

Summary
The Cdk-Rb-E2F pathway integrates external and internal signals to control progression at the G1/S transition of the mammalian cell cycle. Alterations in this pathway are found in the vast majority of human cancers and specific Cdk4/6 inhibitors are approved or in clinical trials for the treatment of diverse cancers. In the long-standing paradigm for G1/S control, Cdks inactivate Rb through phosphorylation, which releases E2F transcription factors to drive cell-cycle progression from G1 to S. However, recent observations in the laboratory and clinic challenge central tenets of the current paradigm and demonstrate that our understanding of the Rb pathway and G1/S control is still incomplete. Here, we integrate these recent findings with the previous paradigm to synthesize a current molecular and cellular view of the mammalian G1/S transition. A more complete and accurate understanding of G1/S control will ultimately lead to improved therapeutic strategies targeting the cell cycle in cancer.
Introduction
The cellular decision to transition from G1 to S phase of the cell cycle is critical to normal development and is necessarily misregulated in cancer. During G1, cells decide to either enter the cell cycle, initiate DNA replication, and divide, or to exit the cell cycle and enter quiescence, senescence or differentiation. The G1/S transition is controlled by Cyclin-dependent kinases (Cdks), the retinoblastoma tumor suppressor (Rb), and E2F transcription factors (Figure 1A). These components are typically assembled into a linear pathway, in which the upstream Cdk inhibits Rb to drive E2F-dependent transcription and cell-cycle progression. Abrogation of the G1/S control point is a hallmark of cancer and typically occurs through loss of Rb or through hyper-activation of Cdks (Burkhart and Sage, 2008; Kent and Leone, 2019; Malumbres and Barbacid, 2009; Otto and Sicinski, 2017). Importantly, the linear understanding of how the G1/S transition is controlled by the Cdk-Rb-E2F pathway has led to the development of Cdk4 and Cdk6 (Cdk4/6) kinase inhibitors for cancer therapy (O’Leary et al., 2016; Otto and Sicinski, 2017; Sherr et al., 2016). These Cdk4/6 ATP-competitive inhibitors, which were developed to arrest proliferating cells by inhibiting Rb phosphorylation and inactivation, are the first clinically approved drugs targeting the G1/S transition.
Figure 1: Components of the Cdk-Rb-E2F pathway controlling the G1/S transition.

(A) Simplified model for pathway. (B) Inactive and active states of the key players in the Rb pathway. Cdk4 and Cdk6 (Cdk4/6) have relatively high sequence homology among Cdks. They are inactive as monomers, bound to p16 family proteins, or bound by unphosphorylated p21 and p27 (p21/p27) proteins. Cdk4/6 are activated by association with Cyclin D (CycD) family proteins, but full activity also requires a phosphorylated from of p27 in the complex and phosphorylation on the kinase activation loop. Cdk2 is inactive as a monomer or in complex with p21/p27 family proteins, and it is activated by CycE binding in G1 (or CycA later in the cell cycle) and activation loop phosphorylation. Rb is considered active when hypo-or monophosphorylated; in this state it binds and inhibits E2F. Hyperphosphorylation of Rb leads to its inactivation, dissociation from E2F and subsequent E2F activation. While Rb binds E2F when unphosphorylated during quiescence, it may also be considered inactive in that it cannot perform monophosphorylation-dependent functions.
Despite the progress over the past several decades, important questions remain. While the canonical G1/S pathway is understood as linear (Figure 1A), regulation is still quite complex. There are multiple homologous proteins for each pathway component, each protein is subject to post-translational modifications, and there are critical positive and negative feedback loops that connect the different pathway components (Figure 1B) (for past reviews see (Bertoli et al., 2013; Burkhart and Sage, 2008; Dick and Rubin, 2013; Kent and Leone, 2019; Sherr and Roberts, 1999). As a consequence, the field still struggles to define the critical molecular interactions and activities leading to Rb inactivation and the timing of those mechanisms relative to passage through G1 and into S phase. Moreover, it remains unclear how upstream growth signals feed into the pathway to coordinate cell growth with division, one of the earliest identified functions of G1/S regulation. Finally, there is a need to understand the variable responses to Cdk4/6 inhibitors observed both in the partial arrest of cells cultured in vitro and in the diverse outcomes of patients in clinical trials. According to the textbook linear model, inhibition of the upstream Cdk complexes should arrest cell division. That it often does not implies that there is a lot we do not know about the G1/S transition. Here, we review recent progress in understanding these important aspects of G1/S control in mammalian cells.
Mechanisms and consequences of Rb inactivation
The most critical event that marks the transition from G1 to S is the activation of a transcription program regulated by E2F transcription factors (Kent and Leone, 2019). Toward the end of G1, activating members of the E2F family stimulate expression of a set of hundreds of genes that promote DNA replication and enforce irreversible progression into the cell cycle (Bertoli et al., 2013; DeGregori et al., 1995). This E2F activity driving entry into S phase is restricted by Rb family proteins, which form repressive complexes with E2F proteins on the promoters of the S phase genes. Repression is relieved by Cdk phosphorylation of Rb proteins, which takes place on up to 15 sites and drives structural changes in Rb that lead to E2F dissociation (Dick and Rubin, 2013). Accordingly, a hallmark of the G1/S transition is the hyperphosphorylation of Rb that necessarily precedes DNA synthesis.
The requirement of Rb inactivation for G1/S progression has motivated studies to define the mechanisms by which Cdk complexes phosphorylate Rb and by other means regulate the G1/S transition. In the prevailing model, Cdk4/6-CycD is first to phosphorylate Rb. Then, later in G1, Cdk2-CycE completes phosphorylation to release and fully activate E2F (Figure 2, Model I). This model is supported by the timing of cyclin expression through G1, some reported evidence for specificity of the kinases for non-overlapping phosphorylation sites in Rb, and experiments inhibiting Cdk4/6 or Cdk2 activity (Harbour et al., 1999; Lundberg and Weinberg, 1998). The understanding that Rb phosphorylation is ordered in this manner also fits with models for how Cdk4/6 and then Cdk2 activity gradually increase throughout G1. Two mechanisms explain how Cdk4/6-CycD itself induces the increase in Cdk2-CycE activity. First, the gene encoding CycE is an E2F target that could be expressed if Cdk4/6 phosphorylation of Rb partially inactivated it (DeGregori et al., 1995). Second, evidence suggests that increasing the abundance of Cdk4/6-CycD complexes leads to Cdk2 activation by sequestering the Cdk inhibitors p21 and p27 away from Cdk2 (Sherr and Roberts, 1999). Somewhat paradoxically, p21 and p27 are generally considered cell-cycle inhibitors, but also play a role in Cdk4/6 assembly and activation (Guiley et al., 2019; Sherr and Roberts, 1999).
