcompeting recombination. The fact that low O2 evolution is observed in cyanobacterial and higher-plant PSII excited with far-red light43,49 is consistent with this scenario.
4. Conclusions
We presented large scale MM–MD and QM/MM results on a complete membrane embedded PSII monomer, focusing on excitation energies of single and paired reaction center chromophores. The quantum mechanical level of theory in the QM/MM calculation of excitation energies include long-range corrected TD-DFT and the DLPNO implementation of the similarity transformed equation of motion coupled cluster theory with single and double excitations, STEOM-CCSD. The approach presented here serves as a reference for future studies of the reaction center. Any deviations of past reports from the results of the present work for either single or multiple chromophores can be directly traced to neglect of one or more of the methodological pillars defined in the present study.
Our results demonstrate explicitly that the excitation asymmetry in the reaction center of PSII is not an intrinsic property of RC chromophores and does not originate from their distinct geometric distortion or coordination. Asymmetry is not observed, and cannot be understood, in the absence of the protein environment. It arises exclusively through the electrostatic effect of the protein matrix. We demonstrated that the electrostatic field of the protein acts by shifting the intrinsic site energies of the chlorophylls and pheophytins in opposite directions. Red-shifting of chlorophylls versus blue-shifting of pheophytins creates transverse asymmetry with respect to the membrane normal. Preferential lowering of site energies and most importantly of charge-transfer excited states along the D1 side of the Photosystem II creates lateral asymmetry. In the presence of the protein matrix the pigment with the lowest site energy is ChlD1 and the lowest excited state within the reaction center is a state of charge transfer character localized at the ChlD1–PheoD1 pair. Therefore, the present results support assigning this pair as the site of initial charge separation in PSII. The central PD1–PD2 chlorophylls do not present low-energy charge transfer excitations internally, while the possibility of charge transfer excitation from this pair to ChlD1 is excluded. Finally, protein dynamics have only weak influence on the localization of low-energy excitations, but enable charge transfer excitations within the ChlD1–PheoD1 pair to occur with far-red light.
Acknowledgments
The authors gratefully acknowledge support by the Max Planck Society. We thank Dr. Ragnar Björnsson for his valuable assistance with the QM/MM computations. The authors thank the Max Planck Computing and Data Facility (Garching) for computational resources. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 2033–390677874–RESOLV.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c08526.
Figures S1–S11 and Tables S1–S20, overview of site energies of the PSII RC chromophores; excited state properties of interacting pigment pairs with ωB97X-D3(BJ); excited state properties of interacting pigment pairs with LC-BLYP; comparison of TDDFT and DLPNO-STEOM-CCSD; excited states of the PD1–PD2–ChlD1 trimer; low-lying excited states of interacting chromophores from additional protein configurations; D1 and D2 sequence alignment from different photosynthetic organisms; Cartesian coordinates of optimized chromophores; and additional references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Blankenship R. E.Molecular Mechanisms of Photosynthesis; 2nd ed.; Wiley: Chichester, 2014; p 312. [Google Scholar]
- Photosystem II. The Light-Driven Water:Plastoquinone Oxidoreductase; Wydrzynski T., Satoh K., Eds.; Springer: Dordrecht, 2005; Vol. 22. [Google Scholar]
- Shevela D.; Björn L. O.; Govindjee Photosynthesis: Solar Energy for Life; World Scientific: Singapore, 2017; p 204. [Google Scholar]
- Ferreira K. N.; Iverson T. M.; Maghlaoui K.; Barber J.; Iwata S. Architecture of the Photosynthetic Oxygen-Evolving Center. Science 2004, 303, 1831–1838. 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Barber J. Photosystem II: the water splitting enzyme of photosynthesis and the origin of oxygen in our atmosphere. Q. Rev. Biophys. 2016, 49, e14. 10.1017/S0033583516000123. [DOI] [PubMed] [Google Scholar]
- Krewald V.; Retegan M.; Pantazis D. A. Principles of Natural Photosynthesis. Top. Curr. Chem. 2015, 371, 23–48. 10.1007/128_2015_645. [DOI] [PubMed] [Google Scholar]
- Dekker J. P.; Van Grondelle R. Primary charge separation in Photosystem II. Photosynth. Res. 2000, 63, 195–208. 10.1023/A:1006468024245. [DOI] [PubMed] [Google Scholar]
- Holzwarth A. R.; Müller M. G.; Reus M.; Nowaczyk M.; Sander J.; Rögner M. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: Pheophytin is the primary electron acceptor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6895–6900. 10.1073/pnas.0505371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford A. W.; Osyczka A.; Rappaport F. Back-Reactions, Short-Circuits, Leaks and Other Energy Wasteful Reactions in Biological Electron Transfer: Redox Tuning to Survive Life in O2. FEBS Lett. 2012, 586, 603–616. 10.1016/j.febslet.2011.12.039. [DOI] [PubMed] [Google Scholar]
- Raszewski G.; Renger T. Light Harvesting in Photosystem II Core Complexes Is Limited by the Transfer to the Trap: Can the Core Complex Turn into a Photoprotective Mode?. J. Am. Chem. Soc. 2008, 130, 4431–4446. 10.1021/ja7099826. [DOI] [PubMed] [Google Scholar]
- Renger T.; Schlodder E. Optical properties, excitation energy and primary charge transfer in photosystem II: theory meets experiment. J. Photochem. Photobiol., B 2011, 104, 126–141. 10.1016/j.jphotobiol.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Cardona T.; Sedoud A.; Cox N.; Rutherford A. W. Charge Separation in Photosystem II: A Comparative and Evolutionary Overview. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 26–43. 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Saito K.; Ishida T.; Sugiura M.; Kawakami K.; Umena Y.; Kamiya N.; Shen J.-R.; Ishikita H. Distribution of the Cationic State over the Chlorophyll Pair of the Photosystem II Reaction Center. J. Am. Chem. Soc. 2011, 133, 14379–14388. 10.1021/ja203947k. [DOI] [PubMed] [Google Scholar]
- Narzi D.; Bovi D.; De Gaetano P.; Guidoni L. Dynamics of the special pair of chlorophylls of Photosystem II. J. Am. Chem. Soc. 2016, 138, 257–264. 10.1021/jacs.5b10523. [DOI] [PubMed] [Google Scholar]
- Müh F.; Glöckner C.; Hellmich J.; Zouni A. Light-induced quinone reduction in photosystem II. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 44–65. 10.1016/j.bbabio.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Reimers J. R.; Biczysko M.; Bruce D.; Coker D. F.; Frankcombe T. J.; Hashimoto H.; Hauer J.; Jankowiak R.; Kramer T.; Linnanto J.; Mamedov F.; Müh F.; Rätsep M.; Renger T.; Styring S.; Wan J.; Wang Z.; Wang-Otomo Z.-Y.; Weng Y.-X.; Yang C.; Zhang J.-P.; Freiberg A.; Krausz E. Challenges facing an understanding of the nature of low-energy excited states in photosynthesis. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 1627–1640. 10.1016/j.bbabio.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Mamedov M.; Nadtochenko V.; Semenov A. Primary electron transfer processes in photosynthetic reaction centers from oxygenic organisms. Photosynth. Res. 2015, 125, 51–63. 10.1007/s11120-015-0088-y. [DOI] [PubMed] [Google Scholar]
- Durrant J. R.; Klug D. R.; Kwa S. L.; Van Grondelle R.; Porter G.; Dekker J. P. A multimer model for P680, the primary electron donor of photosystem II. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 4798–4802. 10.1073/pnas.92.11.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raszewski G.; Diner B. A.; Schlodder E.; Renger T. Spectroscopic properties of reaction center pigments in photosystem II core complexes: revision of the multimer model. Biophys. J. 2008, 95, 105–119. 10.1529/biophysj.107.123935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renger T.; Schlodder E. Primary photophysical processes in photosystem II: bridging the gap between crystal structure and optical spectra. ChemPhysChem 2010, 11, 1141–1153. 10.1002/cphc.200900932. [DOI] [PubMed] [Google Scholar]
- Shibata Y.; Nishi S.; Kawakami K.; Shen J.-R.; Renger T. Photosystem II does not possess a simple excitation energy funnel: time-resolved fluorescence spectroscopy meets theory. J. Am. Chem. Soc. 2013, 135, 6903–6914. 10.1021/ja312586p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müh F.; Plöckinger M.; Renger T. Electrostatic Asymmetry in the Reaction Center of Photosystem II. J. Phys. Chem. Lett. 2017, 8, 850–858. 10.1021/acs.jpclett.6b02823. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J.; Epp O.; Miki K.; Huber R.; Michel H. X-ray structure analysis of a membrane protein complex: Electron density map at 3 Å resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Mol. Biol. 1984, 180, 385–398. 10.1016/S0022-2836(84)80011-X. [DOI] [PubMed] [Google Scholar]
- Woodbury N. W.; Allen J. P. In Anoxygenic Photosynthetic Bacteria; Springer: New York, 1995; pp 527–557. [Google Scholar]
- Romero E.; Van Stokkum I. H.; Novoderezhkin V. I.; Dekker J. P.; Van Grondelle R. Two different charge separation pathways in photosystem II. Biochemistry 2010, 49, 4300–4307. 10.1021/bi1003926. [DOI] [PubMed] [Google Scholar]
- Romero E.; Novoderezhkin V. I.; van Grondelle R. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 2017, 543, 355–365. 10.1038/nature22012. [DOI] [PubMed] [Google Scholar]
- Nadtochenko V.; Semenov A. Y.; Shuvalov V. Formation and decay of P680 (PD1–PD2)+ PheoD1– radical ion pair in photosystem II core complexes. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 1384–1388. 10.1016/j.bbabio.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Shelaev I. V.; Gostev F. E.; Vishnev M. I.; Shkuropatov A. Y.; Ptushenko V. V.; Mamedov M. D.; Sarkisov O. M.; Nadtochenko V. A.; Semenov A. Y.; Shuvalov V. A. P680 (PD1PD2) and ChlD1 as alternative electron donors in photosystem II core complexes and isolated reaction centers. J. Photochem. Photobiol., B 2011, 104, 44–50. 10.1016/j.jphotobiol.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Shelaev I. V.; Gostev F. E.; Nadtochenko V. A.; Shkuropatov A. Y.; Zabelin A. A.; Mamedov M. D.; Semenov A. Y.; Sarkisov O. M.; Shuvalov V. A. Primary light-energy conversion in tetrameric chlorophyll structure of photosystem II and bacterial reaction centers: II. Femto- and picosecond charge separation in PSII D1/D2/Cyt b559 complex. Photosynth. Res. 2008, 98, 95. 10.1007/s11120-008-9371-5. [DOI] [PubMed] [Google Scholar]