Figure 2: Proposed models for Rb inactivation during G1.

(A) The three models differ with respect to whether Cdk4/6 and Cdk2 activate (arrow) or inhibit (cross) Rb and in the timing of their activity relative to the G1/S transition. (B) Roles of Cdk complexes in modulating Rb function. In the canonical Model I, Cdk4/6 partially phosphorylates Rb, resulting in some E2F activity. Transcription of genes such as CycE then activates Cdk2 for full Rb hyperphosphorylation and inactivation for S phase. In Model II, Cdk4/6 monophosphorylates Rb, and the different active Rb species inhibit E2F and form functional protein complexes in G1. Cdk4/6 induces Cdk2 activity through several possible mechanisms, including sequestration of p21/p27 inhibitors and phosphorylation of other targets that may indirectly activate Cdk2. Active Cdk2 then hyperphosphorylates Rb, leading to Rb inactivation and S phase entry. In Model III, Cdk4/6 hyperphosphorylates Rb in G1, which is sufficient for Rb inactivation and S phase entry. Cdk2 activity is required to maintain Rb hyperphosphorylation during S phase.
New models for cell-cycle progression at the G1/S transition
The canonical model, in which both Cdk4/6 and Cdk2 activity contribute to Rb phosphorylation and inactivation during G1, has been challenged by a number of recent studies that support two distinct alternative models (Figure 2). By careful quantification of Rb phosphorylation patterns in synchronized cells, it was found that Cdk4/6-CycD activity only induces Rb monophosphorylation, whereas Rb hyperphosphorylation is co-incident with the onset of Cdk2 activity (Narasimha et al., 2014). E2F-dependent gene expression remained low while Rb was monophosphorylated by Cdk4/6-CycD, leading to the conclusion that only Cdk2 directly inactivates Rb (Figure 2, Model II). These observations also change our conception of Rb phosphorylation during G1. Rather than existing in a continuum of phosphorylation states, which results from progressive and successive phosphorylation by the G1 Cdks, Rb is present in discrete states. Rb is either unphosphorylated in cells that exit the cell cycle and lack CycD, monophosphorylated when active in G1 in the presence of CycD, or hyperphosphorylated once Cdk2 is activated (Figure 2B, Model II). Through use of phosphospecific antibodies, monophosphorylation was observed at each of the Rb Cdk consensus sites that were examined, suggesting that there are multiple monophosphorylated forms of Rb. The possibility of different functions for the different monophosphorylated states was further explored by identifying Rb complexes in G1 that contain a particular monophosphorylated species (Sanidas et al., 2019). Specific phosphorylation events assembled Rb with factors, for example the NuRD chromatin remodeling complex, which may induce unique transcriptional outputs. This mass spectrometry-based proteomics study also found that CycD association with several of the monophosphorylated forms of Rb was poor compared to association with unphosphorylated Rb, which could account for how Cdk4/6-CycD activity toward Rb is limited to a single phosphorylation event. If CycD no longer interacts with monophosphorylated Rb, then it could not be responsible for hyperphosphorylation. The mechanism by which Cdk4/6 monophosphorylates various Cdk sites in Rb needs further exploration. Moreover, additional corroborating research is needed to support the conclusion that Cdk4/6-CycD strictly activates Rb and to reconcile this model with other observations, several of which are described below, implicating Cdk4/6-CycD as an Rb-inactivating kinase.
That Cdk4/6-CycD monophosphorylates Rb and that this monophosphorylated Rb inhibits E2F raises the question of whether or not Cdk4/6-CycD inactivates Rb, a central tenet of the current paradigm. If Cdk4/6 activity does not inactivate Rb, then why is Cdk4/6 activity important for cell-cycle progression and proliferation? If Cdk2 is the inactivating kinase, why do Cdk4/6 specific inhibitors like palbociclib result in loss of hyperphosphorylated Rb and cell-cycle arrest in G1? These questions are further motivated by recent evidence affirming that Rb-directed Cdk4/6-CycD activity is critical for Rb hyperphosphorylation and G1/S progression. It was found that mutation of a Cdk4/6-specific docking site in Rb results in impaired Rb hyperphosphorylation, increased G1 arrest, and enhanced Rb tumor suppressor activity in vivo (Topacio et al., 2019). There are several potential explanations for how Cdk4/6 activity leads to Cdk2 activity, and inversely how Cdk4/6 inhibition may indirectly lead to Cdk2 inhibition. In such mechanisms, Cdk4/6 is necessary for the ultimate inactivation of Rb, even if Cdk4/6 activity alone does not directly hyperphosphorylate Rb (Figure 2, Model II). One possibility is that Cdk4/6 monophosphorylation is required for priming Rb for Cdk2 hyperphosphorylation. Such priming mechanisms for Cdk hyperphosphorylation have been observed in other cell-cycle substrates in yeast (Koivomagi et al., 2013), though they have not yet been implicated in the mechanism of Rb hyperphosphorylation. A second possibility is that additional Cdk4/6 targets currently being identified, such as enzymes controlling metabolism (Caillot et al., 2020; Wang et al., 2017), can explain the role of Cdk4/6 in promoting cell-cycle progression and proliferation. In this model, Cdk2 activation would result from Cdk4/6 targeting these other substrates. Importantly, both of these mechanisms are not mutually exclusive and are consistent with the observation that upon expression of CycE, Cdk4/6-CycD is dispensable for G1/S progression (Keenan et al., 2004).
A third possibility for how Cdk4/6 impacts G1/S progression without hyperphosphorylating Rb invokes the idea that Cdk4/6 regulates Cdk2 through their interactions with the network of Cdk protein inhibitors. For example, increasing CycD levels through G1 could increase Cdk2 activity to inactivate Rb by sequestering p21 and p27. Recent studies of the effects of palbociclib support the importance of these connections between the G1 Cdks. For example, it was found that palbociclib does not directly inhibit cellular Cdk4/6 activity toward Rb, but may indirectly inhibit Cdk2 by increasing the abundance of p21 in Cdk2-CycE complexes (Guiley et al., 2019). Other evidence demonstrates that palbociclib still arrests the cell cycle even when Cdk4 with impaired kinase activity is expressed (Persky et al., 2020; Schade et al., 2019). These results highlight the importance of non-catalytic Cdk4-CycD functions and suggest that small molecule Cdk4/6 inhibitors may both directly inhibit Cdk4/6 and also free p21 and p27 to inhibit Cdk2.
In contrast to the body of work suggesting mechanisms through which Cdk4/6-CycD and Cdk2-CycE cooperate in Rb phosphorylation and the G1/S transition, some other recent studies have challenged the conclusion that Cdk2-CycE plays any significant role in inactivating Rb during G1 (Figure 2, Model III) (Chung et al., 2019; Yang et al., 2020). Rather, it is proposed that Cdk4/6-CycD is exclusively responsible for hyperphosphorylating Rb to activate E2F. These studies, which primarily used fluorescent reporters of putative Cdk4/6 and Cdk2 activity in live cells, corroborated the canonical model that Cdk4/6 activity precedes Cdk2 activity and that Cdk2 activity precedes the onset of S phase (Yang et al., 2020). However, upon manipulating activities, primarily by using chemical inhibitors, it was concluded that Cdk4/6 and not Cdk2 is necessary and sufficient for Rb phosphorylation during G1 (Chung et al., 2019; Yang et al., 2020). Then, only once cells were in S phase, Cdk2 function was required to maintain Rb phosphorylation and positive feedback-driven E2F expression.
The observation that Cdk2 is dispensable for Rb hyperphosphorylation contrasts with other experiments demonstrating that acute Cdk2 inhibition results in G1 accumulation (Merrick et al., 2011; Narasimha et al., 2014). This discrepancy may be due to how observations were made, for example using different markers of S phase entry and different approaches to manipulating Cdk activities. There are also limitations of the Cdk activity sensors that need be considered when interpreting those data. In particular, the engineered live-cell reporters likely detect a combination of Cdk activities due to the cross binding of cyclins to non-canonical Cdk partners and the presence of multiple cyclin docking sites on the sensors (Jirawatnotai et al., 2011; Schwarz et al., 2018; Topacio et al., 2019). In addition, the chemical Cdk inhibitors used in these experiments may lack specificity and have off-target effects that have not been properly considered or discovered.
Yet, even if interpreted cautiously, the experiments analyzing Cdk reporters in single cells have reopened important questions regarding the timing of cell-cycle events through the G1/S transition and highlight the acute need for studies that integrate or explain the seemingly contradictory results leading to the different models for Rb inactivation. Considering the complexity of the Cdk network, the precise manner in which perturbations are made and subtle differences in cellular model system may impact outcomes. For example, requirements for traversing G1-S are known to be different in cells re-entering the cell cycle from quiescence compared to continuously cycling cells (Matson and Cook, 2017).
Revisiting the restriction point
The study of the G1/S transition and the Rb pathway has been motivated by a desire to understand commitment to cell division, a process that is necessarily misregulated in cancer. Classically, the restriction point in mammalian cells has been defined as the irreversible point of commitment to division whose traversal requires growth factor signaling (Johnson and Skotheim, 2013; Pardee et al., 1978; Pennycook and Barr, 2020). Beyond the restriction point, cells progress through to division even if growth factors, typically serum, are removed from the media. The restriction point was originally determined to occur in late G1 just prior to the initiation of DNA replication (Pardee et al., 1978). This classical picture fits well with the consensus molecular model for the G1/S transition (Figure 2, Model I) (Bertoli et al., 2013). Once initially activated by Cdk4/6-CycD, the CycE-Rb-E2F positive feedback loop can maintain its activity without CycD, which explains how cells become insensitive to decreases in upstream growth factor signaling in the classic restriction point analyses (Barr et al., 2016).
While the CycE-Rb-E2F feedback loop is an elegant molecular mechanism that explains the physiology of an irreversible G1/S transition, this model has now been challenged by recent single-cell analyses revealing a greater complexity in how cells commit to cell division. Primary fibroblast cell lines exhibit the classical G1 restriction point, but many commonly cultured immortal cell lines do not (Martinsson et al., 2005; Naetar et al., 2014; Schwarz et al., 2018; Spencer et al., 2013). Many cells are already committed to division and insensitive to growth factor removal before completing mitosis in the previous cell cycle (Spencer et al., 2013). These pre-committed cells are born with initially high and rising Cdk activity and phosphorylated Rb, showing that signaling events regulating the G1/S transition take place in the previous cell cycle and are remembered in the subsequent generation (Moser et al., 2018). One contribution to this cellular memory of the previous cell cycle is DNA damage, which, if present, leads to reduced Cdk activity and a longer G1 in the next cell cycle (Arora et al., 2017; Barr et al., 2017; Yang et al., 2020; Yang et al., 2017). In addition, global protein synthesis in the previous cell cycle is required for a rapid G1 in the subsequent cell cycle through its effect on CycD synthesis and possibly through its effect on cell growth and thereby on the size of newborn cells (Min et al., 2020). Taken together, these studies show that signaling events from the previous cell cycle can be integrated to control CycD expression and cell-cycle commitment in the following cell cycle. That signaling history and not just current activity impacts the decision to divide was anticipated by a series of yeast studies looking at how the pheromone-activated MAPK pathway integrates its activity over time to restrict cell division (Doncic et al., 2015; Doncic and Skotheim, 2013). It is likely that such integrated signaling responses may be a common feature of MAPK pathways across eukaryotes to promote accurate cellular decision making.
While cells clearly can integrate signals and pre-commit to division, it is not clear when they do so. Certainly, we anticipate the response to growth factor dynamics to be both context-dependent and cell type specific. Inhibiting PP2A phosphatase signaling results in commitment to cell division in the preceding cell cycle, but such signaling is likely present in wild-type cells (Naetar et al., 2014). In general, active dephosphorylation during mitosis would be expected to reset the cell cycle so that there is little memory of past signaling and the cell cycle would have the modular structure some have observed (Araujo et al., 2016; Chao et al., 2019). One possibility to explain the pre-commitment phenomenon in immortal cell lines is that culturing cells selects for alterations upregulating the Cdk4 activity that has been associated with the pre-commitment phenomenon. Indeed, likely due to its crucial role in regulating cell proliferation, mutations weakening or advancing the restriction point to the previous cell cycle have been associated with cancer-derived cell lines (Pardee et al., 1978; Sherr and DePinho, 2000; Zetterberg et al., 1995). Rapid evolution of lab cultures has revealed that lineages within a yeast population rapidly generated mutations that were then selected for increased proliferation (Levy et al., 2015). While it is easy to imagine something similar happening during the long-term culture of human cell lines, an analogous evolutionary study is needed to test this hypothesis. In addition to the possibility of lab generated mutations affecting the restriction point, we do not know the in vivo context in which these signaling pathways operate that can affect the signaling dynamics. For example, the pulsatile and wave-like dynamics of MAPK signaling observed in mouse epidermis (Hino et al., 2020; Hiratsuka et al., 2015) highlights the importance of understanding the dynamics of growth factor signaling in vivo across diverse cell types. A better understanding of signaling dynamics in vivo could situate the restriction point studies of cultured cells in their physiologically relevant context. One possible outcome is that the location of the restriction point is developmentally regulated to take place in the prior cell cycle or in G1 phase depending on cell type.