- Groot M. L.; Pawlowicz N. P.; van Wilderen L. J. G. W.; Breton J.; van Stokkum I. H. M.; van Grondelle R. Initial electron donor and acceptor in isolated Photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13087–13092. 10.1073/pnas.0503483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhorenko V. I.; Holzwarth A. R. Primary Processes and Structure of the Photosystem II Reaction Center: A Photon Echo Study. J. Phys. Chem. B 2000, 104, 11563–11578. 10.1021/jp002323n. [DOI] [Google Scholar]
- Acharya K.; Zazubovich V.; Reppert M.; Jankowiak R. Primary electron donor (s) in isolated reaction center of photosystem II from Chlamydomonas reinhardtii. J. Phys. Chem. B 2012, 116, 4860–4870. 10.1021/jp302849d. [DOI] [PubMed] [Google Scholar]
- Stewart D. H.; Nixon P. J.; Diner B. A.; Brudvig G. W. Assignment of the Q y absorbance bands of Photosystem II chromophores by low-temperature optical spectroscopy of wild-type and mutant reaction centers. Biochemistry 2000, 39, 14583–14594. 10.1021/bi001246j. [DOI] [PubMed] [Google Scholar]
- Årsköld S. P.; Masters V. M.; Prince B. J.; Smith P. J.; Pace R. J.; Krausz E. Optical spectra of synechocystis and spinach photosystem II preparations at 1.7 K: identification of the D1-pheophytin energies and stark shifts. J. Am. Chem. Soc. 2003, 125, 13063–13074. 10.1021/ja034548s. [DOI] [PubMed] [Google Scholar]
- Novoderezhkin V. I.; Dekker J. P.; Van Grondelle R. Mixing of exciton and charge-transfer states in photosystem II reaction centers: modeling of stark spectra with modified redfield theory. Biophys. J. 2007, 93, 1293–1311. 10.1529/biophysj.106.096867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E.; Diner B. A.; Nixon P. J.; Coleman W. J.; Dekker J. P.; van Grondelle R. Mixed exciton–charge-transfer states in photosystem II: stark spectroscopy on site-directed mutants. Biophys. J. 2012, 103, 185–194. 10.1016/j.bpj.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokvist F.; Sjöholm J.; Mamedov F.; Styring S. The Photochemistry in Photosystem II at 5 K Is Different in Visible and Far-Red Light. Biochemistry 2014, 53, 4228–4238. 10.1021/bi5006392. [DOI] [PubMed] [Google Scholar]
- Pavlou A.; Jacques J.; Ahmadova N.; Mamedov F.; Styring S. The wavelength of the incident light determines the primary charge separation pathway in Photosystem II. Sci. Rep. 2018, 8, 2837. 10.1038/s41598-018-21101-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapper A.; Mamedov F.; Mokvist F.; Hammarström L.; Styring S. Defining the far-red limit of photosystem II in spinach. Plant Cell 2009, 21, 2391–2401. 10.1105/tpc.108.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Blankenship R. E. Expanding the solar spectrum used by photosynthesis. Trends Plant Sci. 2011, 16, 427–431. 10.1016/j.tplants.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Wolf B. M.; Blankenship R. E. Far-red light acclimation in diverse oxygenic photosynthetic organisms. Photosynth. Res. 2019, 142, 349–359. 10.1007/s11120-019-00653-6. [DOI] [PubMed] [Google Scholar]
- Pettai H.; Oja V.; Freiberg A.; Laisk A. Photosynthetic activity of far-red light in green plants. Biochim. Biophys. Acta, Bioenerg. 2005, 1708, 311–321. 10.1016/j.bbabio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Pettai H.; Oja V.; Freiberg A.; Laisk A. The long-wavelength limit of plant photosynthesis. FEBS Lett. 2005, 579, 4017–4019. 10.1016/j.febslet.2005.04.088. [DOI] [PubMed] [Google Scholar]
- Hughes J. L.; Smith P.; Pace R.; Krausz E. Charge separation in photosystem II core complexes induced by 690–730 nm excitation at 1.7 K. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 841–851. 10.1016/j.bbabio.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Greenbaum N. L.; Mauzerall D. Effect of irradiance level on distribution of chlorophylls between PS II and PS I as determined from optical cross-sections. Biochim. Biophys. Acta, Bioenerg. 1991, 1057, 195–207. 10.1016/S0005-2728(05)80102-1. [DOI] [Google Scholar]
- Morton J.; Akita F.; Nakajima Y.; Shen J.-R.; Krausz E. Optical identification of the long-wavelength (700–1700 nm) electronic excitations of the native reaction centre, Mn4CaO5 cluster and cytochromes of photosystem II in plants and cyanobacteria. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 153–161. 10.1016/j.bbabio.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Langley J.; Morton J.; Purchase R.; Tian L.; Shen L.; Han G.; Shen J. R.; Krausz E. The deep red state of photosystem II in Cyanidioschyzon merolae. Photosynthetica 2018, 56, 275–278. 10.1007/s11099-017-0760-x. [DOI] [Google Scholar]
- Nürnberg D. J.; Morton J.; Santabarbara S.; Telfer A.; Joliot P.; Antonaru L. A.; Ruban A. V.; Cardona T.; Krausz E.; Boussac A.; Fantuzzi A.; Rutherford A. W. Photochemistry beyond the red limit in chlorophyll f–containing photosystems. Science 2018, 360, 1210–1213. 10.1126/science.aar8313. [DOI] [PubMed] [Google Scholar]
- Mascoli V.; Bersanini L.; Croce R. Far-red absorption and light-use efficiency trade-offs in chlorophyll f photosynthesis. Nat. Plants 2020, 6, 1044–1053. 10.1038/s41477-020-0718-z. [DOI] [PubMed] [Google Scholar]
- Danielius R. V.; Satoh K.; van Kan P. J. M.; Plijter J. J.; Nuijs A. M.; van Gorkom H. J. The primary reaction of photosystem II in the D1-D2-cytochrome b-559 complex. FEBS Lett. 1987, 213, 241–244. 10.1016/0014-5793(87)81498-9. [DOI] [Google Scholar]
- Mimuro M.; Tomo T.; Nishimura Y.; Yamazaki I.; Satoh K. Identification of a photochemically inactive pheophytin molecule in the spinach D1-D2-cyt b559 complex. Biochim. Biophys. Acta, Bioenerg. 1995, 1232, 81–88. 10.1016/0005-2728(95)00116-2. [DOI] [Google Scholar]
- Montoya G.; Yruela I.; Picorel R. Pigment stoichiometry of a newly isolated D1—D2—Cyt b559 complex from the higher plant Beta vulgaris L. FEBS Lett. 1991, 283, 255–258. 10.1016/0014-5793(91)80601-X. [DOI] [PubMed] [Google Scholar]
- Satoh K.; Nakane H. In Current Research in Photosynthesis: Proceedings of the VIIIth International Conference on Photosynthesis Stockholm, Sweden, August 6–11, 1989; Baltscheffsky M., Ed.; Springer: Dordrecht, 1990; pp 271–274. [Google Scholar]
- Nanba O.; Satoh K. Isolation of a photosystem II reaction center consisting of D1 and D2 polypeptides and cytochrome b559. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 109–112. 10.1073/pnas.84.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoderezhkin V. I.; Romero E.; Dekker J. P.; van Grondelle R. Multiple charge-separation pathways in photosystem II: modeling of transient absorption kinetics. ChemPhysChem 2011, 12, 681–688. 10.1002/cphc.201000830. [DOI] [PubMed] [Google Scholar]
- Fuller F. D.; Pan J.; Gelzinis A.; Butkus V.; Senlik S. S.; Wilcox D. E.; Yocum C. F.; Valkunas L.; Abramavicius D.; Ogilvie J. P. Vibronic coherence in oxygenic photosynthesis. Nat. Chem. 2014, 6, 706–711. 10.1038/nchem.2005. [DOI] [PubMed] [Google Scholar]
- Duan H.-G.; Prokhorenko V. I.; Wientjes E.; Croce R.; Thorwart M.; Miller R. D. Primary charge separation in the Photosystem II reaction center revealed by a global analysis of the two-dimensional electronic spectra. Sci. Rep. 2017, 7, 12347. 10.1038/s41598-017-12564-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz E.; Cox N.; Årsköld S. P. Spectral characteristics of PS II reaction centres: as isolated preparations and when integral to PS II core complexes. Photosynth. Res. 2008, 98, 207–217. 10.1007/s11120-008-9328-8. [DOI] [PubMed] [Google Scholar]
- Hughes J. L.; Prince B. J.; Peterson Årsköld S.; Smith P. J.; Pace R. J.; Riesen H.; Krausz E. The Native Reaction Centre of Photosystem II: A New Paradigm for P680. Aust. J. Chem. 2004, 57, 1179–1183. 10.1071/CH04140. [DOI] [Google Scholar]
- Sindra P. Å.; Prince B. J.; Krausz E.; Smith P. J.; Pace R. J.; Picorel R.; Seibert M. Low-temperature spectroscopy of fully active PSII cores. Comparisons with CP43, CP47, D1/D2/cyt b559 fragments. J. Lumin. 2004, 108, 97–100. 10.1016/j.jlumin.2004.01.023. [DOI] [Google Scholar]
- Frankcombe T. J. Explicit calculation of the excited electronic states of the photosystem II reaction centre. Phys. Chem. Chem. Phys. 2015, 17, 3295–3302. 10.1039/C4CP04468A. [DOI] [PubMed] [Google Scholar]
- Ivashin N.; Larsson S. Excitonic states in photosystem II reaction center. J. Phys. Chem. B 2005, 109, 23051–23060. 10.1021/jp0581734. [DOI] [PubMed] [Google Scholar]
- Vasil’ev S.; Bruce D. A protein dynamics study of photosystem II: the effects of protein conformation on reaction center function. Biophys. J. 2006, 90, 3062–3073. 10.1529/biophysj.105.076075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomivuori C.-M.; Winter N. O.; Hättig C.; Sundholm D.; Kaila V. R. Exploring the Light-Capturing Properties of Photosynthetic Chlorophyll Clusters Using Large-Scale Correlated Calculations. J. Chem. Theory Comput. 2016, 12, 2644–2651. 10.1021/acs.jctc.6b00237. [DOI] [PubMed] [Google Scholar]
- Gelzinis A.; Abramavicius D.; Ogilvie J. P.; Valkunas L. Spectroscopic properties of photosystem II reaction center revisited. J. Chem. Phys. 2017, 147, 115102. 10.1063/1.4997527. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Silva D.-A.; Zhang H.; Yue A.; Yan Y.; Huang X. Dynamic protein conformations preferentially drive energy transfer along the active chain of the photosystem II reaction centre. Nat. Commun. 2014, 5, 4170. 10.1038/ncomms5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y.; Matsuda K.; Hasegawa J.-y. Theoretical study of the excited states of the photosynthetic reaction center in photosystem II: Electronic structure, interactions, and their origin. Biophys. Chem. 2011, 159, 227–236. 10.1016/j.bpc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Palencar P.; Prudnikova T.; Vacha F.; Kuty M. The effects of light-induced reduction of the photosystem II reaction center. J. Mol. Model. 2009, 15, 923–933. 10.1007/s00894-008-0448-z. [DOI] [PubMed] [Google Scholar]
- Kawashima K.; Ishikita H. Energetic insights into two electron transfer pathways in light-driven energy-converting enzymes. Chem. Sci. 2018, 9, 4083–4092. 10.1039/C8SC00424B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König C.; Neugebauer J. Quantum Chemical Description of Absorption Properties and Excited-State Processes in Photosynthetic Systems. ChemPhysChem 2012, 13, 386–425. 10.1002/cphc.201100408. [DOI] [PubMed] [Google Scholar]
- Thompson M. A.; Zerner M. C. A theoretical examination of the electronic structure and spectroscopy of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Am. Chem. Soc. 1991, 113, 8210–8215. 10.1021/ja00022a003. [DOI] [Google Scholar]