APCCdh1 and p21/p27 inactivation and the irreversible commitment to S phase
While Rb inactivation is required for the G1/S transition, cells that have hyperphosphorylated Rb can still arrest before S phase entry in response to a stress that activates p53, such as DNA damage (Cappell et al., 2016; Yang et al., 2020). This observation suggests the presence of additional processes that trigger irreversible entry into S phase, and recent evidence implicates inactivation of the anaphase promoting complex (APC) and degradation of the Cdk inhibitors p21 and p27 (p21/p27) as such mechanisms (Barr et al., 2016; Cappell et al., 2016; Cappell et al., 2018; Heldt et al., 2018) (Figure 3). The APC is a ubiquitin ligase that, with the adaptor subunit Cdh1, promotes degradation of cell-cycle substrates during G1 (Schrock et al., 2020). Several of these substrates, including the Cdk2-activator CycA and the ubiquitin ligase adaptor Skp2, play important regulatory roles during S phase. The development of a live-cell reporter for APCCdh1 activity led to the observation that Cdh1 inactivation follows Rb inactivation but precedes DNA replication (Cappell et al., 2016). Notably, Cdh1 inactivation could not be reversed through Cdk inhibition or cellular stress including DNA damage. Two mechanisms have been described for APCCdh1 inactivation, including phosphorylation by Cdk2 (Keck et al., 2007; Lukas et al., 1999), which directly inhibits the ubiquitylation reaction, and binding of the inhibitor protein Emi1 (Figure 3) (Cappell et al., 2018; Miller et al., 2006). The gene encoding Emi1 is a transcriptional target of E2F, which places Rb inactivation upstream of Cdh1 inactivation. Emi1 is an APCCdh1 substrate that becomes an inhibitor when its protein concentration increases and Cdh1 is phosphorylated. This dual function of Emi1 has been invoked as a mechanism for how Cdh1 inactivation acts as an irreversible switch (Cappell et al., 2018).
Figure 3: APCCdh1 and p21/p27 inactivation are required for S phase entry.
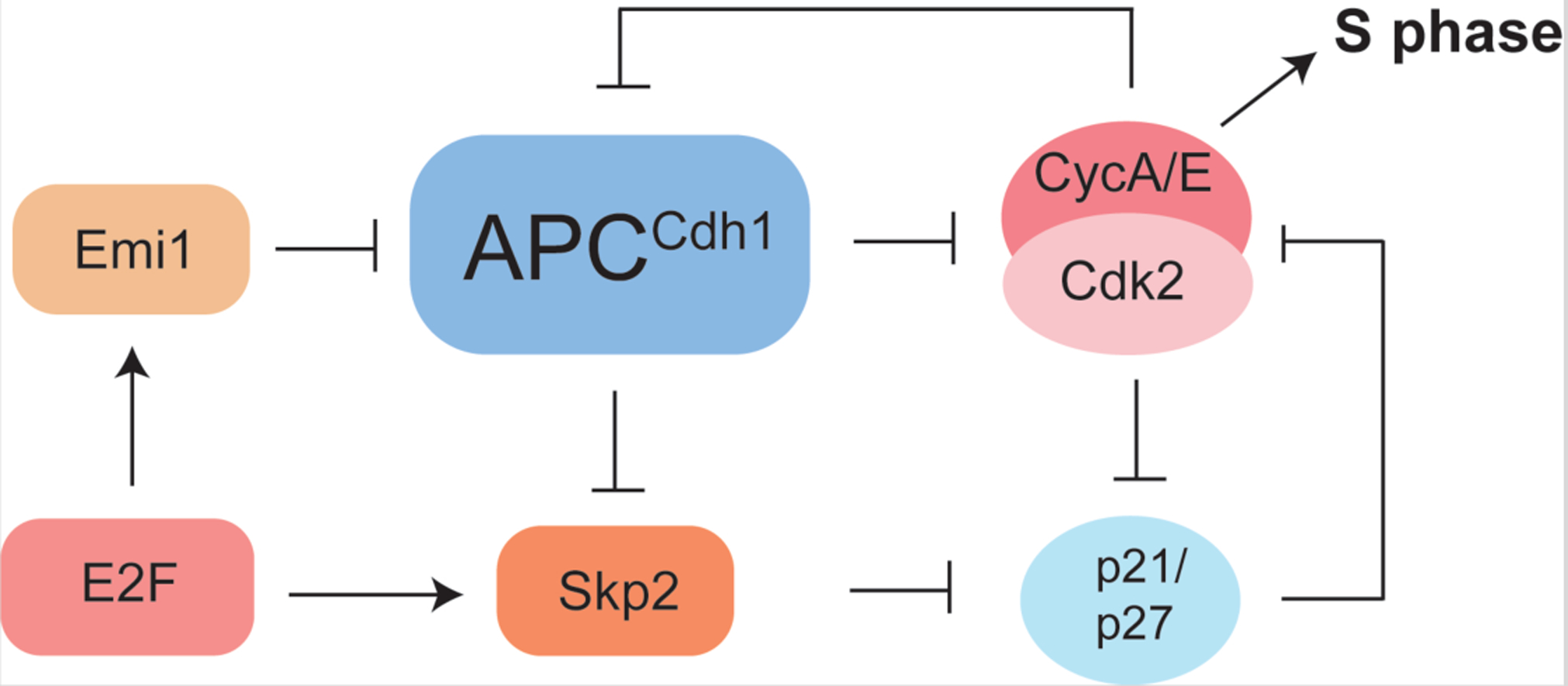
S phase activity of Cdk2 is inhibited by p21/p27 and APCCdh1, which is a ubiquitin ligase that stimulates CycA degradation. APCCdh1 is inactivated by Cdk2 phosphorylation and the protein Emi1. p21/p27 degradation is induced by Cdk2 phosphorylation and the ubiquitin ligase Skp2, which is in turn degraded by APCCdh1. Both Emi1 and Skp2 are transcriptional targets of E2F, which places these inactivation events downstream of Rb inactivation. These two connected double negative feedback loops involved in Cdk2 activation are thought to render S phase entry irreversible.
Another important event at the G1/S transition is the degradation of the p21/p27 inhibitors, which is required for activation of Cdk2 in S phase (Barr et al., 2016; Heldt et al., 2018). Skp2, a substrate adaptor of the cullin ubiquitin ligase that targets p21/p27 for degradation, is itself a Cdh1 target (Schrock et al., 2020), and therefore p21/p27 inactivation likely occurs downstream of Cdh1 inactivation (Figure 3). Other parallels between Cdh1 and p21/p27 inactivation are apparent. p21/p27 degradation is stimulated by Cdk2 activity (Sheaff et al., 1997), which provides positive feedback as p21/p27 inhibits Cdk2 (Figure 3). p21 is a p53 target that is induced upon DNA damage, and so p21 degradation, like Cdh1 inactivation, also commits the cell to S phase despite subsequent stress. The degradation of p21/p27 at the onset of S phase may have implications for why Cdk4/6 inhibitors are limited in their efficacy to G1. Once levels of p21/p27 decrease, there is no pathway to indirectly inhibiting Cdk2 activity through reshuffling of the trimer Cdk complexes. As these additional mechanisms of Cdh1 and p21/p27 inactivation implicate, the critical determinant of S phase progression is the complete activation of Cdk2 (Figure 3).