- Parson W. W.; Warshel A. Spectroscopic properties of photosynthetic reaction centers. 2. Application of the theory to Rhodopseudomonas viridis. J. Am. Chem. Soc. 1987, 109, 6152–6163. 10.1021/ja00254a040. [DOI] [Google Scholar]
- Warshel A.; Parson W. W. Spectroscopic properties of photosynthetic reaction centers. 1. Theory. J. Am. Chem. Soc. 1987, 109, 6143–6152. 10.1021/ja00254a039. [DOI] [Google Scholar]
- Blomberg M. R. A.; Siegbahn P. E. M.; Babcock G. T. Modeling Electron Transfer in Biochemistry: A Quantum Chemical Study of Charge Separation in Rhodobacter sphaeroides and Photosystem II. J. Am. Chem. Soc. 1998, 120, 8812–8824. 10.1021/ja9805268. [DOI] [Google Scholar]
- Tamura H.; Saito K.; Ishikita H. Acquirement of water-splitting ability and alteration of the charge-separation mechanism in photosynthetic reaction centers. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 16373–16382. 10.1073/pnas.2000895117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmayer T. J.; de Groot H. J. M.; van de Wetering E.; Neugebauer J.; Buda F. Mechanism and Reaction Coordinate of Directional Charge Separation in Bacterial Reaction Centers. J. Phys. Chem. Lett. 2012, 3, 694–697. 10.1021/jz201695p. [DOI] [PubMed] [Google Scholar]
- Gelzinis A.; Valkunas L.; Fuller F. D.; Ogilvie J. P.; Mukamel S.; Abramavicius D. Tight-binding model of the photosystem II reaction center: application to two-dimensional electronic spectroscopy. New J. Phys. 2013, 15, 075013. 10.1088/1367-2630/15/7/075013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksu H.; Schubert A.; Geva E.; Dunietz B. D. Explaining Spectral Asymmetries and Excitonic Characters of the Core Pigment Pairs in the Bacterial Reaction Center Using a Screened Range-Separated Hybrid Functional. J. Phys. Chem. B 2019, 123, 8970–8975. 10.1021/acs.jpcb.9b07646. [DOI] [PubMed] [Google Scholar]
- Aksu H.; Schubert A.; Bhandari S.; Yamada A.; Geva E.; Dunietz B. D. On the Role of the Special Pair in Photosystems as a Charge Transfer Rectifier. J. Phys. Chem. B 2020, 124, 1987–1994. 10.1021/acs.jpcb.9b11431. [DOI] [PubMed] [Google Scholar]
- Curutchet C.; Mennucci B. Quantum Chemical Studies of Light Harvesting. Chem. Rev. 2017, 117, 294–343. 10.1021/acs.chemrev.5b00700. [DOI] [PubMed] [Google Scholar]
- Artiukhin D. G.; Eschenbach P.; Neugebauer J. Computational Investigation of the Spin-Density Asymmetry in Photosynthetic Reaction Center Models from First Principles. J. Phys. Chem. B 2020, 124, 4873–4888. 10.1021/acs.jpcb.0c02827. [DOI] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- Lin H.; Truhlar D. G. QM/MM: what have we learned, where are we, and where do we go from here?. Theor. Chem. Acc. 2007, 117, 185–199. 10.1007/s00214-006-0143-z. [DOI] [Google Scholar]
- Mennucci B. Modeling environment effects on spectroscopies through QM/classical models. Phys. Chem. Chem. Phys. 2013, 15, 6583–6594. 10.1039/c3cp44417a. [DOI] [PubMed] [Google Scholar]
- Dreuw A.; Harbach P. H. P.; Mewes J. M.; Wormit M. Quantum chemical excited state calculations on pigment–protein complexes require thorough geometry re-optimization of experimental crystal structures. Theor. Chem. Acc. 2010, 125, 419–426. 10.1007/s00214-009-0680-3. [DOI] [Google Scholar]
- Wanko M.; Hoffmann M.; Strodel P.; Koslowski A.; Thiel W.; Neese F.; Frauenheim T.; Elstner M. Calculating Absorption Shifts for Retinal Proteins: Computational Challenges. J. Phys. Chem. B 2005, 109, 3606–3615. 10.1021/jp0463060. [DOI] [PubMed] [Google Scholar]
- Dreuw A.; Head-Gordon M. Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev. 2005, 105, 4009–4037. 10.1021/cr0505627. [DOI] [PubMed] [Google Scholar]
- Dunietz B. D.; Dreuw A.; Head-Gordon M. Initial Steps of the Photodissociation of the CO Ligated Heme Group. J. Phys. Chem. B 2003, 107, 5623–5629. 10.1021/jp0226376. [DOI] [Google Scholar]
- Beglov D.; Roux B. An Integral Equation To Describe the Solvation of Polar Molecules in Liquid Water. J. Phys. Chem. B 1997, 101, 7821–7826. 10.1021/jp971083h. [DOI] [Google Scholar]
- Imai T.; Hiraoka R.; Kovalenko A.; Hirata F. Locating missing water molecules in protein cavities by the three-dimensional reference interaction site model theory of molecular solvation. Proteins: Struct., Funct., Genet. 2007, 66, 804–813. 10.1002/prot.21311. [DOI] [PubMed] [Google Scholar]
- Kovalenko A.; Hirata F. Three-dimensional density profiles of water in contact with a solute of arbitrary shape: a RISM approach. Chem. Phys. Lett. 1998, 290, 237–244. 10.1016/S0009-2614(98)00471-0. [DOI] [Google Scholar]
- Sindhikara D. J.; Hirata F. Analysis of Biomolecular Solvation Sites by 3D-RISM Theory. J. Phys. Chem. B 2013, 117, 6718–6723. 10.1021/jp4046116. [DOI] [PubMed] [Google Scholar]
- Sindhikara D. J.; Yoshida N.; Hirata F. Placevent: An algorithm for prediction of explicit solvent atom distribution—Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. 10.1002/jcc.22984. [DOI] [PubMed] [Google Scholar]
- Schott-Verdugo S.; Gohlke H. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein–Lipid-Bilayer System Building. J. Chem. Inf. Model. 2019, 59, 2522–2528. 10.1021/acs.jcim.9b00269. [DOI] [PubMed] [Google Scholar]