Coupling cell growth to cell-cycle progression in G1/S
An important function of the G1/S transition in animal cells is to link cell growth to division (Ginzberg et al., 2015; Zatulovskiy and Skotheim, 2020). This connection manifests as an inverse correlation between how big a cell is when it is born and how long it spends in the G1 phase of the cell cycle. The inverse correlation allows cells that are initially born smaller to catch up in size before entering the cell division cycle. Recent single-cell studies confirmed that cell size at cell birth negatively correlates with the time cells spend in the G1 phase of the cell cycle, although the quantitative relationship between cell size, growth, and G1/S progression depended on the particular cell line (Cadart et al., 2018; Ginzberg et al., 2018; Varsano et al., 2017). Importantly, the G1/S transition of epidermal stem cells in vivo was more size-dependent than the G1/S transition of cultured cells (Xie and Skotheim, 2020), supporting the role of the G1/S transition in coupling cell growth to division
One mechanism human cells use to link cell growth to the G1/S transition is intimately related to the size-dependent scaling of cellular biosynthesis that generally maintains cellular components at constant concentrations (Schmoller and Skotheim, 2015). Differential patterns of biosynthetic scaling enable cells to measure their size through size-dependent concentration changes of particular proteins and to use these concentration changes to regulate G1/S in a size-dependent manner. This possibility was first discovered in budding yeast, where the synthesis of cell-cycle activators is roughly proportional to cell size and thereby follows the scaling pattern typical of most proteins, but the synthesis of the cell-cycle inhibitor Whi5 is independent of cell size (Schmoller and Skotheim, 2015). This means Whi5 concentration is lower in larger cells, which reduces its inhibitory effect to trigger earlier cell-cycle entry compared to smaller cells. Strikingly, the same phenomenon of dilution during G1 was discovered in human cells for Rb, which is the functional ortholog of Whi5 (Zatulovskiy et al., 2020) (Figure 4). Thus, it is possible that in both yeast and human cells, growth triggers cell division by diluting a protein that inhibits cell cycle-dependent transcription, while activators remain at constant concentration.
Figure 4: Cell growth and size signals drive the G1/S transition.
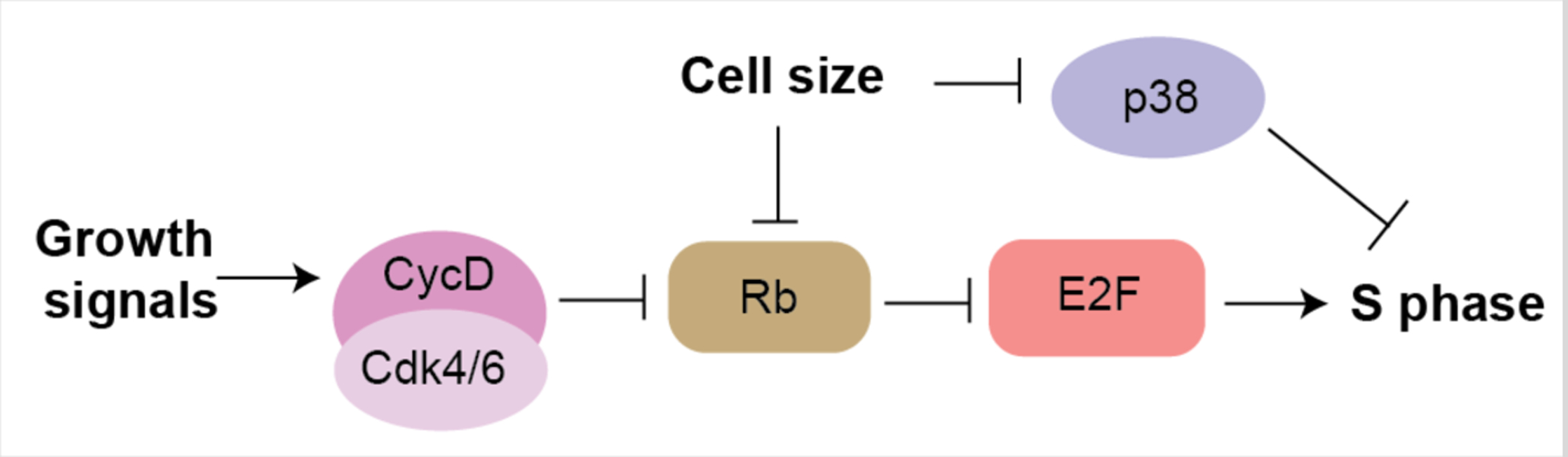
Growth factors trigger the synthesis of CycD and Cdk4/6-dependent activity. Cell growth in G1 dilutes Rb and leads to a decrease in activity of the p38 stress-activated kinase to trigger cell-cycle progression in larger cells.
In addition to Rb dilution, cell size likely impacts the G1/S transition through the activity of the p38 stress activated kinase. In the p38 model, small cell size activates p38 to restrict the G1/S transition until cells grow sufficiently large (Liu et al., 2018). However, the molecular mechanism through which cell size regulates p38 activity, and how such a size-activated p38 functions to inhibit the G1/S transition is unknown. Moreover, the effect on cell-cycle progression of p38 activity is context-dependent and may also be isoform-dependent. While p38γ was recently shown to directly phosphorylate Rb to drive G1/S progression in hepatocytes in vivo (Tomás-Loba et al., 2019), p38β and p38δ also contributed to the size-dependent G1/S control in cultured cells (Liu et al., 2018). In general, the p38 family has been linked to inhibition of the G1/S and G2/M transitions through activation of p53 and p21 ((Campbell et al., 2011) and reviewed in (Thornton and Rincon, 2009)). It will be interesting to learn the mechanism through which small cell size activates p38, and how size-dependent p38 activation regulates the G1/S transition.
Thus, while cell size and growth have long been linked to the G1/S transition, only now are we beginning to uncover the underlying molecular mechanisms. The recent mechanistic p38 and Rb studies mark a turning point in the field, and the next step is to understand if and how these mechanisms operate in vivo. Importantly, Rb dilution and p38 activation are not mutually exclusive mechanisms, and it is likely that there are additional mechanisms linking cell growth to division at the G1/S transition depending on cell type. In addition, these mechanisms may work hand in hand with the classic view that growth factors activate CycD expression. For example, both Cdk activity, which weakens Rb-E2F affinity, and Rb concentrations can modulate the capacity of Rb to bind and inhibit E2F. Finally, a full accounting of how growth regulates the G1/S transition will also likely require a more detailed understanding of the mTOR signaling network and its mechanistic links to G1 control (Liu and Sabatini, 2020). We anticipate the nexus of cell size, growth, and the G1/S transition will be a very active area of research in the coming years.