- Martinez L.; Andrade R.; Birgin E. G.; Martinez J. M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Duan Y.; Wu C.; Chowdhury S.; Lee M. C.; Xiong G.; Zhang W.; Yang R.; Cieplak P.; Luo R.; Lee T.; Caldwell J.; Wang J.; Kollman P. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role Of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Sigfridsson E.; Ryde U. Comparison of methods for deriving atomic charges from the electrostatic potential and moments. J. Comput. Chem. 1998, 19, 377–395. . [DOI] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Li P.; Merz K. M. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. 10.1021/acs.chemrev.6b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M.; Procacci P.; Marchi M. An ab initio force field for the cofactors of bacterial photosynthesis. J. Comput. Chem. 2003, 24, 129–142. 10.1002/jcc.10198. [DOI] [PubMed] [Google Scholar]
- Giammona D. A.Ph.D. Thesis; University of California at Davis: Davis, CA, 1984. [Google Scholar]
- Sakashita N.; Watanabe H. C.; Ikeda T.; Saito K.; Ishikita H. Origins of Water Molecules in the Photosystem II Crystal Structure. Biochemistry 2017, 56, 3049–3057. 10.1021/acs.biochem.7b00220. [DOI] [PubMed] [Google Scholar]
- Guerra F.; Siemers M.; Mielack C.; Bondar A.-N. Dynamics of Long-Distance Hydrogen-Bond Networks in Photosystem II. J. Phys. Chem. B 2018, 122, 4625–4641. 10.1021/acs.jpcb.8b00649. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–74. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Dickson C. J.; Madej B. D.; Skjevik Å. A.; Betz R. M.; Teigen K.; Gould I. R.; Walker R. C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjevik Å. A.; Madej B. D.; Walker R. C.; Teigen K. LIPID11: A Modular Framework for Lipid Simulations Using Amber. J. Phys. Chem. B 2012, 116, 11124–11136. 10.1021/jp3059992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Roberts B. P.; Chakravorty D. K.; Merz K. M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for + 2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. 10.1021/ct400146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Song L. F.; Merz K. M. Parameterization of Highly Charged Metal Ions Using the 12–6-4 LJ-Type Nonbonded Model in Explicit Water. J. Phys. Chem. B 2015, 119, 883–895. 10.1021/jp505875v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. 10.1021/jp902584c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- Ben-Shalom I. Y.; Lin C.; Kurtzman T.; Walker R. C.; Gilson M. K. Simulating Water Exchange to Buried Binding Sites. J. Chem. Theory Comput. 2019, 15, 2684–2691. 10.1021/acs.jctc.8b01284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S.; Götz A. W.; Walker R. C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. III; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Belfon K.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Giambasu G.; Giese T.; Gilson M .K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Krasny R.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Man V.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wilson L.; Wolf R. M.; Wu X.; Xiao L.; Xiong Y.; York D.M.; Kollman P. A.. AMBER 2019; University of California: San Francisco, 2019. [Google Scholar]
- Shao J.; Tanner S. W.; Thompson N.; Cheatham T. E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. III Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J. Comput. Chem. 2018, 39, 2110–2117. 10.1002/jcc.25382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Metz S.; Kästner J.; Sokol A. A.; Keal T. W.; Sherwood P. ChemShell—a modular software package for QM/MM simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 101–110. 10.1002/wcms.1163. [DOI] [Google Scholar]
- Sherwood P.; de Vries A. H.; Guest M. F.; Schreckenbach G.; Catlow C. R. A.; French S. A.; Sokol A. A.; Bromley S. T.; Thiel W.; Turner A. J.; Billeter S.; Terstegen F.; Thiel S.; Kendrick J.; Rogers S. C.; Casci J.; Watson M.; King F.; Karlsen E.; Sjøvoll M.; Fahmi A.; Schäfer A.; Lennartz C. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct.: THEOCHEM 2003, 632, 1–28. 10.1016/S0166-1280(03)00285-9. [DOI] [Google Scholar]
- Benediktsson B.; Bjornsson R. QM/MM Study of the Nitrogenase MoFe Protein Resting State: Broken-Symmetry States, Protonation States, and QM Region Convergence in the FeMoco Active Site. Inorg. Chem. 2017, 56, 13417–13429. 10.1021/acs.inorgchem.7b02158. [DOI] [PubMed] [Google Scholar]
- Kästner J.; Carr J. M.; Keal T. W.; Thiel W.; Wander A.; Sherwood P. DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations. J. Phys. Chem. A 2009, 113, 11856–11865. 10.1021/jp9028968. [DOI] [PubMed] [Google Scholar]
- Ramos F. C.; Nottoli M.; Cupellini L.; Mennucci B. The molecular mechanisms of light adaption in light-harvesting complexes of purple bacteria revealed by a multiscale modeling. Chem. Sci. 2019, 10, 9650–9662. 10.1039/C9SC02886B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk D.; Dym O.; Prabahar V.; Peleg Y.; Pike D. H.; Noy D. Fine Tuning of Chlorophyll Spectra by Protein-Induced Ring Deformation. Angew. Chem., Int. Ed. 2016, 55, 6901–6905. 10.1002/anie.201512001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchelli G.; Brogioli D.; Casazza A. P.; Garlaschi F. M.; Jennings R. C. Chlorophyll Ring Deformation Modulates Qy Electronic Energy in Chlorophyll-Protein Complexes and Generates Spectral Forms. Biophys. J. 2007, 93, 2240–2254. 10.1529/biophysj.107.104554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Kendall R. A.; Früchtl H. A. The Impact of the Resolution of the Identity Approximate Integral Method on Modern ab Initio Algorithm Development. Theor. Chem. Acc. 1997, 97, 158–163. 10.1007/s002140050249. [DOI] [Google Scholar]