Response of tumors to Cdk4/6 inhibitors and mechanisms of resistance
The Cdk4/6 inhibitor palbociclib was approved in 2016 for the treatment of estrogen receptor-positive breast cancer and was followed by the similar molecules abemaciclib and ribociclib (Finn et al., 2016; Im et al., 2019; Johnston et al., 2019; Sherr et al., 2016). Positive clinical trial results for pallbociclib (PALOMA 3), ribociclib (MONALEESA-7), and other inhibitors have validated Cdk4/6 as therapeutic targets in breast cancer (Im et al., 2019; Sledge et al., 2019; Spring et al., 2020; Turner et al., 2018), and it is likely that Cdk4/6 inhibitors will demonstrate efficacy in other cancer types currently being tested (clinicaltrials.gov). However, a number of questions remain, including why only some patients benefit from treatment with these drugs (Spring et al., 2020). Our lack of an answer arises from our current inability to predict short- and long-term response to Cdk4/6 inhibitors. As an example, there was a puzzling lack of response in the PALOMA-3 trial in a majority of breast tumors that continue to express Rb (Turner et al., 2018), even though Rb is thought to be the main target of Cdk4/6 in the control of cell-cycle progression. Why don’t Cdk4/6 inhibitors inhibit cancer more effectively in patients and what are the mechanisms that dictate the initial response to these molecules and that mediate resistance to their inhibitory effects?
A growing number of studies have been using tumor samples from breast cancer patients to identify pathways that mediate the intrinsic or selected resistance to Cdk4/6 inhibitors. These analyses identified expected alterations in the core Rb pathway and also in other pathways linked to the Rb pathway and cell-cycle control, including RAS/AKT/PTEN/PI3K/MTOR signaling (Costa et al., 2020; Wander et al., 2020), Hippo signaling (Li et al., 2018), p53 signaling (Wander et al., 2020), and molecules involved in the G2/M transition of the cell cycle (Wander et al., 2020). These studies in patients samples have been corroborated by studies in pre-clinical models (Álvarez-Fernández and Malumbres, 2020).
While the overall picture drawn from these studies support an on-target activity of the Cdk4/6 inhibitors, a number of new questions have arisen. First, because Rb is a key target of Cdk4/6 and Rb inactivation promotes tumorigenesis in multiple contexts, loss of Rb would be expected to be a frequent event in tumors that become resistant to Cdk4/6 inhibitors. However, the events leading to the functional inactivation of Rb (e.g., inactivating mutations in the RB1 gene) are relatively rare in clinical samples from tumors resistant to Cdk4/6 inhibitors (Condorelli et al., 2018; Costa et al., 2020; Li et al., 2018; Wander et al., 2020). This rarity raises the question of whether Rb is really the key target of Cdk4/6 in cell-cycle control or whether loss of Rb is actually detrimental to cancer cells - both these ideas challenge the current Rb pathway paradigm.
A second set of observations from clinical samples have shown that elevated levels of Cdk4 (Finn et al., 2020) or Cdk6 (Li et al., 2018) are also associated with increased resistance to Cdk4/6 inhibitors. One interpretation of these data is that, surprisingly, the levels of Cdk4/6 inhibitors that reach cancer cells are not enough to inhibit these kinases even with a few-fold increase in the levels of the drug target. However, data indicating that increased levels of D-type cyclins is linked with increased sensitivity of some cancer cells to Cdk4/6 inhibition (Gong et al., 2017) are at odds with the simplest hypothesis that high levels of CycD-Cdk4/6 complexes drive resistance to treatment in tumors. More active Cdk4/6-CycD complexes could mean that cancer cells have become more dependent on the kinase activity of Cdk4/6. Alternatively, increased expression of particular kinase subunits could lead to changes in protein complexes that render them less amenable to inhibition by small molecule inhibitors. It is also possible that CycD proteins have Cdk4/6-independent effects (Bienvenu et al., 2001; Jirawatnotai et al., 2011; Neuman et al., 1997), including reshuffling of protein partners that also impinge upon Cdk2 activity (Guiley et al., 2019). In support of increased Cdk2 activity as a mechanism of resistance, high CycE levels are associated with decreased response to palbociclib in breast tumors (Turner et al., 2019; Wander et al., 2020). But if increased Cdk2 activity is a major mechanism of resistance to Cdk4/6 inhibitors, why are Rb loss of function events not more frequent? One possible, but unattractive, answer is that Rb is not the key target of Cdk4/6 and Cdk2 in cancer.
Collectively, these analyses underscore our incomplete understanding of how cancer cells respond to the inhibition of Cdk4 and Cdk6 and, more generally, our incomplete understanding of the Rb pathway in G1/S control.
Concluding remarks
Decades of research have identified most, if not all the major regulators of the G1/S transition in mammalian cells. This work has led to the successful development of Cdk4/6 inhibitors now used in the clinic, and Cdk2 inhibitors are currently being tested in cancer patients (clinicaltrials.gov). However, the results of clinical studies with Cdk4/6 inhibitors show that we only partly understand the mechanisms underlying a durable response to these inhibitors. Recent laboratory studies, including the analysis of the G1/S transition at the single cell level, further reveal our incomplete grasp of how the decision to replicate DNA and progress towards mitosis is made at the molecular level.
Some of these gaps in our understanding will be filled by the analysis of cancer specimens from patients treated with Cdk4/6 and Cdk2 inhibitors. The identification of mechanisms of resistance to these inhibitors will lead to new clinical trials with combination therapies and will instruct new studies to better inform how different signaling pathways regulate cell-cycle progression in G1. An important aspect of these clinical studies will be to distinguish how various treatment strategies directly regulate the cell cycle of cancer cells relative to other cells in the tumor microenvironment, including immune cells, which have already been shown to be directly and indirectly impacted by treatment with Cdk4/6 inhibitors (Zhang et al., 2018).
It is likely that our understanding of the restriction point and G1/S regulators will further change as more investigators examine the mechanisms regulating cell-cycle progression in multiple contexts, including in vivo (Cuitino et al., 2019; Sakaue-Sawano et al., 2017; Yano and Hoffman, 2018). In most organs and tissues, cell density, cell-cell interactions, and metabolic activities are different than in cell culture. We anticipate that these differences between cells in culture and in vivo likely influence how the decision to progress from G1 to S is made. For example, the complete loss of Rb family proteins blocks the ability of these cells in culture to arrest in G1 in all contexts examined, but the same Rb family deletion in the mouse liver is not sufficient to block cell-cycle arrest (Ehmer et al., 2014). Furthermore, the definition of the restriction point and the mechanisms of cell-cycle progression in G1/S may differ between quiescent stem cells re-entering the cell cycle from G0 and stem/progenitor cells that cycle frequently and transition from M to G1 directly. It is also possible that differences exist between human cells and mouse cells. The identification of key regulators of G1/S in different in vivo contexts may considerably improve our understanding of cell-cycle progression and give us insight into the plasticity of this molecular network.
We find it striking that even with recent advances in genetic engineering, single cell analyses, and structural studies, so little has been done to investigate the molecular mechanisms underlying how key players in the extended Rb pathway function. For example, post-translational modifications of Rb pathway components (phosphorylation and others) have only been superficially investigated and similarities and differences between family members at every level in the pathway are still poorly understood. The mechanisms underlying protein-protein interactions and the dynamic nature of protein complexes in the pathway are also still mostly unclear. It is likely that further research in the next decade will not only refine our understanding of the molecular mechanisms of the G1/S transition but will also uncover new paradigms.
Acknowledgements
Related research in the authors’ laboratories is supported by grants from the National Institutes of Health (R01GM124148 to S.M.R., R35CA231997 to J.S., R01CA228413 to S.M.R. and J.S., and R35GM134858 to J.M.S.) and the California Tobacco Related Disease Research Program (28IR-0046 to S.M.R. and J.S.).
References
- Álvarez-Fernández M, and Malumbres M (2020). Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 37, 514–529. [DOI] [PubMed] [Google Scholar]
- Araujo AR, Gelens L, Sheriff RS, and Santos SD (2016). Positive Feedback Keeps Duration of Mitosis Temporally Insulated from Upstream Cell-Cycle Events. Mol Cell 64, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora M, Moser J, Phadke H, Basha AA, and Spencer SL (2017). Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep 19, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, and Bakal C (2017). DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Heldt FS, Zhang T, Bakal C, and Novak B (2016). A Dynamical Framework for the All-or-None G1/S Transition. Cell Syst 2, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, and de Bruin RA (2013). Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu F, Gascan H, and Coqueret O (2001). Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. Journal of Biological Chemistry 276, 16840–16847. [DOI] [PubMed] [Google Scholar]
- Burkhart DL, and Sage J (2008). Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 8, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadart C, Monnier S, Grilli J, Sáez PJ, Srivastava N, Attia R, Terriac E, Baum B, Cosentino-Lagomarsino M, and Piel M (2018). Size control in mammalian cells involves modulation of both growth rate and cell cycle duration. Nature communications 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillot M, Bourgeais J, Dakik H, Coste E, Mazure NM, Lelievre E, Coqueret O, Herault O, Mazurier F, and Sola B (2020). Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JS, Argast GM, Yuen SY, Hayes B, and Fausto N (2011). Inactivation of p38 MAPK during liver regeneration. Int J Biochem Cell Biol 43, 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, and Meyer T (2016). Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 166, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Mark KG, Garbett D, Pack LR, Rape M, and Meyer T (2018). EMI1 switches from being a substrate to an inhibitor of APC/C(CDH1) to start the cell cycle. Nature 558, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HX, Fakhreddin RI, Shimerov HK, Kedziora KM, Kumar RJ, Perez J, Limas JC, Grant GD, Cook JG, Gupta GP, et al. (2019). Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol Syst Biol 15, e8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M, Liu C, Yang HW, Köberlin MS, Cappell SD, and Meyer T (2019). Transient hysteresis in CDK4/6 activity underlies passage of the restriction point in G1. Molecular cell 76, 562–573. e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli R, Spring L, O’shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy R, Lanman R, and Iafrate A (2018). Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Annals of Oncology 29, 640–645. [DOI] [PubMed] [Google Scholar]
- Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS, Huynh T, Healy C, Peterson R, and Yanase S (2020). PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer discovery 10, 72–85. [DOI] [PubMed] [Google Scholar]
- Cuitino MC, Pecot T, Sun D, Kladney R, Okano-Uchida T, Shinde N, Saeed R, Perez-Castro AJ, Webb A, Liu T, et al. (2019). Two Distinct E2F Transcriptional Modules Drive Cell Cycles and Differentiation. Cell Rep 27, 3547–3560 e3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, and Nevins JR (1995). Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes [published erratum appears in Mol Cell Biol 1995 Oct;15(10):5846–7]. Mol Cell Biol 15, 4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, and Rubin SM (2013). Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Atay O, Valk E, Grande A, Bush A, Vasen G, Colman-Lerner A, Loog M, and Skotheim JM (2015). Compartmentalization of a bistable switch enables memory to cross a feedback-driven transition. Cell 160, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, and Skotheim JM (2013). Feedforward regulation ensures stability and rapid reversibility of a cellular state. Mol Cell 50, 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmer U, Zmoos AF, Auerbach RK, Vaka D, Butte AJ, Kay MA, and Sage J (2014). Organ Size Control Is Dominant over Rb Family Inactivation to Restrict Proliferation In Vivo. Cell reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Diéras V, Im S-A, Gelmon KA, Harbeck N, and Lu DR (2020). Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clinical Cancer Research 26, 110–121. [DOI] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al. (2016). Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 375, 1925–1936. [DOI] [PubMed] [Google Scholar]
- Ginzberg MB, Chang N, D’Souza H, Patel N, Kafri R, and Kirschner MW (2018). Cell size sensing in animal cells coordinates anabolic growth rates and cell cycle progression to maintain cell size uniformity. Elife 7, e26957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginzberg MB, Kafri R, and Kirschner M (2015). On being the right (cell) size. Science 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Litchfield LM, Webster Y, Chio L-C, Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, and Torres R (2017). Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776. e766. [DOI] [PubMed] [Google Scholar]
- Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, Bunch KL, Tripathi S, Knudsen ES, Witkiewicz AK, et al. (2019). p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, and Dean DC (1999). Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869. [DOI] [PubMed] [Google Scholar]
- Heldt FS, Barr AR, Cooper S, Bakal C, and Novak B (2018). A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells. Proc Natl Acad Sci U S A 115, 2532–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino N, Rossetti L, Marin-Llaurado A, Aoki K, Trepat X, Matsuda M, and Hirashima T (2020). ERK-Mediated Mechanochemical Waves Direct Collective Cell Polarization. Dev Cell 53, 646–660 e648. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T, Fujita Y, Naoki H, Aoki K, Kamioka Y, and Matsuda M (2015). Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. Elife 4, e05178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, Chow L, Sohn J, Lee KS, Campos-Gomez S, et al. (2019). Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med 381, 307–316. [DOI] [PubMed] [Google Scholar]
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, et al. (2011). A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 474, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A, and Skotheim JM (2013). Start and the restriction point. Current opinion in cell biology 25, 717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, Frenzel M, Hardebeck MC, Cox J, Barriga S, et al. (2019). MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang LC, Jackson PK, and Reed SI (2007). Cyclin E overexpression impairs progression through mitosis by inhibiting APC(Cdh1). J Cell Biol 178, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan SM, Lents NH, and Baldassare JJ (2004). Expression of cyclin E renders cyclin D-CDK4 dispensable for inactivation of the retinoblastoma tumor suppressor protein, activation of E2F, and G1-S phase progression. J Biol Chem 279, 5387–5396. [DOI] [PubMed] [Google Scholar]