- Sierka M.; Hogekamp A.; Ahlrichs R. Fast Evaluation of the Coulomb Potential for Electron Densities Using Multipole Accelerated Resolution of Identity Approximation. J. Chem. Phys. 2003, 118, 9136–9148. 10.1063/1.1567253. [DOI] [Google Scholar]
- Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–290. 10.1016/0009-2614(95)00621-A. [DOI] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. 10.1063/1.2834918. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. 10.1039/c3cp54374a. [DOI] [PubMed] [Google Scholar]
- Najibi A.; Goerigk L. The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches. J. Chem. Theory Comput. 2018, 14, 5725–5738. 10.1021/acs.jctc.8b00842. [DOI] [PubMed] [Google Scholar]
- Cupellini L.; Calvani D.; Jacquemin D.; Mennucci B. Charge transfer from the carotenoid can quench chlorophyll excitation in antenna complexes of plants. Nat. Commun. 2020, 11, 662. 10.1038/s41467-020-14488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Nieman R.; Aquino A. J. A.; Lischka H.; Tretiak S. Comparison of LC-TDDFT and ADC(2) Methods in Computations of Bright and Charge Transfer States in Stacked Oligothiophenes. J. Chem. Theory Comput. 2014, 10, 3280–3289. 10.1021/ct500072f. [DOI] [PubMed] [Google Scholar]
- Izsák R.; Neese F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. 10.1063/1.3646921. [DOI] [PubMed] [Google Scholar]
- Neese F.; Wennmohs F.; Hansen A.; Becker U. Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. 10.1016/j.chemphys.2008.10.036. [DOI] [Google Scholar]
- Tawada Y.; Tsuneda T.; Yanagisawa S.; Yanai T.; Hirao K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. 10.1063/1.1688752. [DOI] [PubMed] [Google Scholar]
- Cai Z.-L.; Sendt K.; Reimers J. R. Failure of density-functional theory and time-dependent density-functional theory for large extended π systems. J. Chem. Phys. 2002, 117, 5543–5549. 10.1063/1.1501131. [DOI] [Google Scholar]
- Dahlbom M. G.; Reimers J. R. Successes and failures of time-dependent density functional theory for the low-lying excited states of chlorophylls. Mol. Phys. 2005, 103, 1057–1065. 10.1080/00268970412331333528. [DOI] [Google Scholar]
- Dreuw A.; Head-Gordon M. Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin–bacteriochlorin and bacteriochlorophyll–spheroidene complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. 10.1021/ja039556n. [DOI] [PubMed] [Google Scholar]
- Neese F. A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry. JBIC, J. Biol. Inorg. Chem. 2006, 11, 702–711. 10.1007/s00775-006-0138-1. [DOI] [PubMed] [Google Scholar]
- Linnanto J.; Korppi-Tommola J. Quantum chemical simulation of excited states of chlorophylls, bacteriochlorophylls and their complexes. Phys. Chem. Chem. Phys. 2006, 8, 663–687. 10.1039/B513086G. [DOI] [PubMed] [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- Loos P.-F.; Scemama A.; Jacquemin D. The Quest for Highly Accurate Excitation Energies: A Computational Perspective. J. Phys. Chem. Lett. 2020, 11, 2374–2383. 10.1021/acs.jpclett.0c00014. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Mei Y.; Sundholm D.; Kaila V. R. I. Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores. J. Chem. Theory Comput. 2020, 16, 587–600. 10.1021/acs.jctc.9b00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J. J.; Sauer S. P. A.; Mikkelsen K. V.; Christiansen O.; Jensen H. J. A.; Kongsted J. Failures of TDDFT in describing the lowest intramolecular charge-transfer excitation in para-nitroaniline. Mol. Phys. 2013, 111, 1235–1248. 10.1080/00268976.2013.793841. [DOI] [Google Scholar]
- Lischka H.; Nachtigallová D.; Aquino A. J. A.; Szalay P. G.; Plasser F.; Machado F. B. C.; Barbatti M. Multireference Approaches for Excited States of Molecules. Chem. Rev. 2018, 118, 7293–7361. 10.1021/acs.chemrev.8b00244. [DOI] [PubMed] [Google Scholar]
- Izsák R. Single-reference coupled cluster methods for computing excitation energies in large molecules: The efficiency and accuracy of approximations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, e1445. 10.1002/wcms.1445. [DOI] [Google Scholar]
- Dutta A. K.; Neese F.; Izsák R. Towards a pair natural orbital coupled cluster method for excited states. J. Chem. Phys. 2016, 145, 034102. 10.1063/1.4958734. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Nooijen M.; Neese F.; Izsák R. Automatic active space selection for the similarity transformed equations of motion coupled cluster method. J. Chem. Phys. 2017, 146, 074103. 10.1063/1.4976130. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Nooijen M.; Neese F.; Izsák R. Exploring the Accuracy of a Low Scaling Similarity Transformed Equation of Motion Method for Vertical Excitation Energies. J. Chem. Theory Comput. 2018, 14, 72–91. 10.1021/acs.jctc.7b00802. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Saitow M.; Demoulin B.; Neese F.; Izsák R. A domain-based local pair natural orbital implementation of the equation of motion coupled cluster method for electron attached states. J. Chem. Phys. 2019, 150, 164123. 10.1063/1.5089637. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Saitow M.; Riplinger C.; Neese F.; Izsák R. A near-linear scaling equation of motion coupled cluster method for ionized states. J. Chem. Phys. 2018, 148, 244101. 10.1063/1.5029470. [DOI] [PubMed] [Google Scholar]