- Kent LN, and Leone G (2019). The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 19, 326–338. [DOI] [PubMed] [Google Scholar]
- Koivomagi M, Ord M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ER, Rubin SM, and Loog M (2013). Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol 20, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, and Sherlock G (2015). Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature 519, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, Hsieh W, Sanchez-Vega F, Brown DN, and Paula AFDC (2018). Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell 34, 893–905. e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews Molecular Cell Biology, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ginzberg MB, Patel N, Hild M, Leung B, Li Z, Chen Y-C, Chang N, Wang Y, and Tan C (2018). Size uniformity of animal cells is actively maintained by a p38 MAPK-dependent regulation of G1-length. Elife 7, e26947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Sorensen CS, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters JM, Bartek J, and Lukas J (1999). Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, and Weinberg RA (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Martinsson HS, Starborg M, Erlandsson F, and Zetterberg A (2005). Single cell analysis of G1 check points-the relationship between the restriction point and phosphorylation of pRb. Exp Cell Res 305, 383–391. [DOI] [PubMed] [Google Scholar]
- Matson JP, and Cook JG (2017). Cell cycle proliferation decisions: the impact of single cell analyses. FEBS J 284, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick KA, Wohlbold L, Zhang C, Allen JJ, Horiuchi D, Huskey NE, Goga A, Shokat KM, and Fisher RP (2011). Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol Cell 42, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, and Jackson PK (2006). Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev 20, 2410–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Rong Y, Tian C, and Spencer SL (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science 368, 1261–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J, Miller I, Carter D, and Spencer SL (2018). Control of the Restriction Point by Rb and p21. Proc Natl Acad Sci U S A 115, E8219–E8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naetar N, Soundarapandian V, Litovchick L, Goguen KL, Sablina AA, Bowman-Colin C, Sicinski P, Hahn WC, DeCaprio JA, and Livingston DM (2014). PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol Cell 54, 932–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, and Brown M (1997). Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Molecular and Cellular Biology 17, 5338–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B, Finn RS, and Turner NC (2016). Treating cancer with selective CDK4/6 inhibitors. Nature Reviews Clinical Oncology 13, 417–430. [DOI] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB, Dubrow R, Hamlin JL, and Kletzien RF (1978). Animal cell cycle. Annu Rev Biochem 47, 715–750. [DOI] [PubMed] [Google Scholar]
- Pennycook BR, and Barr AR (2020). Restriction point regulation at the crossroads between quiescence and cell proliferation. FEBS Lett 594, 2046–2060. [DOI] [PubMed] [Google Scholar]
- Persky N, Hernandez D, Do Carmo M, Brenan L, Cohen O, Kitajima S, Nayar U, Walker A, Pantel S, and Lee Y (2020). Defining the landscape of ATP-competitive inhibitor resistance residues in protein kinases. Nature Structural & Molecular Biology 27, 92–104. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Yo M, Komatsu N, Hiratsuka T, Kogure T, Hoshida T, Goshima N, Matsuda M, Miyoshi H, and Miyawaki A (2017). Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol Cell 68, 626–640 e625. [DOI] [PubMed] [Google Scholar]
- Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, Ting DT, Lawrence MS, Haas W, and Dyson NJ (2019). A Code of Mono-phosphorylation Modulates the Function of RB. Mol Cell 73, 985–1000 e1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade AE, Oser MG, Nicholson HE, and DeCaprio JA (2019). Cyclin D-CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene 38, 4962–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoller KM, and Skotheim JM (2015). The biosynthetic basis of cell size control. Trends in cell biology 25, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock MS, Stromberg BR, Scarberry L, and Summers MK (2020). APC/C ubiquitin ligase: Functions and mechanisms in tumorigenesis. Semin Cancer Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz C, Johnson A, Koivomagi M, Zatulovskiy E, Kravitz CJ, Doncic A, and Skotheim JM (2018). A Precise Cdk Activity Threshold Determines Passage through the Restriction Point. Mol Cell 69, 253–264 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, and Clurman BE (1997). Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 11, 1464–1478. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Beach D, and Shapiro GI (2016). Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discovery 6, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, and DePinho RA (2000). Cellular senescence: mitotic clock or culture shock? Cell 102, 407–410. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, and Roberts JM (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al. (2019). The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring LM, Wander SA, Andre F, Moy B, Turner NC, and Bardia A (2020). Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. The Lancet 395, 817–827. [DOI] [PubMed] [Google Scholar]
- Thornton TM, and Rincon M (2009). Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci 5, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Loba A, Manieri E, González-Terán B, Mora A, Leiva-Vega L, Santamans AM, Romero-Becerra R, Rodríguez E, Pintor-Chocano A, and Feixas F (2019). p38γ is essential for cell cycle progression and liver tumorigenesis. Nature 568, 557–560. [DOI] [PubMed] [Google Scholar]
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, Sage J, Koivomagi M, and Skotheim JM (2019). Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol Cell 74, 758–770 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, André F, and Bayar MA (2019). Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. Journal of Clinical Oncology 37, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et al. (2018). Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med 379, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Varsano G, Wang Y, and Wu M (2017). Probing mammalian cell size homeostasis by channel-assisted cell reshaping. Cell reports 20, 397–410. [DOI] [PubMed] [Google Scholar]
- Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, Kim D, Luo F, Mao P, and Helvie K (2020). The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor positive metastatic breast cancer. Cancer Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, and Suski JM (2017). The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, and Skotheim JM (2020). A G1 Sizer Coordinates Growth and Division in the Mouse Epidermis. Current Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Cappell SD, Jaimovich A, Liu C, Chung M, Daigh LH, Pack LR, Fan Y, Regot S, and Covert M (2020). Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. Elife 9, e44571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Chung M, Kudo T, and Meyer T (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, and Hoffman RM (2018). Real-Time Determination of the Cell-Cycle Position of Individual Cells within Live Tumors Using FUCCI Cell-Cycle Imaging. Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatulovskiy E, S. Z, Berenson DF, Topacio BR, and Skotheim JM (2020). Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatulovskiy E, and Skotheim JM (2020). On the Molecular Mechanisms Regulating Animal Cell Size Homeostasis. Trends in Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, Larsson O, and Wiman KG (1995). What is the restriction point? Curr Opin Cell Biol 7, 835–842. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]