- Riplinger C.; Neese F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- Sirohiwal A.; Berraud-Pache R.; Neese F.; Izsák R.; Pantazis D. A. Accurate Computation of the Absorption Spectrum of Chlorophyll a with Pair Natural Orbital Coupled Cluster Methods. J. Phys. Chem. B 2020, 10.1021/acs.jpcb.0c05761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma B.; Tajti A.; Demoulin B.; Izsák R.; Nooijen M.; Szalay P. G. A New Benchmark Set for Excitation Energy of Charge Transfer States: Systematic Investigation of Coupled Cluster Type Methods. J. Chem. Theory Comput. 2020, 16, 4213–4225. 10.1021/acs.jctc.0c00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llansola-Portoles M. J.; Li F.; Xu P.; Streckaite S.; Ilioaia C.; Yang C.; Gall A.; Pascal A. A.; Croce R.; Robert B. Tuning antenna function through hydrogen bonds to chlorophyll a. Biochim. Biophys. Acta, Bioenerg. 2020, 1861, 148078. 10.1016/j.bbabio.2019.148078. [DOI] [PubMed] [Google Scholar]
- Agostini A.; Meneghin E.; Gewehr L.; Pedron D.; Palm D. M.; Carbonera D.; Paulsen H.; Jaenicke E.; Collini E. How water-mediated hydrogen bonds affect chlorophyll a/b selectivity in Water-Soluble Chlorophyll Protein. Sci. Rep. 2019, 9, 1–10. 10.1038/s41598-019-54520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raszewski G.; Saenger W.; Renger T. Theory of Optical Spectra of Photosystem II Reaction Centers: Location of the Triplet State and the Identity of the Primary Electron Donor. Biophys. J. 2005, 88, 986–998. 10.1529/biophysj.104.050294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Bommer M.; Chatterjee R.; Hussein R.; Yano J.; Dau H.; Kern J.; Dobbek H.; Zouni A. Structural Insights into the Light-Driven Auto-Assembly Process of the Water-Oxidizing Mn4CaO5-Cluster in Photosystem II. eLife 2017, 6, e26933. 10.7554/eLife.26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadova N.; Mamedov F. Formation of Tyrosine Radicals in Photosystem II Under Far-Red Illumination. Photosynth. Res. 2018, 136, 93–106. 10.1007/s11120-017-0442-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H.; Burnap R. L.. Photoactivation: The Light-Driven Assembly of the Water Oxidation Complex of Photosystem II. Front. Plant Sci. 2016, 7 10.3389/fpls.2016.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen M. A.; Lao K.; Boxer S. G. Dielectric Asymmetry in the Photosynthetic Reaction Center. Science 1994, 264, 810–816. 10.1126/science.264.5160.810. [DOI] [PubMed] [Google Scholar]
- Saggu M.; Fried S. D.; Boxer S. G. Local and Global Electric Field Asymmetry in Photosynthetic Reaction Centers. J. Phys. Chem. B 2019, 123, 1527–1536. 10.1021/acs.jpcb.8b11458. [DOI] [PubMed] [Google Scholar]
- Renger T.; Müh F. Understanding photosynthetic light-harvesting: a bottom up theoretical approach. Phys. Chem. Chem. Phys. 2013, 15, 3348–3371. 10.1039/C3CP43439G. [DOI] [PubMed] [Google Scholar]
- Kavanagh M. A.; Karlsson J. K. G.; Colburn J. D.; Barter L. M. C.; Gould I. R. A TDDFT investigation of the Photosystem II reaction center: Insights into the precursors to charge separation. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 19705. 10.1073/pnas.1922158117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedringhaus A.; Policht V. R.; Sechrist R.; Konar A.; Laible P. D.; Bocian D. F.; Holten D.; Kirmaier C.; Ogilvie J. P. Primary processes in the bacterial reaction center probed by two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3563–3568. 10.1073/pnas.1721927115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ratés M.; Neese F. Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020, 41, 922–939. 10.1002/jcc.26139. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Miyashita H.; Ikemoto H.; Kurano N.; Adachi K.; Chihara M.; Miyachi S. Chlorophyll d as a major pigment. Nature 1996, 383, 402–402. 10.1038/383402a0. [DOI] [Google Scholar]
- Chen M.; Schliep M.; Willows R. D.; Cai Z.-L.; Neilan B. A.; Scheer H. A Red-Shifted Chlorophyll. Science 2010, 329, 1318–1319. 10.1126/science.1191127. [DOI] [PubMed] [Google Scholar]
- Renger T.; Schlodder E. The Primary Electron Donor of Photosystem II of the Cyanobacterium Acaryochloris marina Is a Chlorophyll d and the Water Oxidation Is Driven by a Chlorophyll a/Chlorophyll d Heterodimer. J. Phys. Chem. B 2008, 112, 7351–7354. 10.1021/jp801900e. [DOI] [PubMed] [Google Scholar]
- Chen M.; Telfer A.; Lin S.; Pascal A.; Larkum A. W.; Barber J.; Blankenship R. E. The nature of the photosystem II reaction centre in the chlorophyll d-containing prokaryote, Acaryochloris marina. Photochem. Photobiol. Sci. 2005, 4, 1060–1064. 10.1039/b507057k. [DOI] [PubMed] [Google Scholar]
- Judd M.; Morton J.; Nürnberg D.; Fantuzzi A.; Rutherford A. W.; Purchase R.; Cox N.; Krausz E. The primary donor of far-red photosystem II: ChlD1 or PD2?. Biochim. Biophys. Acta, Bioenerg. 2020, 1861, 148248. 10.1016/j.bbabio.2020.148248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.